
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Mitochondrial Genome Analysis of Myricaria laxiflora, a Protected Endangered Plant
1 School of Culture and Tourism, Chongqing City Management College, Chongqing, 401331, China
2 Chongqing Urban Ecosystem National Positioning Observation and Research Station, Chongqing Academy of Forestry Sciences, Chongqing, 400036, China
3 College of Agronomy and Biotechnology, Southwest University, Chongqing, 400715, China
4 Key Laboratory of Eco-Environments in the Three Gorges Reservoir Region (Ministry of Education), School of Life Sciences, Southwest University, Chongqing, 400715, China
* Corresponding Author: Qian Wang. Email:
Phyton-International Journal of Experimental Botany 2024, 93(8), 1981-1993. https://doi.org/10.32604/phyton.2024.050099
Received 27 January 2024; Accepted 10 July 2024; Issue published 30 August 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Myricaria laxiflora, which grows along the Yangtze River in China, holds ornamental, ecological, and medicinal value. However, its wild population is threatened and currently designated protected as a national priority. The present research was the first to sequence and assemble M. laxiflora’s mitochondrial genome and examine its structural characteristics and phylogenetic relationships with other sequenced Caryophyllales species. The mitochondrial double-stranded closed-ring genome of M. laxiflora was found to be 389,949 bp in length, containing numerous repetitive sequences and RNA editing sites, with 34 protein encoding, 21 tRNA, and 3 rRNA genes. Although there are 22 fragments in the mitochondrial genome of M. laxiflora that are homologous to its chloroplast genome, they are incomplete gene fragments. Phylogenetic analysis demonstrated evolutionary associations with related populations and was in agreement with findings on the chloroplast genome. These findings not only lay a foundation for its preservation but also offer valuable insights for evolutionary analysis and plant breeding research.Keywords
Supplementary Material
Supplementary Material FileMyricaria laxiflora (Tamaricaceae) is a perennial shrub with an elegant tree shape adorned with dense flowers, rendering it an optimal candidate for landscaping applications and possessing significant ornamental value [1]. This species grows abundantly along the banks of the Yangtze River, demonstrating robust resilience to flooding conditions, thereby qualifying it as an exceptional tree species for soil conservation [2]. Furthermore, M. laxiflora serves as a traditional medicinal herb, exhibiting therapeutic potential in the treatment of burns and scalds [2]. This species has been added to China’s list of national vital protected wild plants, due to its small natural population and vulnerability to human activities. Currently, research on this species is mainly focused on morphology, physiology, ecology, breeding techniques, etc. [3–5]. While both the nuclear and chloroplast genomes have recently been analyzed [6,7], there have been no reports on its mitochondrial genome.
Mitochondria are the sites of respiratory metabolism in eukaryotic cells and act as the “power plants” that provide energy for cellular activities. Mitochondria are regarded as semi-autonomous genetic organelles and, like chloroplasts, contain separate genomes [8]. Most mitochondrial genomes of plants are circular, with sizes between 200 and 2500 kb. Significant size and structural variations, numerous repetitive sequences, frequent sequence migration from nuclear and chloroplast genes, and other traits make plant mitochondrial genomes unique [9]. Repetitive sequences in mitochondrial genes are responsible for most differences in the sizes of mitochondrial genomes among plant species. This contradiction in the relationship between genome size and gene number, known as the C-value paradox, suggests that an increase in mitochondrial genome size is not directly linked to an increase in the number of genes [9]. One major explanation for the dynamic alterations observed within plant mitochondrial genomes is the common occurrence of horizontal gene transfer in plants [10]. This transfer allows for the exchange of genes between plants and other organisms [11,12]. Individuals within the same species can differ in the size of their genomes due to differences in repetitive sequence numbers [13]. Furthermore, plant mitochondrial genomes are mostly maternally inherited [14]. Despite their large size, these genomes exhibit core gene conservation, high gene density, few introns, and efficient utilization of genetic information. As a result, plant mitochondrial genomes serve as effective genetic markers for species identification and are commonly used in phylogenetic analyses to elucidate genetic relationships, evolutionary levels, and species characteristics [13].
Here, the mitochondrial genome of M. laxiflora was sequenced, assembled, and analyzed. Additionally, we investigated the phylogenetic link between this species and its closely related counterparts. The research results can provide important data support for the germplasm conservation and genetic research of M. laxiflora.
2.1 Material Gathering and Genomic Sequencing
In Jiangjin District, Chongqing City, cuttings of M. laxiflora were collected from a wild individual and successfully cultivated under artificial conditions to produce fresh leaves. DNA was extracted using a modified cetyltrimethylammonium bromide (CTAB) method and a short-read library (350 bp) was created and sequenced on Illumina Novaseq 6000 plantform (Illumina Inc., San Diego, CA, USA). Furthermore, PacBio Sequel II sequencing was performed on the same DNA sample (Pacific Biosciences, Menlo Park, CA, USA).
2.2 Mitochondrial Genome Annotation and Assembly
First, the BBmap software (https://github.com/BioInfoTools/BBMap, accessed on 10 March 2023) was utilized to perform deep filtering of the second-generation reads (kmer = 51), removing those with a depth of less than 50×. Subsequently, SPAdes 3.11.0 was employed to assemble the filtered second-generation reads [15]. Then, Bandage software was utilized to visualize the assembly graph, and through in-depth hierarchical filtering, a complete mitochondrial graph was identified. The unitig sequence was extracted from this mitochondrial graph, and then BLAT was used to align it with the third-generation data (-minScore = 500 -minIdentity = 70) [16], enriching mitochondrial third-generation reads. These third-generation reads were corrected using the NECAT program (https://github.com/xiaochuanle/NECAT, accessed on 10 March 2023) and subsequently assembled using Flye software [17]. The Bandage software was applied again for visualization, thus enabling the selection of mitochondria. After mitochondria extraction, the second-generation reads were used for sequence correction with pilon (https://github.com/broadinstitute/pilon, accessed on 10 March 2023).
Annotation of the sequenced genome was performed with the web tool GeSeq (https://chlorobox.mpimp-golm.mpg.de/geseq.html, accessed on 10 March 2023). Apollo software was used for determining the positions of the start and stop codons and boundaries between introns and exons [18]. tRNA genes were annotated using tRNAscan-SE v2.0.7 [19]. Ultimately, OrganellarGenomeDRAW was used to generate a circular mitochondrial genome map [20]. The information has been uploaded to NCBI with the accession number PP093696.
RNA editing sites in protein-coding genes were predicted using the Plant RNA Editing Prediction Analysis Computer Tool (PREPACT 2.0) (http://www.prepact.de, accessed on 10 April 2024).
2.4 Identification of Repeat Elements
MISA (https://webblast.ipk-gatersleben.de/misa/, accessed on 10 May 2023) was applied for identification of Simple Repeat Sequences (SSRs) [21] using thresholds of 10, 5, 4, and 3 for mononucleotide, dinucleotide, trinucleotide, and tetra-, penta-, and hexanucleotide repeats, respectively. Tandem repeats were investigated with the Tandem Repeats Finder (https://tandem.bu.edu/trf/trf.html, accessed on 10 May 2023), using default options [22].
REPuter (https://bibiserv.cebitec.uni-bielefeld.de/reputer, accessed on 10 May 2023) was used for identifying long repeats, including direct repetitions and reverse, palindromic, and complementary repeats. The specific parameters are listed below: Hamming distance is set to 3, maximum number of repeats to calculate is 5000, and minimum number of repeats is 30 [23].
In addition, we selected a representative plant from each of the other five families of Caryophyllales for comparative analysis with M. laxiflora. They are Fallopia aubertii (Polygonaceae), Tetragonia tetragonoides (Aizoaceae), Agrostemma githago (Caryophyllaceae), Chenopodium quinoa (Amaranthaceae), and Mirabilis himalaica (Nyctaginaceae). Their sequences were downloaded from NCBI (Table 1).

2.5 Analysis of Migration Sequence from Chloroplasts to Mitochondria
The genome of M. laxiflora’s chloroplasts was obtained from the NCBI using accession number MN867948.1. BLASTN [24] was used to evaluate homologous segments in the genomes, between chrolorplast and mitochondrial, and the Circos software program was used to show the results [25].
Nicotiana tabacum (Solanaceae) was utilized as an outgroup for phylogenetic analysis, and we gathered mitochondrial genome sequences of 13 Caryophyllales species from NCBI (Table 1) to examine the systematic position of M. laxiflora within the Caryophyllales. Using PhyloSuite (v1.2.1), a total of 26 shared mitochondrial genes were retrieved [26]. IQ-TREE v1.6.5 [27] was utilized for tree construction with the maximum-likelihood method utilizing the best-fit GTR+F+R2 model and 1000 bootstrap iterations. To perform Bayesian phylogenetic inference, we employed MrBayes v3.2.127 [28] and configured its parameters as follows: mcmc ngen = 20,000, samplefreq = 1000, printfreq = 100, and diagnfreq = 1000.
3.1 Features of the Mitochondrial Genome of M. laxiflora
The overall length of M. laxiflora mitochondrial genome is 389,949 bp. It has a double-stranded closed ring structure (Fig. 1). It has 21 tRNA genes (trnC, trnM, trnN, and trnS with multiple copies), 34 protein-coding genes (PCGs), 3 rRNA (26S rRNA and 5S rRNA each have two copies), and 21 tRNA genes (Table 2). There were spacer and overlapping sections between the genes; the 57 spacer portions were between 49 and 23,182 bp, while the lengths of the three gene overlapping regions were 9, 14, and 757 bp, respectively (Table S1).
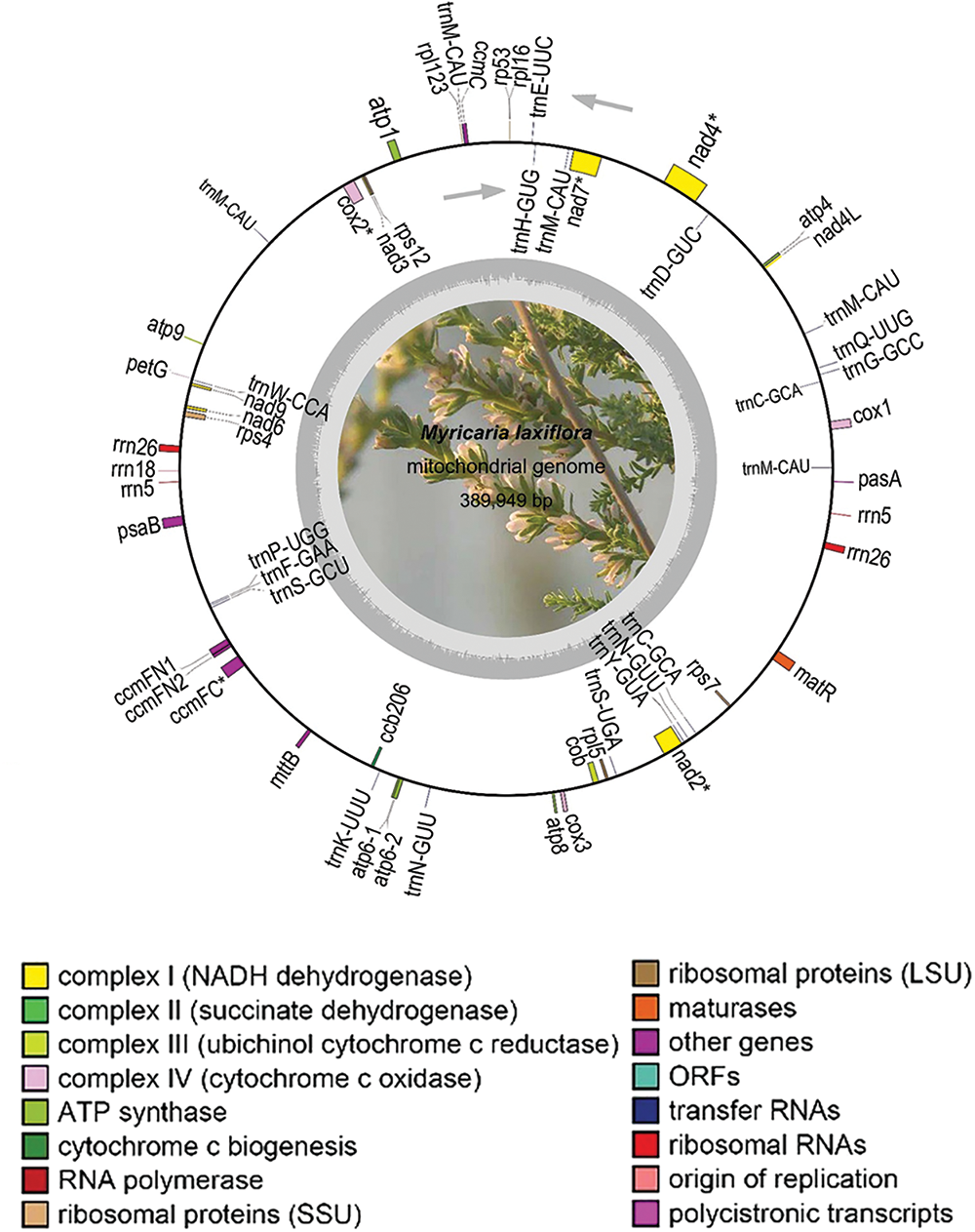
Figure 1: Structure of M. laxiflora’s mitochondrial genome. The inner gray circle represents the GC content, with the circle indicating the 50% barrier. Colors in the legend indicate functional classifications. * indicates genes with introns. Gray arrows indicate the transcription direction, with counterclockwise transcription of genes situated outside the ring and clockwise transcription of those inside the ring
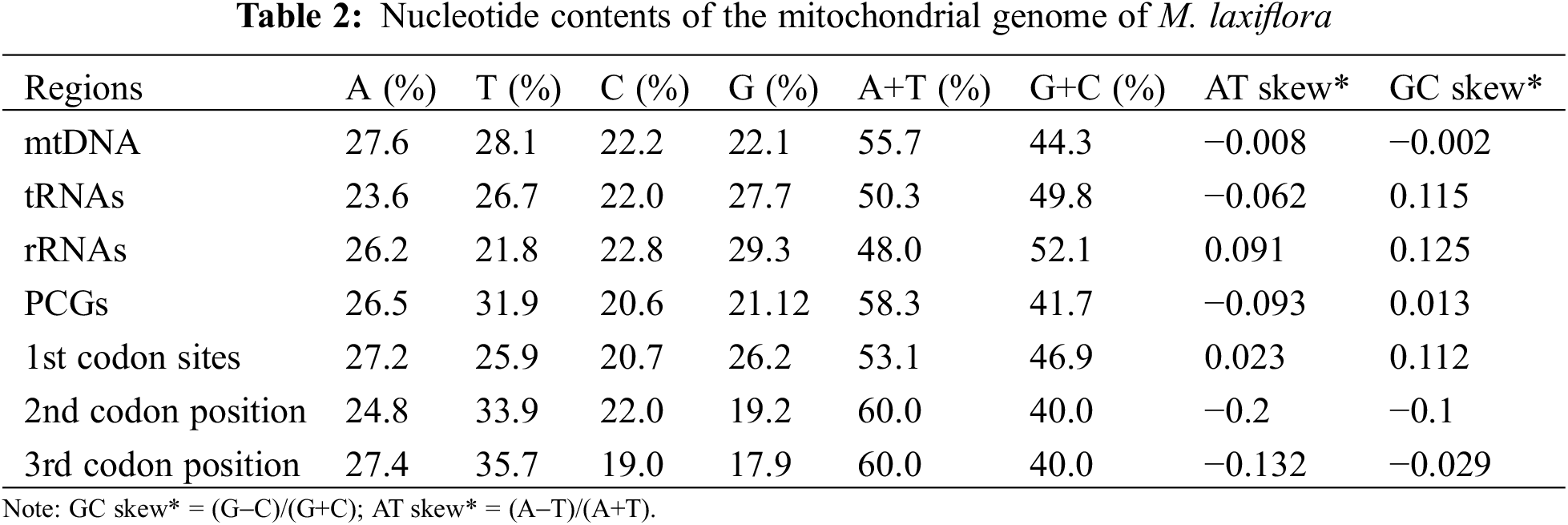
The total G + C content of mitochondrial coding genes in M. laxiflora is 44.3% (A 27.6%, T 28.1%, C 22.2%, G 22.1%), which indicates a higher abundance of A/T bases. Specifically, the G + C contents were 49.8% for tRNA, 52.1% for rRNA, and 41.7% for protein-coding genes. Moreover, it is important to note that the G + C content of the first codon was 46%, higher than that overall, with the contents at the second and third bases remaining constant at 40%, slightly lower than that overall (Table 2).
3.2 Genes and RNA Editing Sites
It was found that the genome contained 34 genes coding for protein, of which 7 coded for Complex I, 1 for Complex III, 3 for Complex IV, and 6 for Complex V. Additionally, there are 5 genes associated with Cytochrome c synthesis. It also includes three genes encoding ribosomal large subunits and four genes encoding ribosomal small subunits, as well as three genes related to photosynthesis (petG, psaA, psaB), one gene related to mitochondrial mRNA processing, and one gene related to intracellular protein transmembrane transport. The total length of PCGs was 41,937 bp, accounting for 10.75% of the overall length of the M. laxiflora genome. The longest gene is nad4 (7073 bp) and the shortest is petG (42 bp) (Table 3).

Twenty-one tRNA genes were observed (Table 3). The overall length of these genes is 1604 bp, with the longest being trnN1 (101 bp) and the shortest trnC2 (71 bp). There are 3 rRNA genes (rrn5, rrn18, rrn26) in the mitochondrial genome of M. laxiflora. Among these, the rrn26 gene is the longest, at 1280 bp, while the rrn18 gene is the shortest, at 81 bp.
In the mitochondrial genome of M. laxiflora, 332 RNA editing sites were observed in 26 genes coding for protein, with the highest number in nad4 (Fig. 2B). Of the 14 amino acid conversions that were seen, of which the most common was replacement of serine with leucine, followed by the replacement of proline with leucine (Fig. 2A).
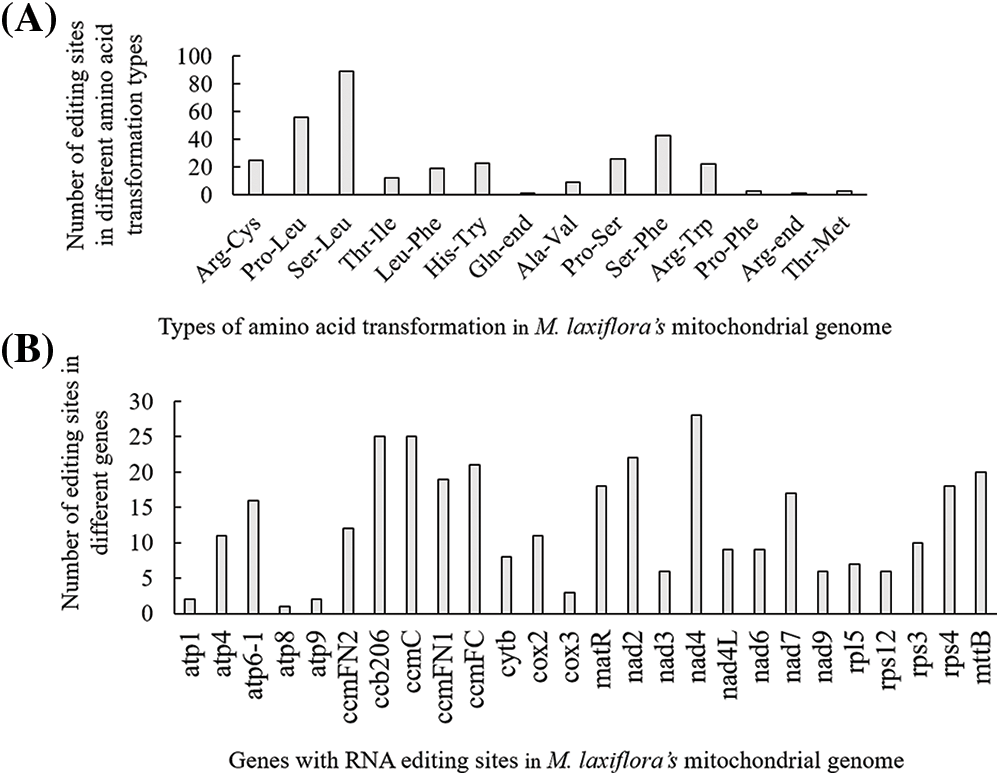
Figure 2: RNA-editing sites within protein-encoding genes. (A) RNA editing sites associated with amino acid replacements. (B) RNA editing sites within the 26 genes encoding protein
3.3 Analysis of Repeated Sequences
There are a total of 97 SSR sites, including 37 repeats of single nucleotides, 16 of dinucleotides, 11 of trinucleotides, and 33 of tetranucleotides (Fig. 3). There are no pentanucleotide or hexanucleotide repeat sequences present (Fig. 4). Out of the SSRs analyzed, thymine (T) monomer repeats constitute 64.86% (24 out of 37) of the monomer SSRs. TTTC repeats were the most frequent tetramer SSR at 15.15% (5 out of 33).
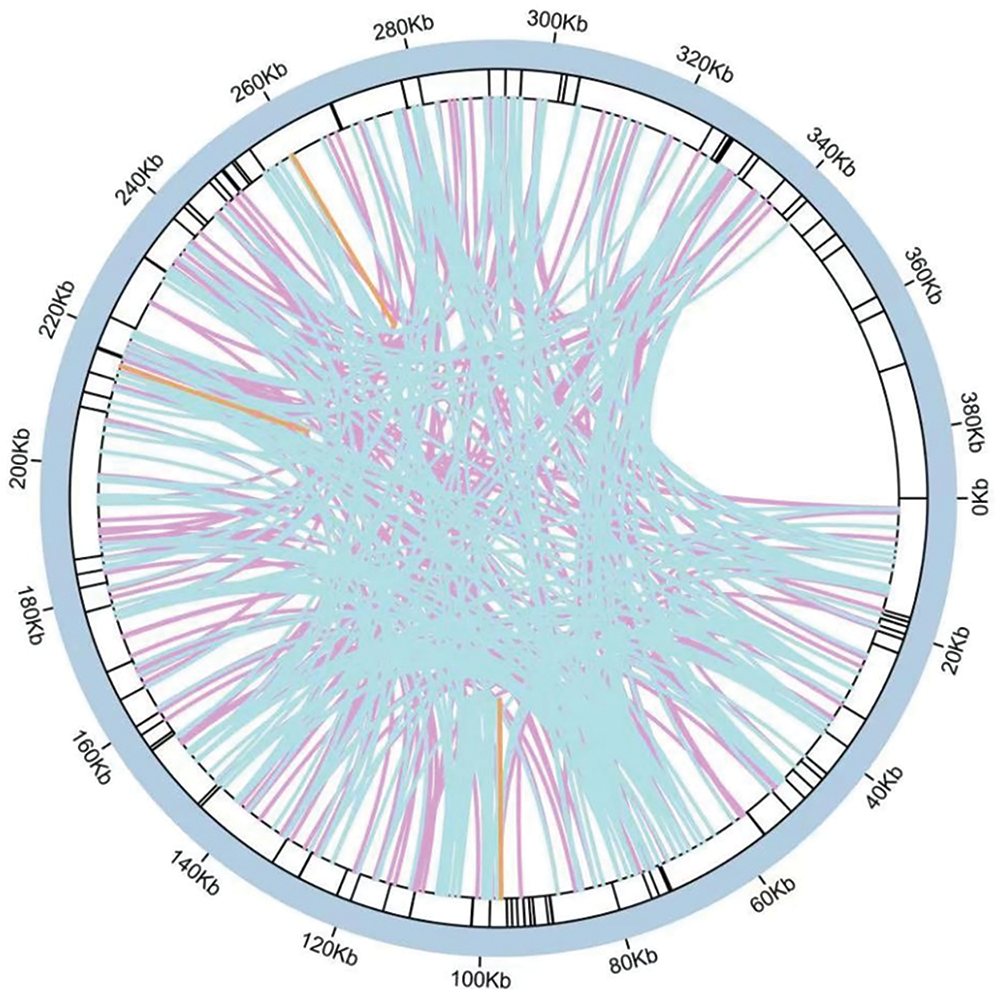
Figure 3: Sequence repeats in the mitochondrial genome of M. laxiflora
Note: The inner loop displays scattered repeats that are 50 base pairs or longer. The outside ring symbolizes the presence of both tandem repetitions and simple repeats. Purple lines indicate palindromic, blue lines forward, and orange lines reverse repeats.
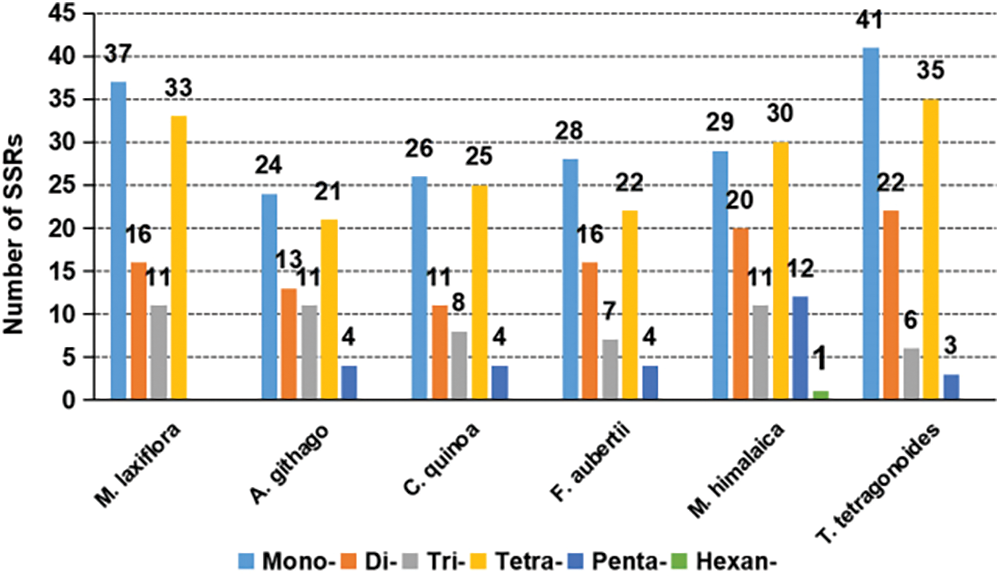
Figure 4: Simple sequence repeat types and contents in six plant mitochondrial genomes
Among the representative plants of various families in Caryophyllales, except for M. laxiflora (of Tamaricaceae) and M. himalaica (of Nyctaginaceae), most species show the highest frequency of mononucleotide repeats, followed by tetranucleotide repeats, and lower frequencies of dinucleotide and trinucleotide repeats, with pentanucleotide repeats being the least frequent. Notably, M. laxiflora is the only species among the representatives that lacks pentanucleotide repeats. Additionally, M. himalaica is the only species with a higher frequency of tetranucleotide repeats than mononucleotide repeats and also exhibits hexanucleotide repeats (Fig. 4).
A total of 575 scattered repeated sequences were found in the M. laxiflora mitochondrial genome. There are no complement repetitions (C) in these sequences, which consist of 292 palindromic repeats (P), 282 forward repeats (F), and 1 reverse repeat (R). Among the six representative plants selected in this study, palindromic and forward repeats are the main types, while reverse repeats are fewer or even absent. Complement repeats are only found in M. himalaica, and the number is relatively small (Fig. 5).
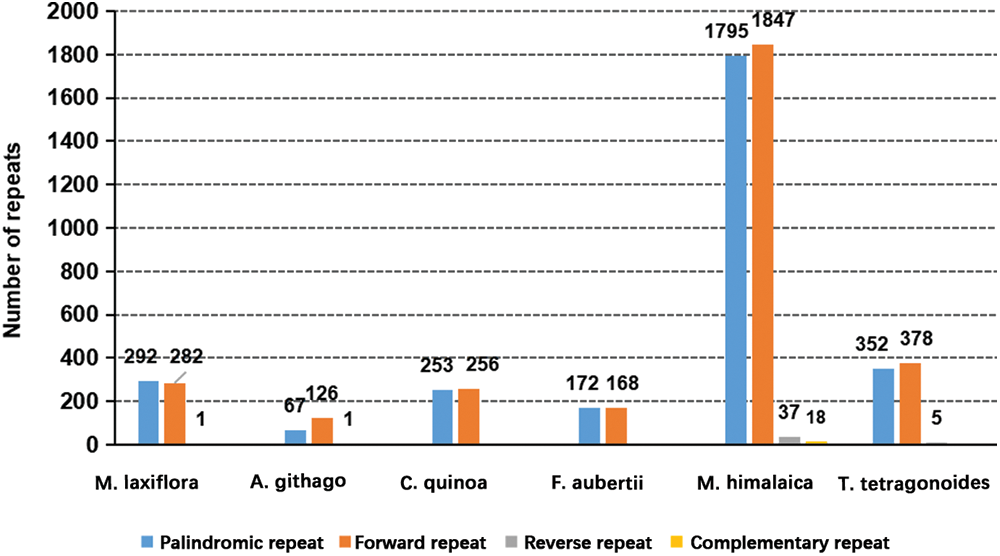
Figure 5: Types and content of scattered repeats in the mitochondrial genomes of six plants
3.4 Gene Migration from Chloroplasts to Mitochondria
Sequence similarity study indicates that 22 segments, totaling 15,439 bp and making up 3.96% of the mitochondrial genome length, in M. laxiflora’s mitochondrial genome are related to its chloroplast genome (accession number: MN867948.1). Four of these fragments are longer than 1000 base pairs, the longest of which is 3347 base pairs. These homologous sequences were annotated, indicating the presence of the psaA, psaB, petG, rrn23, rrn16, and rpl23 genes, and some tRNA genes. However, all of these genes were found to be incomplete (Fig. 6 and Table S2).
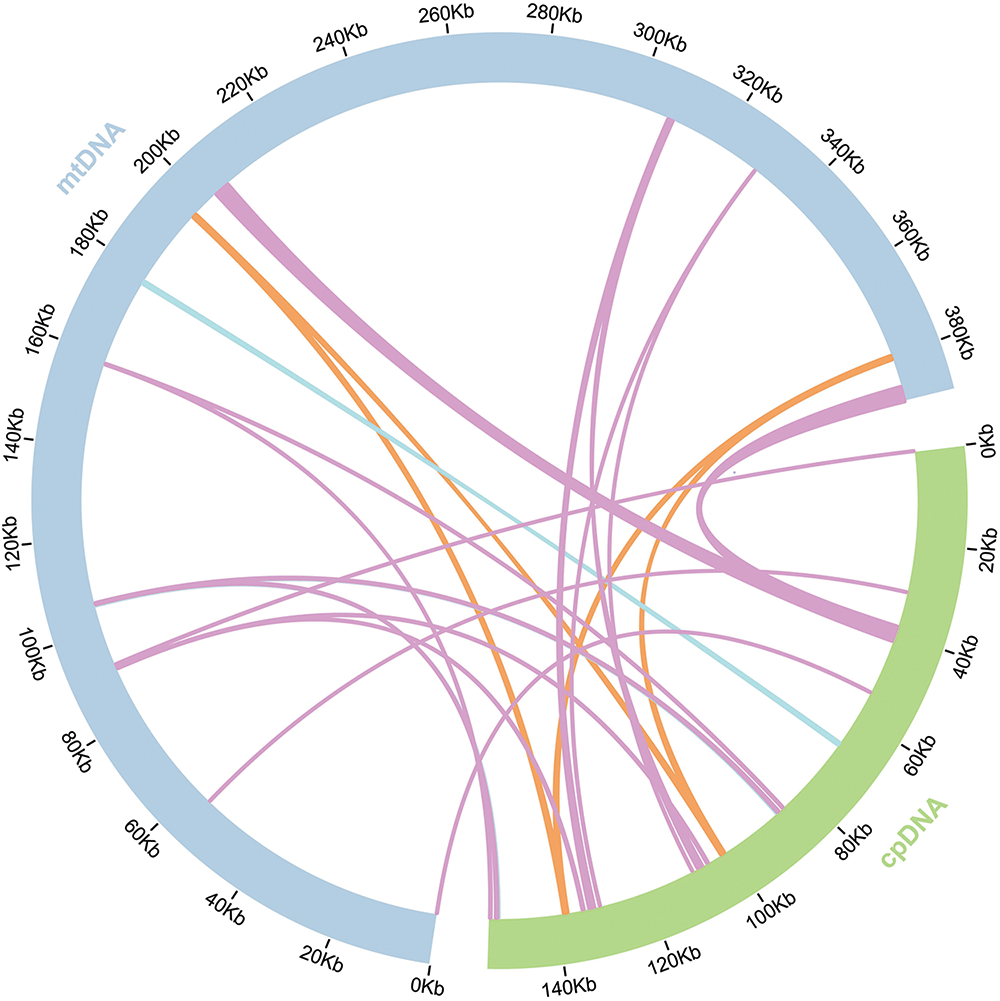
Figure 6: Diagram of M. laxiflora chloroplast-mitogenome gene transfers
A systematic phylogenetic study comparing the mitochondrial genome of M. laxiflora to those of 14 other reported plant species was conducted to investigate the evolutionary processes (Table 1). The findings demonstrate that the same tree topology was produced with high credibility by both the Bayesian inference (BI) tree and the maximum likelihood (ML) tree (Fig. 7). All ML bootstrap percentages are greater than 80%, while BI posterior probabilities are all 1. As Fig. 7 illustrates, Tamaricaceae and Polygonaceae constitute a clade, at the base of the Caryophyllales order. In addition, Aizoaceae and Nyctaginaceae cluster together, while Caryophyllaceae and Amaranthaceae form another clade, and these two clades are sister groups.
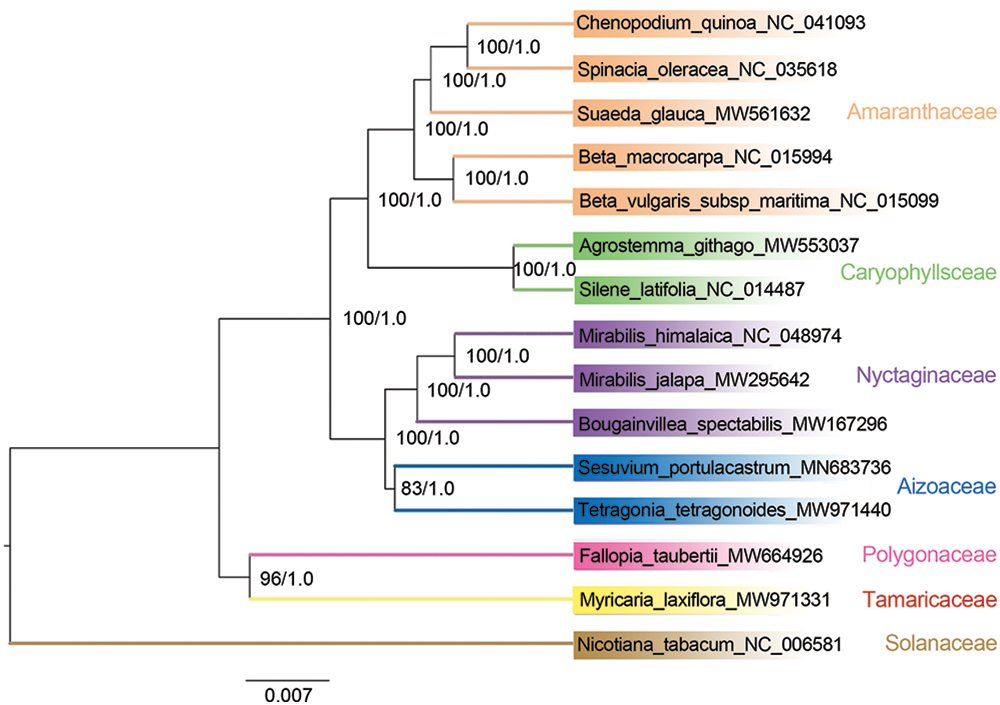
Figure 7: Phylogeny of Caryophyllale according to 26 shared mitochondrial genes. The numbers at nodes represent the BI posterior probabilities and the ML bootstrap percentages (1000 replicates), respectively. The NCBI GenBank has all of the sequences, with the accession numbers indicated beside each scientific name
Mitchondrial genomes tend to be more complex in plants than in animals due to their wide size variation, high number of repetitions, and highly conserved coding regions [29,30]. Studying the structures, evolution, functions, and inheritance of the mitochondrial genome requires an understanding of its genomic structure [30]. In the present investigation, we examined M. laxiflora’s mitochondrial genome properties and compared them with those of representative plants from different Caryophyllales families. The mitochondrial genomes of about 30 species from 13 genera of Caryophyllales have been assembled, annotated, and uploaded to the NCBI database for public access. Among the Caryophyllales, M. laxiflora’s mitochondrial genome is medium-sized, measuring 389,949 bp in total length. Overall, 34 protein-coding, 21 tRNA, and 3 rRNA genes were identified. Relative to annotations of mitochondrial genomes in other Caryophyllales plants, shown in Table 1, the four genes of petG, psaA, psaB, and rpl23 in the mitochondrial genome of M. laxiflora are less common in other species. The collinearity analysis indicated that these four genes in the mitochondrial genome of M. laxiflora originated from its chloroplast genome, but their sequence lengths are shorter than the corresponding genes in the chloroplast genome (Table S2). This suggests the likelihood of these four genes being pseudogenes.
With a few exceptions, RNA editing commonly occurs in terrestrial plant mitochondria, and the frequency of this process varies greatly between species [31]. A total of 332 RNA editing sites were found within the 26 identified protein-coding genes, with the greatest number of sites observed in nad4, and serine to leucine representing the most frequent replacement (Fig. 2). We found more RNA editing sites in the mitochondrial genome of M. laxiflora than in plants of Colobanthus [32] and Suaeda [29], which are also members of the Caryophyllales. The most common kind of alteration in these plants is the serine-to-leucine conversion. Nonetheless, there is variability in these genes with high numbers of editing sites with Colobanthus showing more sites in ccmB and nad5 [32] and Suaeda glauca having more sites in ccmB [29].
Repeated sequences are a common occurrence in the mitochondrial genomes of plants [33]. These have important functions in determining the structure and development of the mitochondrial DNA [34]. Here, SSRs and scattered repetitive sequences were intensively evaluated, and compared with representative plants from different families of Caryophyllales. Consequently, the mitochondrial genome of M. laxiflora had 575 dispersed repeats and 97 SSRs. Compared with representatives of other families in the Caryophyllales, M. laxiflora has more SSRs and scattered repeats than A. githago, C. quinoa, and F. aubertii, but fewer than M. himalaica and T. tetragonoides. This may indicate that the molecular recombination frequency of the mitochondrial genome in M. laxiflora is at a moderate level in Caryophyllales. Overall, plants in Caryophyllales exhibit similar patterns in repetitive types, with SSRs mostly consisting of single nucleotide and tetranucleotide repeats, while scattered repeats are primarily palindromic and forward repeats.
During mitochondrial evolution, certain gene fragments originating from chloroplasts can migrate to mitochondrial genomes. These migrating pieces vary in both sequence and length across species [35]. This investigation identified 22 pieces that showed homology between the two genome types, all of which were incomplete gene fragments. Migration of tRNA genes from the chloroplast to the mitochondrion is common in angiosperms [35].
Furthermore, the mitochondrial genome information was utilized to examine the evolutionary connections between M. laxiflora and selected plants from different Caryophyllales families. Subsequently, phylogenetic trees were generated using the protein-coding gene sequences. The resulting phylogenetic trees reflected taxonomic relationships among various Caryophyllales groups, indicating that P. polygonum from the Polygonaceae family was most closely related to M. laxiflora. This phylogenetic relationship aligns with the research results based on the chloroplast genome by Yao et al. [36], providing further evidence for determining the systematic position of Tamaricaceae. Previous morphological studies have classified Tamaricaceae into Violales, Parietales, Guttiferales, and Rutiflorae [37]. Nevertheless, under the APG system, the family Tamaricaceae falls within the Caryophyllales [38,39], a perspective that is further substantiated by studies conducted on chloroplast genomes [6], mitochondrial genomes, and whole-genomes [7].
In summary, the present investigation compiled and annotated the M. laxiflora mitochondrial genome, conducting a comprehensive evaluation of its structural and functional features. The M. laxiflora mitochondrial genome was found to have an overall length of 389,949 bp with a double-stranded closed-ring structure and included 3 rRNA, 21 tRNA, and 34 protein-coding genes. Sequence repeats and RNA editing sites were also analyzed. Furthermore, there was evidence of gene transfer between chloroplasts and mitochondria. The phylogenetic analysis indicates that M. laxiflora also holds significant value in studying the evolutionary relationships of related groups. As a plant resource with protective value, the analysis of the M. laxiflora mitochondrial genome not only lays a foundation for its preservation but also serves as a reference for evolutionary analysis and plant breeding research.
Acknowledgement: We thank Biomarker Technologies Corporation (Beijing, China), for providing us with sequencing services.
Funding Statement: This work was funded by the Youth Project of the Science and Technology Research Program of the Chongqing Education Commission (No. KJQN202303329). This work was also supported by the Scientific Research Project of Promoting Forestry Development through Science and Technology in Chongqing (No. YLKYQN2024-3).
Author Contributions: Conceptualization, Chaoying Wang, Bo Ding and Qian Wang; methodology, Chaoying Wang, Yingzan Xie, Bo Ding and Qian Wang; investigation, Lihui Ma and Yingzan Xie; writing—original draft preparation, Chaoying Wang, Bo Ding, Jingsi Huang and Qian Wang; writing—review and editing, Jingsi Huang and Qian Wang; supervision, Qian Wang. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: All the data supporting the findings of this study are included in this article.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Supplementary Materials: The supplementary material is available online at https://doi.org/10.32604/phyton.2024.050099.
References
1. Guan SP. Ecological study on regeneration of residual populations and plant growth restriction mechanism of Myricaria laxiflora. Three Gorges University: Chongqing, China; 2020 (In Chinese). [Google Scholar]
2. Chen F, Wang C. The ecological conservation of the endangered and rare plant Myricaria laxiflora in the Three Gorges region. China: Science Press; 2015 (In Chinese). [Google Scholar]
3. Liu W, Liu XQ, Sun Z. Investigation and conservation of M. laxiflora community. Hubei Forest Sci and Technol. 2017;46:25–31 (In Chinese). [Google Scholar]
4. Leng JM, Huang SL. Analysis of M. laxiflora research status based on literature statistics. Forest and Environ Sci. 2017;33:102–6 (In Chinese). [Google Scholar]
5. Wang T. Study on the anatomical structure and histochemistry of rare plant M. laxiflora adapted to the riverbank environment in the Three Gorges. Yangtze University: Hubei, China; 2021(In Chinese). [Google Scholar]
6. Wang Q, Zhang S, Ding B, Zhu X, Deng HP. The complete chloroplast genome of Myricaria laxiflora (Tamaricaceaean endemic and endangered species from China. Mitochondrial DNA B. 2020;5:1153–4. doi:10.1080/23802359.2020.1730266. [Google Scholar] [CrossRef]
7. Wang Q, Ding B, Wang CY, Xie YZ, Ma LH, Deng HP. Chromosome-scale genome assembly clarifies the mechanism of flooding tolerance and evolutionary history of Myricaria laxiflora. Ind Crops & Prod. 2024;215:118595. doi:10.1016/j.indcrop.2024.118595. [Google Scholar] [CrossRef]
8. Su AG. Sequencing and preliminary sequence analysis of mitochondrial genome of cotton. China Agricultural University: Beijing, China; 2013 (In Chinese). [Google Scholar]
9. Lei BB, Li SS, Liu GZ, Wang YM, Su AG, Hua JP. Analysis of mitochondrial genome evolution in higher plants. Molec Plant Breed. 2012;10:490–500. [Google Scholar]
10. Li SS. Preliminary study on mitochondrial genome sequencing and cytoplasmic male sterility mechanism of cotton. China Agricultural University: Beijing, China; 2013 (In Chinese). [Google Scholar]
11. Bock R. The give-and-take of DNA: horizontal gene transfer in plants. Trends Plant Sci. 2013;15(1):11–22. doi:10.1016/j.tplants.2009.10.001. [Google Scholar] [PubMed] [CrossRef]
12. Archibald JM, Richards TA. Gene transfer: anything goes in plant mitochondria. BMC Biol. 2010;8(1):147. doi:10.1186/1741-7007-8-147. [Google Scholar] [PubMed] [CrossRef]
13. Li YQ, Zhao HK, Tan H, Liu XD, Zhang CB, Dong YS. Analysis and comparison of mitochondrial genome structure of eight plants. Bullet of Biotech. 2011;231:156–62 (In Chinese). [Google Scholar]
14. Munasinghe M, Ågren JA. When and why are mitochondria paternally inherited? Curr Opin Genet Dev. 2023;80:102053. doi:10.1016/j.gde.2023.102053. [Google Scholar] [PubMed] [CrossRef]
15. Bankevich A, Nurk S, Antipov D. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencin. J Comput Biology. 2012;19(5):455–77. doi:10.1089/cmb.2012.0021. [Google Scholar] [PubMed] [CrossRef]
16. Kent WJ. BLAT-the BLAST-like alignment tool. Genome Res. 2002;12(4):656–64. [Google Scholar] [PubMed]
17. Kolmogorov M, Yuan J, Lin Y. Assembly of long error-prone reads using repeat graphs. Nature Biotech. 2019;37(5):540–6. doi:10.1038/s41587-019-0072-8. [Google Scholar] [PubMed] [CrossRef]
18. Lee E, Harris N, Gibson M, Chetty R, Lewis S. Apollo: a community resource for genome annotation editing. Bioinformatics. 2009;25:1836–7. doi:10.1093/bioinformatics/btp314. [Google Scholar] [PubMed] [CrossRef]
19. Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64. doi:10.1093/nar/25.5.955. [Google Scholar] [PubMed] [CrossRef]
20. Lohse M, Drechsel O, Kahlau S, Bock R. OrganellarGenomeDRAW–a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013;41:575–81. doi:10.1093/nar/gkt289. [Google Scholar] [PubMed] [CrossRef]
21. Beier S, Thiel T, Munch T, Scholz U, Mascher M. MISAweb: a web server for microsatellite prediction. Bioinformatics. 2017;33:2583–5. doi:10.1093/bioinformatics/btx198. [Google Scholar] [PubMed] [CrossRef]
22. Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–80. doi:10.1093/nar/27.2.573. [Google Scholar] [PubMed] [CrossRef]
23. Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R, et al. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001;29:4633–42. doi:10.1093/nar/29.22.4633. [Google Scholar] [PubMed] [CrossRef]
24. Chen Y, Ye W, Zhang Y, Xu Y. High speed BLASTN: an accelerated MegaBLAST search tool. Nucleic Acids Res. 2015;43:7762–8. doi:10.1093/nar/gkv784. [Google Scholar] [PubMed] [CrossRef]
25. Zhang H, Meltzer P, Davis S. RCircos: an R package for Circos 2D track plots. BMC Bioinformatics. 2013;14:244. doi:10.1186/1471-2105-14-244. [Google Scholar] [PubMed] [CrossRef]
26. Zhang D, Gao F, Jakovlić I, Zou H, Zhang J, Li WX, et al. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour. 2020;20(1):348–55. doi:10.1111/men.v20.1. [Google Scholar] [CrossRef]
27. Nguyen LT, Schmidt HA, Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Molec Biol Evolution. 2015;32:268–74. doi:10.1093/molbev/msu300. [Google Scholar] [PubMed] [CrossRef]
28. Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–42. doi:10.1093/sysbio/sys029. [Google Scholar] [PubMed] [CrossRef]
29. Cheng Y, He X, Priyadarshani SVGN, Wang Y, Qin Y. Assembly and comparative analysis of the complete mitochondrial genome of Suaeda glauca. BMC Genomics. 2020;22:167. [Google Scholar]
30. Kozik A, Rowan BA, Lavelle D, Berke L, Christensen AC. The alternative reality of plant mitochondrial DNA: one ring does not rule them all. PLoS Genet. 2019;15:e1008373. doi:10.1371/journal.pgen.1008373. [Google Scholar] [PubMed] [CrossRef]
31. Wendel JF, Greilhuber J, Dolezel J. Plant genome diversity. Gen Rev. 2012;1:123–44. [Google Scholar]
32. Androsiuk P, Paukszto Ł., Jastrzębski JP, Milarska SE, Okorski A, Pszczółkowska A. Molecular diversity and phylogeny reconstruction of genus Colobanthus (Caryophyllaceae) based on mitochondrial gene sequences. Genes. 2022;13(6):1060. doi:10.3390/genes13061060. [Google Scholar] [PubMed] [CrossRef]
33. Gualberto JM, Mileshina D, Wallet C, Niazi AK, Weber-Lotfi F, Dietrich A. The plant mitochondrial genome: dynamics and maintenance. Biochimie. 2014;100:107–20. doi:10.1016/j.biochi.2013.09.016. [Google Scholar] [PubMed] [CrossRef]
34. Dong S, Zhao C, Chen F, Liu Y, Zhang S, Wu H, et al. The complete mitochondrial genome of the early flowering plant Nymphaea colorata is highly repetitive with low recombination. BMC Genomics. 2018;19(1):1–12. [Google Scholar]
35. Liu D, Guo H, Zhu J, Qu K, Chen Y, Guo YT, et al. Complex physical structure of complete mitochondrial genome of Quercus acutissima (Fagaceaea significant energy plant. Genes. 2022;13:1321. doi:10.3390/genes13081321. [Google Scholar] [PubMed] [CrossRef]
36. Yao G, Jin JJ, Li HT, Yang JB, Mandala VS, Croley M, et al. Plastid phylogenomic insights into the evolution of Caryophyllales. Mol Phylogenet Evol. 2019;134:74–86. doi:10.1016/j.ympev.2018.12.023. [Google Scholar] [PubMed] [CrossRef]
37. Zhang DY. Discuss on some systematical problems of Tamaricaceae. Acta Botanica Yunnanica. 2019;27(5):471–8 (In Chinese). [Google Scholar]
38. Group TAP. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: aPG III. Botanical J Linnean Soc. 2009;161(2):105–21. doi:10.1111/(ISSN)1095-8339. [Google Scholar] [CrossRef]
39. Group TAP. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: aPG IV. Botanical J Linnean Soc. 2016;181(1):1–20. doi:10.1111/boj.2016.181.issue-1. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools