
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE
Transcription Factors and Retained Intron Act Vital Roles in Cadmium Stress Response of Medicinal Model Plant Salvia miltiorrhiza
1 College of Traditional Chinese Medicine (College of Life Sciences), Jiangxi University of Chinese Medicine, Nanchang, 330004, China
2 School of Agricultural Sciences, Jiangxi Agricultural University, Nanchang, 330045, China
3 Research Center for Traditional Chinese Medicine Resources and Ethnic Minority Medicine, Jiangxi University of Chinese Medicine, Nanchang, 330004, China
4 School of Pharmacy, Jiangxi University of Chinese Medicine, Nanchang, 330004, China
* Corresponding Authors: Xiaoyun Wang. Email: ; Haihui Fu. Email:
Phyton-International Journal of Experimental Botany 2024, 93(9), 2267-2284. https://doi.org/10.32604/phyton.2024.055338
Received 24 July 2024; Accepted 16 August 2024; Issue published 30 September 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Cadmium (Cd) has seriously affected the quality of traditional Chinese medicinal material Salvia miltiorrhiza in recent years, threatening human health. The physiological and metabolic profiles of S. miltiorrhiza in response to Cd stress have been revealed in previous studies. However, transcriptional and post-transcriptional regulation in response to different degrees of Cd (0, 25, 50, and 100 mg/kg) stress in S. miltiorrhiza remains unclear. Here, transcriptome atlas in S. miltiorrhiza under different degrees of Cd Stress was unveiled using RNA sequencing (RNA-seq). These results showed that the profiles of gene expression were different in the response to Cd treatment. Defense response-related biological processes were involved in differentially expressed genes (DEGs). In total, 1966 genes were identified as transcription factors (TFs) with seven expressed trends. Retained intron (RI) was the major phenomenon. Targeted genes of intron splicing factors were identified via weighted gene co-expression network analysis (WGCNA). All of these indicated that transcriptional and post-transcriptional regulations were involved in response to Cd stress in S. miltiorrhiza. Our study will provide the most comprehensive resource for studying heavy metals in traditional Chinese medicine plants.Keywords
Supplementary Material
Supplementary Material FileList of Abbreviations
| Cd | Cadmium |
| RNA-seq | RNA sequencing |
| DEGs | Differentially expressed genes |
| TFs | Transcription factors |
| RI | Retained intron |
| WGCNA | Weighted gene co-expression network analysis |
| AS | Alternative splicing |
| SE | Skipped exon |
| A5SS | Alternative 5′ splice site |
| A3SS | Alternative 3′ splice site |
| MXE | Mutually exclusive exon |
| GGPPS | Geranylgeranyl diphosphate synthase |
| UV-B | Ultraviolet-B |
| JXUCM | Jiangxi University of Chinese Medicine |
| N | Nitrogen |
| P | Phosphorus |
| K | Potassium |
| CdCl2·2.5H2O | Cadmium chloride hemi (pentahydrate) |
| SE | Skipped exon |
| FDR | False discovery rate |
| GO | Gene Ontology |
| GC | Guanine and cytosine |
| PCA | Principal component analysis |
| PC | Principal component |
| UTR | Untranslated region |
| FPKM | Fragments per kilobase of exon model per million mapped fragments |
| RPKM | Reads per kilobase per million mapped reads |
Salvia miltiorrhiza belongs to the commonly used traditional Chinese medicinal materials with a wide range of medicinal functions and has been approved by the Chinese Ministry of Health for use as health food, on the one hand. On the other hand, S. miltiorrhiza is a model medicinal plant with a wide range of ecological adaptations and belongs to the Lamiaceae (Labiatae) family [1]. Unreasonable operations in industrial and agricultural production processes have resulted in heavy metals pollution in soils [2]. The botanical drug substances, including S. miltiorrhiza, have faced the potential threat of heavy metal pollution [3–5]. Considering the great pharmacological value and market demand for herbal medicines, it is of great imperative to ensure the safety of botanical drug substances.
Researchers have reported that the Cd content of S. miltiorrhiza from some production areas in China was higher than the limits established by the Chinese Pharmacopoeia Committee of P. R. China [6–8]. The accumulated Cd could enhance the Cd residues of S. miltiorrhiza [9], and constitute a threat to human health by the food chain [9]. Concurrently, residual Cd could affect the physiological characteristics (e.g., malondialdehyde, soluble protein, proline), and some important metabolites (e.g., amino acids, fatty acids, and organic acids), coupled with antioxidant enzymes (e.g., catalase and superoxide dismutase) acted vital roles in Cd resistance in S. miltiorrhiza [9]. Even though much attention has been put on the problem of Cd contamination in S. miltiorrhiza [10,11], little has been known as relates to the molecular mechanisms of S. miltiorrhiza response to Cd stress.
The plant response to stress is based on the interactions and crosstalk among lots of mechanisms, which are involved in the regulation of gene expression at different stages, from transcriptional to post-translational levels [12]. Alternative splicing (AS) and transcription factors (TFs) are master regulators of gene expression at the post-translational level and transcriptional level, respectively [13]. TFs, the transcriptional regulators, work by binding to specific cis-regulatory elements which present in the promoters of target genes [14]. AS refers to a pre-mRNA generated multiple different mRNA isoforms through alternative splicing sites, and the common types of it include retain intron (RI), skipped exon (SE), alternative 3′ splice site (A3SS), alternative 5′ splice site (A5SS), and mutually exclusive exon (MXE) [15]. Moreover, both TFs and AS are involved in lots of physiological processes, as well as plant response to biotic and abiotic stresses [12,13,16].
Based on transcriptome sequencing analysis, abiotic stress (e.g., drought, abscisic acid, and H2O2 treatment) could up-regulate the SmWD40-170 gene of S. miltiorrhiza and regulate the resistance to abiotic stress [17]; Cd could down-regulate geranylgeranyl diphosphate synthase (GGPPS) gene of S. miltiorrhiza, and reduce the concentration of tanshinones [18]; Ultraviolet-B (UV-B) radiation could up-regulate SmNAC1 gene of S. miltiorrhiza and promote the synthesis of salvianolic acids [19]. Hence, under biotic and abiotic stresses, the changes in active component contents in S. miltiorrhiza are regulated by the co-expression of their biosynthesis-related genes [20]. These studies revealed the molecular basis of the stress effect on the synthesis of active components in S. miltiorrhiza. The present study is intended to elucidate the Cd effect on the transcription of S. miltiorrhiza to provide a theoretical basis for revealing molecular mechanisms of Cd stress affecting the synthesis of the active components in S. miltiorrhiza. This could provide theoretical bases for the improvement of the Cd stress resistance in the breeding of S. miltiorrhiza and the safe cultivation of medicinal plant resources.
The top soil of Shennong Garden in the Jiangxi University of Chinese Medicine (JXUCM) in Jiangxi, China was collected in the study. After air-dried, the soil was sieved and mixed thoroughly. The physical and chemical properties of the topsoil, including available nitrogen (N), total N, available phosphorus (P), total P, available potassium (K), total K, organic matter content, pH value, and total Cd concentration, were presented in Table S1 [21]. The total Cd content was 0.95 mg/kg, which was smaller than the critical value of Cd (1.0 mg/kg) to ensure normal plant growth.
According to studies of Zhang et al. [9] and Wang et al. [22], Cadmium chloride hemi (pentahydrate) (CdCl2·2.5H2O, Sinopharm Chemical Reagent Co., Ltd., China) was mixed with the topsoil, obtaining soils of four Cd treatment degrees (e.g., 0, 25, 50 and 100 mg/kg) as our previous study [21]. Then 2 kg of soils were added to pots (16 cm × 17 cm), watered every 5 d, and incubated for 30 d.
2.2 Plant Material and Cd Treatment
S. miltiorrhiza seedlings with about four leaves were bought from the S. miltiorrhiza plantation of Pingyi Country, Shandong Province, which were cultivated by the annual S. miltiorrhiza roots. These seedlings were identified by Prof. Xiaoyun Wang of JXUCM, and planted in Shennong Garden. Then the seedlings were cultivated into appropriate pots. A pot with two seedlings were combined as one replicate, and three replicates were sampled for every treatment. Seedlings of each treatment were named CK (0 mg/kg Cd), TR (25 mg/kg Cd), TS (50 mg/kg Cd), and TT (100 mg/kg Cd), respectively, and watered once a week. After 15 d, root samples were harvested to conduct transcriptome analysis (Fig. 1a). Voucher specimens were named No. DS-001, and kept in the public herbarium of Research Center for Traditional Chinese Medicine Resources and Ethnic Minority Medicine of JXUCM.
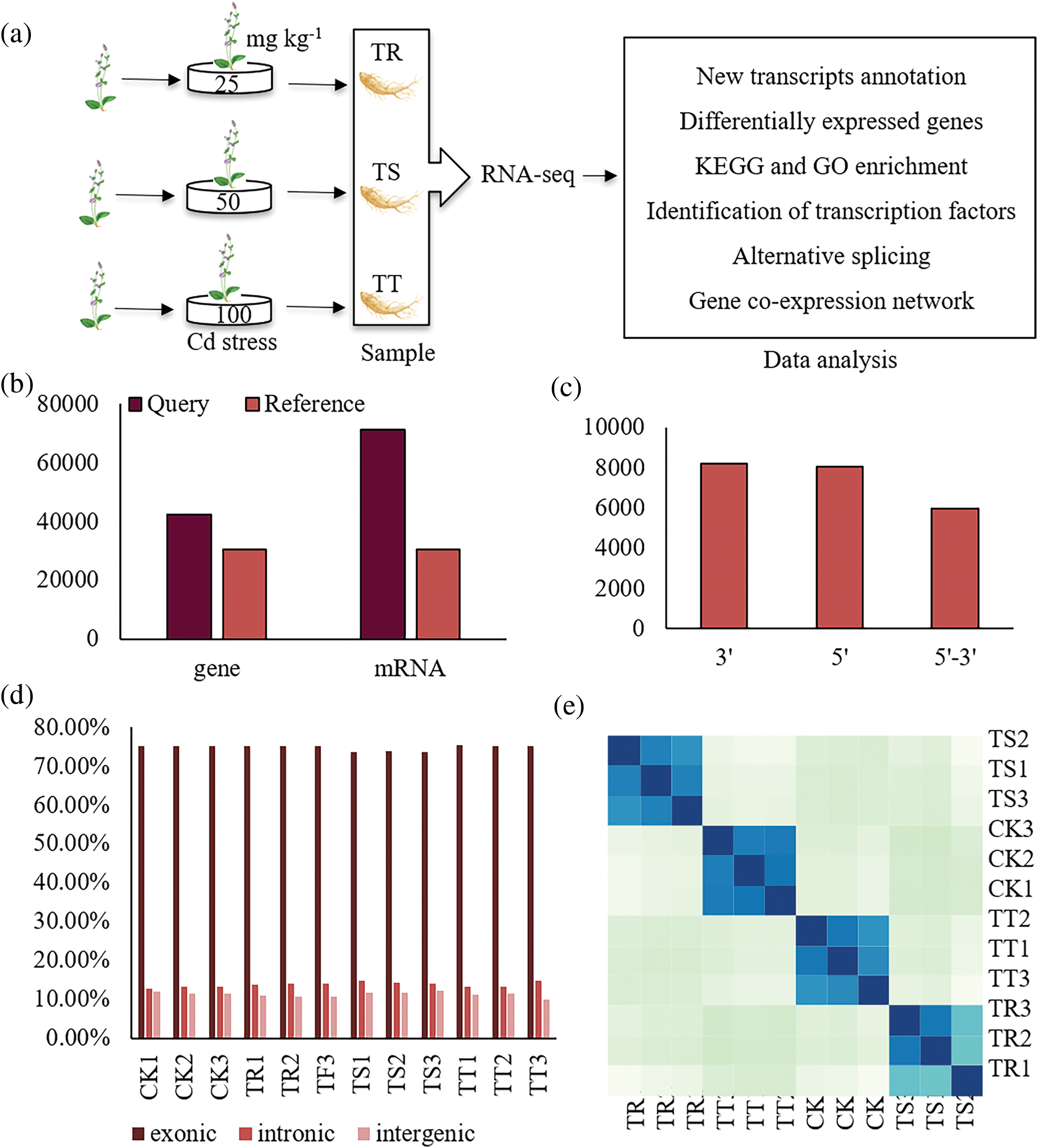
Figure 1: Transcriptomic analysis of Cd stress in S. Miltiorrhizae. (a) The outline of this study. TR represents sample of root with 25 mg/kg Cd stress, and TS is 25 mg/kg Cd stress, and TT is 100 mg/kg Cd stress. CK is 0 mg/kg Cd stress. (b) Transcript reconstruction result statistics. Y axis is number of gene and transcripts that annotated. (c) Gene structure optimization. Y axis is number of different region of gene. (d) The ratio of the coverage area of each sample reads. (e) Heatmap of the sample-to-sample distances
2.3 Total RNA Sample Extraction and Purification
Total RNA was extracted with the RNA prep Pure Plant Kit (Invitrogen, Carlsbard, CA, USA). The integrity and purity of RNA samples were evaluated with Agilent 2100 (Agilent Technologies, Santa Clara, CA, USA) and Nanodrop (Westlake Village, CA, USA), respectively. The samples that meet the requirements of the transcriptome sequencing were frozen and stored for the further analysis.
Library construction was performed according to the study of Liu et al. [23].
2.5 Transcriptome Data Analysis
After obtaining the original reading fastq file from the Illumina sequencing platform, use Fastx_Toolkit (0.0.14) cutting joints and low-quality sequencing reads. The software parameter was the default value, and clean reads was used for subsequent analysis. Then use Hisat2 to compare clean reads in the study to the reference genome of S. miltiorrhiza and reconstruct the transcript. After the reconstruction of the transcript, a comparison would be made with the annotation of the previous reference sequence. The establishment of the correspondence between the two versions, the optimization of the annotation of the original gene structure, and the selection of new transcripts and new genes were performed sequentially. This sentence was placed in the original text. The feature count was used to assess the reads mapped to each gene. The total reading of each gene was normalized by DESeq2, and the normalized reading of all samples was used to cluster with default parameters, and principal component analysis (PCA) was performed by DESeq2 and ggplot2. According to the amount of gene expression in the sample, the correlation between two samples was conducted using the R language coreplot. Due to the high sensitivity of transcriptome data in detecting gene expression, the expression distribution density map and expression boxplot were drawn by ggplot2.
The differentially expressed genes (DEGs) between different groups were calculated by the DESeq2 R software package. Generally, padj < 0.05 and | log2foldchange | > 1 were considered as DEGs, and then the volcano map was drawn by ggplot2, and the heat map of DEGs was drawn by the R software package pheatmap function. Venny analysis of DEGs was performed using online software Venn. The R software package cluster profiler was used for GO enrichment analysis, and Benjamin Hochberg multiple test adjusted p < 0.05 as the threshold. Use STEM (version 1.3.13) to analyze the trend of gene expression value RPKM (Reads Per Kilobase per Million mapped reads) of different groups across samples.
2.6 Identification of Transcription Factors (TF)
Plant transcription factor prediction was conducted using iTAK software. Its basic principle was to identify TF by hmmscan using TF family and rules defined in database.
2.7 Alternative Splicing Analysis
Through rMATS (v4.0.2) software, the alternative splicing prediction of the HISAT2 comparison results was carried out, and the alternative splicing analysis of the differences between samples was carried out. Count the number of five alternative splice types, such as skipped exon (SE), RI, A5SS, A3SS, and MXE. The standard was that the IncLevelDifference was greater than 0.1 and the false discovery rate (FDR) was less than 0.05. Wayne analysis and Gene Ontology (GO) analysis were carried out for alternative splicing genes and differential genes that occurred significantly across samples. We only conducted follow-up analysis on the event that an intron reservation occurred in a gene, and divided the RI event into RI up and RI down.
2.8 Regulation Analysis of the Co-Expression Network of Intron Splicing Factor and Interacting Genes
Gene co-network regulation analysis was carried out with taking the RPKM of genes in different samples as the data set, taking CK, TF, TS, TT as the sample characteristics, and using the R scripts WGCNA for the power. β Parameter determination was conducted according to the calculation, and the power value was selected at the inflection point of the R2 and power diagram.
2.9 Gene Expression Confirmation
Real-time quantitative reverse transcription PCR of five genes was executed using SYBR green PCR master mix. All data analysis of five genes was normalized to actin. Primer sequences were shown in Table S2.
3.1 Transcriptomic Profile in S. miltiorrhiza
We sequenced 12 cDNA libraries constructed for each treatment (Fig. 1b). Sequencing of the libraries generated averagely 23,124,944 pair-end reads and 6.81 Gb clean data in each Cd stressed condition. The clean reads had above 46.01% guanine and cytosine (GC) content with Q20 above 97.86% and Q30 above 93.83% (Table S3). These revealed that, in the study, the quality of data was acceptable, and met the requirements of subsequent bioinformatics analysis. At the same time, cluster analysis showed that each sample biologically clustered on the same branch (Fig. 1e), and the PCA result presented that differences in gene expression profiles were significantly among root samples under different degrees of Cd stress, and the principal component (PC)1 and PC2 explaining 44% variance and 30% variance, respectively (Fig. S1). These results illustrated high reliability of the transcriptome data among duplicate samples in the study.
Among the clean reads, 85.04%–87.86% were mapped to the reference genome of S. miltiorrhiza (Table S4). Gene expression levels of these samples ranged from 10−3 to 103, and presented overall consistency (Figs. 2 and 3). And in each sample, above 73.56% reads matched to the exons, above 12.84% to introns, and above 10% to the intergenic sequences (Fig. 1d). The large number of intron regions indicates the occurrence of potential IR events. New gene structure annotation file was generated (Table S5). Compared with the original annotation results, number of genes and transcripts increased 28% and 57%, respectively (Fig. 1b). The optimization of the original gene structure annotation was carried out by extending the untranslated region (UTR) to up and backward positions with mapped reads (Table S6). There were more than 8000 genes extended both in 3′ end and 5′ end, and more than 5000 genes in 5′-3′ (Fig. 1c; Table S6).
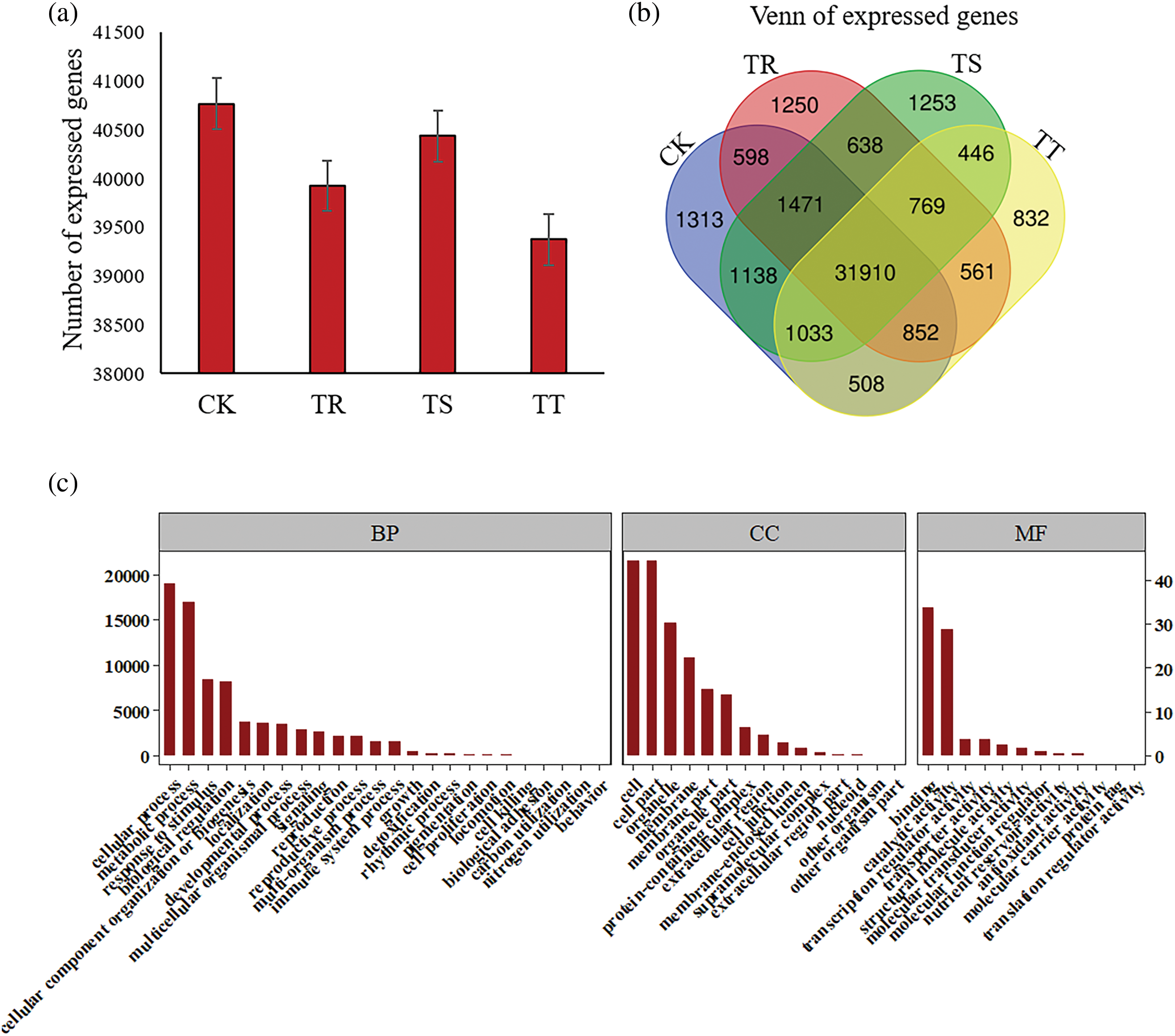
Figure 2: Analysis of global gene expression across samples. (a) Number of the genes expressed among samples. (b) Venn analysis of the genes detected among samples. (c) GO annotation of the genes expressed across samples
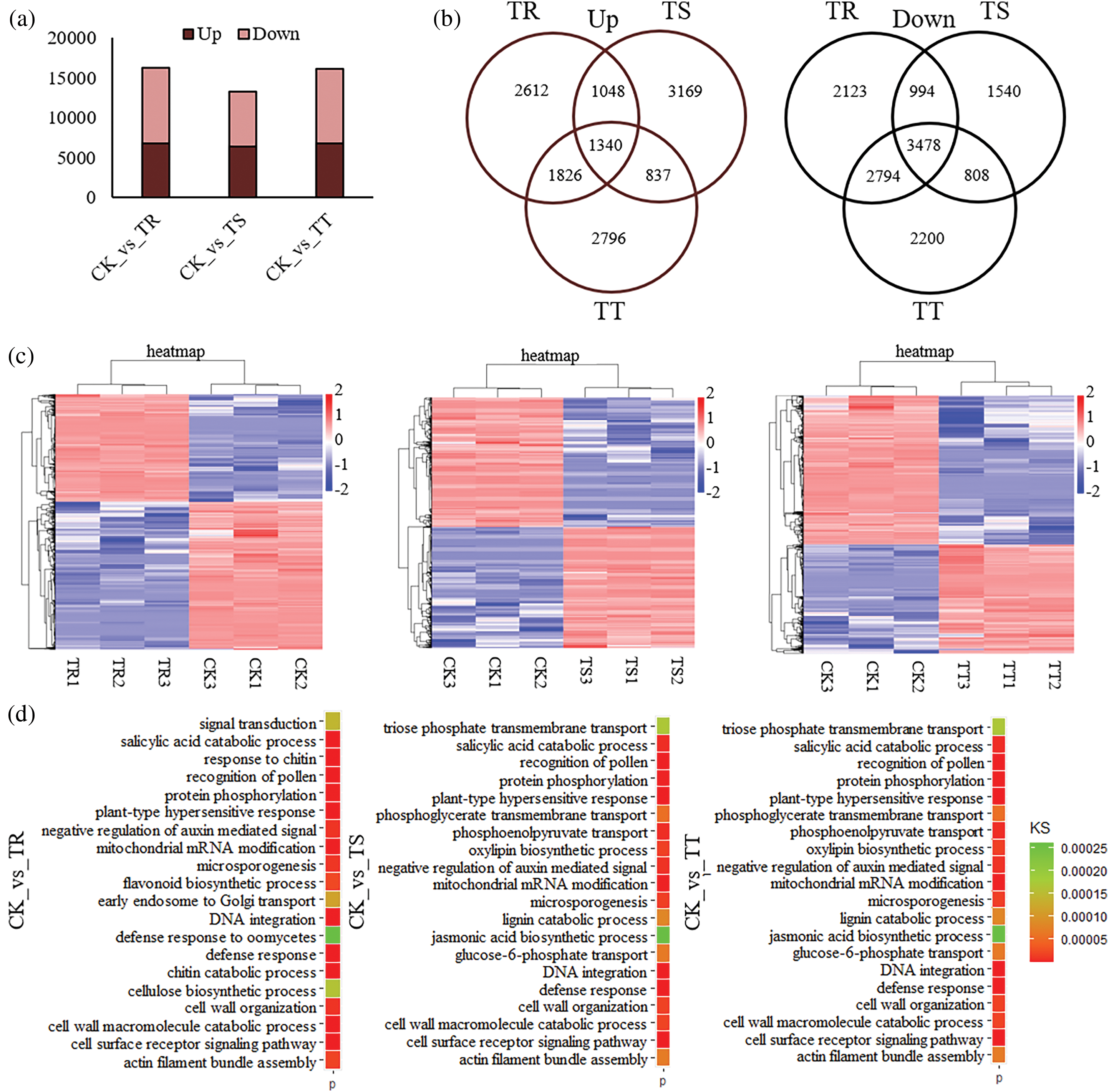
Figure 3: Analysis of differential expressed gene (DEGs) across samples. (a) Number of DEGs among samples. (b) Venn analysis of DEGs among samples. (c) Heatmap of log2 (RPKM+1) of DEGs among samples. (d) Top 20 GO annotation of DEGs across samples
The levels of gene expression were obtained according to FPKM (Fragments Per Kilobase of exon model per Million mapped fragments) across all the samples, and the positions of gene expression density maps in different samples overlap, and the peaks are the same (Figs. S2 and S3; Table S7). The number of gene expression in CK was 40768 ± 260.57, which was higher than that in Cd-stressed groups (Fig. 2a). Among all the genes, 31910 genes were expressed in all the groups (Fig. 2b), and 1313, 1250, 1253 and 832 genes expressed only in CK, TR, TS and TT, respectively (Fig. 2b; Table S8). All expressed genes are mainly enriched in the biological stress-related biological process, including response to stimulus, biological regulation as well as detoxification, etc. (Fig. 2c).
Analysis of DEGs was conducted to examine genes displaying significant expression changes subjected to four treatments, and in total, 16,215, 13,214, and 16,078 DEGs were isolated from comparisons between CK and TR (DEG&1), CK and TS (DEG&2), and CK and TT (DEG&3), respectively (Table S7; Fig. 3a,b). Of 16215 DEGs in DEG&1, 6826 DEGs were significantly up-related by 25 mg/kg Cd and 9389 DEGs were significantly down-related. In DEGs&2, there were 6394 up-regulated DEGs and 6820 down-regulated DEGs. And in DEG&3, there were 6799 up-regulated DEGs and 9280 down-regulated DEGs (Fig. 3b). Among the DEGs, 1340 up-regulated DEGs and 3478 down-regulated DEGs were identified in all the treatment groups (Fig. 3b).
In addition, heatmap analysis of gene expression was conducted to evaluate the expression profile of DEGs in every group (Fig. 3c). Samples in each group were clustered together according to Cd treatment degrees, whereas expression patterns of DEGs between different Cd-stressed groups presented significant differences (Fig. 3c). Defense response, and cell wall metabolism (e.g., cell wall organization, and cell wall macromolecule catabolic process) were involved in top 20 Go annotations of DEGs (Fig. 3d).
Transcriptome data were validated by qRT-PCR with DEGs with most significant differences related Cd stress. SMil_00009208, SMil_00026794, and MSTRG.9110 genes decreased by Cd stress compared with CK, and transcripts of SMil_00009221, and SMil_00013650 were enhanced in TR and TS, while inhibited in TT (Fig. 4). The expression profiles of DEGs were consistent between the qRT-PCR data and RNA-seq data.
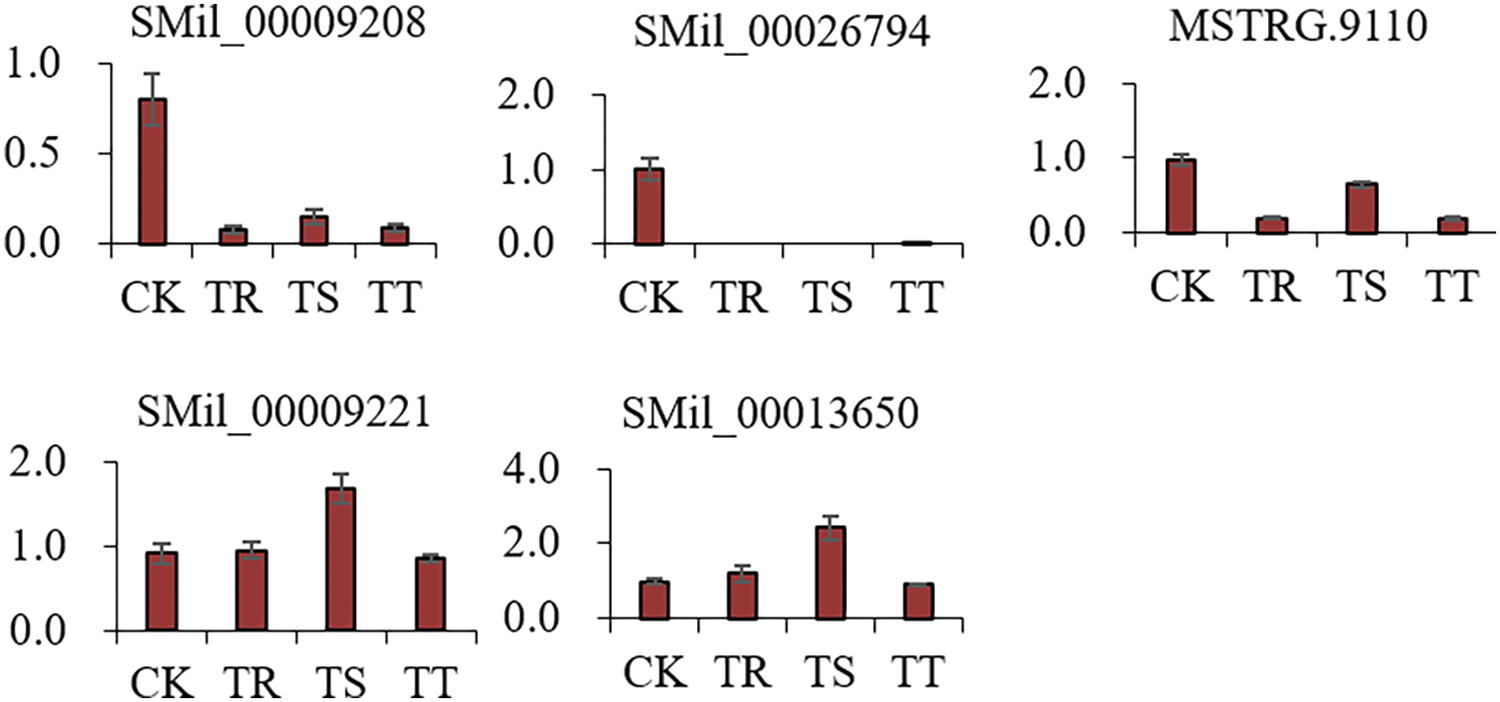
Figure 4: Verification of the expression level of some genes by real-time quantitative reverse transcription PCR
Transcription factor analysis was conducted to identify the TFs mediating transcriptional regulation in response to Cd stress of S. miltiorrhiza. A total of 1966 genes were identified as TF (Fig. 5a). With the increase of Cd level, TF change trends were different (Fig. 5b). Besides, the top three were MYB, AP2/ERF, and bHLH, with 229, 182 and 147 genes identified, respectively (Fig. 5a,b).
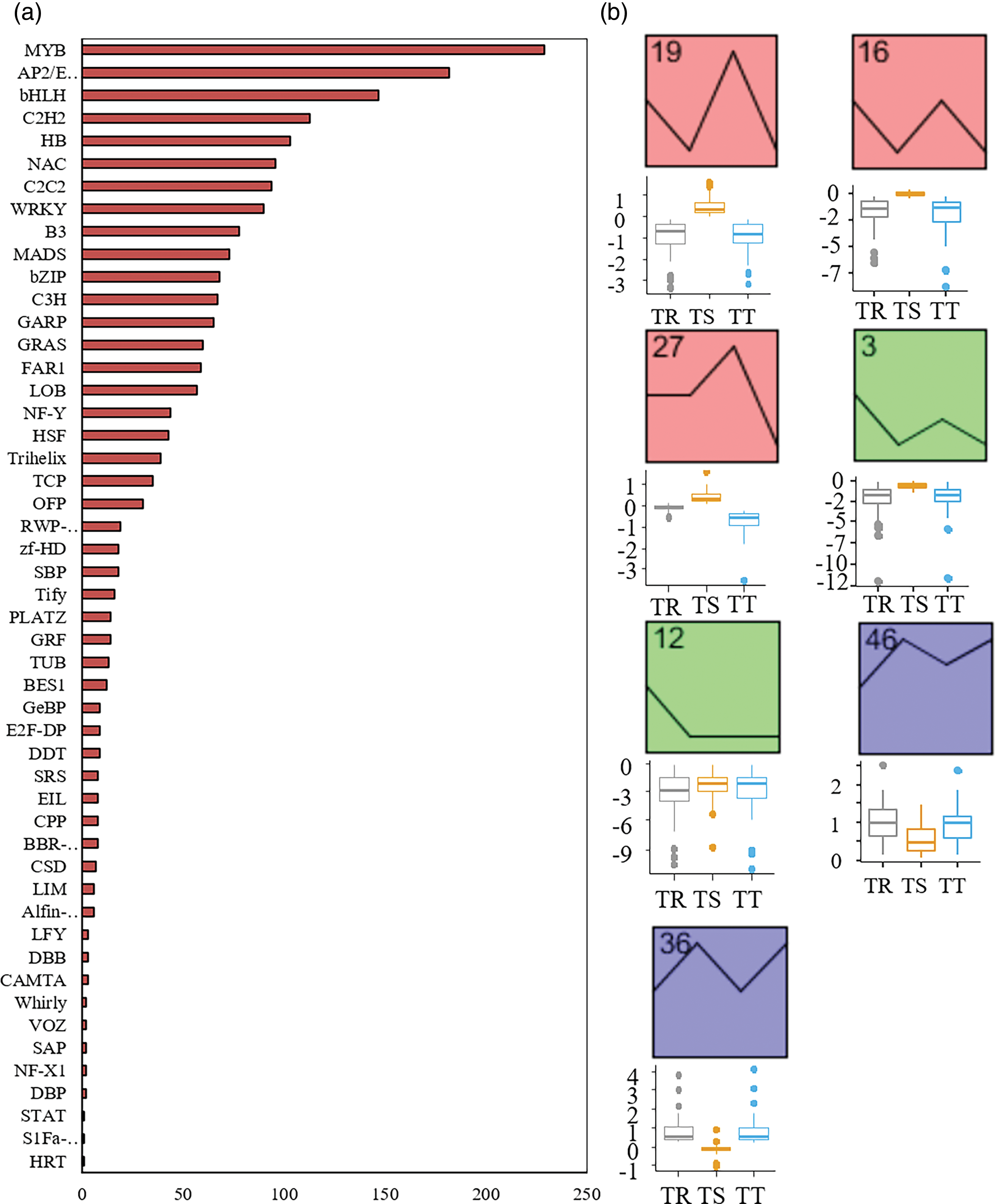
Figure 5: Analysis of transcription factor (TF) across samples. (a) Analysis of transcription factor (TF) detected across samples. (b) Trend analysis of transcription factor expressed. Upper is trend analysis of transcription factor expressed. Lower is the fold change of the corresponding expressed transcription factor
3.4 AS Events in S. miltiorrhiza
Further comprehensive profiles of AS events in Cd stress conditions were identified and quantified with the total RNA-seq data sets. Five AS types (e.g., RI, SE, MXE, A3SS, and A5SS) were identified in S. miltiorrhiza roots with Cd treatments (Fig. 6a). In total, 2367, 2295, and 2484 differential AS events occurred in TR, TS, and TT, respectively, with FDR below 0.05 and IncLevel Difference above 0.1 (Fig. 6b). Between different degrees of Cd treatment groups, the proportion of these five AS types showed no significant difference (Fig. 6a). In detail, RI-type accounted for the highest proportion (about 38%) out of the five AS types, followed by A3SS (about 24%); the proportion of SE and A3SS events were about 19%, and 24%, respectively, out of these AS events, while the proportion of MXE events (about 5%) was far fewer than that of the other AS types (Fig. 6a). Besides, a total of 248, 197, and 268 AS genes were overlapped with DEGs in TR, TS, and TT, respectively (Fig. 6c).
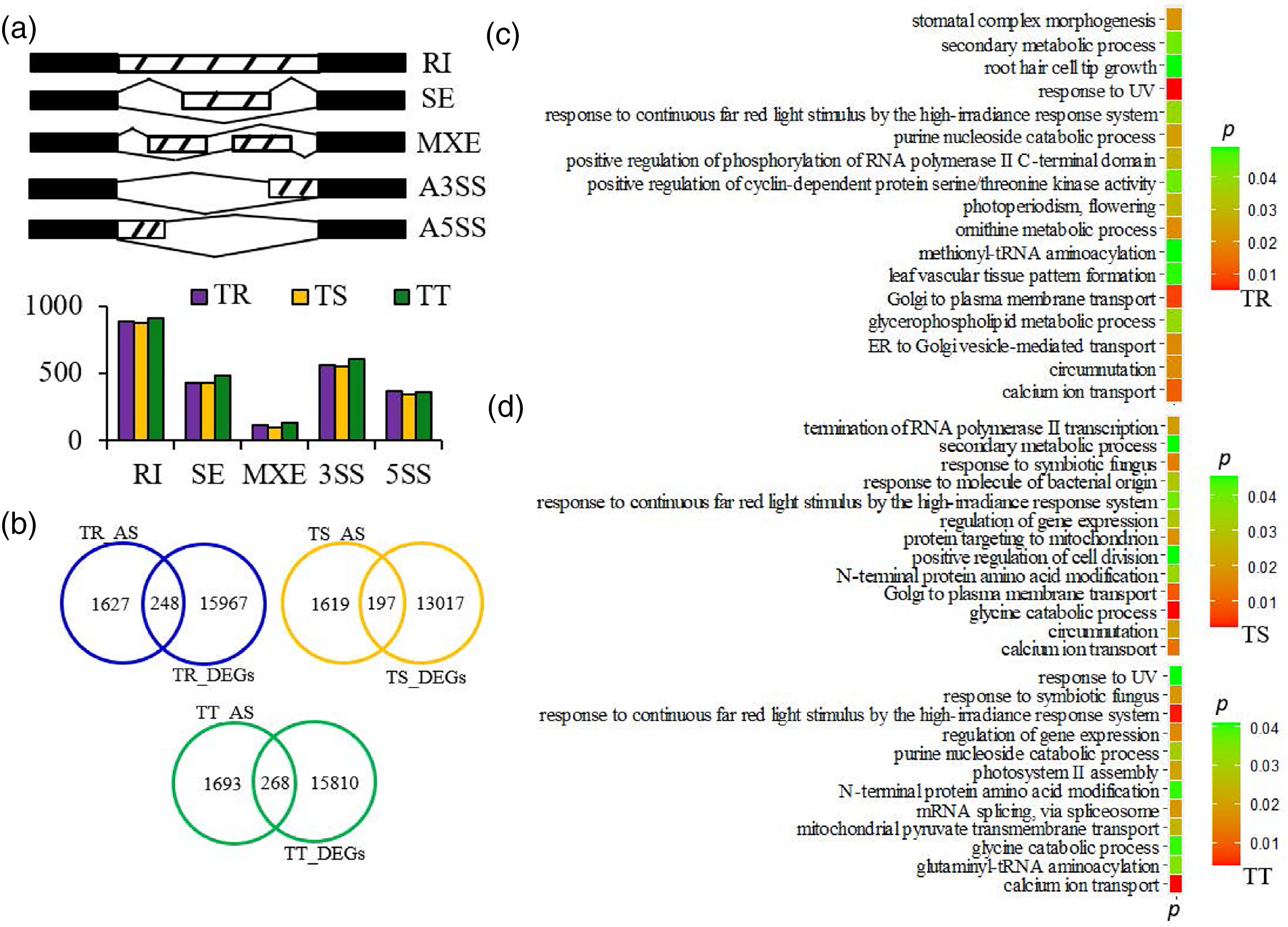
Figure 6: Analysis of alternative splicing (AS) among samples. (a) Number of AS event among samples. Skipped exon (SE), Mutually exclusive exons (MXE), Retained intron (RI), Alternative 5′ splice site (A5SS), and Alternative 3′ splice site (A3SS). FDR is less than 0.05 and IncLevel difference is more than 0.1. (b) Venn analysis of AS and DEGs among samples. (c) GO enrichment of AS gene across samples. p ≤ 0.05
What’s more, to investigate the biological functions changed by significant differential AS events of S. miltiorrhiza under Cd stress, GO enrichment analysis was applied with all genes with significantly differential AS events. A total of 17, 13, and 12 GO terms of significantly enrichment were obtained (p < 0.05). In TR and TS, the significant enriched biological functions included secondary metabolic process, calcium ion transport, and vesicle transport (e.g., golgi to plasma membrane transport, and ER to golgi vesicle-mediated transport), while in TT, they were calcium ion transport, glycine catabolic process, and mitochondrial pyruvate transmembrane transport (Fig. 6c).
To reveal deeply the effect of Cd stress on S. miltiorrhiza, RI-only genes were selected and analysed (Fig. 7). Compared to CK, genes in each Cd treatment group with RI-type event increasing and with RI event decreasing were named RI_up and RI_down, respectively. From TS to TT, the RI_up increased and then decreased, which was opposite to the change of RI_down (Fig. 7a). And in each condition, the number of RI_up was higher than that of RI_down (Fig. 7a), and RI also slightly affects gene expression levels (Fig. 7b).
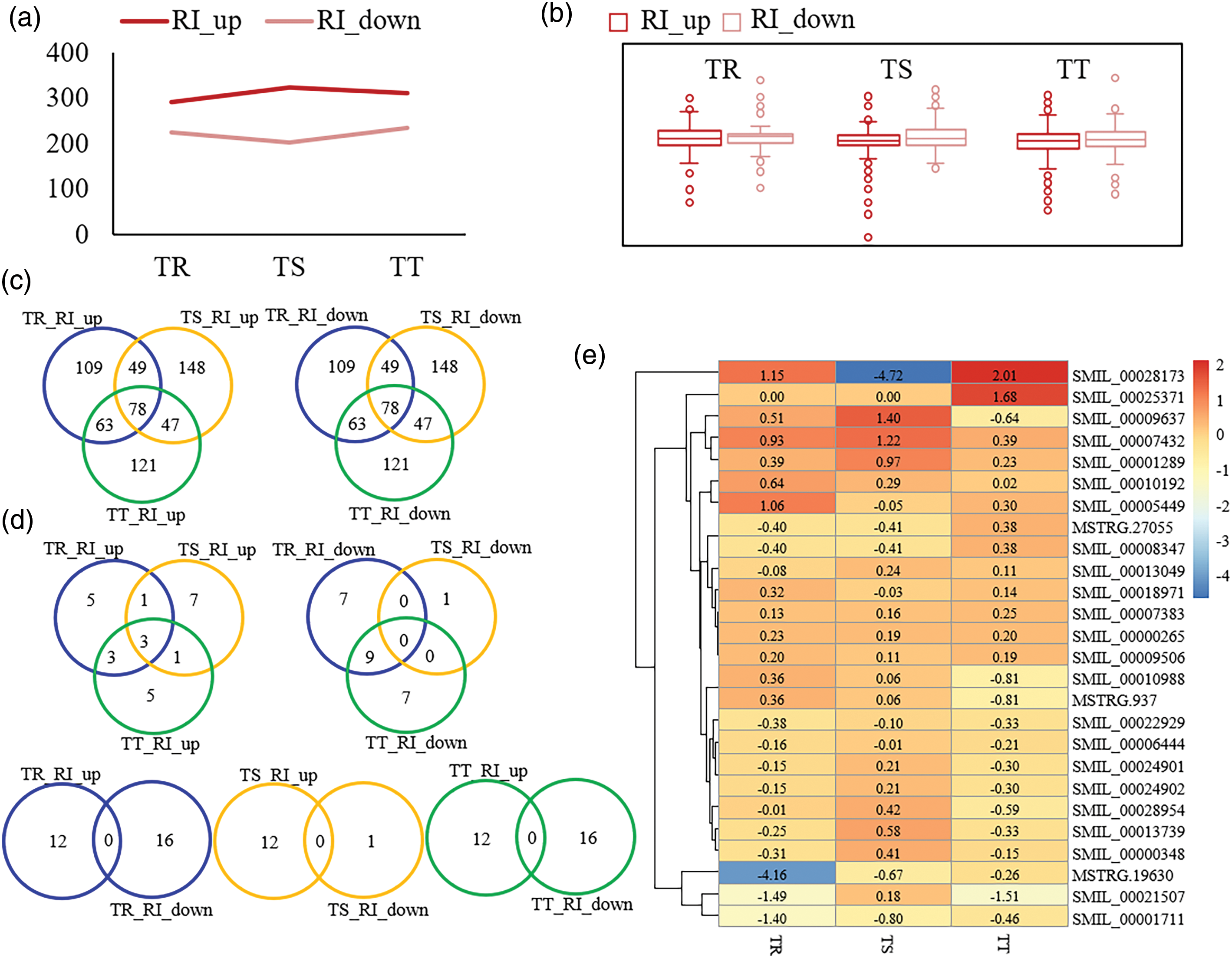
Figure 7: Analysis of RI events among samples. (a) Number statistics of RI increased and RI decreased event among samples. (b) The relationship of RI genes and expression level among samples. (c) Venn analysis of RI increased and RI decreased genes among samples. (d) GO venny analysis of RI increased and RI decreased genes among samples. (e) Fold change of expression level of intron splicing factor among samples. p ≤ 0.01
Venn analysis showed that intron retention of some genes continued to increase under different concentrations of Cd stress, while some genes continued to decrease (Fig. 7c). Venn analysis of RI_up and RI_down annotating significantly to the Go terms showed that 12% (3) RI_up overlapped in the three conditions, and 20% (5), 28% (7), and 20% (5) RI_up was unique in samples of TR, TS, and TT, respectively; there was no overlap in RI_down among these conditions, and 29.2% (7), 4.2% (1), and 29.2% (7) RI_down was peculiar to samples in TR, TS, and TT, respectively; in each condition, RI_up and RI_down presented no overlap (Fig. 7d).
To investigate further, intron special factors in response to Cd of different degrees were obtained, and they were different expression pattern between different conditions (Fig. 7e). In TR, the intron special factors with |log2(fold change)| above 1 included MSTRG.19630, SMIL_00021507, SMIL_00001711, SMIL_00005449, and SMIL_00028173; in TS, these intron special factors were SMIL_00028173, SMIL_00007432, and SMIL_00009637; in TT, they were SMIL_00021507, SMIL_00025371, SMIL_00028173 (Fig. 7e). It was indicated that intron special factors acted an important role in Cd response and regulated the selection of intron in the downstream genes.
In order to further explore the intron retention genes potentially regulated by these factors, we conducted analysis of gene co-expression network regulation using WGCNA (Fig. S4). All expressed genes are clustered in 33 modules, among which the green module, turquoise module, and red module have the strongest correlation with TR (r = 0.99 and p value = 5e−10), TS (r = 0.99 and p value = 4e−10), and TT (r = 0.99 and p value = 1e−10) samples, respectively (Fig. S5). Of the 26 intron special factors, two factors are in the green module, which acts on 63 genes with the changes of RI. Similarly, nine factors are in the turquoise module which acts on 145 genes and two factors are in the red module which acts on 35 genes (Figs. 8 and S4).
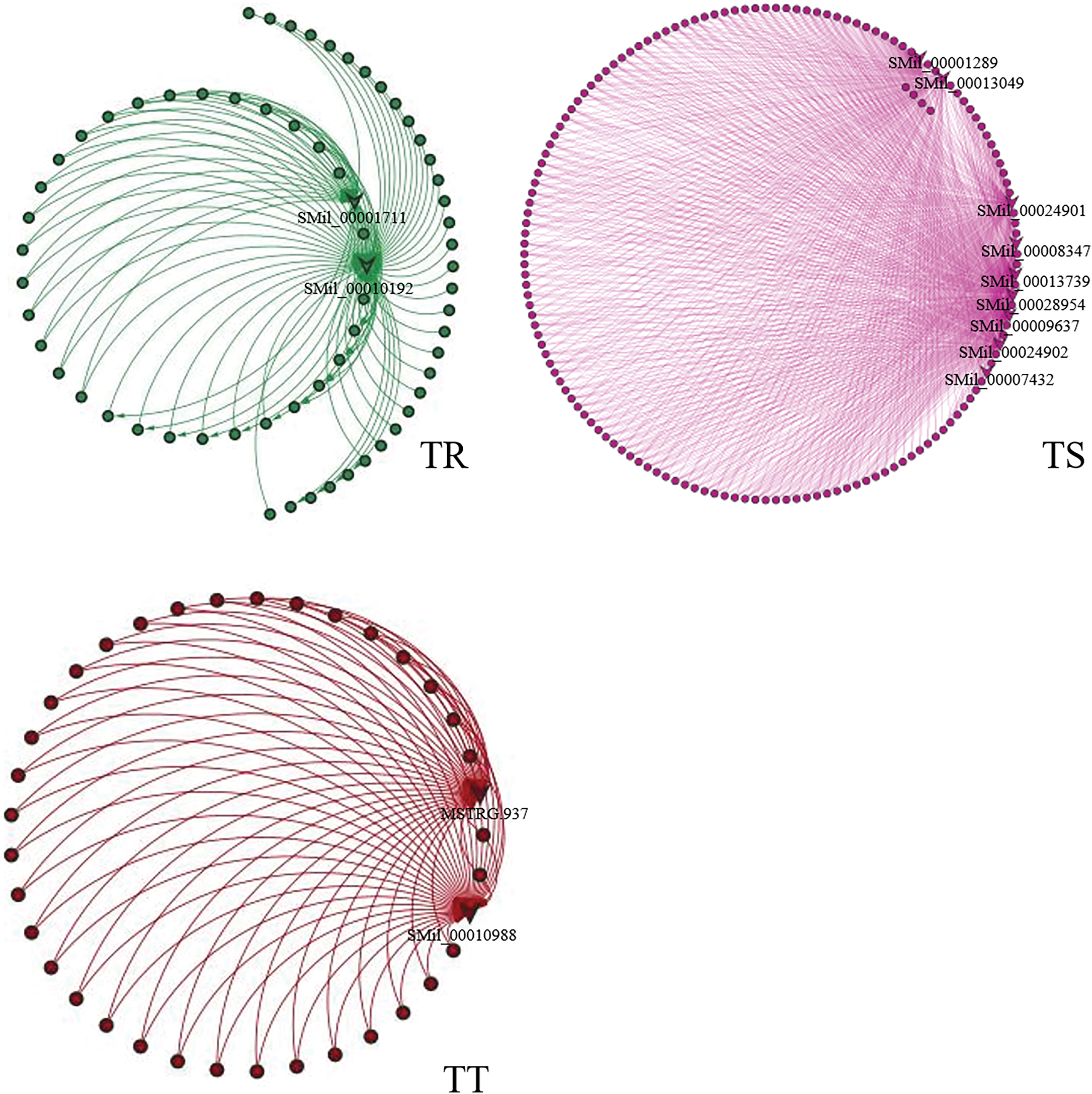
Figure 8: Targeted gene of intron splicing factor across samples. Sharp corners represent intron splicing factor and round is targeted genes
As we know, the study was the first to demonstrate, in terms of transcriptome-based gene profiling, how medicinal plant S. miltiorrhiza resists Cd stress at transcriptional and post-transcriptional levels. Based on our results, the discussions focused on DEGs, TF, and AS in transcriptional regulation of S. miltiorrhiza under Cd stress.
4.1 DEGs Involved in Defense Response, and Cell Wall Metabolism Functioned Importantly in Regulating Cd Stress Response
DEGs associated with defense response, and cell wall metabolism played vital roles in regulating Cd-resistance of S. miltiorrhiza. Firstly, our study showed that samples in control group and each Cd-stressed group could be separated significantly (Fig. 1a) with large numbers of DEGs involving defense response (Figs. 3d and 6c). Under Cd treatment, plants would adjust physiological and biochemical parameters to defense Cd toxicity, which were regulated by co-regulation of various related gene expression [20,24,25]. For instance, maize (Zea mays) roots genes in response to stress (e.g., cold, drought, salt) were up-regulated post Cd treatment [26], and DEGs involving response to stimulus, and defense response were enriched in rice (Oryza sativa), collectively [22]. The result might be related to that a considerable number of the total expressed genes were involved in response to stimulus in S. miltiorrhiza under Cd stress (Figs. 1a, 2a,c). Besides, considering that metabolic processes in plants were regulated by expression of various metabolic pathway related-genes [25,27], to a certain extent, the result was also related to that, when exposed to Cd stress, S. miltiorrhiza would regulate metabolic process (e.g., amino acid metabolism, fatty acid metabolism) for defense against Cd stress [21].
Secondly, our study also clearly revealed that, following Cd exposure, a significant number of DEGs were annotated to cell wall metabolism (e.g., cell wall macromolecule catabolic process, and cell wall organization) in S. miltiorrhiza (Figs. 3d and 6c). Similar to the result, when exposed to Cd, Salix matsudana and maize would regulate up-regulated expression of genes associated with cell wall metabolism (e.g., callose deposition in the cell wall, cell wall thickening) in roots [26,28]. That was because cell wall played an important role in improving plant heavy metal tolerance. As the first barrier of plant cells, cell wall could not only combine with heavy metal ions to reduce heavy metal content to alleviate the toxicity, but also have sites of metabolic and functional signal molecules responding to heavy metal stress [16,29].
4.2 Transcriptional (TFs) and Post-Transcriptional Regulation (AS) Involved in Cd Stress Response
The top three transcriptional regulators in TFs were MYB, AP2/ERF, and bHLH, which were associated with the regulation of Cd stress response in S. miltiorrhiza. In the study, there were 147, 182, and 229 genes identified as bHLH, AP2/ERF, and MYB, respectively, which were the top three TF families (Fig. 4). As reported by others, bHLH, AP2/ERF, and MYB belonged to the main TF families that related to stresses in plants [16,29]. They would be activated to start the expression of specific genes and led to metabolic and phenotypic changes in response to stress of plants [16,29]. Differently, under Cd stress, WRKY, bZIP, ERF, and MYB were the key TFs that related to Cd stress in creeping bentgrass (Agrostis stolonifera) [30], and MYB, bHLH, and NAC were the top three key TFs in Potentilla sericea [31]. The difference might be related to that plants hold greatly different tolerance to Cd stress due to plant species and Cd-contaminated degrees [32]. Besides, the three TFs took part in the regulation of the active ingredient synthesis in S. miltiorrhiza [33]. Hence, in part, results of Zhang et al. [9] and our unpublic study, which clearly showed that Cd stress could inhibit synthesis of the main active ingredients in S. miltiorrhiza roots, illuminated that, MYB, AP2/ERF, and bHLH played vital roles in S. miltiorrhiza Cd-resistance.
The post-transcriptional regulation, AS (especially RI), acted vital roles in the biological regulation of S. miltiorrhiza responding to Cd stress. In the study, a total of 2367, 2295, and 2484 differential AS events occurred in TS, TR, and TT, respectively (Fig. 6a). AS, a mechanism for the regulation of gene expression, was widely involved in the enhancement of plant abiotic stress (e.g., Cd, salt, cold) tolerance [12]. Among AS types, RI was the most common in plants and was an important molecular mechanism to cope with stress [34,35]. Hence, when expose to biotic stress, RI in plants accounted for the highest proportion in plants, and acted important roles in stress resistance (Fig. 6a) [36,37].
Interestingly, AS was served as an independent mechanism for gene regulation in Cd resistance of S. miltiorrhiza. These AS genes were highly enriched in calcium ion transport, vesicle transport (e.g., ER to golgi vesicle-mediated transport, golgi to plasma membrane transport), and regulation of gene expression (Fig. 6c). These suggested that significant AS events occurred in calcium ion transport, vesicle transport, and gene expression in S. miltiorrhiza response to Cd stress. These results differed from the GO analysis results of DEGs (Figs. 3d and 6c). The difference indicated that AS performed different functions from DEGs in S. miltiorrhiza response to Cd stress. Hence, a small number of genes were overlapped between AS genes and DEGs (Fig. 6b). Similarly, sampled Arabidopsis thaliana under different CO2 and O3 concentrations, Huang [38] found that AS genes and DEGs also functioned differently in response to different degrees of O3 and CO2.
4.3 Different Transcriptional and Post-Transcriptional Regulations Existed in Response to Different Degrees of Cd Stress
When stressed with Cd of different degrees, both transcriptional and post-transcriptional regulators functioned differently in S. miltiorrhiza. Firstly, at transcriptional level, the DEGs and TFs changed significantly in S. miltiorrhiza under different degrees of Cd stress. In the study, the Venn diagram of expressed genes showed that only 31910 expressed genes, 1340 up-regulated DEGs, and 3478 down-regulated DEGs were concurrently detected in roots under different degrees of Cd Stress (Figs. 2b and 3b), with the trend of TF genes expressed different among different degrees of Cd stress (Fig. 4). These might result in the remarkably metabolic and physiological changes of S. miltiorrhiza across different degrees of Cd stress in our previous and unpublished study [21]. This might be resulted from that relative quantity of gene expression in plants differed between different degrees of environmental stress (Fig. 2a,b). Coincide with our results, under different degrees of Cd Stress, gene expression profiles of Brassica napus seedlings and rice (Oryza sativa) roots were different [39,40]. Similarly, TFs relative quantity of gene (including TFs genes) expression in ginseng (Panax ginseng) performed a significant change under different degrees of drought stress [41], and the number of DEGs was different in mosses (Hypnum plumaeforme) under different concentrations of cerium stress [42].
Secondly, at post-transcriptional level, AS changed significantly in S. miltiorrhiza stressed with Cd of different degrees. In the study, among genes with significantly differential Cd stress-responsive AS events, the number of genes presenting both significant Cd-responsive expression patterns and AS regulation patterns were different in S. miltiorrhiza of different Cd-stressed groups (Fig. 6b), and both intron splicing factor and targeted gene of them were different between these groups (Figs. 7e and 8). This suggested that different AS regulation patterns existed in S. miltiorrhiza when stressed with different degrees of Cd. This was because that depending on Cd-contaminated degrees, the post-transcriptional and transcriptional response to Cd in plants differed greatly [32,40]. The phenomenon would also occur in model plant A. thaliana with environmental stress. For instance, the number of AS events in A. thaliana would increase with the rise in CO2 concentration [38,43], and the number of AS events in A. thaliana was different between different degrees of NaCl stress [44].
Different degrees of Cd-stressed S. miltiorrhiza showed that both different Cd-responsive expression patterns and different AS regulation patterns in response to different degrees of Cd stress existed in S. miltiorrhiza. Firstly, based on the results of PCA, gene expression profiles, and expression patterns of DEGs were significantly different in S. miltiorrhiza stressed with Cd of different degrees, a total of 31910 genes, 1340 up-regulated DEGs and 3478 down-regulated DEGs overlapped in all the groups with the DEGs number differing among CK, TR, TS, and TT. This suggested that different degrees of Cd treatment performed different effects on the gene expression of S. miltiorrhiza. Secondly, the top 20 Go annotations of DEGs in different groups showed that defense response, and cell wall metabolism (e.g., cell wall organization, and cell wall macromolecule catabolic process) were involved. This illustrated that DEGs associated with defense response, and cell wall metabolism played vital roles in the the Cd stress response of S. miltiorrhiza. Thirdly, a total of 1966 genes were identified as TFs, the main of which belonged to MYB, AP2/ERF, and bHLH. This revealed that, at transcriptional levels, TFs, especially MYB, AP2/ERF, and bHLH, played vital roles in the Cd stress response of S. miltiorrhiza. Fourthly, under Cd stress, five AS types (e.g., RI, SE, MXE, A3SS, and A5SS) were identified with RI-type accounting for the highest, and small numbers of AS genes were overlapped with DEGs in different Cd stressed groups with the GO terms of significant enrichment differing from DEGs’. It indicated that, at post-transcriptional levels, AS events, especially RI, played vital roles in the Cd stress response of S. miltiorrhiza, which could serve as an independent mechanism for gene regulation. Finally, with the increase of Cd level, TF change trends, numbers of differential AS events, changes of genes in Cd treatment groups with RI-type event, and intron special factors in response to Cd were different, with GO enrichment analysis results of genes with significantly differential AS events differing in different Cd treatment groups. These results indicated that the gene expression regulation was different in S. miltiorrhiza under Cd of different degrees of stress both at transcriptional and post-transcriptional levels.
Acknowledgement: We extended our thanks to the Nanjing Genepioneer Biotechnologies Co., Ltd., and Assoc. Prof. Yuye Zhu (School of Pharmacy of Jiangxi University of Traditional Chinese Medicine).
Funding Statement: This work was supported by Jiangxi Provincial Natural Science Foundation (20232BAB216119), Science and Technology Research Project of Jiangxi Provincial Department of Education (GJJ2200978), Jiangxi Provincial Health Commission S&T Plan (SKJP220227132), and Jiangxi Provincial Administration of Traditional Chinese Medicine S&T Program (2022A392).
Author Contributions: Jun Yuan analyzed the data and wrote the manuscript. Jun Yuan and Rongpeng Liu collected the samples. Haihui Fu and Xiaoyun Wang revised the manuscript. Jun Yuan, Haihui Fu and Xiaoyun Wang designed the research study. All authors reviewed the results and approved the final version.
Availability of Data and Materials: The raw sequence data in the paper have been uploaded in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2022), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA012377) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa (accessed on 11 July 2024).
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Supplementary Materials: The supplementary material is available online at https://doi.org/10.32604/phyton.2024.055338.
References
1. Editorial Board of Pharmacopeia of the People’s Republic of China. Pharmacopoeia of the People’s Republic of China (IV). China: The Medicine Science and Technology Press of China; 2020 (In Chinese). [Google Scholar]
2. Protection MoE, Resources MoLa. 2014 national soil pollution status survey bulletin. China Environ Protect Indus. 2014;36(5):1689–92. [Google Scholar]
3. Wang Z. Heavy metal pollution in commercially available Chinese herbal medicines from Kunming market and source-to-end methods for heavy metal removal (Master Thesis). Kunming University of Science and Technology: Kunming, China; 2019. [Google Scholar]
4. Xuan J, Zhao Z, Li Z, Li M, Kong X, Li M, et al. Research status on cadmium content of Chinese traditional medicine. J Liaoning Univ TCM. 2019;21(8):138–41 (In Chinese). [Google Scholar]
5. Guo L, Zhou L, Wang S, Kang C, Hao Q, Yang W, et al. Statistic analysis of heavy metal residues in Chinese crude drugs with the international standards of Chinese medicine-Chinese herbal medicine heavy metal limit. Sci Technol Review. 2017;35(11):91–8 (In Chinese). [Google Scholar]
6. Editorial Board of Pharmacopeia of the People’s Republic of China. Pharmacopoeia of the People’s Republic of China. China: China Medical Science Press; 2015 (In Chinese). [Google Scholar]
7. Yan H, Feng H, Huang W, Li H, Feng C. Evaluation for heavy metal pollution of soil and herb from the main producing area of Salvia miltiorrhiza bge. in China. Chin Agricul Sci Bull. 2012;28(4):288–93 (In Chinese). [Google Scholar]
8. Meng M, Chen T, Chen J, Ma Z, Ma W, Jia M. Determination of Pb, Cd, As, Hg, Cu in radix Salviae miltiorrhizae. Tianjing J Tradit Chin Med. 2009;26(3):248–9. [Google Scholar]
9. Zhang X, Li K, Chen K, Liang J, Cui L. Effects of cadmium stress on seedlings growth and active ingredients in Salvia miltiorrhiza. Plant Sci J. 2013;31(6):583–9. doi:10.3724/SP.J.1142.2013.60583. [Google Scholar] [CrossRef]
10. Efferth T, Kain B. Toxicities by herbal medicines with emphasis to traditional Chinese medicine. Curr Drug Metab. 2011;12(10):989–96. doi:10.2174/138920011798062328. [Google Scholar] [PubMed] [CrossRef]
11. Chen J. State of heavy metals/chemicals pollution of Chinese patent medicines exported to the United States and the violation of FDA regulations. Chin J Inf Tradit Chin Med. 2000;7(8):90–1 (In Chinese). [Google Scholar]
12. Mastrangelol AM, Marone D, Laidò G, De Leonardis AM, De Vita P. Alternative splicing: enhancing ability to cope with stress via transcriptome plasticity. Plant Sci. 2012;185–186:40–9. [Google Scholar]
13. Seo PJ, Park MJ, Park CM. Alternative splicing of transcription factors in plant responses to low temperature stress: mechanisms and functions. Planta. 2013;237:1415–24. doi:10.1007/s00425-013-1882-4. [Google Scholar] [PubMed] [CrossRef]
14. Riechmann JL. Transcription factors of arabidopsis and rice: a genomic perspective in regulation of transcription in plants. Oxford, UK: Blackwell Publishing; 2006. vol. 29. [Google Scholar]
15. Gilbert W. Why genes in pieces? Nature. 1978;271:501. doi:10.1038/271501a0. [Google Scholar] [PubMed] [CrossRef]
16. Alves MS, Dadalto SP, Gonçalves AB, de Souza GB, Barros VA, Fietto LG. Transcription factor functional protein-protein interactions in plant defense responses. Proteomes. 2014;2(1):85–106. [Google Scholar] [PubMed]
17. Liu Y, Ma W, Niu J, Li B, Zhou W, Liu S, et al. Systematic analysis of SmWD40s, and responding of SmWD40-170 to drought stress by regulation of ABA-and H2O2-induced stomal movement in Salvia miltiorrhiza bunge. Plant Physiol Bioch. 2020;153:131–40. doi:10.1016/j.plaphy.2020.05.017. [Google Scholar] [PubMed] [CrossRef]
18. Ali F, Qanmber G, Wei Z, Yu D, Li Y, Gan L, et al. Genome-wide characterization and expression analysis of geranylgeranyl diphosphate synthase genes in cotton (Gossypium spp.) in plant development and abiotic stresses. BMC Plant Biol. 2020;21:561. [Google Scholar]
19. Yin X, Fan H, Chen Y, Li L, Song W, Fan Y, et al. Integrative omic and transgenic analyses reveal the positive effect of ultraviolet-B irradiation on salvianolic acid biosynthesis through upregulation of SmNAC1. Plant J. 2020;104:781–99. doi:10.1111/tpj.v104.3. [Google Scholar] [CrossRef]
20. Kai G, Liao P, Xu J, Wang J, Zhou C, Zhou W, et al. Molecular mechanism of elicitor-induced tanshinone accumulation in Salvia miltiorrhiza hairy root cultures. Acta Physiol Plant. 2012;34:1421–33. doi:10.1007/s11738-012-0940-z. [Google Scholar] [CrossRef]
21. Yuan J, Liu R, Sheng S, Fu H, Wang X. Untargeted LC-MS/MS-based metabolomic profiling for the edible and medicinal plant Salvia miltiorrhiza under different levels of cadmium stress. Front Plant Sci. 2022;13:889370. doi:10.3389/fpls.2022.889370. [Google Scholar] [PubMed] [CrossRef]
22. Wang Y, Huang T, Wu G, Huang S, Zhong Q, Wang P, et al. Transcriptome analysis of rice bud stage in response to cadmium stress. Acta Agric Boreali-Sinica. 2022;37(5):25–35. [Google Scholar]
23. Liu N, Qin L, Miao S. Regulatory mechanisms of L-lactic acid and taste substances in Chinese acid rice soup (rice-acid) fermented with a Lacticaseibacillus paracasei and Kluyveromyces marxianus. Front Microbiol. 2021;12:1–16. [Google Scholar]
24. Gerami Z, Lakzian A, Hemati A, Amirifar A, Lajayer BA, van Hullebusch ED. Effect of cadmium on sorghum root colonization by glomeral fungi and its impact on total and easily extractable glomalin production. Environ Sci Pollut Res. 2021;28:34570–83. doi:10.1007/s11356-021-13205-0. [Google Scholar] [PubMed] [CrossRef]
25. Yuan J, Liu R, Sheng S, Fu H, Wang X. Integrated metabolomic and transcriptomic revealing coping mechanisms of the edible and medicinal homologous plant Plantago asiatica L. cadmium resistance. Open Life Sci. 2022;17(1):1347–59. doi:10.1515/biol-2022-0501. [Google Scholar] [CrossRef]
26. Cheng D, Tan M, Yu H, Li L, Zhu D, Chen Y, et al. Comparative analysis of Cd-responsive maize and rice transcriptomes highlights Cd co-modulated orthologs. BMC Genomics. 2018;19:709. doi:10.1186/s12864-018-5109-8. [Google Scholar] [PubMed] [CrossRef]
27. Wang Y. Integrated transcriptome and metabonomic analysis of key metabolic pathways in response to cadmium stress in winter wheat (Master Thesis). Huangzhong Agricultural University: China; 2021. [Google Scholar]
28. Yang J, Li K, Zheng W, Zhang H, Cao X, Lan Y, et al. Characterization of early transcriptional responses to cadmium in the root and leaf of Cd-resistant Salix matsudana Koidz. BMC Genomics. 2015;16:705. doi:10.1186/s12864-015-1923-4. [Google Scholar] [PubMed] [CrossRef]
29. Wang H, Wang H, Shao H, Tang X. Recent advances in utilizing transcription factors to improve plant abiotic stress tolerance by transgenic technology. Front Plant Sci. 2016;7:67. [Google Scholar] [PubMed]
30. Yuan J, Bai Y, Chao Y, Sun X, He C, Liang X, et al. Genome-wide analysis reveals four key transcription factors associated with cadmium stress in creeping bentgrass (Agrostis stolonifera L.). PeerJ. 2018;6:e5191. doi:10.7717/peerj.5191. [Google Scholar] [PubMed] [CrossRef]
31. Fan W. Transcriptome analysis of Potentilla sericea and preliminary study on PSMYB2 function under cadmium stress (Master Thesis). Northeast Forestry University: China; 2021. [Google Scholar]
32. Shi G, Xia S, Liu C, Zhang Z. Cadmium accumulation and growth response to cadmium stress of eighteen plant species. Environ Sci Pollut Res. 2016;23:23071–80. doi:10.1007/s11356-016-7545-9. [Google Scholar] [PubMed] [CrossRef]
33. Zhan Z. Advances in biosynthesis and regulation of the active ingredient of Salvia miltiorrhiza based on multi-omics approach. Acta Pharm Sin. 2020;55(12):2892–903. [Google Scholar]
34. Li S, Yamada M, Han X, Ohler U, Benfey PN. High resolution expression map of the Arabidopsis root reveals alternative splicing and lincRNA regulation. Dev Cell. 2016;39(4):508–22. doi:10.1016/j.devcel.2016.10.012. [Google Scholar] [PubMed] [CrossRef]
35. Laloum T, Martín M, Duque P. Alternative splicing control of abiotic stress responses. Trends Plant Sci. 2018;23(2):140–50. doi:10.1016/j.tplants.2017.09.019. [Google Scholar] [PubMed] [CrossRef]
36. Feng Y, Xiong Y, Zhang J, Yuan J, Cai A, Ma C. Role of alternative splicing in plant development and abiotic stress responses. J Nucl Agric Sci. 2020;34(1):62–70. [Google Scholar]
37. Ding Y, Wang M, Tang M, Li Z, Xie S. Differential analysis of transcriptome variable splicing in two cotton varieties under high temperature stress. Jiangsu Agric Sci. 2022;50(15):1–11 (In Chinese). [Google Scholar]
38. Huang W. Changes of alternative splicing in Arabidopsis thaliana grown under different CO2 and O3 concentrations (Master Thesis). Zhejiang University: China; 2019. [Google Scholar]
39. Zhang D, Du Y, Wu J, Zhou D, Liu L, Liu Z, et al. Effect of cadmium stress on plant growth and gene expression in Brassica napus seedlings. Chin J Oil Crop Sci. 2020;42(4):613–22. [Google Scholar]
40. He F, Liu Q, Zheng L, Cui Y, Shen Z, Zheng L. RNA-seq analysis of rice roots reveals the involvement of post-transcriptional regulation in response to cadmium stress. Front Plant Sci. 2015;6:1136. [Google Scholar] [PubMed]
41. Liu H. Effects of drought stress on gene expression ability of ginseng transcription factor and saponin synthase. Jilin University: China; 2022. [Google Scholar]
42. Su Y. Adptation study of Hypnum plumaeforme to different concentrations of cerium stress treatment. Inner Mongolia University: China; 2022. [Google Scholar]
43. Huang W, Chen X, Guan Q, Zhong Z, Ma J, Yang B, et al. Changes of alternative splicing in Arabidopsis thaliana grown under different CO2 concentrations. Gene. 2019;689:43–50. doi:10.1016/j.gene.2018.11.083. [Google Scholar] [PubMed] [CrossRef]
44. Ding F, Cui P, Wang Z, Zhang S, Ali S, Xiong L. Genome-wide analysis of alternative splicing of pre-mRNA under salt stress in Arabidopsis. BMC Genomics. 2014;15:431. doi:10.1186/1471-2164-15-431. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools