
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Cu Stress-Induced Transcriptome Alterations in Sorghum and Expression Analysis of the Transcription Factor-Encoding Gene SbWRKY24
1 College of Biological Engineering, Sichuan University of Science & Engineering, Yibin, 644002, China
2 Wuliangye Yibin Co., Ltd., Yibin, 644000, China
* Corresponding Author: Zhenhui Kang. Email:
Phyton-International Journal of Experimental Botany 2024, 93(7), 1503-1521. https://doi.org/10.32604/phyton.2024.051718
Received 13 March 2024; Accepted 07 May 2024; Issue published 30 July 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Sorghum is not only an important bio-energy crop but also a vital raw material for brewing. Exogenous copper affects the growth and metabolism of crops in specific ways. This study identified 8475 differentially expressed genes (DEGs) by high-throughput transcriptome sequencing in the sorghum cultivar ‘Jinnuoliang 2’ after 24 h of treatment with 10 mM CuSO. Using GO analysis, 476 genes were functionally annotated, which were mainly related to catabolism and biosynthetic processes. Additionally, 90 pathways were annotated by employing the KEGG analysis. Among them, glutathione metabolism and peroxisome were induced, while photosynthesis, photosynthesis-antenna protein, and carbon sequestration of photosynthetic organisms were inhibited. Of the DEGs, 399 were identified to encode transcription factors belonging to 49 families. This study also identified a WRKY transcription factor-encoding gene SbWRKY24 from the transcriptome data. For studying its function, the relative expression levels of SbWRKY24 in roots and leaves post-treatment with different growth hormones and exposure to a variety of abiotic stresses were detected by RT-qPCR. SbWRKY24 showed treatment-and tissue-specific expression patterns, indicating its unique role in stress tolerance. This study lays a theoretical basis for the functional exploration of SbWRKY24, elucidating the mechanism of copper resistance, and elaborating on the stress responses in sorghum. It also guides the exploration of the molecular mechanism of copper ions inducing intracellular signal transduction pathways.Keywords
Sorghum bicolor is a monocot grass that serves as a grain and feed crop and is a critical brewing material with high resistance. Sorghum is affected by heavy metals [1], cold-induced injury, high salt, and pathogens, leading to a decline in its productivity. Nowadays, the soils of cultivated lands in China are massively polluted with copper, primarily due to the exploitation of copper mines and the use of insecticides. This pollution causes high biological toxicity, persistence, irreversibility, and hysteresis [2]. Copper plays a concentration-dependent role in plant growth and metabolism. Trace amounts of copper can promote the photosynthesis, respiration, growth, and development of flower organs, along with metabolism [3]. On the contrary, excessive copper can threaten plants, reduce their growth and quality, induce many symptoms, such as dwarfism, chlorosis of leaves, reduction in photosynthetic rates, destruction of cell membranes, and promote the production and accumulation of reactive oxygen species (ROS) in cells, which are harmful to plant growth [4–7]. Excessive copper may also demonstrate some effects on human health through enrichment in the food chain. Cu stress has made great progress in the research of seed germination, plant growth and development, as well as substance metabolism and detoxification [8]. However, there are few studies on multi-omics such as binding transcriptomes, metabolomics, and proteomics. Additionally, research on molecular regulation network technical mechanisms is lacking, such as the resistance mechanism of plants in response to copper stress and the transport and absorption of Cu ions, which should be discussed in combination with multi-omics [9], thereby revealing the mechanism of copper resistance of plants. So far, sorghum’s response to Cu stress at the transcriptome level has not been reported.
Transcriptome sequencing (RNA-seq) has been extensively applied in plant stress resistance research for the mining of genes and their functional identification. Some copper-tolerant plants, such as fengdan, grapes, wheat, and Dactylus canadensis, have been screened by RNA-seq, and their physiological and biochemical aspects have been studied. By analyzing the differentially expressed genes (DEGs) in plants under copper stress, a large number of genes and their related metabolic pathways were screened, which provided a reference for mining copper tolerance-related genes. Thus, it can provide a theoretical basis for screening copper-tolerant plants and breeding copper-tolerant varieties. Research on resistance genes is conducive to agricultural development [10].
Transcription factors play a crucial inducing or inhibiting role in the entire process of plant growth and development. WRKY transcription factors possess two WRKY domains that bind to DNA and are involved in the growth and development, metabolism, senescence, and defense of plant. SPFI was the first WRKY transcription factor isolated from sweet potato (Dioscorea esculenta) [11]. Subsequently, a growing number of WRKY transcription factors have been identified in species such as numbering 72 in Arabidopsis thaliana [12].
WRKY transcription factors can enhance the resistance to excessive heavy metal stress by regulating their chelation [13]. In rice, OsFRDL4, which encodes a citrate transporter, is regulated by the aluminum (Al) resistance transcription factor 1 (ART1), while OsWRKY22 can promote the expression of OsFRDL4 induced by Al [14]. In tomato (Solanum lycopersicum), SlWRKY6 was induced under CdCl2, CuCl2, and HgSO4 stress, improving the resistance to them [15]. TaWRKY74 in wheat regulates the expression of glutathione S-transferase1 (TaGST1) and affects the accumulation of glutathione (GSH) under Cu stress, which may contribute to the tolerance against copper toxicity [16]. The expression of NnWRKY22 in lotus was induced under copper stress, suggesting that it may be involved in stress resistance [17]. Numerous studies have revealed that the WRKY family is involved in responses to biotic and abiotic stresses in plants [18,19]. SbWRKY30 plays a positive regulatory role under drought stress [20]. The CaMV35S-BcWRKY46 expressing tobacco plants revealed a reduced sensitivity to salt and ABA stress [21].
WRKY24 protein has been reported in plants such as rice (Oryza sativa), wheat (Triticum aestivum), kiwifruit (Actinidia chinensis), banana (Musa acuminata), and tomatoes (Solanum lycopersicum). However, WRKY24 protein has not been reported in sorghum [22–26]. Additionally, there is no report on the current research status of WRKY24 in heavy metal stress. This study employed the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses to analyze the DEGs in sorghum after copper stress through RNA-seq. Simultaneously, SbWRKY24 was screened and cloned, and quantitative real-time PCR (RT-qPCR) was performed to investigate its expression characteristics by detecting its relative expression levels in roots and leaves after exposure to different growth hormones and abiotic stresses. These analyses provide a theoretical basis for further functional study of the SbWRKY24 protein, the molecular mechanism of its regulation under stress, and its role in responses to copper stress. They also assist in the mining of candidate copper-tolerance-related genes in sorghum, which are used for breeding varieties that are copper-tolerant but not copper-enriched.
The sorghum variety ‘Jinnuoliang 2’ was selected as the experimental material. The non-damaged and large-sized sorghum seeds were selected. They were soaked in NaClO solution (available chlorine 4%) for 15 min and then washed with distilled water. They were sown at a depth of 1 cm in nursery trays filled with nutrient soil covered with a plastic film after applying sufficient water. The film was later removed, allowing the seedlings to grow. Seedlings with the same growth status at the one leaf and one heart stage were selected and treated with 0, 0.4, 2, 10, and 50 mM CuSO4 for 24 h. Each treatment was performed in three biological replicates. The tissue samples of the seedlings treated with 0 and 10 mM CuSO4 were flash-frozen in liquid N2 (the whole plant).
RNA extraction and transcriptomics of the samples were conducted at the Wuhan Kang Testing Technology Co., Ltd. (Wuhan, China). The original image data were obtained from the Illumina® Hiseq™ 2500 sequencing. The software fastp (version 23.0) was used to remove the linker sequences, low-quality reads (base ratio <Q20 was >0.08), high-N-rate sequences (reads with > five N bases), and short-length sequences to obtain clean reads and maintain the quality of the subsequent analysis.
2.3 Reference Sequence Alignment and Enrichment Analysis
The complete transcript information was obtained by aligning the clean data with the reference genome (NCBIv3) [27]. The RPKM value was used as the index of gene expression, and |logFC| > 1 and P-value < 0.05 were used as the criteria for screening the DEGs. Thus, the functional annotation and classification of the protein was achieved.
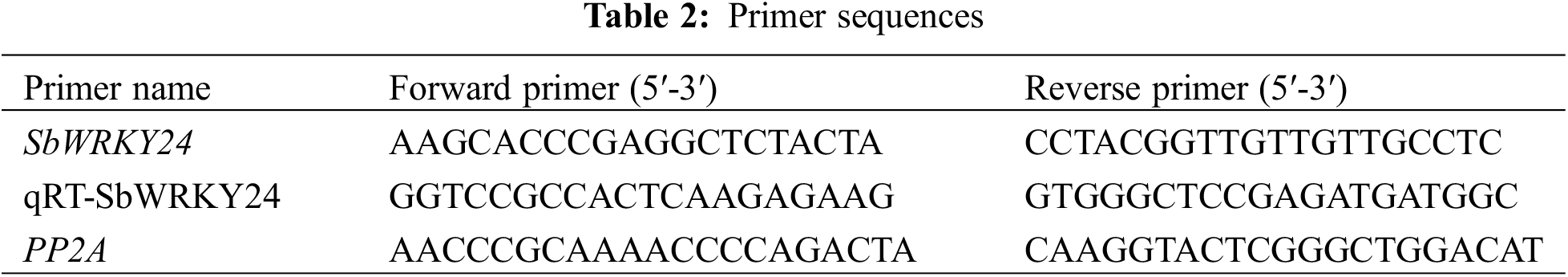
Using sorghum PP2A as an internal reference, seven DEGs were randomly selected and identified by RT-qPCR. The melting curve was applied to verify the specificity of the product. The relative gene expression was calculated using the 2−ΔΔCT method (Table 1).

SbWRKY24, which was repressed by copper stress, was identified from the RNA-seq data, and its sequence was obtained from the NCBI database (https://www.ncbi.nlm.nih.gov/gene/8085397, accessed on 21/04/2024).
2.6 Expression Analysis of SbWRKY24 under Different Treatments
The seedlings were cultured in Hoagland’s nutrient solution until the three-leaf stage and then subjected to varying treatments under the same growth conditions. Salt and drought stresses were simulated using 100 mM NaCl, 20% PEG-6000 (polyethylene glycol), 100 μM abscisic acid (ABA), 100 μM l-aminocyclopropane-l-carboxylic acid (ACC), 100 μM gibberellic acid (GA), and 100 μM jasmonic acid (JA). Each treatment group was set in three biological replicates. Root and leaf samples were collected at 0, 1, 12, 24, 48, and 72 h and were flash-frozen in liquid N2.
The total RNA from the root and leaf tissue samples was extracted separately and then reverse-transcribed using the same kits mentioned in Section 2.4. Each reaction was set in three biological replicates, and fusion curves were used to verify the specificity of the products. The results were plotted using GraphPad Prism 6.0 (https://www.graphpad.com, accessed on 21/04/2024). SPSS software was used for the one-way analysis of variance (Table 2).
2.7 Structure Prediction of the SbWRKY24 Protein
The isoelectric point (pI), molecular weight (MW), and amino acid composition of the SbWRKY24 protein were determined by the ProtParam program, and its hydrophilicity was determined by the ProtScale program. The transmembrane region, protein domain, secondary structure, and tertiary structure were predicted by the DeepTMHMM program, NCBI, SPOMA, and SWISS-MODEL, respectively.
2.8 Subcellular Localization of the SbWRKY24 Protein
The subcellular localization of the SbWRKY24 protein was predicted in silico by the PSORT program and verified in vivo by fusion with green fluorescent protein. The full-length CDS fragments of SbWRKY24 (without the stop codon TAG) containing two restriction endonuclease sites, KpnI and BamHI, were subjected to PCR amplification using sorghum cDNA as a template. After double restriction endonuclease digestion, the fragments were cloned into the pCAMBIA1301-GFP vector and sequenced at BGI. The constructed vectors, pCAMBIA1301-SbWRKY24-GFP and pCAMBIA1301-GFP, were used to transform the Agrobacterium tumefaciens EHA105 cells. The PCR-positive colonies were cultured in the dark at 28°C and 200 rpm till the OD600 was ~1.0. Then, the culture medium was centrifuged, and the pellet was resuspended and used to infect the lower epidermal cells of onions. These cells were cultured on a solid MS medium for two days. Then, the expression of the target protein in the cells was observed by transmission light and green fluorescence under a fluorescence microscope.
2.9 Transcription Activation Ability of the SbWRKY24 Protein
Full-length CDS fragments containing EcoRI and BamHI sites were subjected to PCR amplification using sorghum cDNA as a template and SbWRKY24 primers. After digestion, the CDS fragments were cloned into the pGBKT7 vector and sequenced at BGI. The CDS cloned and empty (control) vectors were used to transform cells of the yeast strain Y2HGold. Their growth phenotype and the intensity of the blue color were observed on media containing tryptophan (Trp) or X-α-Gal (a chromogenic substrate of α-galactosidase) to verify the transcription activation ability of SbWRKY24.
3.1 The Phenotype of Seedlings at Different Concentrations of CuSO4
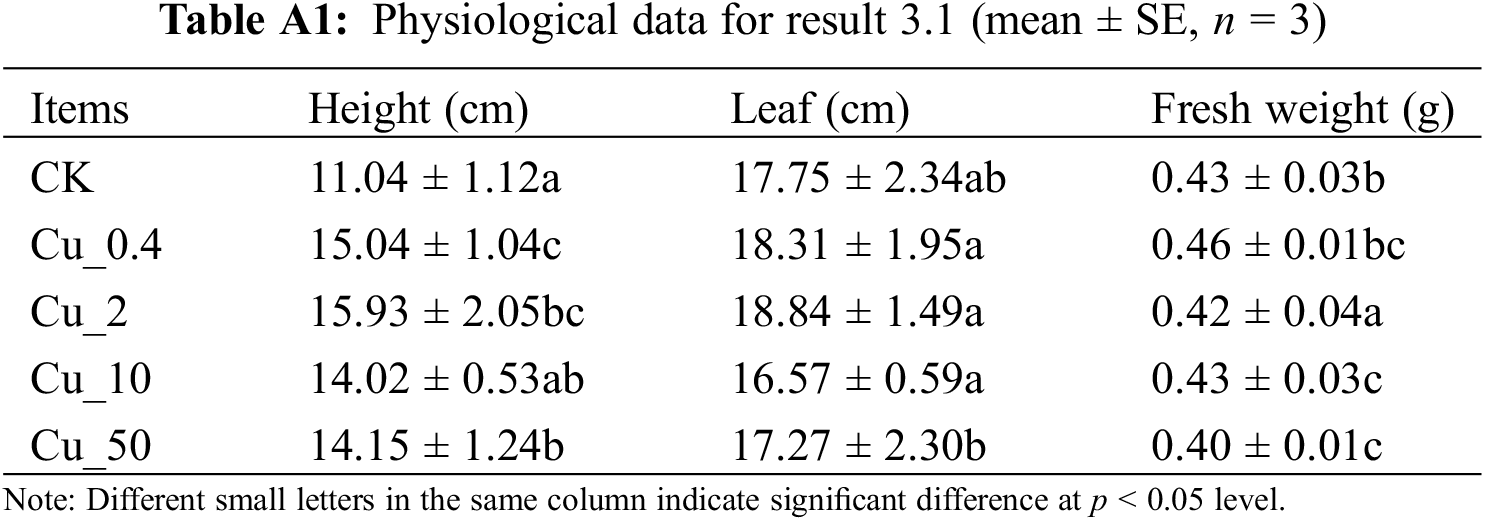
Sorghum seedlings with similar growth were treated with 0, 0.4, 2, 10, and 50 mM CuSO4 for 24 h (each treatment group was set in three biological replicates). Those exposed to 0.4 and 2 mM CuSO4 showed no phenotypic changes. However, they wilted at 10 mM and withered at 50 mM CuSO4. The transcriptomes of those treated with 0 and 10 mM CuSO4 were sequenced (Fig. 1) (the physiological data are shown in the attached Table A1).

Figure 1: Phenotype of sorghum under different concentrations. (A) 0 mM; (B) 0.4 mM; (C) 2 mM; (D) 10 mM; (E) 50 mM
3.2 Quality of Sequencing Data
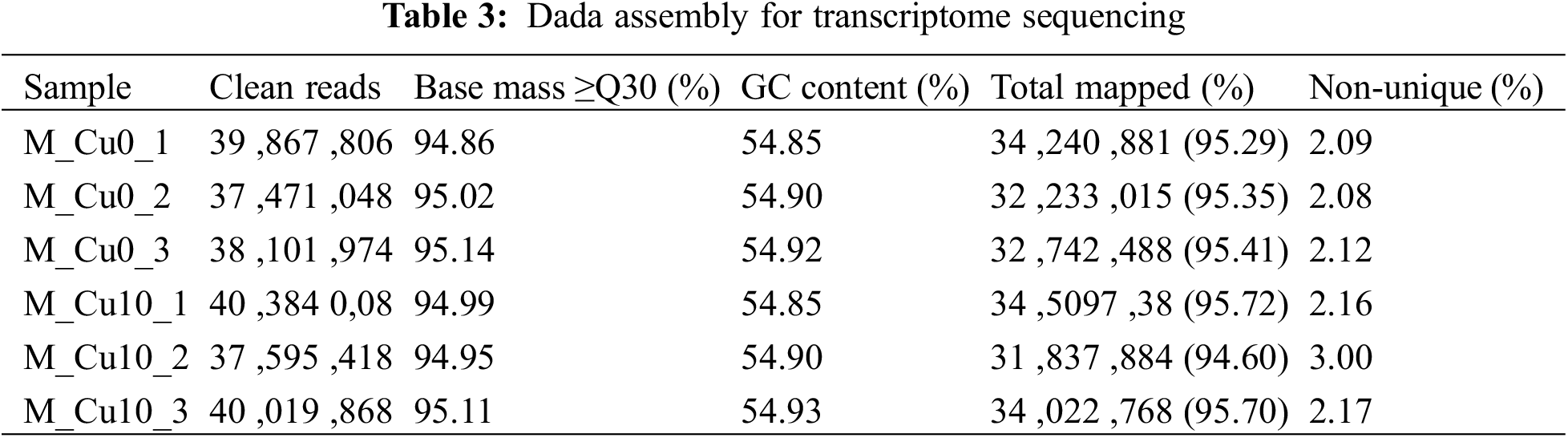
The transcriptomes of seedlings treated with 10 (M_Cu10) and 0 mM CuSO4 (M_Cu0) were sequenced. The clean reads obtained from M_Cu0_1, M_Cu0_2, and M_Cu0_3 (control groups) were 39,867,806, 37,471,048, and 38,101,974, respectively. The GC contents were 54.85%, 54.90%, and 54.92%, respectively. The clean reads obtained from M_Cu10_1, M_Cu10_2, and M_Cu10_3 (treatment groups) were 40,384,008, 37,595,418, and 40,019,868, respectively. The GC contents were 54.85%, 54.90%, and 54.93%, respectively, and the Q30 value was >94%. An alignment of the clean reads with the reference genome indicated that the alignment rate of the control groups was >95%, and that of the treatment groups was >94%. The proportion of non_unique Mapped Reads or Fragments between the control and experimental groups was <3%. These results reflected the high quality of the transcriptome sequencing data, which could be used for further analysis (Table 3).
3.3 The Distribution of Reads in the Different Regions of the Reference Genome
The reads were located in the CDS, intergenic, and intron regions of the reference genome. As sorghum is a higher eukaryote, most of its mRNA is situated within the CDS region. The reads in this study were predominantly distributed in the CDS region, indicating a successful capture that meets the requirements of library construction (Fig. 2).
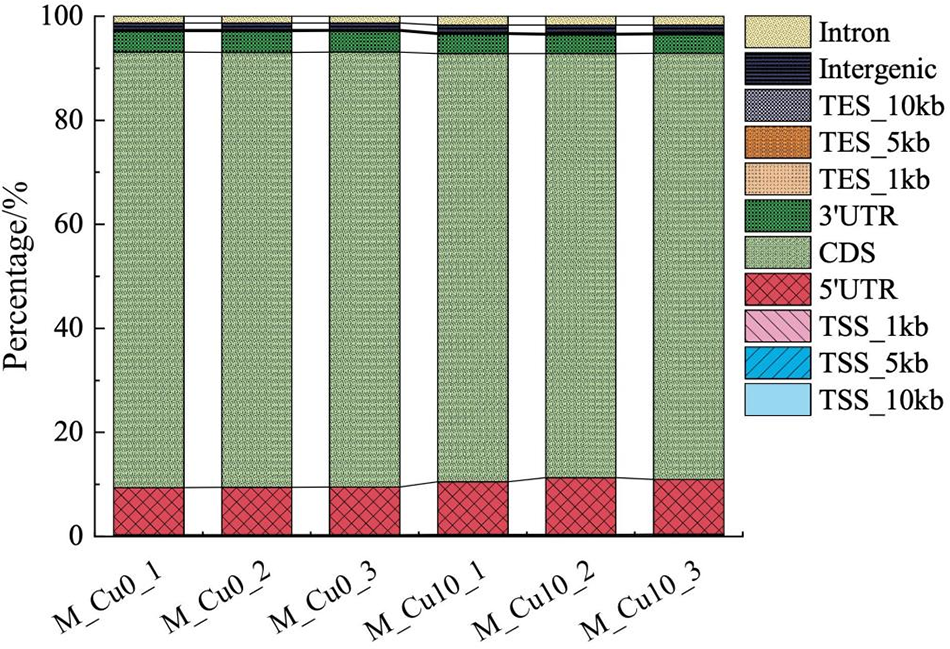
Figure 2: Distribution of reads in different regions on the reference genome
The reference sequences of sorghum genes were obtained from the plant gene database, and the RPKM (Reads per Kilobase per Million Reads) values were used as an index of gene expression. The RPKM value box and density curve distribution plots of the genes of each control and experimental group were generated (Fig. 3). The total gene expression showed a few differences between both groups in the dispersion and distribution, indicating that the expression of genes in sorghum varied under copper stress.
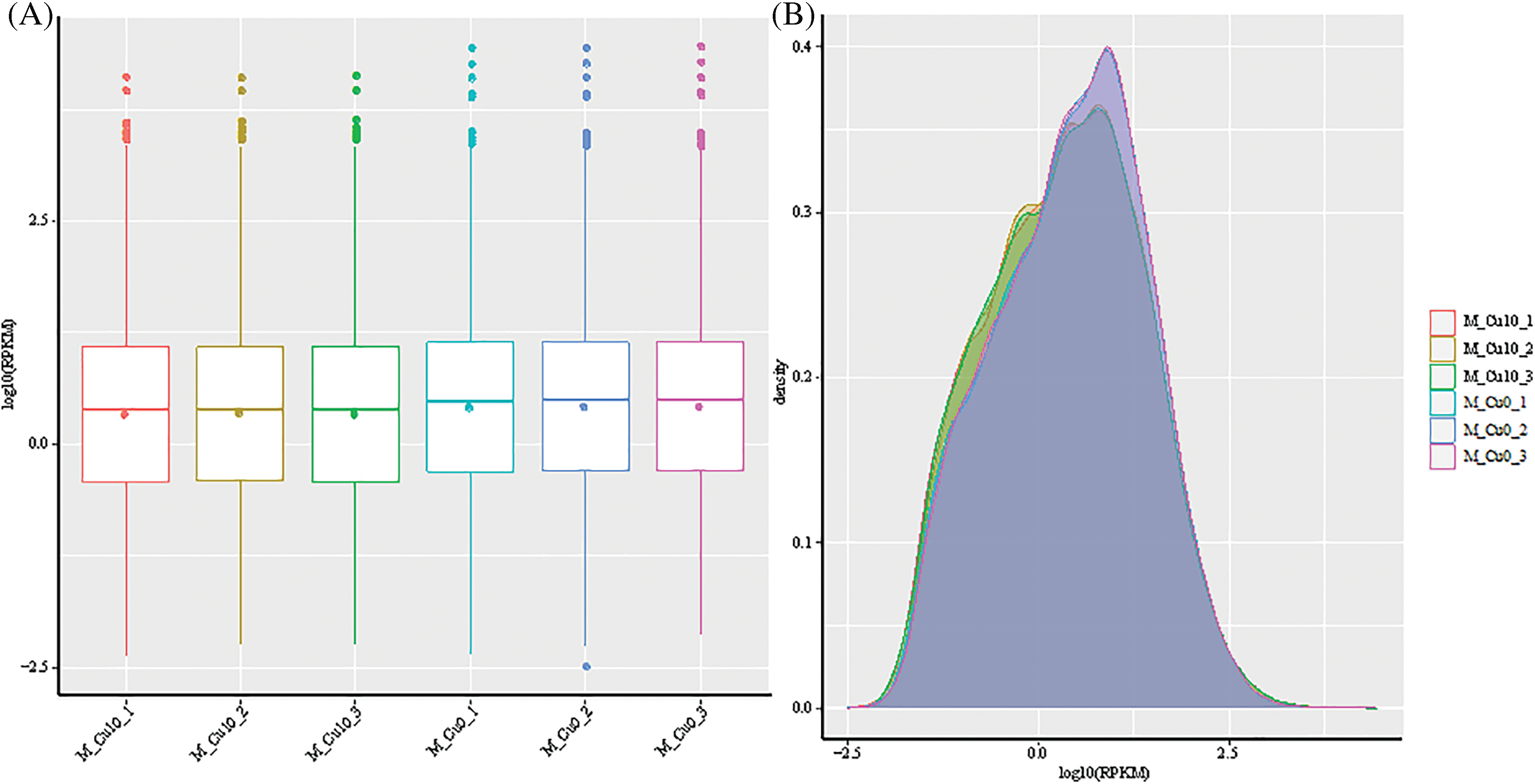
Figure 3: RPKM value box diagram and density distribution diagram. (A) Box diagram of RPKM value. (B) Distribution of RPKM value density
3.5 Screening and Clustering of DEGs
This study screened 8475 DEGs under copper stress, of which 3662 were upregulated and 4813 were downregulated (Fig. 4A). Expression clustering was analyzed according to the RPKM values, with high-expression genes represented in red and low-expression genes in blue. Additionally, different rows and columns represent varying genes and treatment groups, respectively (Fig. 4B). The genes with similar structure and function were classified into one group based on the differential gene cluster map, indicating that they might play similar physiological roles in sorghum after copper stress.
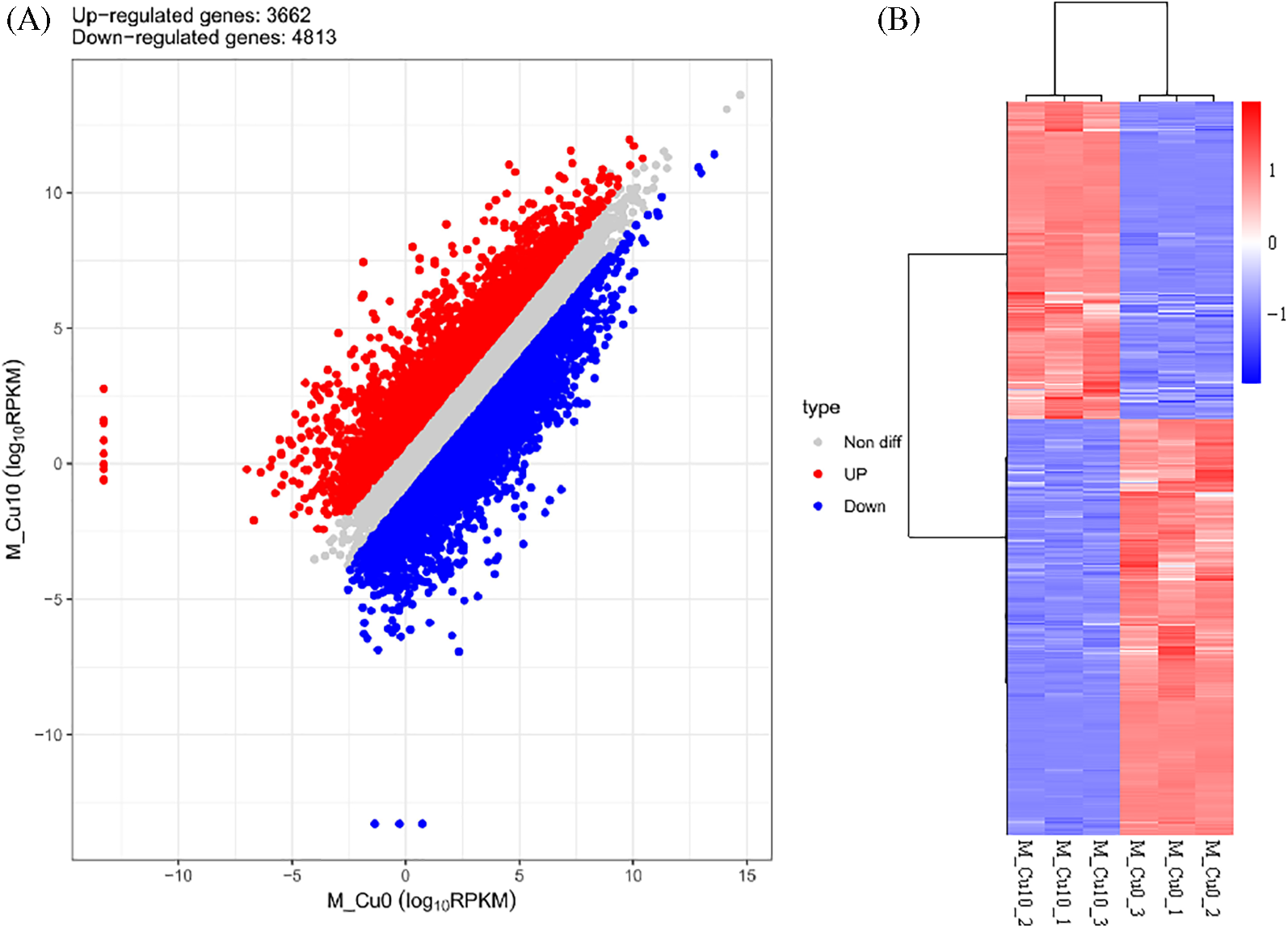
Figure 4: Scatter plot of expression between comparison groups and clustering diagram of DEG. (A) Scatter plot of expression between comparison groups. (B) Clustering diagram of DEGs
3.6 GO Functional Enrichment of the DEGs
The biological significance of the 8475 DEGs was categorized based on the GO database. The 3662 upregulated DEGs were mainly enriched in 142 GO terms, of which 83 were involved in BP, 12 in CC, and 47 in MF. The 83 GO terms in BP were mainly enriched in the catabolic process (GO: 0009056), cellular catabolic process (GO: 0044248), organic substance catabolic process (GO: 1901575), organonitrogen compound catabolic process (GO: 1901565), lipid metabolic process (GO: 0006629), monocarboxylic acid metabolic process (GO: 0032787), etc. GO terms in CC were mainly enriched in membrane (GO: 0016020), membrane part (GO: 0044425), intrinsic component of membrane (GO: 0031224), and others. The MFs of acyltransferase were significantly different across the following pathways: transferase activity, transferring acyl groups (GO: 0016746), cation transmembrane transporter activity (GO: 0008324), transferase activity, transferring hexosyl groups (GO: 0016758), ion transmembrane transporter activity (GO: 0015075), transporter activity (GO: 0005215), transmembrane transporter activity (GO: 0022857), transferase activity, and glycosyl transferring groups (GO: 0016757) (Fig. 5A).
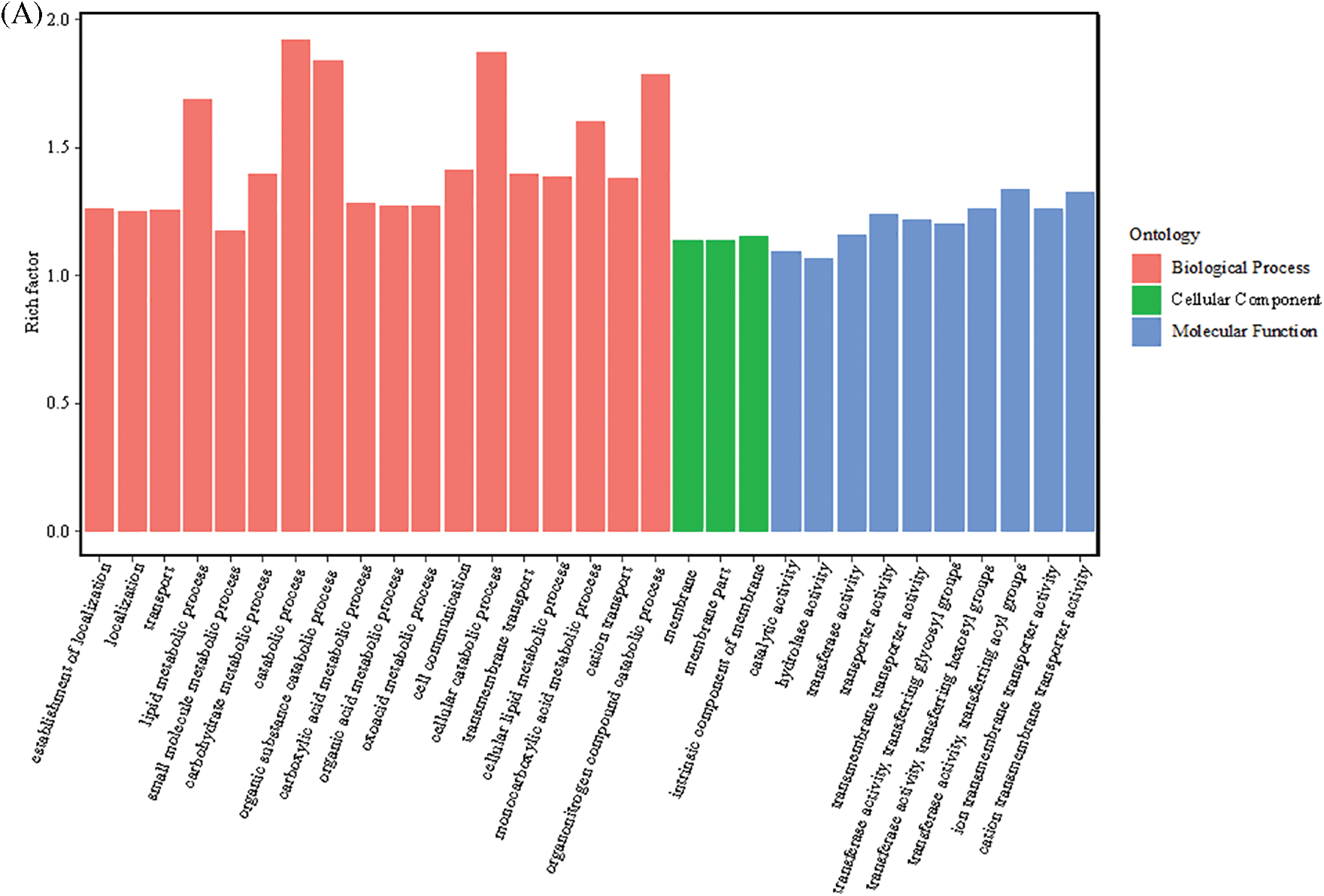
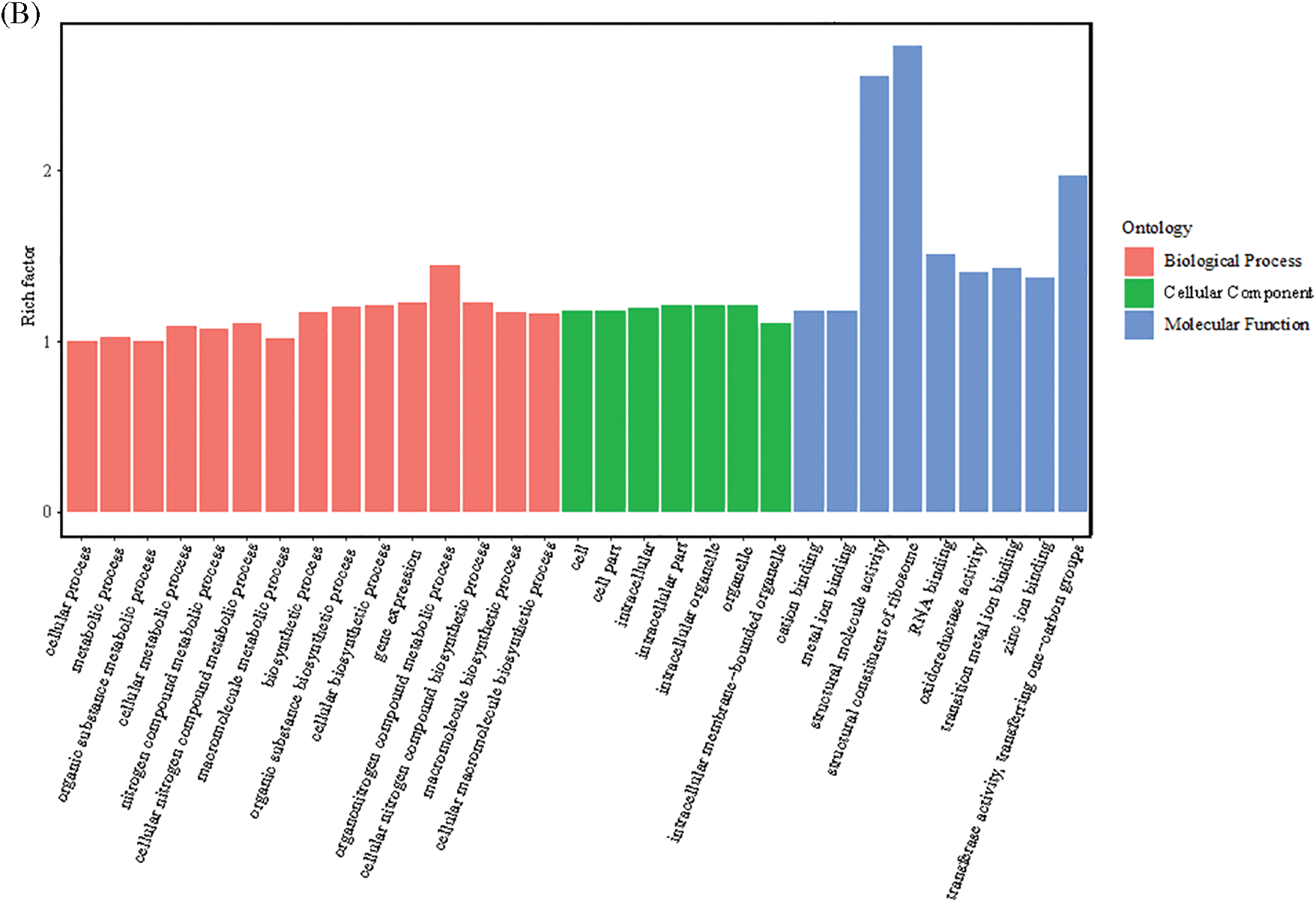
Figure 5: GO functional enrichment of the DEGs. (A) GO annotation classification statistics of upregulated DEGs. (B) GO enrichment map of downregulated DEGs
The 4813 downregulated DEGs were mainly enriched in 334 GO terms, involving 167 BPs, 75 CCs, and 92 MFs. BP terms were primarily enriched in biosynthetic processes (GO: 0009058), organic substance biosynthetic process (GO: 1901576), cellular biosynthetic process (GO: 0044249), gene expression (GO: 0010467), organonitrogen compound metabolic process (GO: 1901564), cellular nitrogen compound biosynthetic process (GO: 0044271), macromolecule biosynthetic process (GO: 0009059), and cellular macromolecule biosynthetic process (GO: 0034645), etc. The CC terms were mainly enriched in cell (GO: 0005623), cell part (GO: 00446464), intracellular (GO (0005622), intracellular part (GO: 0044424), intracellular organelle (GO: 0043229), organelle (GO: 0043226), and others. The MF terms were mainly enriched in the pathways of structural molecule activity (GO: 0005198), structural constituent of ribosome (GO: 0003735), RNA binding (GO: 0003723), oxidoreductase activity (GO: 0016491), transition metal ion binding (GO: 0046914), zinc ion binding (GO: 0008270), transferase activity, and transferring one-carbon groups (GO: 0016741) (Fig. 5B).
GO analysis showed that the DEGs activated were distributed primarily in the membrane, membrane part, and membrane intrinsic component pathway. This observation indicated that the related membrane constituents might be associated with the tolerance of sorghum to copper stress, with the membrane-related genes playing an active role. For example, membrane transporters are stimulated by copper ions to ferry them directly across the membrane to the various parts of the cell, or the transferases are activated, thus maintaining ion balance. In this study, the activities of glycosyl, hexosyl, and acyl transferases, transmembrane ion transporters, and transmembrane cationic transporters were also elevated, which may be involved in the membrane-related detoxification functions.
3.7 KEGG Pathway Enrichment of the DEGs
The results of the KEGG pathway enrichment of the DEGs are shown in Fig. 6. A total of 54 significantly enriched pathway annotations (p < 0.05) were obtained for the upregulated DEGs. The top 20 most significantly enriched pathways were metabolic pathways (sbi01100), biosynthesis of secondary metabolites (sbi01110); valine, leucine, and isoleucine degradation (sbi00280); propanoate metabolism (sbi00640), glutathione metabolism (sbi00480), fatty acid degradation (sbi00071), carbon metabolism (sbi01200), phenylpropanoid biosynthesis (sbi00940), glycerophospholipid metabolism (sbi00564), pyruvate metabolism (sbi00620), autophagy-other (sbi04136), galactose metabolism (sbi00052), and peroxisome (sbi04146) (Fig. 6A).
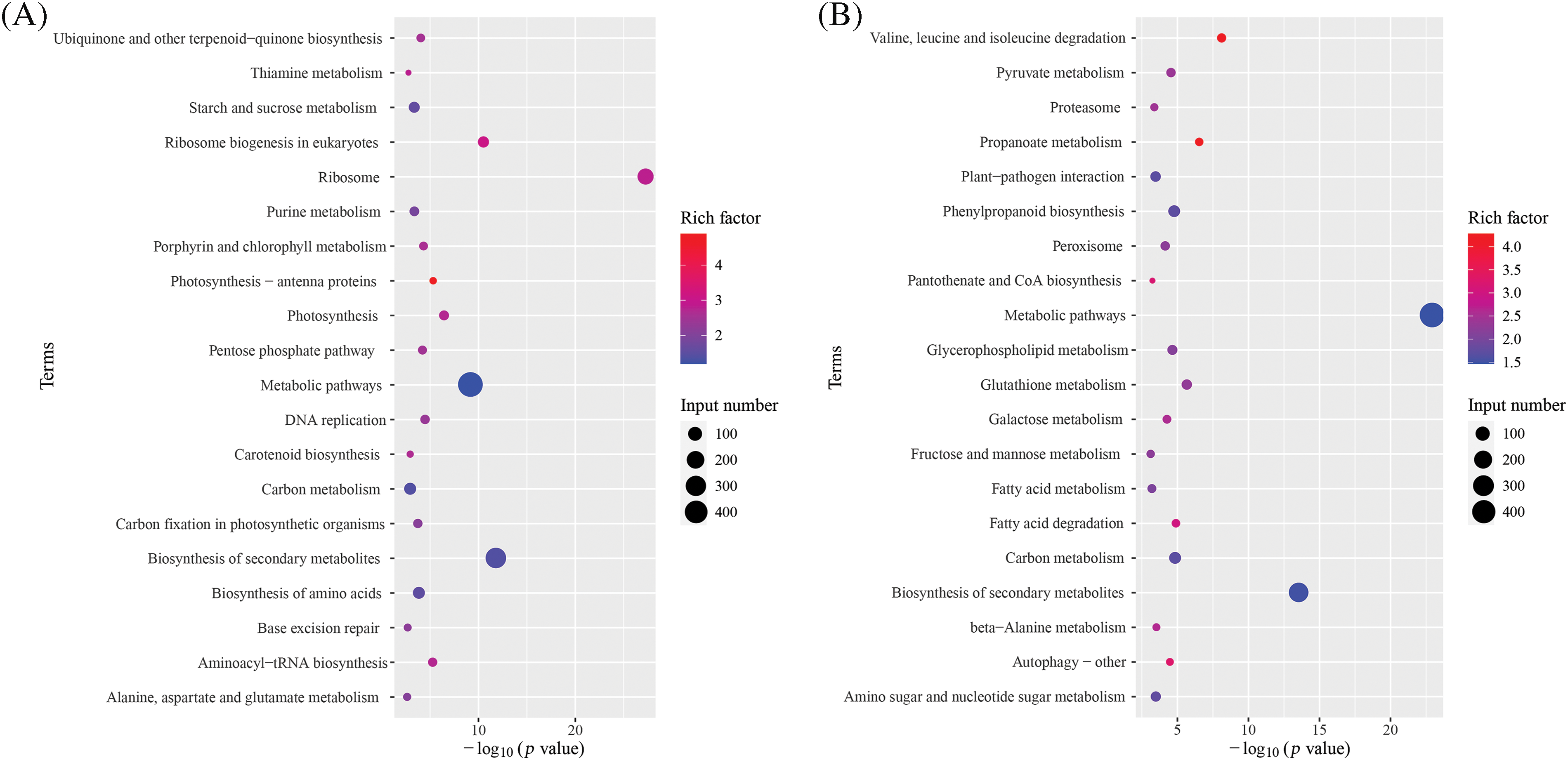
Figure 6: KEGG pathway enrichment of the DEGs. (A) KEGG metabolic pathway diagram of upregulated DEGs. (B) KEGG metabolic pathway diagram of downregulated DEGs
The downregulated DEGs were annotated by 36 KEGG pathways. They included ribosome (sbi03010), biosynthesis of secondary metabolites (sbi01110), ribosome biogenesis in eukaryotes (sbi03008), metabolic pathways (sbi01100), photosynthesis (sbi00195), photosynthesis-antenna proteins (sbi00196), aminoacyl-tRNA biosynthesis (sbi00970), DNA replication (sbi03030), porphyrin and chlorophyll metabolism (sbi00860), pentose phosphate pathway (sbi00030), ubiquinone and other terpenoid-quinone biosynthesis (sbi00130), biosynthesis of amino acids (sbi01230), and carbon fixation in photosynthetic organisms (sbi00710) (Fig. 6B).
KEGG analysis also showed that the GSH metabolic pathway was activated, which played a role in detoxification. GSH acts as an antioxidant, is involved in plant defense against adversity, and scavenges the ROS to reduce damage. Under heavy metal stress, glutathione peroxidase (GSH-Px) and glutathione-S-transferases (GSTs) in plant cells catalyze the binding of GSH to the metal ions, thereby reducing or eliminating their toxicity. Phytochelatins (PCs) are sulfhydryl-containing polypeptides synthesized using GSH as a substrate and catalyzed by PC synthetase. PCs have a high affinity to heavy metal ions and chelate them, forming a heavy metal-PC complex with minimal or no toxicity to plants, thereby preventing damage to the critical enzymes involved in cell metabolism. Copper is also one of the inducers of PC production; glutathione reductase (GR) activity and the mitochondrial content of tolerant plants are markedly enhanced under copper stress [28]. Plants, upon exposure to heavy metals, activate antioxidant mechanisms, such as the activities of peroxidase (POD), GR, and mitochondrial GR and the levels of mitochondrial glutathione (mGSH) to remove excessive ROS. Under physiological conditions, glutathione mainly occurs in plants in the reduced (GSH) and oxidized (GSSG) forms [29]. GSH can directly lessen the ROS to form GSSG. GSSG can be reduced to GSH by GR to decrease the toxic effect of copper and maintain their life activities [28].
3.8 Validation of the Transcriptome Sequencing Results by RT-qPCR
Seven of the DEGs, including LOC8085397, LOC8077113, LOC8082339, LOC8055257, LOC8074454, LOC8065916, and LOC8081225, were randomly selected, and sorghum PP2A was used as an internal reference for RT-qPCR validation. The changes in the relative expression levels of the seven DEGs in sorghum under copper stress were consistent with the RNA-seq results, indicating the high reliability of the transcriptome data (Fig. 7).
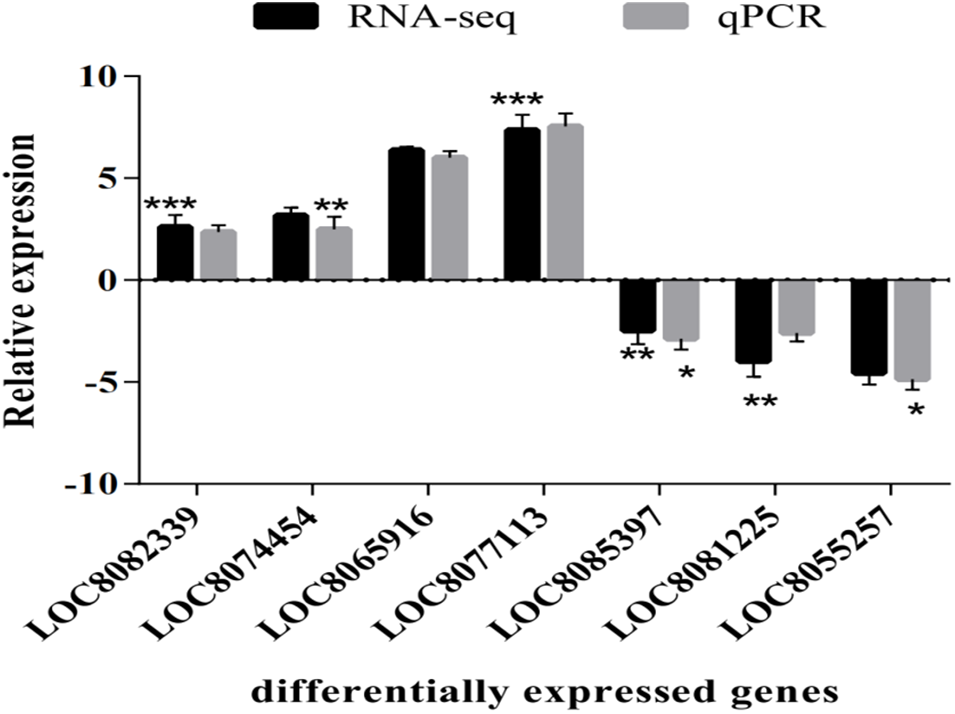
Figure 7: Transcriptome sequencing results were verified by RT-qPCR. *, ** and *** indicate significant differences at p < 0.05, p < 0.01 and p < 0.001, respectively
3.9 Screening of SbWRKY24 in Sorghum under Copper Stress
The RNA-seq data revealed 30 DEGs belonging to the WRKY gene family, 26 of which were upregulated and four were downregulated. Among the latter, the expression of SbWRKY24 was inhibited by copper stress and was downregulated the most, suggesting that it might be involved in the regulation of the response to copper stress in sorghum (Table 4). SbWRKY24 (gene ID: LOC8085397) belongs to Class II of the WRKY family, which is located on chromosome 3 of the sorghum genome. GO functional and KEGG pathway enrichment analyses revealed that SbWRKY24 was enriched in CC and related to intracellular organelle (GO: 0043229), organelle (GO: 0043226), intracellular membrane-bounded organelle (GO: 0043231), and membrane-bounded organelle (GO: 0043227).

3.10 Analysis of the Tissue-Specific Expression of SbWRKY24 under Different Treatments
To explore the putative functions of SbWRKY24, its relative expression levels in roots and leaves under treatment with different growth hormones and exposure to multiple abiotic stresses were detected by RT-qPCR (Fig. 8). The results indicated that SbWRKY24 responded to various treatments in certain trends. The expression of SbWRKY24 induced by ABA or NaCl was the highest at 12 h. However, the degree of induction was lower than that by ABA or 20% PEG-6000, stimulating drought. SbWRKY24 was more remarkably induced in the roots than in the leaves at 12 and 48 h, which improved the tolerance to the drought-like conditions induced by 20% PEG-6000; SbWRKY24 was the most abundant at 24 h. SbWRKY24 was also activated to varying degrees by JA but not by GA. SbWRKY24 was involved in a variety of physiological processes in plants.
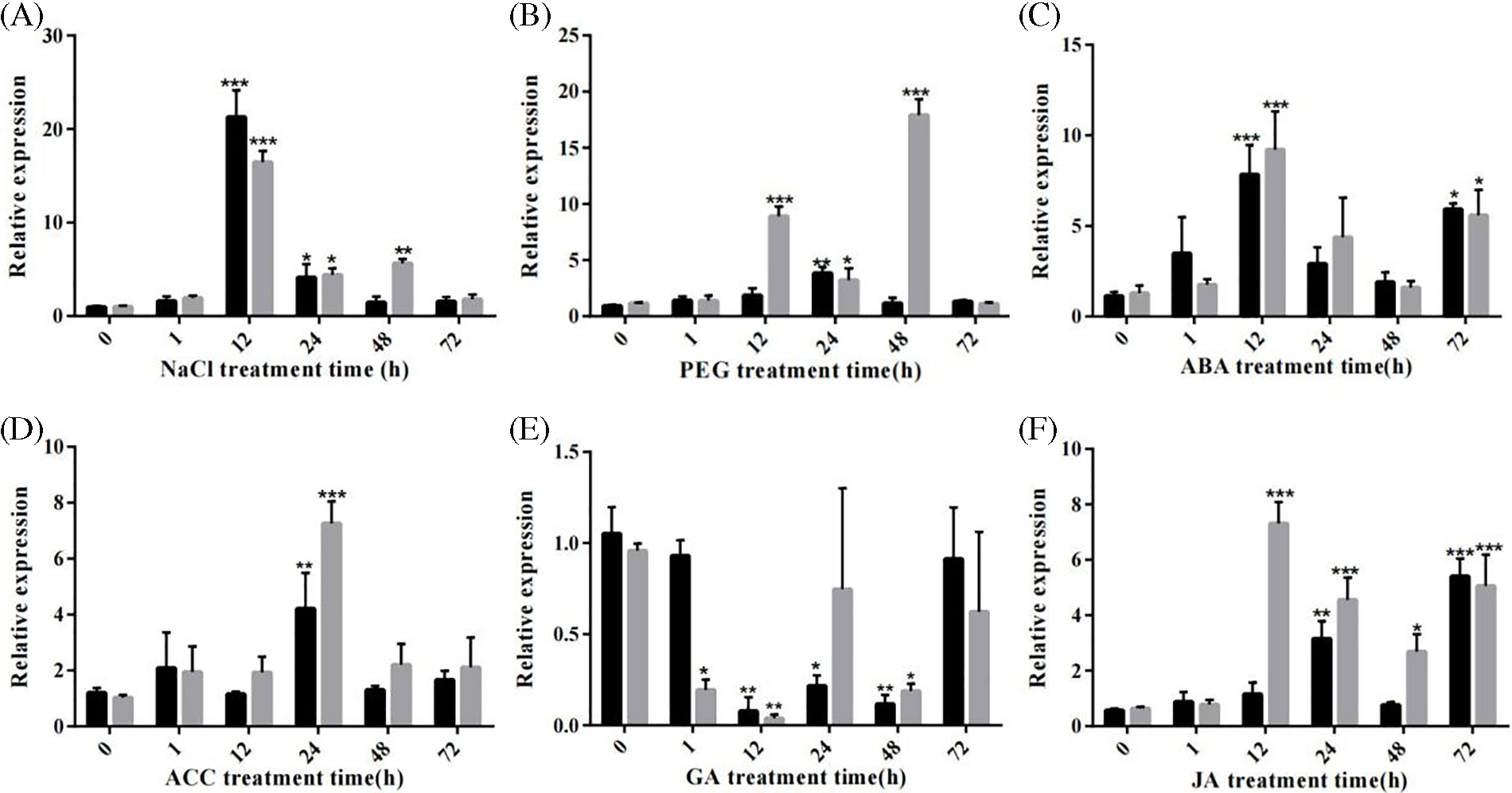
Figure 8: Relative expression of the SbWRKY24 gene under different treatments. (A) 100 mM NaCl. (B) 20% PEG-6000. (C) 100 µM ABA. (D) 100 µM ACC. (E) 100 mM GA. (F) 100 µM JA
Note: *, ** and *** indicate significant differences at p < 0.05, p < 0.01 and p < 0.001, respectively
3.11 Structure Prediction of the SbWRKY24 Protein
The open reading frame of SbWRKY24 was 834 bp long, encoding a 29.64 kD protein of 277 amino acids long. The number of negatively charged residues (Asp and Glu) and positively charged residues (Arg and Lys) was 30 and 24, respectively. The predicted instability coefficient was 48.82, indicating instability. The average hydrophilicity was −0.490, indicating that it was a hydrophilic protein. The highest numerical hydrophobicity (1.933) was observed at residue 12, and the lowest value indicating the highest hydrophilicity (−2.556) was located at residue 155. The protein lacked a transmembrane region, and the conserved DNA binding domain was at positions 116–173. The secondary structure prediction revealed the proportions of α-helices, extended chains, β-folds, and irregular coils to be 34.3%, 10.11%, 4.69%, and 50.9%, respectively. The predicted tertiary structure contained β-folds and irregular coils.
3.12 SbWRKY24 Is Localized to the Nucleus and Has Transcription Activation Ability
After being predicted by TargetP, the SbWRKY24 protein was located in the nucleus. The results of in vivo subcellular localization detected the fluorescence signal of the fusion protein SbWRKY24-GFP only in the nucleus, whereas that of GFP appeared in the plasma membrane, cytoplasm, and nucleus. These results confirmed that SbWRKY24 was confined to the nucleus (Fig. 9).
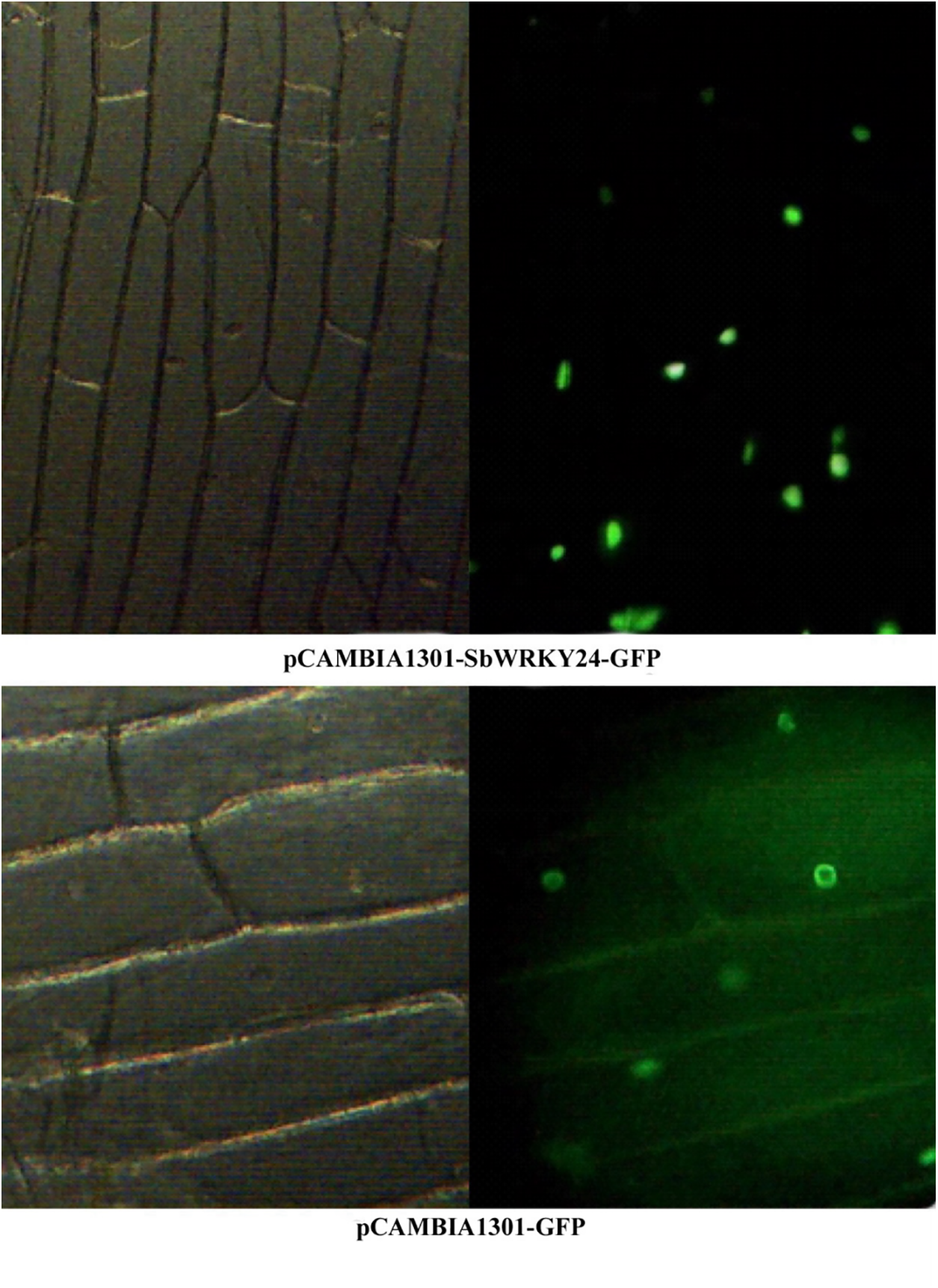
Figure 9: Subcellular localization of SbWRKY24 and GFP fusion protein in onion. Left: Morphology of onion epidermal cells in the light field. Right: GFP fluorescence image in the dark field
The yeast cells transformed with the pGBKT7-SbWRKY24 or pGBKT7 empty vectors grew well on the SD/-Trp medium. However, those transduced with pGBKT7-SbWRKY24 showed blue clones on the X-α-Gal-containing medium (Fig. 10). Based on yeast growth and color formation, SbWRKY24 was confirmed to possess transcription activation ability.

Figure 10: Transcriptional activation activity analysis of SbWRKY24
3.13 Protein Sequence Alignment and Phylogenetic Tree Construction
The amino acid sequences of sorghum were aligned in silico by employing the BLAST program. Multiple sequence alignment was performed based on the WRKY amino acid sequences from 16 species with high sequence similarity to SbWRKY24, and a phylogenetic tree was constructed (Fig. 11). The phylogenetic tree revealed that sorghum, maize, millet, and switchgrass were clustered in the same branch, with sorghum and maize being the closest relatives.
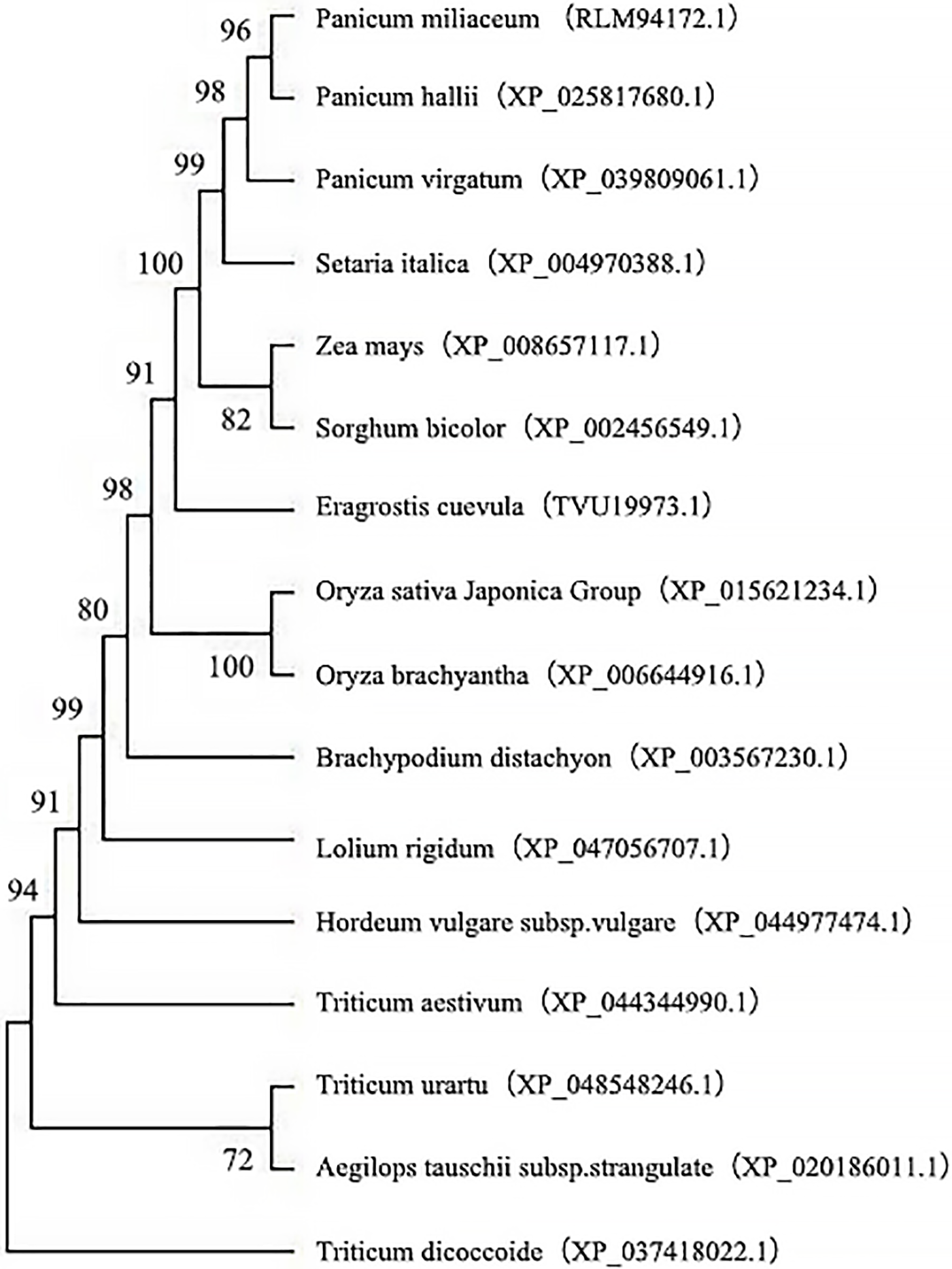
Figure 11: Phylogenetic tree of SbWRKY24 protein and other plant WRKY proteins
Cu2+, involved in the photosynthetic and respiratory electron transport in plants, is a significant constituent of chloroplastic cyanin and an activator of certain enzymes during chloroplast formation. In this study, GO enrichment analysis revealed that although chloroplasts were inhibited, the photosynthesis rate increased proportionally with the concentration of Cu2+. KEGG enrichment analysis showed that photosynthesis, photosynthesis-antenna protein, and carbon sequestration in photosynthetic organisms were also inhibited. These results indicated that excessive copper accumulation and copper pollution inhibited chloroplast synthesis and affected photosynthesis in a concentration-dependent manner. High levels of copper ions inactivated the chlorophyll-associated proteins, altered the ultrastructure of chloroplast, destroyed the structure and inhibited the function of the thylakoid cavity, reduced photosynthetic efficiency, and affected the accumulation of nutrients, thereby producing toxic effects.
Ribosomes are the major sites of protein synthesis. The expression of ribosomal RNA genes is influenced by the environment and is involved in the regulation of stress response, thus playing a crucial role in cell development and metabolism. In this study, KEGG enrichment analysis showed that the expression of ribosomes in sorghum was inhibited in response to copper stress, which was consistent with previous findings [30]. Most ribosomes were continuously downregulated during cell differentiation, with some initially downregulated and then upregulated. Most plants enhance the expression of ribosome-related genes in response to copper stress [31].
Autophagy is an intracellular self-degradation pathway involved in abiotic stress response in plants [32]. Copper stress induced 35 autophagy-related genes in grapes [33]. Many organelles in plant cells, including peroxisomes, mitochondria, endoplasmic reticulum, and ribosomes, have specific autophagic properties. KEGG analysis showed an activation of the autophagy and peroxisome-related pathways, which was consistent with previous studies.
The WRKY transcription factor family plays an essential regulatory role in vascular plants. It is involved not only in plant development and metabolism but also in the response of plants to stresses, such as drought, salt, high and low temperatures, diseases, insect pests, and others [34]. OsWRKY8 and ten other genes were induced under high or low temperatures and PEG-induced stress. OsWRKY45 enhanced the resistance of Arabidopsis plants to salt, drought, and diseases [35,36]. The SbWRKY24 gene identified in this study belonged to Class II of the WRKY family and was located on chromosome 3 of the sorghum genome. The encoded transcription factor possessed one WRKYGQK domain and a C2H2-type zinc finger. Alignment of multiple protein sequences and phylogenetic tree construction indicated that the closest relationship was between sorghum and maize at 82%. SbWRKY24 was characterized as an unstable hydrophilic protein, which was similar to the lotus NnWRKY24 and tomato SlWRKY6. Most WRKY transcription factors are positively regulated in response to stress. However, in this study, SbWRKY24 was repressed by copper stress, which was similar to the repression of WRKY46 by Al in Arabidopsis. The expression of ALMT1, which encodes a malate efflux transporter, was induced by Al stress, suggesting that WRKY46 can regulate its expression and thereby improve Al tolerance [37]. Tobacco plants overexpressing DgWRKY3 suggest that DgWRKY3 plays a positive regulatory role in the tolerance of plants to salt stress [38]. The expression level of OsWRKY11 was elevated in response to heat and drought stress. In rice plants, the overexpression of the OsWRKY11 cDNA fused with the promoter of the rice HSP101, the leaves wilted slowly, the surviving green fraction was damaged less, and the resistance to heat and drought was enhanced [39]. Overexpression of Tawrky10 in tobacco enhanced the tolerance to drought and salt stress due to higher proline and soluble sugar contents but lower MDA and ROS levels [40]. In this study, the expression of SbWRKY24 was induced under NaCl and simulated drought stress, suggesting that it may be involved in the response to these stresses in sorghum, consistent with SbHDT701 was up-regulated under drought (d-mannitol) and salt (NaCl) stresses [41]. Additionally, SbWRKY24 expression was induced by ABA, and the 35S::OsWRKY45 transgenic Arabidopsis plants were less sensitive to ABA. This result may be partially attributed to the closure of stomata during drought due to the induction of stress-related genes. NaCl, 20% PEG-6000, ABA, ACC, and JA could induce the expression of SbWRKY24 in roots and leaves to a limited extent but varying levels. This observation indicated the tissue specificity of SbWRKY24, due to which it can respond to varying stresses through selective expression in different parts of the plant.
So far, progress has been made in understanding the response mechanisms of plants to copper stress and in the breeding of resistant varieties. However, this progress has some limitations, as the functions of genes have not been studied comprehensively. This study provides a theoretical basis for further exploring the role of SbWRKY24 in the response and resistance to copper stress in sorghum. The response of sorghum to copper stress is a complex biological process that involves the regulation and co-expression of multiple stress-related genes and requires a balance in the complex gene network.
Acknowledgement: The Key Planned Projects of the Sichuan Provincial Department of Science & Technology (2020YFN0023); the Cooperation Project of Wuliangye Group Co., Ltd., and Sichuan University of Science & Engineering, China (CXY2021ZR010).
Funding Statement: This research was funded by the Key Planned Projects of the Sichuan Provincial Department of Science & Technology (2020YFN0023); the Cooperation Project of Wuliangye Group Co., Ltd., and Sichuan University of Science & Engineering, China (CXY2021ZR010).
Author Contributions: The authors confirm contribution to the manuscript as follows: study conception and design: Mingchuan Yang, Zhenhui Kang; data collection: Mingchuan Yang, Jia Zheng, Yanghua Li; analysis and interpretation of results: Mingchuan Yang, Wenhui Yu, Zilu Zhang, Zhenhui Kang, Yali Wang; draft manuscript preparation: M.Y., Z.K; All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The statement raw data that have been used are confidential. The other datasets the conclusions of the article included within the article.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Zhan Y, Zhang C, Zheng Q, Huang Z, Yu C. Cadmium stress inhibits the growth of primary roots by interfering auxin homeostasis in Sorghum bicolor seedlings. J Plant Biol. 2017;60:593–603. doi:10.1007/s12374-017-0024-0. [Google Scholar] [CrossRef]
2. Zhao X. Current situation and monitoring analysis of soil environmental pollution. Environ Dev. 2019;31:174–5. [Google Scholar]
3. Demirevska-Kepova K, Simova-Stoilova L, Stoyanova Z, Hölzer R, Feller U. Biochemical changes in barley plants after excessive supply of copper and manganese. Environ Exp Bot. 2004;52:253–66. doi:10.1016/j.envexpbot.2004.02.004. [Google Scholar] [CrossRef]
4. Michaud AM, Chappellaz C, Hinsinger P. Copper phytotoxicity affects root elongation and iron nutrition in durum wheat (Triticum turgidum durum L.). Plant Soil. 2008;310:151–65. doi:10.1007/s11104-008-9642-0. [Google Scholar] [CrossRef]
5. Thomas JC, Malick FK, Endreszl C, Davies EC, Murray KS. Distinct responses to copper stress in the halophyte Mesembryanthemum crystallinum. Physiol Plantarum. 1998;102:360–8. doi:10.1034/j.1399-3054.1998.1020304.x. [Google Scholar] [CrossRef]
6. Adrees M, Ali S, Rizwan M, Zia-Ur-Rehman M, Ibrahim M, Abbas F, et al. Mechanisms of silicon-mediated alleviation of heavy metal toxicity in plants: a review. Ecotox Environ Safe. 2015;119:186–97. doi:10.1016/j.ecoenv.2015.05.011. [Google Scholar] [PubMed] [CrossRef]
7. de Freitas TA, França MGC, de Almeida AF, de Oliveira SJR, de Jesus RM, Souza VL, et al. Morphology, ultrastructure and mineral uptake is affected by copper toxicity in young plants of Inga subnuda subs. Luschnathiana (Benth.) T.D. Penn. Environ Sci Pollut R. 2015;22:15479–94. doi:10.1007/s11356-015-4610-8. [Google Scholar] [PubMed] [CrossRef]
8. Li Q, Chen H, Qi Y, Ye X, Yang L, Huang Z, et al. Excess copper effects on growth, uptake of water and nutrients, carbohydrates, and PSII photochemistry revealed by OJIP transients in Citrus seedlings. Environ Sci Pollut R. 2019;26:30188–205. doi:10.1007/s11356-019-06170-2. [Google Scholar] [PubMed] [CrossRef]
9. Roy SK, Cho S, Kwon SJ, Kamal AHM, Lee D, Sarker K, et al. Proteome characterization of copper stress responses in the roots of sorghum. Biometals. 2017;30:765–85. doi:10.1007/s10534-017-0045-7. [Google Scholar] [PubMed] [CrossRef]
10. Wang J, Gao H, Guo Z, Meng Y, Yang M, Li X, et al. Adaptation responses in C4 photosynthesis of sweet maize (Zea mays L.) exposed to nicosulfuron. Ecotox Environ Safe. 2021;214:112096. doi:10.1016/j.ecoenv.2021.112096. [Google Scholar] [PubMed] [CrossRef]
11. Ishiguro S, Nakamura K. Characterization of a cDNA encoding a novel DNA-binding protein, SPF1, that recognizes SP8 sequences in the 5' upstream regions of genes encoding for sporamin and beta-amylase from sweet potato. Mol Gen Genet. 1994;244:563–71. doi:10.1007/BF00282746. [Google Scholar] [PubMed] [CrossRef]
12. Ulker B, Somssich IE. WRKY transcription factors: from DNA binding towards biological function. Curr Opin Plant Biol. 2004;7:491–8. doi:10.1016/j.pbi.2004.07.012. [Google Scholar] [PubMed] [CrossRef]
13. Shi K, Liu X, Zhu Y, Bai Y, Shan D, Zheng X, et al. MdWRKY11 improves copper tolerance by directly promoting the expression of the copper transporter gene MdHMA5. Hortic Res. 2020;7:105. doi:10.1038/s41438-020-0326-0. [Google Scholar] [PubMed] [CrossRef]
14. Li GZ, Wang ZQ, Yokosho K, Ding B, Fan W, Gong QQ, et al. Transcription factor WRKY22 promotes aluminum tolerance via activation of OsFRDL4 expression and enhancement of citrate secretion in rice (Oryza sativa). New Phytol. 2018;219:149–62. doi:10.1111/nph.2018.219.issue-1. [Google Scholar] [CrossRef]
15. Wang R, Chen C, Yu L, Jin X. Cloning of tomato SlWRKY6 gene and its expression analysis under heavy metal stress. Acta Agriculturae Boreali-Sinica. 2021;36:54–62. [Google Scholar]
16. Li G, Zheng Y, Chen S, Liu J, Wang P, Wang Y, et al. TaWRKY74 participates copper tolerance through regulation of TaGST1 expression and GSH content in wheat. Ecotox Environ Safe. 2021;221:112469. doi:10.1016/j.ecoenv.2021.112469. [Google Scholar] [PubMed] [CrossRef]
17. Wang W, Liu Y, Liu H, He D, Liu Y, Kong D. Cloning and expression of NnWRKY22 in lotus. Fujian J Agric Sci. 2022;37:486–91 (In Chinese) [Google Scholar]
18. He G, Xu J, Wang Y, Liu J, Li P, Chen M, et al. Drought-responsive WRKY transcription factor genes TaWRKY1 and TaWRKY33 from wheat confer drought and/or heat resistance in Arabidopsis. BMC Plant Biol. 2016;16:116. doi:10.1186/s12870-016-0806-4. [Google Scholar] [PubMed] [CrossRef]
19. Song H, Sun W, Yang G, Sun J. WRKY transcription factors in legumes. Bmc Plant Biol. 2018;18:243. doi:10.1186/s12870-018-1467-2. [Google Scholar] [PubMed] [CrossRef]
20. Yang Z, Chi X, Guo F, Jin X, Luo H, Hawar A, et al. SbWRKY30 enhances the drought tolerance of plants and regulates a drought stress-responsive gene, SbRD19, in sorghum. J Plant Physiol. 2020;246-247:153142. doi:10.1016/j.jplph.2020.153142. [Google Scholar] [PubMed] [CrossRef]
21. Wang F, Hou X, Tang J, Wang Z, Wang S, Jiang F, et al. A novel cold-inducible gene from Pak-choi (Brassica campestris ssp. chinensisBcWRKY46, enhances the cold, salt and dehydration stress tolerance in transgenic tobacco. Mol Biol Rep. 2012;39:4553–64. doi:10.1007/s11033-011-1245-9. [Google Scholar] [PubMed] [CrossRef]
22. Xixu P, Ting X, Jiao M, Zong T, Dinggang Z, Xinke T, et al. Differential expression of rice Valine-Qlutamine gene family in response to nitric oxide and regulatory circuit of OsVQ7 and OsWRKY24. Rice Sci. 2020;27:10–20. doi:10.1016/j.rsci.2019.12.002. [Google Scholar] [CrossRef]
23. Liu G, Zeng H, Li X, Wei Y, Shi H. Functional analysis of MaWRKY24 in transcriptional activation of autophagy-related gene 8f/g and plant disease susceptibility to soil-borne Fusarium oxysporum f. sp. cubense. Pathogens. 2019;8:264. doi:10.3390/pathogens8040264. [Google Scholar] [PubMed] [CrossRef]
24. Arhondakis S, Bita CE, Perrakis A, Manioudaki ME, Krokida A, Kaloudas D, et al. In silico transcriptional regulatory networks involved in tomato fruit ripening. Front Plant Sci. 2016;7:1234. doi:10.3389/fpls.2016.01234. [Google Scholar] [PubMed] [CrossRef]
25. Li L, Zang X, Liu J, Ren J, Wang Z, Yang D. Integrated physiological and weighted gene co-expression network analysis reveals the hub genes engaged in nitrate-regulated alleviation of ammonium toxicity at the seedling stage in wheat (Triticum aestivum L.). Front Plant Sci. 2022;13:1012966. doi:10.3389/fpls.2022.1012966. [Google Scholar] [PubMed] [CrossRef]
26. Xie G, Liu N, Zhang Y, Tan S, Xu Y, Luo Z. Postharvest MeJA maintains the shelf quality of kiwifruit after cold storage by regulating antioxidant capacity and activating the disease resistance. Postharvest Biol Technol. 2024;211:112827. doi:10.1016/j.postharvbio.2024.112827. [Google Scholar] [CrossRef]
27. Gladman N, Olson A, Wei S, Chougule K, Lu Z, Tello-Ruiz M, et al. SorghumBase: a web-based portal for sorghum genetic information and community advancement. Planta. 2022;255:35. doi:10.1007/s00425-022-03821-6. [Google Scholar] [PubMed] [CrossRef]
28. Zhou C, Huang MY, Ren HJ, Yu JD, Wu JM, Ma XQ. Bioaccumulation and detoxification mechanisms for lead uptake identified in Rhus chinensis Mill. seedlings. Ecotoxicol Environ Saf. 2017;142:59–68. doi:10.1016/j.ecoenv.2017.03.052. [Google Scholar] [PubMed] [CrossRef]
29. Kawasaki S, Borchert C, Deyholos M, Wang H, Brazille S, Kawai K, et al. Gene expression profiles during the initial phase of salt stress in rice. Plant Cell. 2001;13:889–905. doi:10.1105/tpc.13.4.889. [Google Scholar] [PubMed] [CrossRef]
30. Noctor G, Mhamdi A, Chaouch S, Han YI, Neukermans J, Marquez Garcia B, et al. Glutathione in plants: an integrated overview. Plant Cell Environ. 2012;35:454–84. doi:10.1111/pce.2012.35.issue-2. [Google Scholar] [CrossRef]
31. Ding S, Ma C, Shi W, Liu W, Lu Y, Liu Q, et al. Exogenous glutathione enhances cadmium accumulation and alleviates its toxicity in Populus × canescens. Tree Physiol. 2017;37:1697–712. doi:10.1093/treephys/tpx132. [Google Scholar] [PubMed] [CrossRef]
32. Qi H, Xia FN, Xie LJ, Yu LJ, Chen QF, Zhuang XH, et al. TRAF family proteins regulate autophagy dynamics by modulating AUTOPHAGY PROTEIN6 stability in arabidopsis. Plant Cell. 2017;29:890–911. doi:10.1105/tpc.17.00056. [Google Scholar] [PubMed] [CrossRef]
33. Shangguan L, Fang X, Chen L, Cui L, Fang J. Genome-wide analysis of autophagy-related genes (ARGs) in grapevine and plant tolerance to copper stress. Planta. 2018;247:1449–63. doi:10.1007/s00425-018-2864-3. [Google Scholar] [PubMed] [CrossRef]
34. Jiang J, Ma S, Ye N, Jiang M, Cao J, Zhang J. WRKY transcription factors in plant responses to stresses. J Integr Plant Biol. 2017;59:86–101. doi:10.1111/jipb.12513. [Google Scholar] [PubMed] [CrossRef]
35. Qiu Y. Cloning and analysis of expression profile of 13 WRKY genes in rice. Chin Sci Bull. 2004;49:2159–68. doi:10.1360/982004-183. [Google Scholar] [CrossRef]
36. Qiu Y, Yu D. Over-expression of the stress-induced OsWRKY45 enhances disease resistance and drought tolerance in Arabidopsis. Environ Exp Bot. 2009;65:35–47. doi:10.1016/j.envexpbot.2008.07.002. [Google Scholar] [CrossRef]
37. Ding ZJ, Yan JY, Xu XY, Li GX, Zheng SJ. WRKY46 functions as a transcriptional repressor of ALMT1, regulating aluminum-induced malate secretion in Arabidopsis. Plant J. 2013;76:825–35. doi:10.1111/tpj.12337. [Google Scholar] [PubMed] [CrossRef]
38. Liu Q, Zhong M, Li S, Pan Y, Jiang B, Jia Y, et al. Overexpression of a chrysanthemum transcription factor gene, DgWRKY3, in tobacco enhances tolerance to salt stress. Plant Physiol Bioch. 2013;69:27–33. doi:10.1016/j.plaphy.2013.04.016. [Google Scholar] [PubMed] [CrossRef]
39. Wu X, Shiroto Y, Kishitani S, Ito Y, Toriyama K. Enhanced heat and drought tolerance in transgenic rice seedlings overexpressing OsWRKY11 under the control of HSP101 promoter. Plant Cell Rep. 2009;28:21–30. doi:10.1007/s00299-008-0614-x. [Google Scholar] [PubMed] [CrossRef]
40. Wang C, Deng P, Chen L, Wang X, Ma H, Hu W, et al. A wheat WRKY transcription factor TaWRKY10 confers tolerance to multiple abiotic stresses in transgenic tobacco. PLoS One. 2013;8:e65120. doi:10.1371/journal.pone.0065120. [Google Scholar] [PubMed] [CrossRef]
41. Du Q, Qu Z, Wang L, Jiang J, Fu X, Fang Y, et al. Histone deacetylase SbHDT701 in Sorghum bicolor reveals functions in response to stress factors by enhancing acetylation. Pestic Biochem Phys. 2021;178:104908. doi:10.1016/j.pestbp.2021.104908. [Google Scholar] [PubMed] [CrossRef]
Appendix A
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools