
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Cloning, Characterization and Transformation of Methyltransferase 2a Gene (Zmet2a) in Maize (Zea mays L.)
1 Faculty of Agronomy, Jilin Agricultural University, Changchun, 130118, China
2 Rice Research Center, Jilin Agricultural University, Changchun, 130118, China
* Corresponding Authors: Zhenhui Wang. Email: ; Fenglou Ling. Email:
Phyton-International Journal of Experimental Botany 2024, 93(7), 1767-1779. https://doi.org/10.32604/phyton.2024.052844
Received 17 April 2024; Accepted 17 June 2024; Issue published 30 July 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
DNA methylation is an important epigenetic regulatory mechanism, it regulates gene expression by recruiting proteins involved in gene repression or by inhibiting the binding of transcription factor(s) to DNA. In this study, a novel methyltransferase 2a gene (Zmet2a) was cloned in maize and identified by polymerase chain reaction-base (PCR-base) using a bioinformatics strategy. The Zmet2a cDNA sequence is 2739 bp long and translates to 912 amino acid peptides. The Zmet2a protein revealed that it contains BAH and CHROMO structural domains, is a non-transmembrane protein that is hydrophilically unstable, and has no signal peptide structure. Meanwhile, we verified the biological roles of Zmet2a using transgenic Arabidopsis overexpressing Zmet2a and Zmet2a-knockout maize. Transgenic Zmet2a Arabidopsis thaliana showed highly significant advancement in flowering time, and Zmet2a-knockout maize showed advancement in flowering time, with significant changes in several traits. Altogether, these report the role of Zmet2a in the regulation of flowering time, which will lay a foundation for revealing the biological function and epigenetic regulation mechanism of Zmet2a in the growth, development and flowering of maize.Keywords
Supplementary Material
Supplementary Material FileEpigenetics refers to a molecular modification of DNA that does not depend on the DNA sequence and remains stable during mitosis [1]. All cell in the same organism contains the same genetic information, but not every cell expresses genes at the same time. So, in multicellular organisms, there are different patterns of gene expression in various cells and tissues [2–4]. One major epigenetic mechanism, DNA methylation, involves direct DNA chemical modification. DNA methylation is mediated by DNA methyltransferases (DNMTs), where a methyl group of S-Adenosine methionine (SAM) binds to the fifth carbon of a cytosine residue to form 5mC [5,6].
Many studies have demonstrated that DNA methylation regulates gene expression through DNA-protein interactions that alter the chromatin structure [7]. Moreover, DNA methylation regulates various biochemical processes during the plant life cycle through intercellular interactions. Thus, DNA methylation is important for regulating plant growth and gene expression. An imbalanced gene methylation pattern affects the expression of flowering morphology, resulting in abnormal growth and development [8]. Plant species have different levels of DNA methylation; for example, tobacco has 28% more DNA methylation than Arabidopsis [9,10]. Plant DNA methylation levels vary significantly between developmental cycles and tissues. For example, mature leaves of maize have higher DNA methylation levels than endosperm development [11]. 5-azaC, a DNA methylation inhibitor, reduced the methylation levels in plants [12]. 5-azaC also reduced the DNA methylation levels in Chrysanthemum stem tips, inhibiting the germination of Chrysanthemum bush buds and resulting in improper root system development [13]. Therefore, DNA methylation strictly regulates plant growth and development and is crucial in epistatic traits.
Since the 1980s, several researchers have demonstrated that DNA methylation is involved in gene expression and differentiation [14,15]. Furthermore, DNA methylation plays a critical role in the regulation of gene activity. In plants, cytosine methylation involves three main enzymes, including methyltransferase 1 (MET1), chromomethylase 3 (CMT3), and domains rearranged methylase (DRM). MET1 methylates CG sequences and a homolog of the mammalian DNMT1 [16]. CMT3 methylates CNG and a small amount of CNN sequences. However, CMT3 may rely on direct binding to H3K9me2 to target chromatin and methylation. In addition, DRM, a domain rearrangement methylase homologous to DNMT3 in plants, maintains asymmetric methylation of siRNA and plays an auxiliary role in the methylation process of CNG [6,17]. The three of them (MET1, CMT3, and DRM2) are the major DNA methylation enzymes in plants. For instance, MET1-dependent CG methylation has been implicated in plant regeneration [18]. In Arabidopsis thaliana, MET1 is the homolog of mammalian DNMT1 [16]. Besides, met1 is the only known major CG methylase null mutant that can be found in the whole genome scale of higher eukaryotes [19]. Previous studies have identified two closely related putative MET1 genes in rice, OsMET1-1 and OsMET1-2 [20]. Hu et al. also screened a null mutant of the OsMet1-2 gene, which showed severe plant dysplasia and rapid necrosis and death of all germinated seedlings. Besides, Cao et al. identified maize (Zmet3) and Arabidopsis (DRM) genes encoding proteins closely related to DNMT3 methyltransferases but containing a novel arrangement of the eight diagnostic methyltransferase amino acid motifs.
Photoperiodic reactions exist widely in plants and are important for their survival. Photoperiodic plants regulate nutrition and reproduction through diurnal changes [21]. The timing of flowering greatly affects crop yield, thus many crop domestication and improvement systems have involved variations in photoperiod response extensively [22]. Moreover, the vegetative growth regulation, floral development, and fruiting of the plant are strongly influenced by the fluctuation of day length [23]. Maize is a critical food, feed, industrial raw material and energy crop in the global world today, which plays a huge role in ensuring world food security, economic development and alleviating energy crisis. Moreover, maize is a short-day C4 crop that is sensitive to low-temperature stress [24]. In maize, the regulation of DNA methylation in photoperiod-induced flowering is unknown, yet this approach can provide genetic resources for improving flowering-based DNA-methylation mechanisms. This study revealed the preliminary function of Zmet2a in regulating gene expression in Arabidopsis and maize and the interactions between DNA methylation enzymes and gene expression. Intended to reveal the role of the Zmet2a gene in regulating photoperiod and crop phenotype and characterize the Met2a gene in maize plants. Altogether, this paper provides a theoretical basis for revealing the function of the Zmet2a gene and its important role in maize growth and development.
2.1 Plant Materials and Gene Isolation
Maize Seeds were collected from B73 inbred lines, and cultivated in an artificial climate box at 25°C~18°C for 16/8 h (day/night) condition (Light intensity 12000 LUX, 60% relative humidity). Leaves were collected at 3 leaves (about 30 days after seeding) to extract RNA. Maize, RNA was extracted using TRIZOL RNA extraction reagent following the manufacturer’s procedure, and reverse transcribed into cDNA. The cDNA was used as a template to amplify the CDS sequence. Specific primers (Table S1) were designed from the Zmet2a sequence (Gene ID: Zm00001d026291), and RNA templates were amplified using standard PCR conditions, and the PCR program involved denaturation at 95°C (5 min), 35 cycles of 95°C (30 s), 50°C (30 s), and 72°C (90 s), and a final extension at 72°C (30 s), The PCR products were then sequenced accordingly.
The PCR products of Zmet2a gene and its promoter were respectively linked with pMDTM18-T vector by TA ligase to construct cloning plasmids. After screening, the positive clones were sent to Shanghai Sangong Biological Co., Ltd. (Shanghai, China) for sequencing. The reverse transcription kit and cloning related reagents were purchased from Taraka Reagent Company (Dalian, China).
2.2 Bioinformatics Analysis of the Zmet2a Gene
We used the following websites to make bioinformatics predictions for the Zmet2a gene:
(1) Physico-chemical analysis: https://web.expasy.org/protparam/ (accessed on 9 May 2022); (2) Protein hydrophobicity analysis: https://web.expasy.org/protscale/ (accessed on 13 May 2022); (3) Protein conserved functional domain prediction: https://smart.embl.de (accessed on 22 May 2022); (4) Protein secondary structure: https://npsa-prabi.ibcp.fr/cgi-bin/ (accessed on 22 May 2022); (5) Protein tertiary structure: https://swissmodel.expasy.org/ (accessed on 9 June 2022); (6) Transmembrane structure prediction: http://www.cbs.dtu.dk/services/TMHMM/ (accessed on 9 July 2022); (7) Signal peptide prediction: http://www.cbs.dtu.dk/services/SignalP/ (accessed on 16 July 2022).
2.3 Vector Construction and Arabidopsis Genetic Transformation
Transgenic Arabidopsis plants that overexpress Zmet2a gene were selected as representative of the study on the function of Zmet2a in plants. Amplification of Zmet2a gene was performed with PCR primers carrying specific restriction enzyme sites to construct the plasmid. The amplified Zmet2a cDNA was cloned into a pCamiba3301 vector driven by a 35S promoter. The primers with enzyme sites are provided in Table S1. Agrobacterium strain was transformed into Arabidopsis thaliana by floral dip method to obtain positive plants. Next, seeds of the T0 and T3 generations were selected on 1/2 MS plates supplemented with hygromycin (30 μg/mL) and confirmed by PCR with the T-F (upstream primer): CGGGGGACTCTT GACCATGGTAATGGCGCCGAGCTCC CCGTC and T-R (downstream primer): AGAAATTTACCCTCAGATCTA TTACTGCTCAACTACCTCCCCTG primers. The presence of Zmet2a was verified by qRT-PCR with at least 3 biological replicates per sample to ensure the reliability of the qRT-PCR analysis results. After several generations of culture and screening, the T3 generation homozygous lines were finally obtained for the subsequent phenotypic and physiological and biochemical indicators testing.
2.4 Flowering Time Assessment of Transgenic Arabidopsis
40 plants were randomly selected from each line, and wild type (WT) and transgenic Arabidopsis seedlings were grown in incubators at 22°C (16 h light, 8 h darkness) and 60% relative humidity. At least three plants were counted per line. Here, we observed that Arabidopsis showed different degrees of differences before and after flowering, which confirmed the regulatory role of Zmet2a gene on time of Arabidopsis flowering time.
2.5 RNA Extraction and Quantitative Real-Time PCR Analysis
We conducted qRT-PCR experiments to verify the expression profile of related genes, verifying the biological function of Zmet2a. Total RNA was isolated from three distinct biological leaf samples using the Trizol RNA isolation (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s instructions. The first-strand cDNA was reverse transcribed from 1 µg of the total RNA using the PrimeScriptTM RT reagent Kit (TaKaRa, RR047A, supplied by TAKARA in Japan). Next, cDNA was synthesized and used for quantitative real-time PCR analysis. The qRT-PCR program involved pre-denaturation for 15 min at 95°C, 40 cycles at 95°C for 10 s, 55°C for 30 s and 72°C for 30 s. The gene expression levels were calculated using the 2−∆∆Ct method [25]. Each experiment had at least three technical and biological replicates. The primers used in this study are listed in Table S2.
2.6 Vector Construction and Maize Genetic Transformation
The transgenic maize plant with the CRISPR/Cas9 Zmet2a gene was constructed as a typical representative. Two sgRNA sequences in Zmet2a gene were used to produce the sgRNA-Cas9 binary expression vector (http://crispr.hzau.edu.cn/CRISPR2/, accessed on 9 May 2022 (Table S1)). The secondary structure analysis of the sgRNA sequences was performed using the RNA Folding program (http://unafold.rna.albany.edu/?q=mfold/RNA-Folding-Form2.3, accessed on 9 May 2022). Subsequently, the two sgRNAs were cloned into pBUE411 of the CRISPR/Cas9 system and directly transformed B73 through pollen-mediated transformation [26].
Through herbicide screening and bar gene test strips identification, the knocked out of Zmet2a gene transgenic plants. By directly taking plant materials and using TransDirect Plant Tissue PCR Kit kit (TransGen), and using Zmet2a gene expression primers, PCR amplification of plant tissues is carried out to achieve the purpose of PCR identification of transgenic plants. After several generations of culture and screening, the T1 generation homozygous lines were finally obtained for the subsequent phenotypic and physiological and biochemical indicators testing.
2.7 Physiological Parameter Determination of Transgenic Maize
Plant height, ear position, and stem thickness were measured during the lactation stage at T2. The number of leaves on the main stalk was recorded after the staminode stage. The number of days between seedling emergence and pollen dispersal was also recorded. After threshing and fully dehydrating the maize, the ear length and thickness were measured, and the number of rows, the number of grains in the rows, and the average 100-grain weight of the two surveys were recorded. The control group was maize selfed line B73, and measurements were recorded along with the transgenic maize plants.
SPSS 20.0 software (SPSS Inc., Chicago, IL, USA) was used for one-way ANOVA and statistical analysis. All experiments were conducted in three experimental replicates, compared on mean values using the LSD test at the probability level of 5%.
3.1 Cloning, Identification of Zmet2a Gene in Maize
3.1.1 The RNA Extracted from Maize Leaves was Reverse
Transcribed to obtain cDNA, which was used as a template for PCR amplification with cloning primers (Table S1), and finally a 2739 bp band was obtained (Fig. 1a). After sequencing, its sequence was completely consistent with the with the Zm00001d026291 sequence of maize in NCBI (accessed on 27 December 2022).
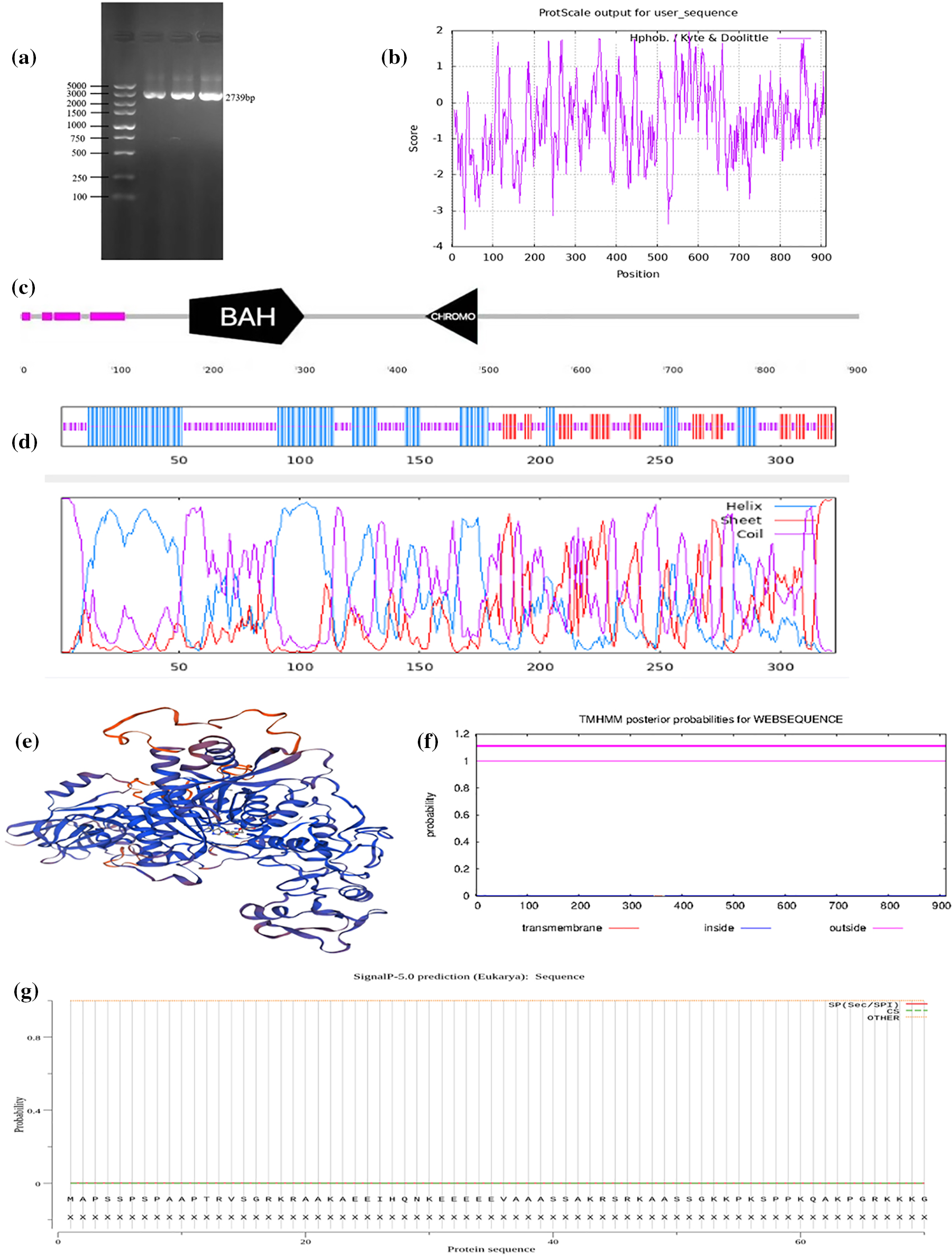
Figure 1: Zmet2a cloning and bioinformatic prediction. (a) PCR identifies. (b) Zmet2a hydrophobicity. (c) The conserved domain of Zmet2a. (d) The secondary structure of Zmet2a. (e) The tertiary structure prediction of Zmet2a. (f) Prediction across the membrane region. (g) Signal peptide prediction
The Zmet2a gene was analyzed using the ExPASy-Protparam online tool, gene rating 912 amino acids of 101045.99 Da (molecular weight) with a theoretical isoelectric point of 5.31. The combined instability coefficient was 44.87, assuming that the Zmet2a protein is unstable. Furthermore, the amino acid hydrophilicity results showed that most of the amino acid numbers on the horizontal axis of the Zmet2a are negative, and the minimum absolute value of the vertical axis is significantly greater than the maximum value on the vertical axis, combined with the GRAVY value of −0.532 (Fig. 1b), Therefore, we hypothesized that the Zmet2a protein is hydrophilic.
The conserved functional domains of the Zmet2a protein have a conserved BAH and a CHROMO domain (Fig. 1c). BAH domains are extensively involved in chromatin biological functions, including protein interactions, methylation recognition, and nucleosome binding [27]. Moreover, CHROMO structural domain proteins are also extensively involved in the regulation of the chromatin structure, mainly through the recognition of methylated lysine sites to regulate gene transcription [28]. Therefore, we speculate that Zmet2a regulates the chromatin structure and biological functions and is crucial in methylation, which contributes to the subsequent studies of the Zmet2a gene.
The secondary structure of the Zmet2a protein was 48.3% random coiled, 34.98% α-helix and 16.72% extended strand (Fig. 1d). Protein hydrophobicity facilitates α-helix formation, and since the secondary structure of Zmet2a has a relatively small alpha-helix percentage, the previous speculation on the hydrophobicity of the Zmet2a protein is tentatively verified. Furthermore, the tertiary structure of Zmet2a is 83.00% similar to DNA (cytosine-5)-methyltransferase 1(DNMT1) (Fig. 1e). The structure of the amino acid transmembrane region encoded by Zmet2a indicated that the Zmet2a protein is a non-transmembrane protein (Fig. 1f). Moreover, the amino acid signal peptide encoded by Zmet2a suggests that the Zmet2a protein has no signal peptide structure (Fig. 1g).
3.2 Phenotypic Analysis of Zmet2a Gene after Genetic Transformation into A. thaliana
The phenotypes of transgenic and WT A. thaliana lines showed significant differences in the leaf phenotypes, mainly the plant size (Fig. 2). Transgenic Arabidopsis had significantly higher lengths and widths of leaves and petioles than the WT. However, the number of leaves and primary branches were not significantly different (Table 1). Transgenic Arabidopsis flowered approximately 7 d earlier than the WT, a significant change (Table 1).

Figure 2: Blade size comparison between transgenic and wild-type (WT) Arabidopsis. The ruler is 1 cm

Quantitative analysis of flowering-related genes in two Arabidopsis species showed that SOC1 gene expression significantly promotes flowering, evident by the higher flowering transgenic than WT Arabidopsis. However, the expression of the FLC gene, which inhibits flowering, was significantly lower in the transgenic than the WT Arabidopsis. The expression of MF, MAF1, MAF2, MAF3, MAF4, and MAF5 genes, which regulate the flowering pathway, did not change significantly in transgenic and WT Arabidopsis (Fig. 3). Therefore, overexpressing Zmet2a regulates flowering through the flowering pathway, promoting earlier flowering in Arabidopsis.
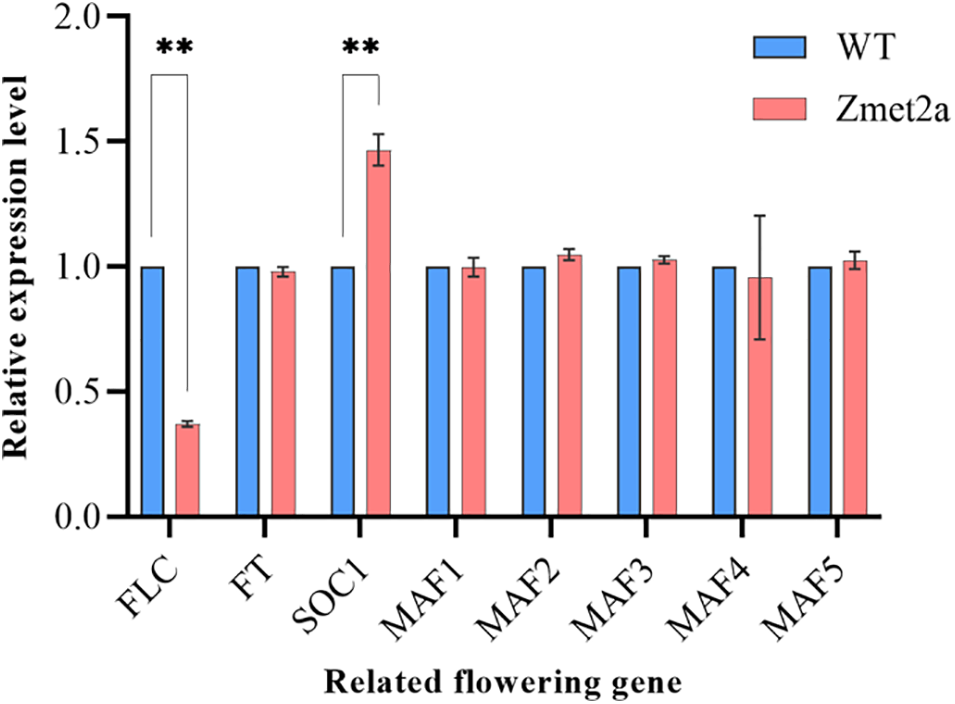
Figure 3: Expression of flowering genes between transgenic and WT Arabidopsis **p < 0.01
Note: WT: wild-type Arabidopsis.
3.3 Identification of Zmet2a Gene Editing Mutants
The four T2 generation positive plants T2-1-1, T2-1-2, T2-2-1, T2-2-2 were identified. 30 samples of each T2-1 and T2-2 were selected for PCR. The cloning and sequencing results obtain three different mutation types, with a variable number of base deletions accounting for most of the mutations, very few single base insertion edits, and only one single base deletion and insertion at different loci (Fig. 4). After sequencing and analyzing the blast, the deletion occurred in exon of the gene. The number of edited plants in samples T2-1-1 and T2-1-2 were 18 and 9, with an editing efficiency of 60% and 30%, respectively. However, T2-2-1 and T2-2-2 had 13 and 19 edited plants, with an editing efficiency of approximately 45% and 65%, respectively. The occurrence of mutations in the plants indicates that the CRISPR/Cas9 system successfully edited the genes.
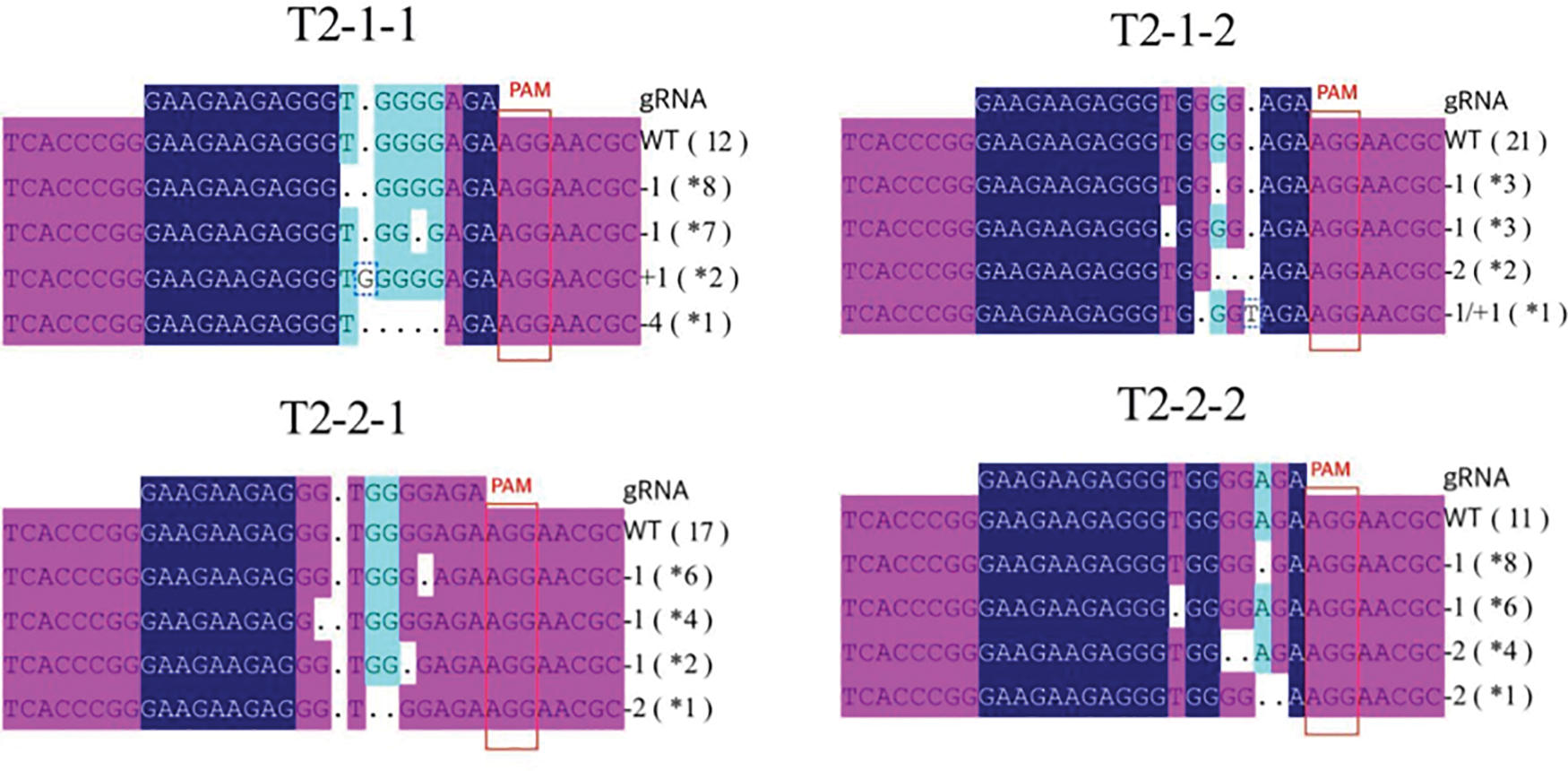
Figure 4: Target mutation results of CRISPR/Cas9-mediated mutations on the Zmet2a target site in transgenic plants. *: “−”: Base deletion, “+”: Base insertion, “−1/1”: Both base deletion and base insertion, *: Sample quantity
3.4 Analysis of Agronomic Traits in Transgenic Maize
Plant height comparisons between the four mutant plants and the self-cross line control (B73) at the lactation stage revealed that T2-1-1 and T2-2-2 were significantly shorter than the B73 (Fig. 5a). The spike height trend was similar to that of plant height (Fig. 5b). However, stem thickness and the number of leaves after the drawing stage were inversely correlated (Fig. 5c,d). The number of days from seedling emergence to pollen dispersal was significantly shorter between T2-1-1 and T2-2-2 than the B73 (Fig. 5e).
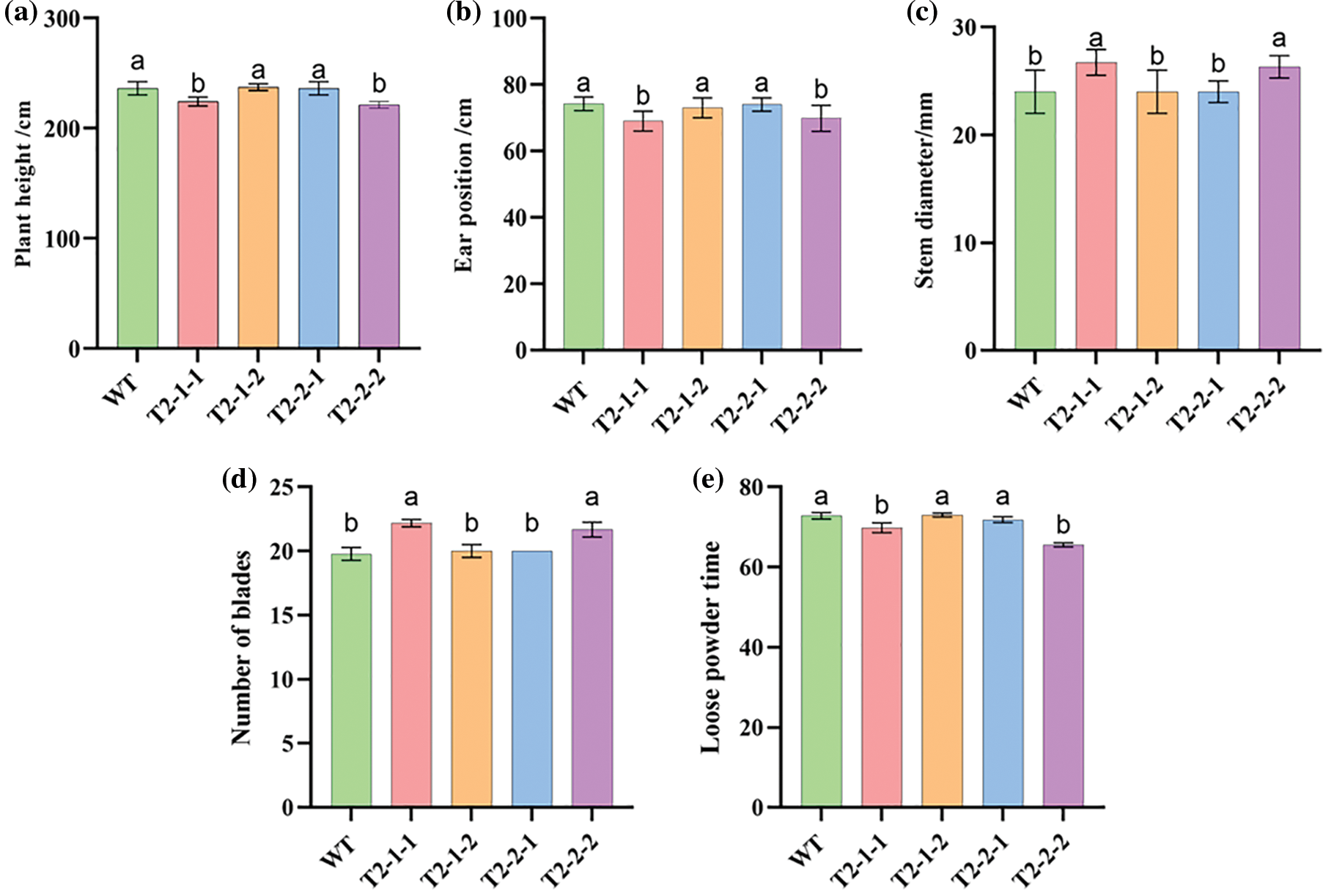
Figure 5: Agronomic trait performance of transgenic maize. (a) Plant-height comparison between transgenic and WT maize. (b) Ear heights of transgenic and WT maize. (c) Stem diameters of transgenic and WT maize. (d) Number of blades between transgenic and WT maize. (e) Loose powder timing between transgenic and WT maize. WT: Maize self-cross line control (B73)
3.5 Differences in Yield Components of Maize
Transgenic maize had significantly smaller ear lengths, ear thicknesses, and 100-grain weights compared to the WT (Fig. 6a–c). The number of rows of ears and the number of grains per row were not significantly different from the control (Fig. 6d,e).
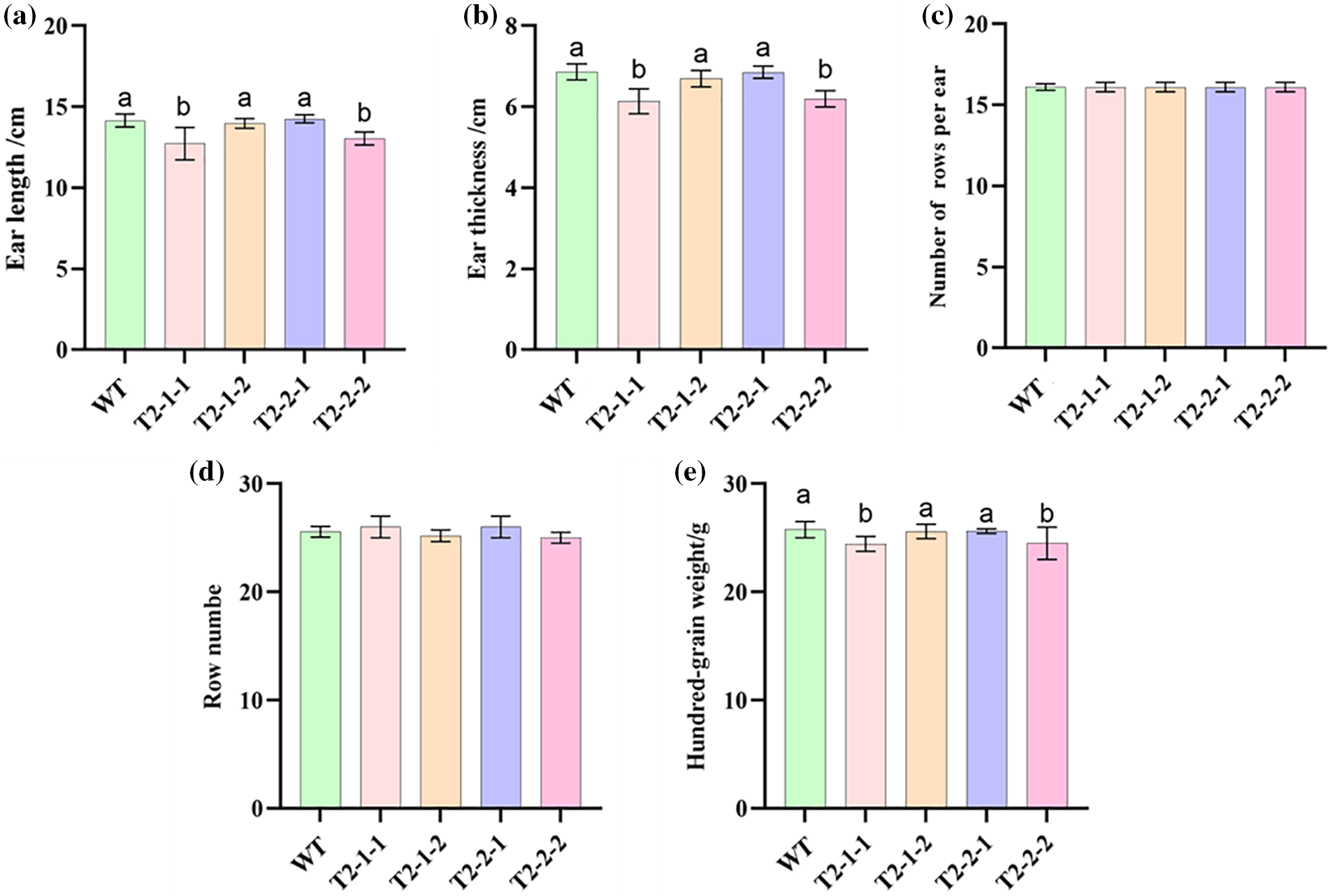
Figure 6: Yield component comparisons between transgenic and WT maize. (a) Ear length. (b) Ear thickness. (c) 100-grain weight. (d) Number of rows per ear. (e) Row number. WT: Maize self-cross line control (B73)
Day length (photoperiod) is an important avenue for plants to perceive environmental cues and is essential for regulating plant development and flowering time [29]. DNA methylation reportedly regulates photoperiodic flowering [30] and is significant in the domestication of important agricultural traits and adaptation of worldwide cultivated crops. Thus, this study reports a novel maize methyltransferase 2a gene (Zmet2a) obtained through a combination of bioinformatics and PCR-based gene mining strategies. The results are the first report on the role of the Zmet2a in regulating flowering time in plants. The results will also provide an important theoretical basis to further unravel the functional and epigenetic regulation mechanism of Zmet2a in growth and flowering time control in maize.
Qian et al.’s study identified and investigated the structure and expression of eight MET genes that encode a MET family protein in maize [31]. Zmet2a is a member of the DNA methyltransferase family maintained in maize. Therefore, we performed a detailed bioinformatics analysis of Zmet2a and hypothesized that the Zmet2a protein, mainly found in the nucleus, is a hydrophilic, unstable, non-transmembrane protein with a non-signaling peptide structure. Furthermore, the Zmet2a protein contained BAH and CHROMO domains, consistent with the Qian et al.’s study. Both BAH and CHROMO domains influence gene response and regulation, but the BAH module is also critical for linking DNA methylation, replication, and transcriptional regulation. Additionally, the CHROMO domain is also widely involved in chromatin structure regulation, mainly through the recognition of methylated lysine sites for gene transcription [32]. In contrast, different MET genes identified in Arabidopsis, rice, Ganoderma, and blueberry contain BAH structural domains in their protein structures and play different roles in different plants. For instance, MET1 decreased in Arabidopsis during leaf senescence and declined significantly before the leaves were senesced [33]. Salt and osmotic stresses down-regulate MET1-2, a gene expressed in all rice tissues [34]. The VcMET1 and VcCMT3 proteins of blueberry contain BAH, DcM, and the BAH, DcM, and CHROMO structural domains, respectively. However, the expression of these two genes in fruit development is inconsistent, with VcMET1 being up-regulated ward and then down-regulated in fruit development, while VcCMT3 was up-regulated, then down-regulated, and later up-regulated [35]. Thus, we hypothesized that Zmet2a, which contains BAH and CHROMO structural domains, can be expressed and play a regulatory role in maize development.
Plant-specific CMT3 maintains CHG methylation [36], and the BAH structural domain of CMT3 regulates plant growth and development by participating in methylation at the H3K9me2 methylation site [37,38]. Moreover, CMT3 and DRM2 redundantly control CHG methylation at specific sites [39]. We hypothesized that Zmet2a, a homolog of CMT3, also possesses the BAH domain and regulates plant growth and development through methylation at specific loci. The present experiment, the difference in traits between transgenic Arabidopsis plants and wild-type Arabidopsis plants under the same culture conditions initially verified the effect of Zmet2a on Arabidopsis. Transgenic Arabidopsis leaves had significantly higher leaf length and width, petiole length, and leaf size than the WT. We, therefore, hypothesize that the Zmet2a gene is involved in the regulation of leaf traits in Arabidopsis.
Previous experiments showed that the Zmet2a homolog, CMT3, promotes earlier flowering in rice regardless of the critical day length of light [40–42]. In this experiment, Zmet2a transgenic Arabidopsis flowered earlier than wild-type Arabidopsis. Multiple pathways control flowering in Arabidopsis, and the key flowering gene, FLC, regulates flowering by combining FT and SOC1 [43–46]. Thus, overexpressing the Zmet2a gene reduced the endogenous flowering gene, FLC, via fluorescence quantitative PCR analysis. The upstream gene, FLC, regulates the downstream gene, SOC1, and decreasing FLC increases SOC1 expression [47]. However, whether overexpressing Zmet2a acts directly on FLC or on both to regulate flowering time remains unknown.
In this study, the Zmet2a gene was knocked out of maize using the CRISPR/Cas9 system, and then the constructed knockout vector pBUE411 was transferred into a self-incompatible maize line, B73, by the ultrasound-mediated method. The Zmet2a gene function was preliminarily verified by identifying the trait differences between the four transgenic positive plants with obvious nested peaks and the maize self-incompatible line B73. PCR fragment cloning and sequencing identified three different mutations with a variable number of base deletions (accounting for the majority), a few single base insertions (the minority), and one single base deletion and insertion at different loci. Several factors, including PAM sequence position, Cas protein, and gene specificity of the target site, governed the editing efficiency. Thus, we speculate that the higher the editing efficiency, the more pronounced the phenotype. The less efficient mutant plants, T2-1-2 and T2-2-1, were not significantly different from the control maize, B73. Therefore, low editing efficiency is partly responsible for this editing inefficiency, while the small trait differences may be related to complete editing did not occur.
Arabidopsis transfected with the Zmet2a-overexpression vector had a significantly earlier flowering period than the wild-type Arabidopsis. The early flowering phenotype produced by transgenic Arabidopsis is a result of Zmet2a overexpression, which reduced FLC expression through the endogenous flowering regulatory pathway in Arabidopsis. The result is an increased expression of the flowering-promoting gene SOC1 and, consequently, an early flowering phenotype. Maize transduced with the Zmet2a knockout vector also flowered earlier than the untransformed maize (WT). Therefore, the early flowering behavior of transgenic maize against Zmet2a knockout vectors may be due to the deletion of the DNA methylation enzyme, Zmet2a, which reduced the DNA methylation levels of the flowering repression-associated genes in maize. The reduction increased the expression of flowering promotion-associated genes in maize, ultimately causing the early flowering phenotype. The effect of flowering on agronomic traits in higher plants is a key point. Zmet2a resulted in the early flowering behavior in transgenic maize. Moreover, transgenic maize had a slightly lower 100-grain weight, which did not achieve the expected yield increase and has implications for future genetic engineering of crop variety traits to achieve high yield and quality.
Acknowledgement: We thank to researcher of maize seed resource library of Jilin Agricultural University (JLAU) for providing experiment materials.
Funding Statement: This research was financially supported by Jilin Province Science and Technology Development Program (20220202014NC) and the National Natural Science Foundation of China (#31471565 and #31170259).
Author Contributions: Xin Qi and Yu Wang conducted the experiments and wrote the manuscript. Xinyang Liu, Xing Zhang, Xiaoshuang Wei and Zhenhui Wang validated and interpreted the data. Fenglou Ling and Zhenhui Wang conducted software analysis and performed formal analysis. Supervised and support the funding acquisition. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The datasets used and analyzed during the current study are available in the manuscript and its supplementary materials.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Supplementary Materials: The supplementary material is available online at https://doi.org/10.32604/phyton.2024.052844.
References
1. Li Y. Modern epigenetics methods in biological research. Methods. 2021;187(2):104–13. doi:10.1016/j.ymeth.2020.06.022. [Google Scholar] [PubMed] [CrossRef]
2. Wang SY, Pollina EA, Wang IH, Pino LK, Bushnell HL, Takashima K, et al. Role of epigenetics in unicellular to multicellular transition in Dictyostelium. Genome Biol. 2021;22(1):134. doi:10.1186/s13059-021-02360-9. [Google Scholar] [PubMed] [CrossRef]
3. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(S3):245–54. doi:10.1038/ng1089. [Google Scholar] [PubMed] [CrossRef]
4. Maeji H, Nishimura T. Epigenetic mechanisms in plants. Adv Bot Res. 2018;88:21–47. doi:10.1016/bs.abr.2018.09.014. [Google Scholar] [CrossRef]
5. Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38(1):23–38. doi:10.1038/npp.2012.112. [Google Scholar] [PubMed] [CrossRef]
6. Yang J, Zhang X, Blumenthal RM, Cheng X. Detection of DNA modifications by sequence-specific transcription factors. J Mol Biol. 2020;432(6):1661–73. doi:10.1016/j.jmb.2019.09.013. [Google Scholar] [PubMed] [CrossRef]
7. Sánchez-Romero MA, Casadesús J. The bacterial epigenome. Nat Rev Microbiol. 2020;18(1):7–20. doi:10.1038/s41579-019-0286-2. [Google Scholar] [PubMed] [CrossRef]
8. Yang Z, Yan H, Wang J, Nie G, Feng G, Xu X, et al. DNA hypermethylation promotes the flowering of orchardgrass during vernalization. Plant Physiol. 2022;190(2):1490–505. doi:10.1093/plphys/kiac335. [Google Scholar] [PubMed] [CrossRef]
9. Wagner I, Capesius I. Determination of 5-methylcytosine from plant DNA by high-performance liquid chromatography. Biochim Biophys Acta. 1981;654(1):52–6. doi:10.1016/0005-2787(81)90135-0. [Google Scholar] [PubMed] [CrossRef]
10. Messeguer R, Ganal MW, Steffens JC, Tanksley SD. Characterization of the level, target sites and inheritance of cytosine methylation in tomato nuclear DNA. Plant Mol Biol. 1991;16(5):753–70. doi:10.1007/BF00015069. [Google Scholar] [PubMed] [CrossRef]
11. Hu Y, Li Y, Weng J, Liu H, Yu G, Liu Y, et al. Coordinated regulation of starch synthesis in maize endosperm by microRNAs and DNA methylation. Plant J. 2021;105(1):108–23. doi:10.1111/tpj.15043. [Google Scholar] [PubMed] [CrossRef]
12. Li Z, Hu Y, Chang M, Kashif MH, Tang M, Luo D, et al. 5-azacytidine pre-treatment alters DNA methylation levels and induces genes responsive to salt stress in kenaf (Hibiscus cannabinus L.). Chemosphere. 2021;271:129562. doi:10.1016/j.chemosphere.2021.129562. [Google Scholar] [PubMed] [CrossRef]
13. Wang ZC, Nie LJ, He YX. The effect of 5-azacytidine to the DNA methylation and morphogenesis character of chrysanthemum during in vitro growth. Acta Horticulturae Sinica. 2009;36:1783–90. [Google Scholar]
14. Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. 1975;187(4173):226–32. doi:10.1126/science.187.4173.226. [Google Scholar] [CrossRef]
15. Compere SJ, Palmiter RD. DNA methylation controls the inducibility of the mouse metallothionein-I gene lymphoid cells. Cell. 1981;25(1):233–40. doi:10.1016/0092-8674(81)90248-8. [Google Scholar] [PubMed] [CrossRef]
16. He L, Huang H, Bradai M, Zhao C, You Y, Ma J, et al. DNA methylation-free Arabidopsis reveals crucial roles of DNA methylation in regulating gene expression and development. Nat Commun. 2022;13(1):1335. [Google Scholar] [PubMed]
17. Zubko E, Gentry M, Kunova A, Meyer P. De novo DNA methylation activity of methyltransferase 1 (MET1) partially restores body methylation in Arabidopsis thaliana. Plant J. 2012;71(6):1029–37. [Google Scholar] [PubMed]
18. Shim S, Lee HG, Seo PJ. MET1-dependent DNA methylation represses light signaling and influences plant regeneration in Arabidopsis. Mol Cells. 2021;44(10):746–57. [Google Scholar] [PubMed]
19. Hu L, Li N, Xu C, Zhong S, Lin X, Yang J, et al. Mutation of a major CG methylase in rice causes genome-wide hypomethylation, dysregulated genome expression, and seedling lethality. Proc Natl Acad Sci U S A. 2014;111(29):10642–47. [Google Scholar] [PubMed]
20. Pavlopoulou A, Kossida S. Plant cytosine-5 DNA methyltransferases: structure, function, and molecular evolution. Genomics. 2007;90(4):530–41. [Google Scholar] [PubMed]
21. Yu B, He X, Tang Y, Chen Z, Zhou L, Li X, et al. Photoperiod controls plant seed size in a CONSTANS-dependent manner. Nat Plants. 2023;9(2):343–54. [Google Scholar] [PubMed]
22. Sun H, Guo Z, Gao L, Zhao G, Zhang W, Zhou R, et al. DNA methylation pattern of Photoperiod-B1 is associated with photoperiod insensitivity in wheat (Triticum aestivum). New Phytol. 2014;204(3):682–92. [Google Scholar] [PubMed]
23. Saad M, Mary H, Amjid U, Shabir G, Aslam K, Shah SM, et al. Photoperiodic stress induces genotype-specific shift in DNA methylation in Tartary buckwheat. Biol Futur. 2019;70(4):278–85. [Google Scholar] [PubMed]
24. Ma Y, Tan R, Zhao J. Chilling tolerance in maize: insights into advances-toward physio-biochemical responses’ and QTL/Genes’ identification. Plants. 2022;11(6):2082. [Google Scholar] [PubMed]
25. Rao X, Huang X, Zhou Z, Lin X. An improvement of the 2–△△CT method for quantitative real-time polymerase chain reaction data analysis. Biostat Bioinforma Biomath. 2013;3(3):71–85. [Google Scholar] [PubMed]
26. Yang L, Cui G, Wang Y, Hao Y, Du J, Zhang H, et al. Expression of foreign genes demonstrates the effectiveness of pollen-mediated transformation in zea mays. Front Plant Sci. 2017;8(635):383. doi:10.3389/fpls.2017.00383. [Google Scholar] [PubMed] [CrossRef]
27. Yang N, Xu RM. Structure and function of the BAH domain in chromatin biology. Crit Rev Biochem Mol Biol. 2013;48(3):211–21. doi:10.3109/10409238.2012.742035. [Google Scholar] [PubMed] [CrossRef]
28. Cavalli G, Paro R. Chromo-domain proteins: linking chromatin structure to epigenetic regulation. Curr Opin Cell Biol. 1998;10(3):354–60. doi:10.1016/S0955-0674(98)80011-2. [Google Scholar] [PubMed] [CrossRef]
29. Song YH, Shim JS, Kinmonth-Schultz HA, Imaizumi T. Photoperiodic flowering: time measurement mechanisms in leaves. Annu Rev Plant Biol. 2015;66(1):441–64. doi:10.1146/annurev-arplant-043014-115555. [Google Scholar] [PubMed] [CrossRef]
30. Shi M, Wang C, Wang P, Yun F, Liu Z, Ye F, et al. Role of methylation in vernalization and photoperiod pathway: a potential flowering regulator? Hortic Res. 2023;10(10):uhad174. doi:10.1093/hr/uhad174. [Google Scholar] [PubMed] [CrossRef]
31. Qian Y, Xi Y, Cheng B, Zhu S. Genome-wide identification and expression profiling of DNA methyltransferase gene family in maize. Plant Cell Rep. 2014;33(10):1661–72. doi:10.1007/s00299-014-1645-0. [Google Scholar] [PubMed] [CrossRef]
32. Wiley EA, Horrell S, Yoshino A, Schornak CC, Bagnani C, Chalker DL. Diversification of HP1-like chromo domain proteins in tetrahymena thermophila. J Eukaryot Microbiol. 2018;65(1):104–16. doi:10.1111/jeu.12443. [Google Scholar] [PubMed] [CrossRef]
33. Yuan L, Wang D, Cao L, Yu N, Liu K, Guo Y, et al. Regulation of leaf longevity by DML3-mediated DNA demethylation. Mol Plant. 2020;13(8):1149–61. doi:10.1016/j.molp.2020.06.006. [Google Scholar] [PubMed] [CrossRef]
34. Tan MP. Analysis of DNA methylation of maize in response to osmotic and salt stress based on methylation-sensitive amplified polymorphism. Plant Physiol Biochem. 2010;48(1):21–6. doi:10.1016/j.plaphy.2009.10.005. [Google Scholar] [PubMed] [CrossRef]
35. Yu L, Ya Z, Yi HZ, Wei L, Yong QL, Mei L, et al. DNA methylation balance is involved in anthocyanin accumulation during Vaccinium corymbosum fruit ripening. J Berry Res. 2020;10(4):651–63. doi:10.3233/JBR-200553. [Google Scholar] [CrossRef]
36. Nozawa K, Chen J, Jiang J, Leichter SM, Yamada M, Suzuki T, et al. DNA methyltransferase CHROMOMETHYLASE3 prevents ONSEN transposon silencing under heat stress. PLoS Genet. 2021;17(8):e1009710. doi:10.1371/journal.pgen.1009710. [Google Scholar] [PubMed] [CrossRef]
37. Fang J, Jiang J, Leichter SM, Liu J, Biswal M, Khudaverdyan N, et al. Mechanistic basis for maintenance of CHG DNA methylation in plants. Nat Commun. 2022;13(1):3877. doi:10.1038/s41467-022-31627-3. [Google Scholar] [PubMed] [CrossRef]
38. Cui X, Cao X. Epigenetic regulation and functional exaptation of transposable elements in higher plants. Curr Opin Plant Biol. 2014;21:83–8. doi:10.1016/j.pbi.2014.07.001. [Google Scholar] [PubMed] [CrossRef]
39. Cao X, Jacobsen SE. Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc Natl Acad Sci U S A. 2002;99(suppl_4):16491–98. doi:10.1073/pnas.162371599. [Google Scholar] [PubMed] [CrossRef]
40. Tan F, Zhou C, Zhou Q, Zhou S, Yang W, Zhao Y, et al. Analysis of chromatin regulators reveals specific features of Rice DNA methylation pathways. Plant Physiol. 2016;171(3):2041–54. doi:10.1104/pp.16.00393. [Google Scholar] [PubMed] [CrossRef]
41. Li X, Zhu B, Lu Y, Zhao F, Liu Q, Wang J, et al. DNA methylation remodeling and the functional implication during male gametogenesis in rice. Genome Biol. 2024;25(1):84. [Google Scholar] [PubMed]
42. Hu D, Yu Y, Wang C, Long Y, Liu Y, Feng L, et al. Multiplex CRISPR-Cas9 editing of DNA methyltransferases in rice uncovers a class of non-CG methylation specific for GC-rich regions. Plant Cell. 2021;33(9):2950–64. [Google Scholar] [PubMed]
43. Zong W, Zhao B, Xi Y, Bordiya Y, Mun H, Cerda NA, et al. DEK domain-containing proteins control flowering time in Arabidopsis. New Phytol. 2021;231(1):182–92. [Google Scholar] [PubMed]
44. Amara U, Hu J, Cai J, Kang H. FLK is an mRNA m6A reader that regulates floral transition by modulating the stability and splicing of FLC in Arabidopsis. Mol Plant. 2023;16(5):919–29. [Google Scholar] [PubMed]
45. Yu Y, Wang S, Wang Z, Gao R, Lee J. Arabidopsis thaliana: a powerful model organism to explore histone modifications and their upstream regulations. Epigenetics. 2023;18(1):2211362. [Google Scholar] [PubMed]
46. Qi HD, Lin Y, Ren QP, Wang YY, Xiong F, Wang XL. RNA splicing of FLC modulates the transition to flowering. Front Plant Sci. 2019;10:1625. doi:10.3389/fpls.2019.01625. [Google Scholar] [PubMed] [CrossRef]
47. Surkova SY, Samsonova MG. Mechanisms of vernalization-induced flowering in legumes. Int J Mol Sci. 2022;23(17):9889. doi:10.3390/ijms23179889. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools