
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Response Mechanisms to Flooding Stress in Mulberry Revealed by Multi-Omics Analysis
1 College of Biology and Food Engineering, Chongqing Three Gorges University, Chongqing, 404100, China
2 Chongqing Three Gorges Academy of Agricultural Sciences, Chongqing Three Gorges University, Chongqing, 404155, China
* Corresponding Author: Liping Gan. Email:
(This article belongs to the Special Issue: Multi-omics Approach to Understand Plant Stress Tolerance)
Phyton-International Journal of Experimental Botany 2024, 93(2), 227-245. https://doi.org/10.32604/phyton.2024.046521
Received 05 October 2023; Accepted 21 December 2023; Issue published 27 February 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Abiotic stress, including flooding, seriously affects the normal growth and development of plants. Mulberry (Morus alba), a species known for its flood resistance, is cultivated worldwide for economic purposes. The transcriptomic analysis has identified numerous differentially expressed genes (DEGs) involved in submergence tolerance in mulberry plants. However, a comprehensive analyses of metabolite types and changes under flooding stress in mulberry remain unreported. A non-targeted metabolomic analysis utilizing liquid chromatography-tandem mass spectrometry (LC-MS/MS) was conducted to further investigate the effects of flooding stress on mulberry. A total of 1,169 metabolites were identified, with 331 differentially accumulated metabolites (DAMs) exhibiting up-regulation in response to flooding stress and 314 displaying down-regulation. Pathway enrichment analysis identified significant modifications in many metabolic pathways due to flooding stress, including amino acid biosynthesis and metabolism and flavonoid biosynthesis. DAMs and DEGs are significantly enriched in the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways for amino acid, phenylpropanoid and flavonoid synthesis. Furthermore, metabolites such as methyl jasmonate, sucrose, and D-mannose 6-phosphate accumulated in mulberry leaves post-flooding stress. Therefore, genes and metabolites associated with these KEGG pathways are likely to exert a significant influence on mulberry flood tolerance. This study makes a substantial contribution to the comprehension of the underlying mechanisms implicated in the adaptation of mulberry plants to submergence.Keywords
Supplementary Material
Supplementary Material FileGlobal climate change is expected to increase both the frequency and magnitude of floods, intensifying the impact on crop yield and quality [1,2]. Additionally, plants in water-level fluctuation zones near artificial dams experience periodic flooding stress, contributing to environmental issues like soil erosion and biodiversity loss [3,4]. Flooding and the induced hypoxia environment have serious effects on the normal growth of intolerant plants. The low-oxygen environment limits oxygen uptake and cellular respiration, resulting in energy shortages in plants. Plants modulate this energy shortage by decomposing sucrose and starch, glycolysis, and fermenting ethanol [5]. The occurrence of flooding stress typically enhances the expression of genes involved in these physiological processes [5–9]. Plants can also manage flooding-induced hypoxia and energy shortages by developing aerenchyma, adventitious roots, or rapid stem elongation [10,11]. Furthermore, the composition and content of biomolecules in plant cells significantly change due to flooding stress.
The plant metabolome encompasses the entirety of low molecular weight metabolites, including primary and secondary metabolites. Primary metabolites play an important role in the biosynthesis of lipids, sugars, and amino acids in plants. They influence plant growth and development via the mediation of the tricarboxylic acid and glycolysis cycles during photosynthesis. Alterations in the synthesis of primary metabolites have the potential to impede the process of photosynthesis and disrupt the osmotic pressure in plants. Flavonoids and carotenoids, as secondary metabolites, arise from pathways linked to primary metabolism. Secondary metabolites are not essential for plant survival and are induced under different stress conditions [12,13]. Metabolites play a variety of roles in plant stress response, including regulation of intracellular osmotic pressure and plant protection from microbial infection, herbivore predation, and environmental stress [13–15]. Metabolomics, crucial for studying abiotic stress and pathogen resistance, plays a significant role in crop improvement and metabolic-assisted breeding [12]. Modern metabolomics techniques aid in a comprehensive understanding of the biological processes within plant cells exposed to various stresses [16,17]. Transcriptomic and metabolomic studies have been previously used to reveal the role of related pathways in plant adaptation to flood or hypoxic stress [18–20].
Recent advances have elucidated key aspects of plant responses to flooding stress. Numerous genes and signaling pathways relevant to this response have been identified. Transcriptomic studies showed that the expression of genes related to GA biosynthesis, cell wall modification, alcohol fermentation, ethylene biosynthesis, and trehalose biosynthesis were significantly induced by flooding stress [21,22]. Flooding stress induced differentially expressed genes were mostly found in the regulatory pathways of phenylpropanoid biosynthesis, ROS signal, antioxidant process, carbohydrate metabolism, photosynthesis, hormone biosynthesis, and signal transduction [4,23,24].
Mulberry, an economically significant plant, has a long cultivation history in China. Mulberry fruit can be eaten fresh or processed into fruit wine, juice, and jam [25]. Mulberry leaves can be used to raise silkworms and mulberry bark is commonly used as raw material for papermaking. Various parts of the mulberry tree, including roots, stems, leaves, and fruits, possess medicinal properties [26]. Recent studies have shown that mulberry is an ideal organism to study woody plant resistance to stress since it is highly tolerant to drought, saline-alkali, and waterlogging stress [26–28]. Metabolomics analysis showed that metabolites, which are involved in amino acid metabolism, phospholipids, energy metabolism, biosynthesis of other secondary metabolites, lipid metabolism, cofactor and vitamin metabolism, and metabolism of other amino acid pathways, played a key role in the response of mulberry to abiotic stresses [29–31]. Transcriptome results identified genes related to starch and sucrose metabolism, glycolysis, and ethanol fermentation. Genes related to hormones and phenylpropanoid were also up-regulated to varying degrees following flooding stress [4]. However, the response of mulberry metabolites to flooding stress remains unclear. Based on previous transcriptomic studies, a post-flooding stress metabolomic analysis was conducted in mulberry. Further combined transcriptomic and metabolomic analyses identified key metabolic processes and genes potentially contributing to mulberry’s short-term flood stress response. In summary, this research significantly contributes to the comprehension of the molecular mechanisms that underlie the tolerance of mulberry plants to flooding stress.
2.1 Flooding Treatment on Plants
Mulberry cuttings of the Yu711 variety were planted in pots. The mulberry plants were grown in a greenhouse at 25°C with a 16-h light/8-h dark photoperiod and 75% relative humidity (RH) for two months. Plants with uniform growth patterns were selected for the flooding stress treatment. During flooding treatment, the water level was consistently maintained 10 cm above the soil surface. Leaves were collected 0, 1, 3, and 7 days after flooding treatment (labeled as Y711_0 (control), Y711_1, Y711_3, and Y711_7, respectively). The specimens were rapidly frozen using liquid nitrogen and subsequently preserved in a freezer set at a temperature of −80°C. Three separate biological replicates were obtained at each designated sampling interval.
2.2 Measurement and Analysis of Physiological Indices
The chlorophyll content was determined by a CCM-200 chlorophyll meter (Opti-Sciences, Tyngsboro, MA, USA). Repeated measurements of the middle three mature leaves from each of the three plants were conducted, using the average SPAD readings to represent relative leaf chlorophyll content. The contents of free proline, malondialdehyde (MDA), and Peroxidase (POD) activity in mulberry leaves were determined according to the instructions of the kits (Comin, Suzhou, China). The MDA and free proline contents were estimated according to Hodges et al. [32] and Liang et al. [33], respectively.
2.3 Extraction and Analysis of Metabolites
Leaves collected following flooding treatment were used to measure the metabolic activity. Leaf samples weighing 100 mg were pulverized using liquid nitrogen, and the resulting homogenate was resuspended through vigorous vortexing with pre-cooled 80% methanol. After incubation on ice for 5 min, samples were centrifuged for 20 min at 15,000 g at 4°C. In order to obtain a 53% methanol concentration, a portion of the supernatant was diluted by water of LC-MS quality. Finally, the samples were transferred to a fresh eppendorf tube and centrifuged at 15,000 g at 4°C for 20 min.
The UHPLC-MS/MS analyses were conducted using a Vanquish UHPLC system coupled with an Orbitrap Q ExactiveTM HF-X mass spectrometer (Thermo Fisher, Bremen, Germany). The samples were injected into a Hypesil Gold column (100 × 2.1 mm, 1.9 μm) using a 17-min linear gradient at a flow rate of 0.2 mL/min. Eluents A (0.1% FA in water) and B (methanol) were employed for the positive polarity, while for negative polarity, eluents A (5 mM ammonium acetate, pH 9.0) and B (methanol) were used. Parameters for establishing the solvent gradient were as follows: 2% B for a duration of 1.5 min, subsequently followed by a linear increase from 2% to 100% over a period of 12.0 min, maintaining 100% B for a duration of 14.0 min, followed by a linear decrease from 100% to 2% B over 14.1 min, and finally 2% B for 17 min. The Q ExactiveTM HF-X mass spectrometer was utilized in either positive or negative polarity mode, with a spray voltage of 3.2 kV. Additionally, the capillary temperature was maintained at 320°C, while the sheath gas flow rate was 40 arb and an auxiliary gas flow rate was 10 arb.
2.4 Metabolite Identification and Data Analysis
Compound Discoverer 3.1 (CD3.1, Thermo Fisher, Waltham, MA, USA) was used to process the raw data obtained from UHPLC-MS/MS. The processing encompassed the tasks of aligning peaks, selecting peaks, and quantifying each metabolite. A minimum intensity threshold of 100,000, a mass tolerance of 5 ppm, a signal intensity tolerance of 30%, a retention time tolerance of 0.2 min, and a signal-to-noise ratio of 3 were the key parameters employed in this study. Subsequently, the peak intensities were normalized with respect to the overall spectral intensity. Following data normalization, fragment ions, additive ions, and molecular peaks were considered to predict the molecular formula. In order to obtain quantitative and qualitative outcomes with precision, a comparison of the obtained peaks with the databases of mzVault, mzCloud, and MassList was then conducted [34]. The identification of metabolites was accomplished through cross-referencing with the HMDB, LIPID Maps, and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases. Principal components analysis (PCA) and Partial least squares discriminant analysis (PLS-DA) were conducted using the metaX software (Version 2.68, Shenzhen, China). The metabolites exhibiting variable importance in projection (VIP) score greater than 1, a p-value less than 0.05, and a fold change (FC) greater than 1.5 or less than 0.667 were identified as differentially accumulated metabolites (DAMs). The cluster Profiler R package (Version 3.8.1, Bioconductor) was used to conduct KEGG analysis to assess DAM enrichment and TBtools was used to generate the heat maps [35].
2.5 Metabolome and Transcriptome Co-Expression Analysis
The transcriptome sequencing data used in this study were acquired from prior investigations [4]. DEGs and DAMs from the cohorts Y711_1 vs. Y711_0, Y711_3 vs. Y711_0, and Y711_7 vs. Y711_0 were selected for further correlation analysis. The correlation analysis, using Pearson’s correlation coefficient via the R mixOmics package (version 6.14.1, Bioconductor), determined the correlation (positive when k > 0, negative when k < 0) and p-value of DEGs and DAMs. The DEGs and DAMs obtained from the above comparisons were subjected to mapping in the KEGG to determine the pathways that are commonly shared and to undergo subsequent statistical analysis.
3.1 Physiological Changes of Mulberry after Flooding Stress
MDA, free proline, POD, and chlorophyll levels were measured to assess physiological changes in mulberry seedlings under flooding stress. The results showed that the chlorophyll content (SPAD value) in mulberry leaves decreased significantly after three days of flooding. MDA content increased significantly only after three days of flooding but did not change significantly at other sampling time points. POD and free proline decreased first and then increased with the development of flooding stress. Except for chlorophyll, significant changes in physiological indices were observed after 3 days of flooding, stabilizing or returning to baseline by 7 days (Fig. 1).
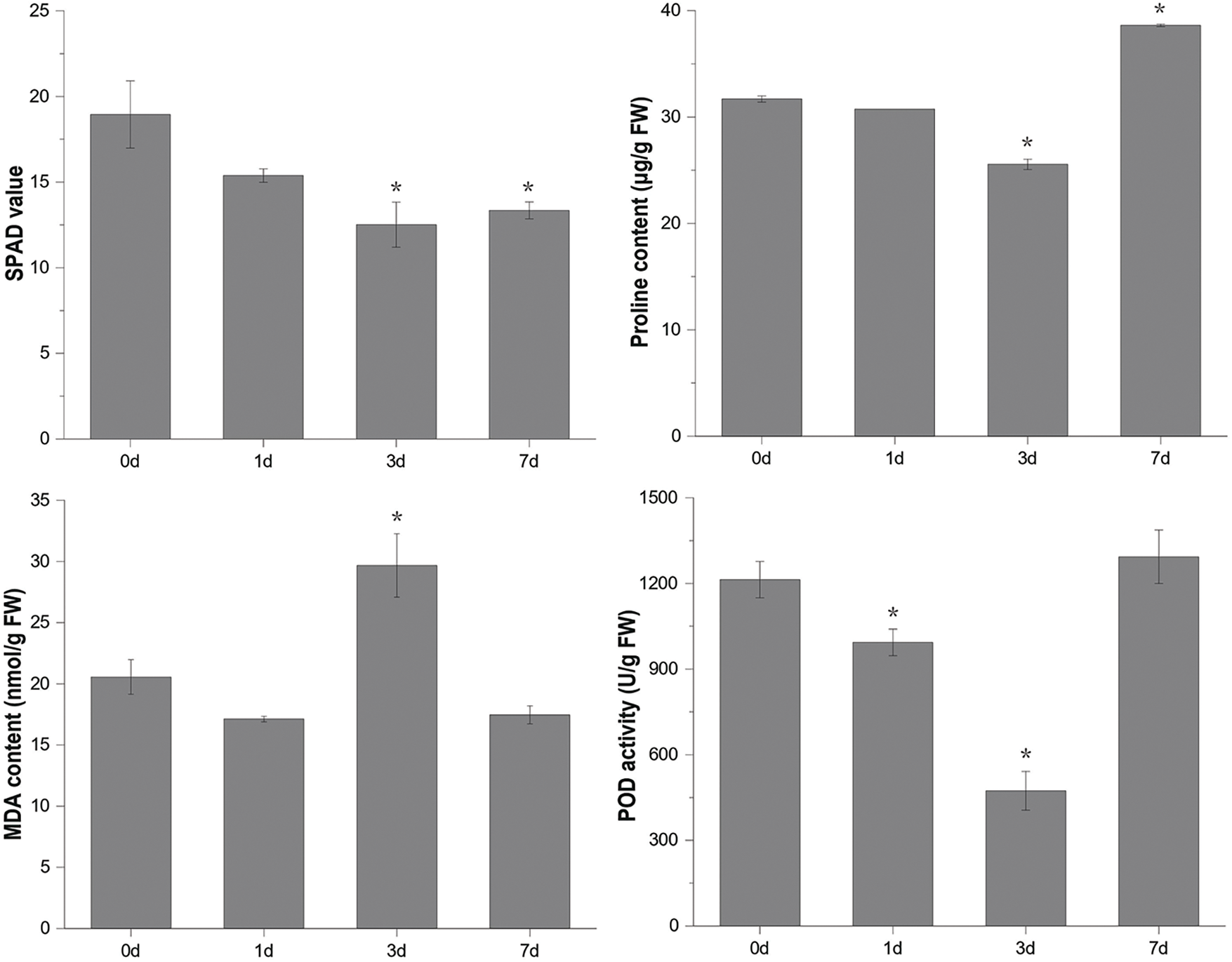
Figure 1: Determination of physiological indexes of mulberry leaves after flooding stress. Values represent mean ± SD (n = 3). *Asterisks indicate a significant difference (p < 0.05) between leaves after flooding stress (1d, 3d, and 7d) and the control (0d)
3.2 Non-Targeted Metabolite Profiling Analysis of the Mulberry after Flooding Stress
Metabolic profiles of mulberry leaves subjected to 0, 1, 3, and 7 days of flooding stress were analyzed using LC-MS. The metabolites of samples, including quality control samples (QC), were analyzed using PCA at the expression level. The results demonstrated that the top three principal components (PC1, PC2, PC3) explained 39.83%, 18.01%, and 14.24% of the variance of all metabolites, respectively. A clear separation was identified in the score plot, suggesting that mulberry leaves at different flooding stress stages exhibited significant metabolic changes (Fig. 2A). In the 12 leaf samples, a total of 1,169 metabolites were detected (Table S1). Among these, 917 metabolites were divided into 13 classes, including 290 (31.62%) lipids and lipid-like molecules, 158 (17.23%) phenylpropanoids and polyketides, 109 (11.89%) organic acids and derivatives, 102 (11.12%) organoheterocyclic compounds, 88 (9.60%) organic oxygen compounds, 78 (8.51%) benzenoids, 43 (4.69%) nucleosides, nucleotides, and analogs, 19 (2.07%) alkaloids and derivatives, 16 (1.74%) lignans, neolignans, and related compounds, 11 (1.20%) organic nitrogen compounds, 1 (0.11%) hydrocarbons, 1 (0.11%) mixed metal/non-metal compounds, and 1 (0.11%) homogeneous non-metal compounds (Fig. 2B). Three biological replicates in each group clustered together, indicating high consistency between materials and reliability of the data (Fig. 2C)
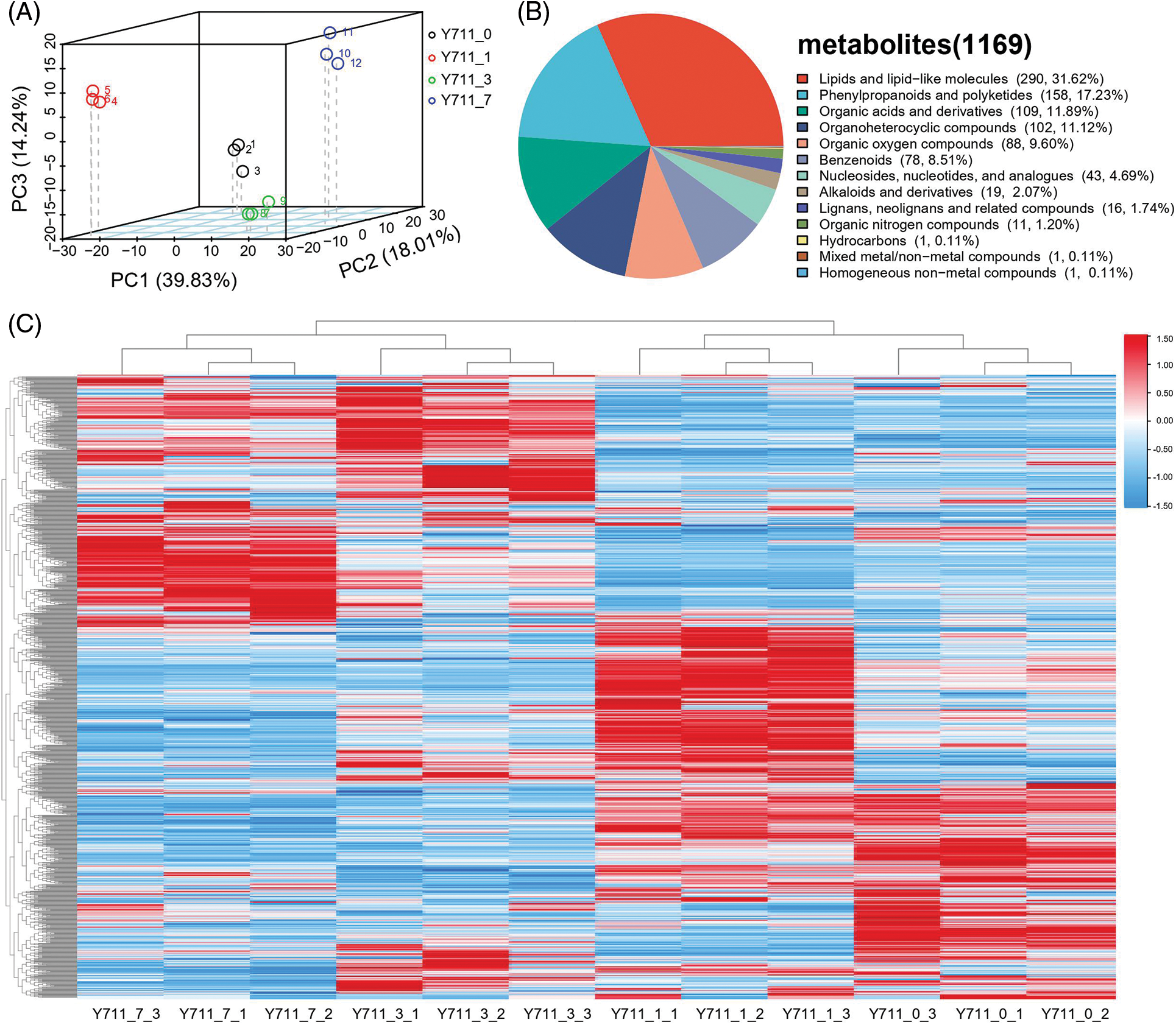
Figure 2: Mulberry leaf sample metabolite analyses. (A) PCA, (B) classification, and (C) hierarchical cluster analysis with individual biological replicates (The color ranges from red to blue, corresponding from large to small)
All identified metabolites were categorized according to the KEGG, HMDB, and LIPID Maps databases. The results demonstrated that within the KEGG database, the metabolites were primarily annotated in the following areas: amino acid metabolism, biosynthesis of other secondary metabolites, carbohydrate metabolism, global and overview maps, and metabolism of cofactors and vitamins (Fig. S1A). The top five significantly enriched pathways in the HMDB database included lipids and lipid-like molecules, phenylpropanoids and polyketides, organic acids and derivatives, organic oxygen compounds, and organoheterocyclic compounds (Fig. S1B). The pathways significantly enriched in the LIPID Maps database were flavonoids, fatty acids and conjugates, isoprenoids, sterols, steroids, and eicosanoids (Fig. S1C).
3.3 Differentially Accumulated Metabolite Screening of Mulberry after Flooding Stress
DAMs were screened using PLS-DA with criteria of VIP > 1.0, FC > 1.5 or < 0.667, and p-value < 0.05. A total of 242 DAMs (124 up-regulated and 118 down-regulated) were identified in the comparison group Y711_1 vs. Y711_0. A comprehensive examination of the Y711_3 vs. Y711_0 groups revealed the presence of 299 DAMs (161 up and 138 down-regulated). Similarly, in the comparison between the Y711_7 vs. Y711_0 groups, a total of 245 DAMs (155 up and 190 down-regulated) were identified (Fig. 3). The observed DAM increase with the increase in flooding stress suggests a substantial reprogramming of the mulberry leaf metabolome induced by submergence. This finding highlights the significant impact of flooding stress on the metabolic profile of mulberry leaves.
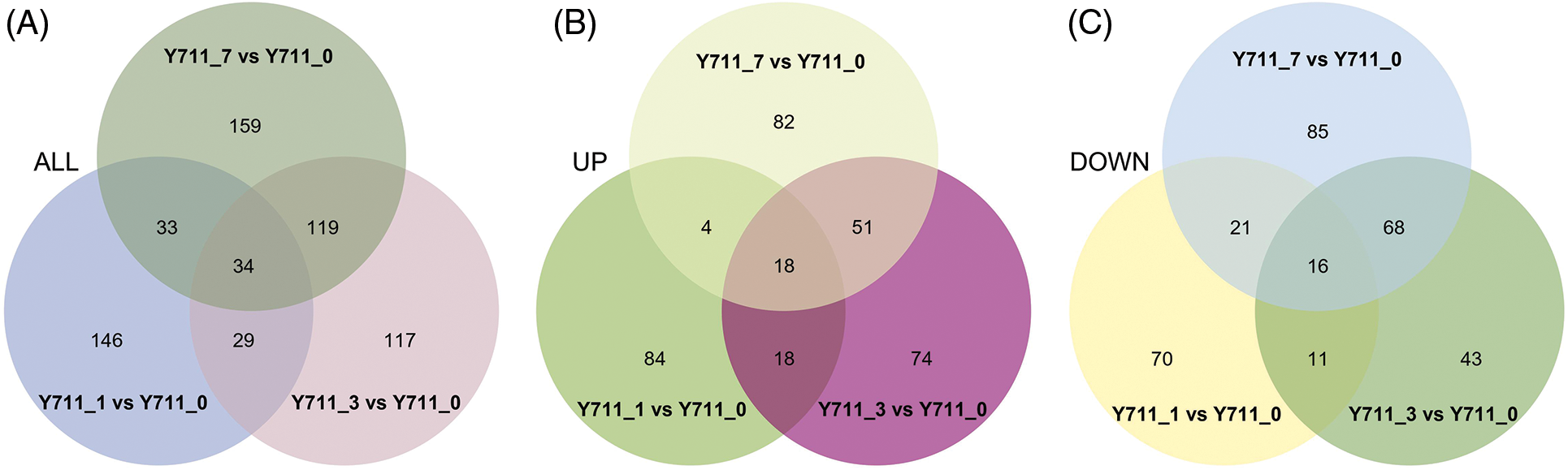
Figure 3: Overview of DAMs identified in mulberry leaves in response to flooding stress. The Venn diagrams represent the quantity of DAMs in mulberry leaves subjected to control conditions and flooding treatment (A), the intersection of all DAMs that were upregulated among all the groups being compared (B), and the intersection of down-regulated DAMs across all the comparison groups (C)
3.4 DAM Analysis in Mulberry Leaves Post Flooding Stress
Table S2 contains the comprehensive list of DAMs across all comparison groups. Among these DAMs, 331 were found to be up-regulated in response to flooding stress, while 314 were down-regulated. These DAMs were then categorized into distinct clusters based on their metabolite expression levels (Fig. 4).
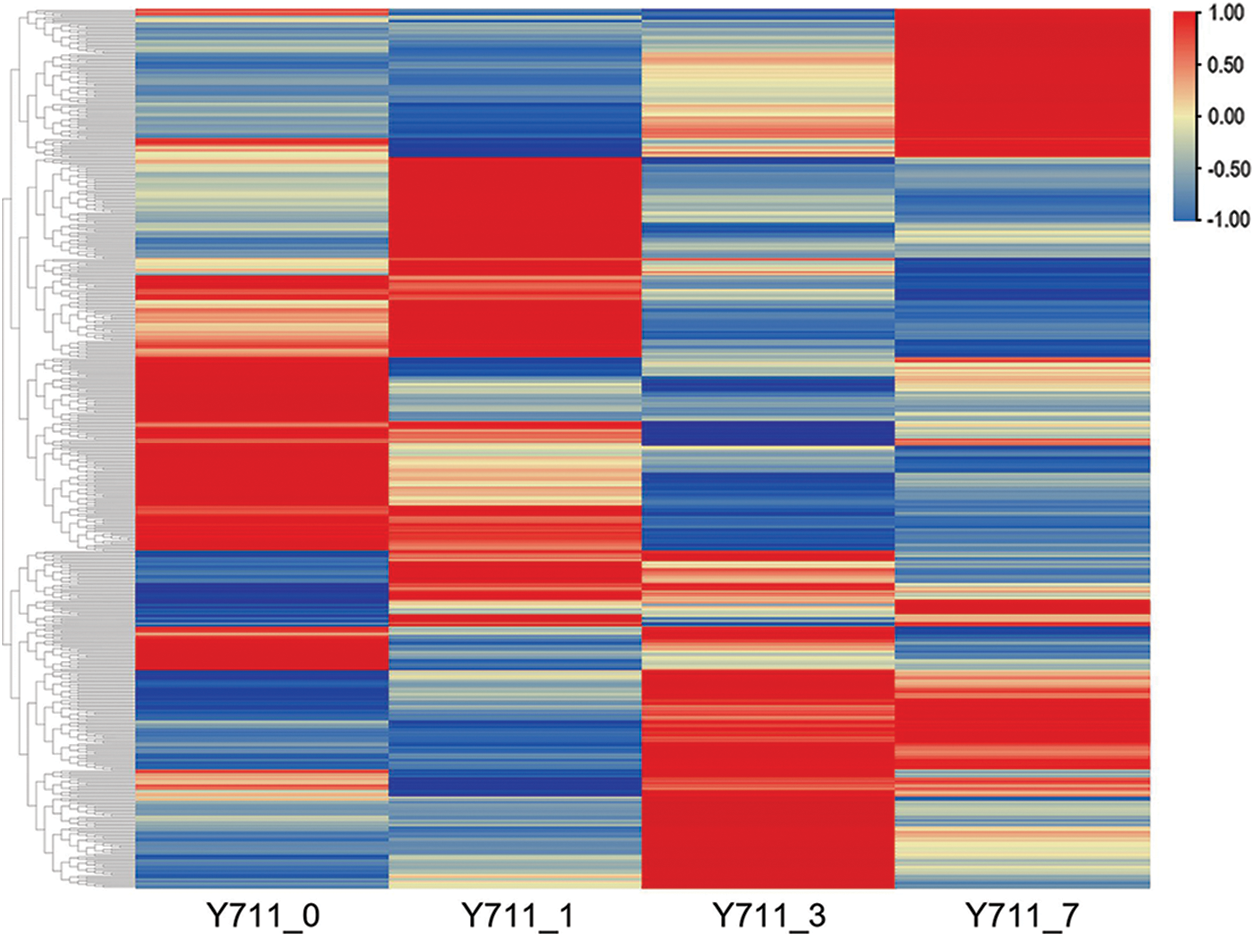
Figure 4: Heatmap depicting the DAMs associated with flooding stress in mulberry plants. The color ranges from red to blue, corresponding to large to small
The KEGG database was used to match the DAMs in mulberry leaves at various stages. The results demonstrated that out of the 242 identified DAMs in the Y711_1 vs. Y711_0 comparison, 43 KEGG pathways were enriched. These metabolites were primarily enriched in the following pathways: citrate cycle (TCA cycle), sphingolipid metabolism, arginine biosynthesis, glyoxylate and dicarboxylate metabolism, as well as alanine, aspartate, and glutamate metabolism. A total of 299 DAMs were identified and assigned to 40 KEGG pathways in the comparison between the Y711_3 vs. Y711_0 groups. The five pathways that exhibited significant enrichment were tryptophan metabolism, aminoacyl-tRNA biosynthesis, phenylalanine, tyrosine, and tryptophan biosynthesis, flavonoid biosynthesis, and flavone and flavonol biosynthesis. In Y711_7 vs. Y711_0, 345 DAMs were found to be enriched in 41 KEGG pathways. Among these pathways, five were enriched in the KEGG pathways, including flavone and flavonol biosynthesis, photosynthesis, valine, leucine, and isoleucine biosynthesis, fatty acid metabolism, and sulfur relay system (Fig. 5).
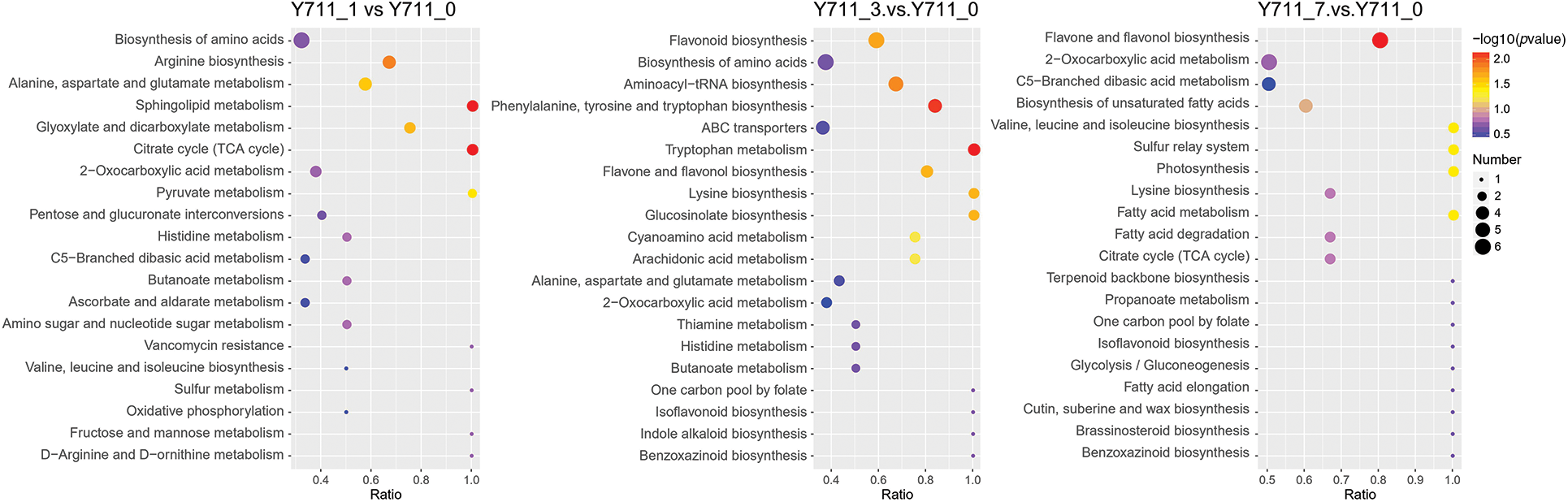
Figure 5: KEGG annotations and enrichment of differentially accumulated metabolites of each pairwise comparison of mulberry. The abscissa is the ratio of the DAMs enriched in the pathway to the number of metabolites annotated in the pathway. The count represents the number of metabolites enriched in the pathway
3.5 DAMs and DEGs Correlation Analysis
The correlation between the top 50 DEGs and top 50 DAMs, based on the p-value, at different flooding stress stages is presented in Figs. S2A–S2C. Integrated transcriptomic and metabolomic analyses demonstrated that many DEGs and DAMs were significantly enriched in the same KEGG pathway in mulberry leaves at different stages. These KEGG pathways included the following: phenylpropanoid biosynthesis, biosynthesis of amino acids, 2-oxocarboxylic acid metabolism, TCA cycle, and flavonoid biosynthesis in the Y711_1 vs. Y711_0 group; flavonoid biosynthesis, phenylpropanoid biosynthesis, biosynthesis of amino acids, phenylalanine metabolism, and tryptophan metabolism in the Y711_3 vs. Y711_0 group; and flavonoid biosynthesis, biosynthesis of unsaturated fatty acids, fatty acid metabolism, flavone and flavonol biosynthesis, and biosynthesis of amino acids in the Y711_7 vs. Y711_0 group (Figs. S2D–S2F). Flavonoid biosynthesis was a common enriched pathway across all groups, while phenylpropanoid biosynthesis was enriched in two (Y711_1 vs. Y711_0 and Y711_3 vs. Y711_0), underscoring their potential roles in flood stress response.
3.6 Combined Transcriptomic and Metabolomic Analyses in the Phenylpropanoid Pathway
The combined analysis identified phenylpropanoid biosynthesis and flavonoid biosynthesis as significantly enriched KEGG pathways during flooding stress. DEGs and DAMs expression in these two pathways under flooding stress conditions was further investigated. Specifically, within the phenylpropanoid pathways, a total of 90 DEGs encoding enzymes such as 4-coumarate-CoA ligase (4 CL), phenylalanine ammonia-lyase (PAL), cinnamate-4-hydroxylase (C4H), and beta-glucosidase (BGLU) were shown to be significantly associated with nine metabolites (Fig. 6). Identified metabolites included caffeoyl quinic acid, coumarin, scopoletin, scopolin, L-tyrosine, L-phenylalanine, sinapyl alcohol, pterostilbene, and piceatannol (Fig. 6B). Among these metabolites, caffeoyl quinic acid belongs to organic oxygen compounds, L-tyrosine and L-phenylalanine belong to organic acids and derivatives, sinapyl alcohol belongs to benzenoids, while the rest of the metabolites belong to phenylpropanoids and polyketides. The expression of 16 out of 19 BGLU and two PAL genes were up-regulated during different flooding stress periods. This was consistent with the results of the large accumulation of coumarin post-flooding stress. The accumulation of phenylalanine indirectly promoted tyrosine and coumarin synthesis (Figs. 6A and 6B). Furthermore, scopoletin, scopolin, pterostilbene, and piceatannol content significantly increased with the persistence of flooding stress (Fig. 6B). In line with this, several DEGs including two genes belonging to the F6H family (112091846 and 21412376), three members of the ALDH2C4 family (21403699, 21403701, and 112094039), and three from the OMT1 family (21387006, 21402556, and 21406195) were also significantly up-regulated by flooding stress.
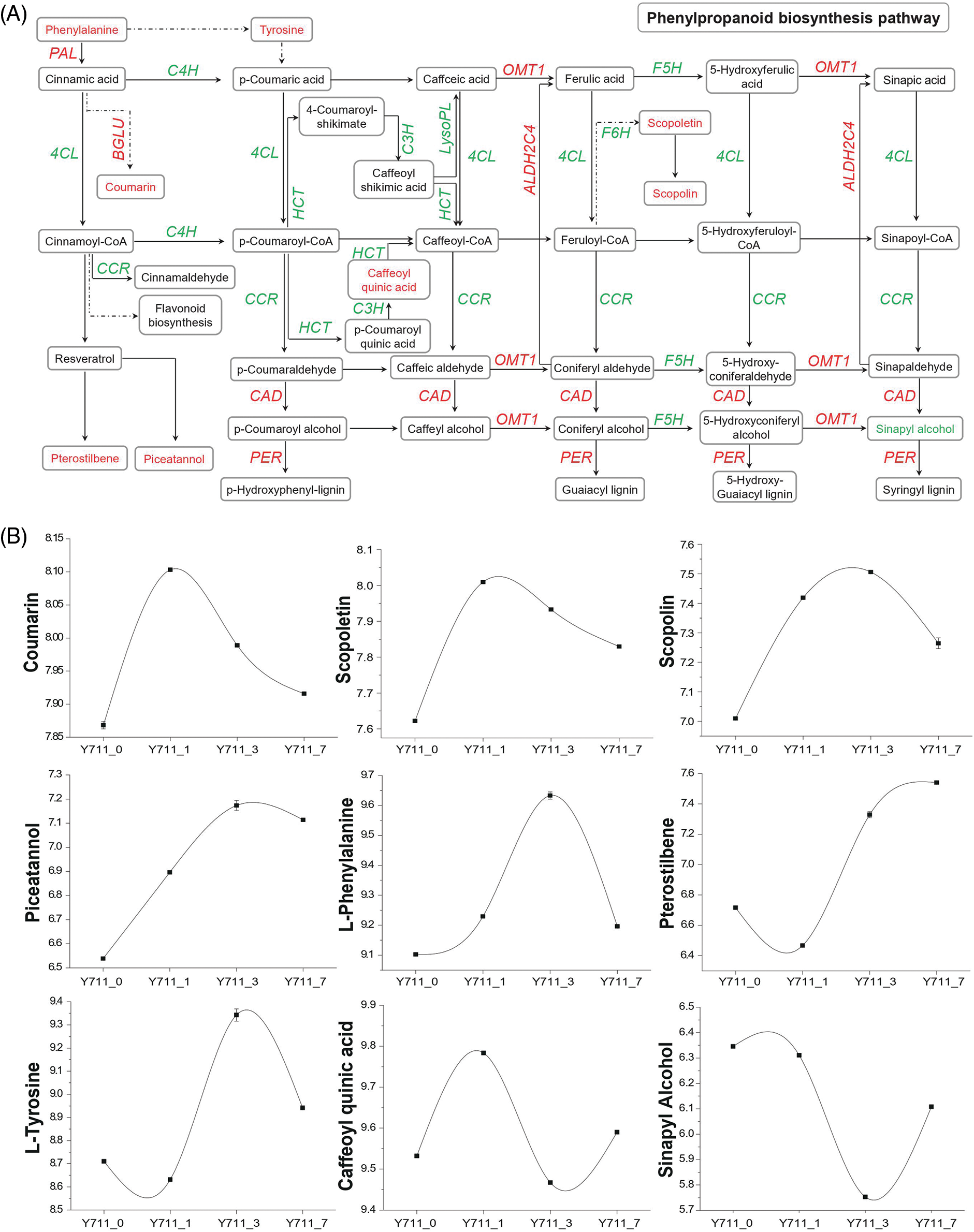
Figure 6: Co-enrichment analysis of DEGs and DAMs in the phenylpropanoid pathway. (A) Overview of phenylpropanoid biosynthesis. PAL, phenylalanine ammonia-lyase; C4H, trans-cinnamate 4-monooxygenase; 4CL, 4-coumarate-CoA ligase; BGLU, beta-glucosidase; OMT1, caffeic acid 3-O-methyltransferase; F5H, ferulate-5-hydroxylase; C3H, coumarate 3-hydroxylase; LysoPL, lysophospholipase; HCT, Hydroxycinnamoyl transferase; CCR, cinnamoyl-CoA reductase; ALDH2C4, aldehyde dehydrogenase 2C4; F6H, feruloyl CoA ortho-hydroxylase; PER, peroxidase; CAD, cinnamyl alcohol dehydrogenase. (B) The DAM trend changes in the phenylpropanoid synthesis pathway with flooding stress. Expression scores are presented as log10(q). Genes (represented by italic capital letters) and metabolites (located in the box) in red font indicate up-regulation after flooding stress, while green fonts indicate down-regulation after flooding stress
In addition, most of the up-regulated metabolites after flooding stress, such as coumarin, scopoletin, scopolin, piceatannol, and L-phenylalanine, began to increase after one day of flooding and reached the highest level after one or three days of flooding. The accumulation level of the down-regulated metabolite sinapyl alcohol also began to decrease after one day of flooding treatment and remained below the accumulation level of 0d until 7 days flooding stress. Other metabolites also showed altered accumulation levels starting one-day post-flooding treatment.
3.7 Combined Transcriptomic and Metabolomic Analyses in the Flavonoid Biosynthesis Pathways
In the flavonoid biosynthesis pathway, a total of 32 DEGs across nine gene families were identified. These families included genes encoding reduced epidermal fluorescence 8 (REF8), cinnamate-4-hydroxylase (C4H), hydroxycinnamoyl-CoA shikimate/quinate hydroxycinnamoyl transferase (HCT), flavanone 3-hydroxylase (F3H), chalcone synthase (CHS), chalcone isomerase (CHI), flavonoid 3′-monooxygenase (F3′H), dihydroflavonol 4-reductase (DFR), and flavonol synthase (FLS). These DEGs were significantly associated with 14 distinct metabolites (Fig. 7A). DAMs such as dihydroquercetin, prunin, caffeoyl quinic acid, kaempferol, catechin, naringin, epigallocatechin, hesperetin, dihydrokaempferol, quercetin, 2′-hydroxygenistein, rutin, trifolin, and laricitrin showed varying alteration trends with increased flooding stress. Among these DAMs, epigallocatechin and hesperetin increased gradually with the persistence of flooding stress. The levels of prunin and dihydroquercetin were also significantly up-regulated in Y711_1 and Y711_3 following flooding stress. Similarly, the expression of several related DEGs, including eight CHS genes (112091300, 21411756, 21412880, 21412237, 21412879, 21412156, 112091301 and 21412323), two CHI genes (21394508, 112095175), one F3′H (21410017), and one C4H (21391595), was also significantly up-regulated by flooding stress. However, quercetin, dihydrokaempferol, 2′-hydroxygenistein, rutin, and trifolin demonstrated a continuous decrease. Under prolonged flooding stress, the accumulation of five other metabolites varied with the duration of stress (Fig. 7B). Consistent with this, the expression levels of DFR (21404779), C4H (21391595), and FLS (21396296) also exhibited fluctuating changes.
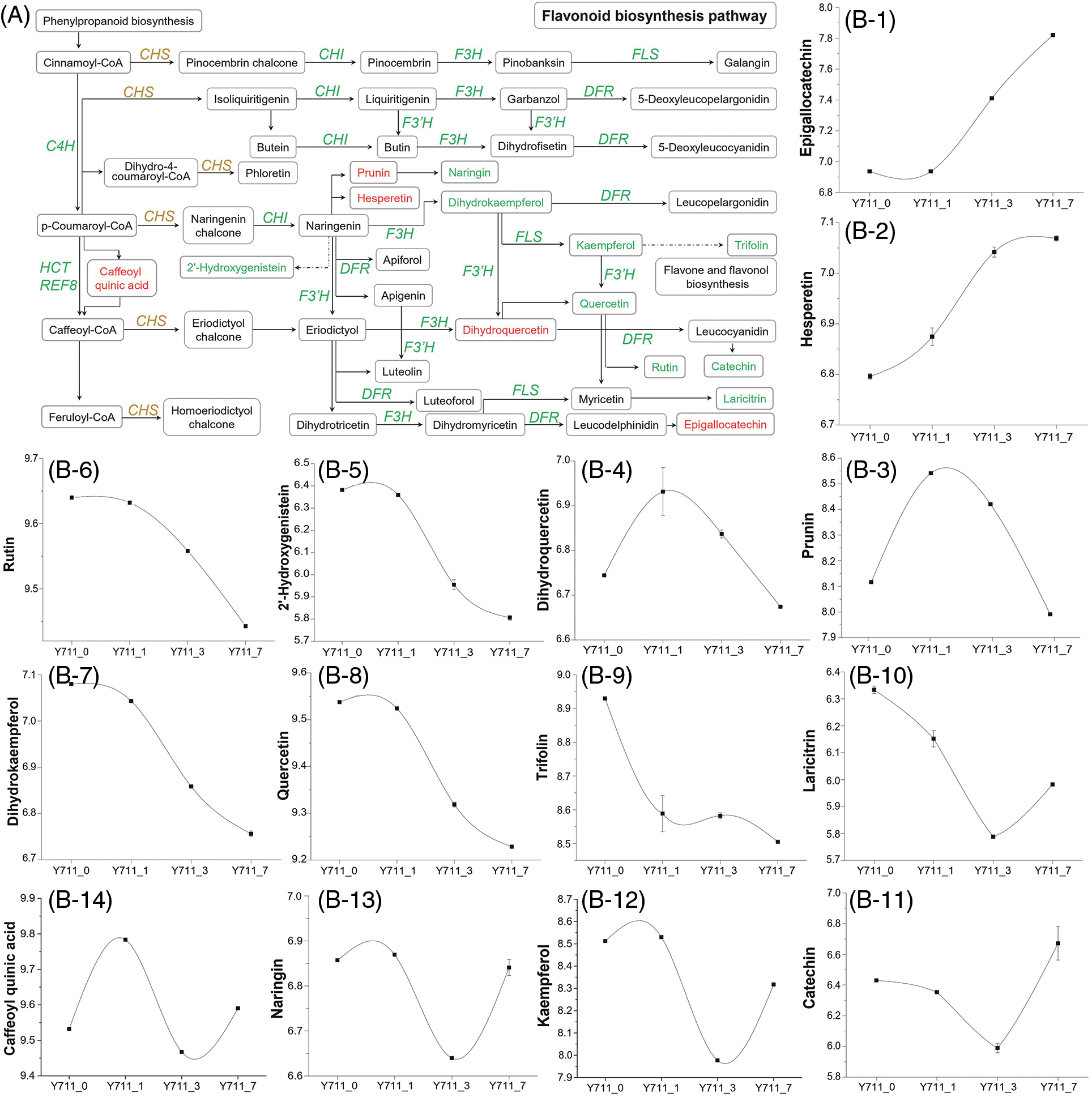
Figure 7: Co-enrichment analysis of DAMs and DEGs in the flavonoid pathway. (A) Overview of flavonoid biosynthesis pathway. CHI, chalcone isomerase; FLS, flavonol synthase; CHS, chalcone synthase; F3H, flavanone 3-hydroxylase; DFR, dihydroflavonol 4-reductase; F3′H, flavonoid 3′-monooxygenase; C3H, coumarate 3-hydroxylase; C4H, cinnamate-4-hydroxylase; HCT, hydroxycinnamoyl transferase. Genes (represented by italic capital letters) and metabolites (located in the box) in yellow font indicate up- and down-regulation after flooding stress, while green fonts indicate down-regulation after flooding stress. (B) The DAM trend changes in the flavonoid biosynthesis pathway with flooding stress. Expression scores are presented as log10(q)
For down-regulated DAMs, including rutin and dihydrokaempferol, accumulation levels were lowest after seven days of flooding. In the up-regulated DAMs, some of the accumulation levels reached the peak after 3 days of flooding, while others did not reach the maximum until 7 days after flooding. A common feature among all DAMs was a significant change in metabolite levels after one or three days of flooding. Except for caffeoyl quinic acid, which belongs to organic oxygen compounds, all metabolites of this pathway belong to phenylpropanoids and polyketides.
3.8 Other Metabolic Pathways Involved in Mulberry Flooding Tolerance
The metabolism of starch and sugar significantly contributes to the plant’s ability to withstand abiotic stress. In our study, metabolomics analysis demonstrated that soluble sugar (including sucrose and D-mannose 6-phosphate) content was significantly up-regulated after one day of flooding stress (Figs. 8A and 8B). Combined transcriptional and metabolic analyses demonstrated significant up-regulation of four genes in fructose and mannose metabolism following flooding stress. The findings of this study indicate that the presence of soluble sugar metabolites, specifically sucrose and mannose, significantly contribute to the ability of mulberry plants to withstand flooding-induced stress.
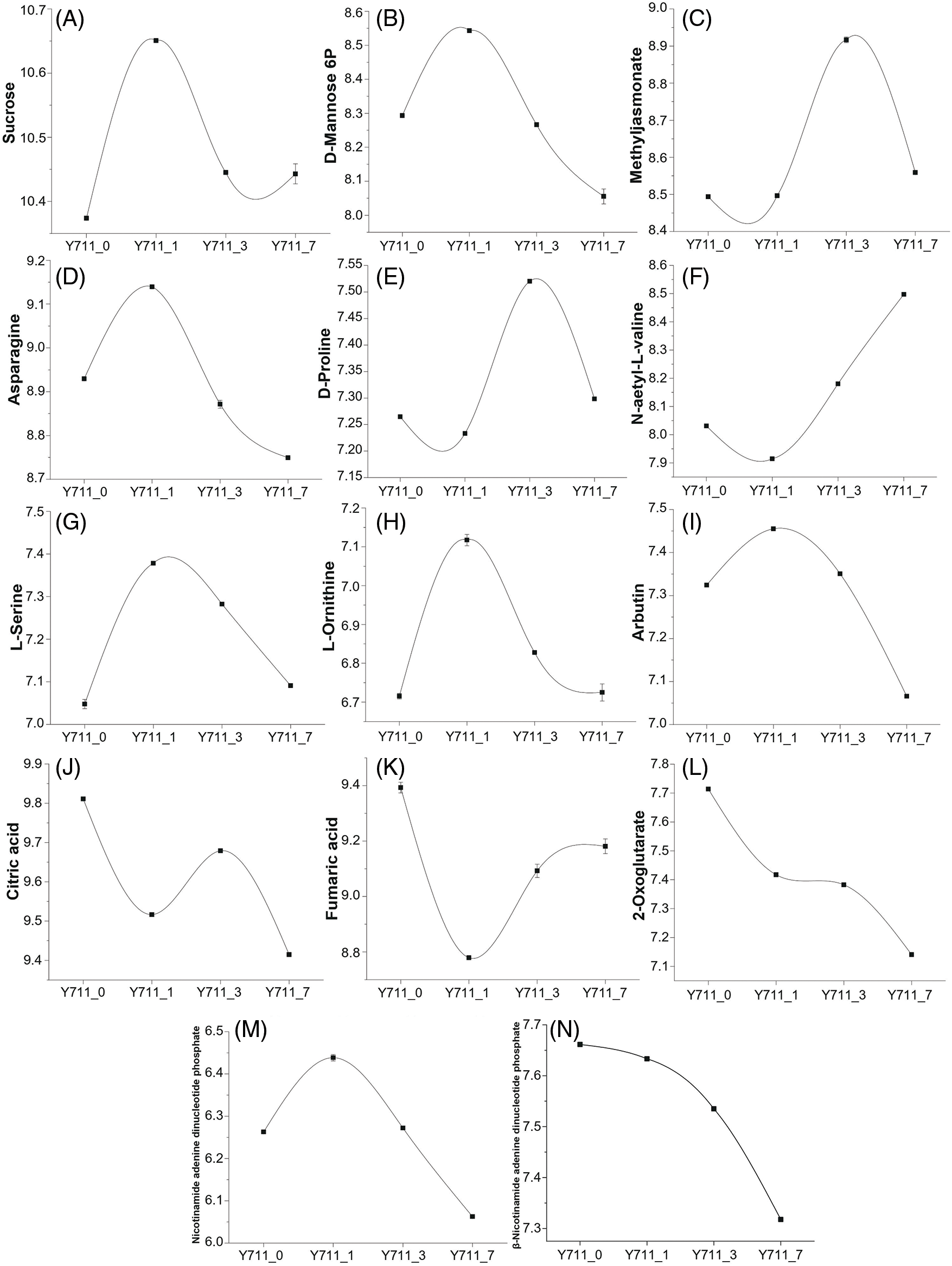
Figure 8: The DAM trend changes in other co-enriched KEEG pathways. A, sucrose; B, D-mannose 6-phosphate; C, methyl jasmonate; D, asparagine; E, D-proline; F, N-acetyl-L-valine; G, L-serine; H, L-Ornithine; I, arbutin; J, citric acid; K, fumaric acid; L, 2-oxoglutarate; M, nicotinamide adenine dinucleotide phosphate; N, beta-nicotinamide adenine dinucleotide phosphate
The application of exogenous methyl jasmonate can significantly improve plant resistance to abiotic stresses. In the present study, methyl jasmonate content significantly increased following three days of flooding stress. In line with this, the expression of five genes involved in alpha-linolenic acid metabolism was also significantly up-regulated post-flooding stress (Fig. 8C).
Metabolites, including asparagine, D-proline, L-serine, N-acetyl-L-valine, L-Ornithine, and L-phenylalanine, derived from glycolysis and the citric acid cycle intermediates, were also induced by flooding stress (Figs. 8D–8H, 6B). Furthermore, arbutin (involved in glycolysis), citric acid, fumaric acid, and 2-oxoglutarate (derived from the citric acid cycle) content significantly decreased following flooding stress. Three citric acid cycle metabolites significantly decreased in the Y711_1 vs. Y711_0 group and remained low throughout the flooding stress. In line with this, the transcription levels of many citric acid cycle pathway genes were also significantly down-regulated in different flooding periods (Figs. 8I–8L). These results suggest that the citric acid cycle metabolic pathway in mulberry leaf cells was significantly inhibited by water flooding stress in the early stage.
Flooding stress usually leads to photosynthesis inhibition. Metabolomics analysis determined that after seven days of flooding stress, metabolites in the photosynthesis pathway (i.e., nicotinamide adenine dinucleotide phosphate and beta-nicotinamide adenine dinucleotide phosphate) were significantly reduced. Furthermore, a number of genes related to photosynthesis were significantly inhibited by flooding stress in the late phase (Figs. 8M and 8N).
Metabolites such as sucrose, D-mannose 6-phosphate, and arbutin are part of the organic oxygen compounds’ metabolic pathways. Methyl jasmonate belongs to lipids and lipid-like molecules pathway, while L-tyrosine, asparagine, D-proline, L-serine, N-acetyl-L-valine, L-Ornithine, L-phenylalanine, citric acid, fumaric acid, and 2-oxoglutarate belong to organic acids and derivatives. Nicotinamide adenine dinucleotide phosphate and beta-nicotinamide adenine dinucleotide phosphate are involved in the photosynthesis pathway.
Metabolites, fundamental to biological phenotypes, are crucial for understanding biological processes. To date, metabolomics techniques have been applied to study plant responses to abiotic stresses such as salt, drought, and flooding [36–38]. This study identified 1,169 metabolites in mulberry leaves post-flooding stress via non-targeted metabolomics. This study identified more metabolites than previous similar studies [18,39].
There were 331 and 314 differentially accumulated metabolites up-regulated and down-regulated by flooding stress, respectively. According to KEGG enrichment analysis, these DAMs were primarily involved in the TCA cycle, sphingolipid metabolism, tryptophan metabolism, phenylalanine, flavone and flavonol biosynthesis, photosynthesis, etc. The observed pattern of metabolite accumulation in our study exhibits partial similarity to the findings of prior research [40,41]. Combined analysis of transcriptomic and metabolomic confirmed that DEGs and DAMs were predominantly enriched in several key KEGG pathways including the biosynthesis of phenylpropanoid, amino acids, flavonoid, flavone and flavonol. This result is similar to that of other plants responding to abiotic stresses [41,42].
4.1 Metabolites Change in Phenylpropanoid Pathway
The phenylpropanoid pathway is one of the most important pathways for the majority of secondary metabolite synthesis in plants. Prior research has demonstrated that the phenylpropanoid metabolic pathways are susceptible to abiotic stresses, including flooding and low temperatures [17,30,39,43]. Plant phenylpropanoids, derived from the phenylalanine carbon skeleton, are key secondary metabolites [44]. PAL is an important enzyme in the phenylpropanoid pathway, which catalyzes phenylalanine to form 4-coumaroyl-CoA. Then, via a series of catalytic reactions, 4-coumaryl-CoA is converted into a variety of phenylpropanoid compounds, such as flavonoids, anthocyanins, and naringin [45]. In this study, the combined metabolomic and transcriptomic analyses demonstrated that DAMs and DEGs were highly enriched in the phenylpropanoid biosynthesis and phenylalanine metabolic pathways. The content of many metabolites involved in phenylpropanoid biosynthesis, including coumarin, pterostilbene, piceatannol, scopoletin, and scopoline, either increased or decreased. Coumarins, which are abundant in diverse plant organs including roots, leaves and fruits, are prominent secondary metabolites. Previous research has shown that coumarin enhances tomato salt tolerance by improving ion homeostasis and enhancing antioxidant defenses [46]. Scopolin and scopoletin, both coumarin derivatives, play significant roles in reactive oxygen species (ROS) scavenging and pathogen defense in plants. Notably, scopolin expression in Arabidopsis is triggered by osmotic and low-temperature stresses [47]. This study observed an increase in coumarin, scopoletin, and scopolin levels at various points following flooding stress. These findings suggest a potential key role for these metabolites in the plant’s response to flooding stress (Fig. 6).
4.2 Metabolites Change in Flavonoids Metabolism Pathway
Flavonoids are vital secondary metabolites that play significant roles in the growth, development, and abiotic stress resistance of plants [48]. As the first enzyme in flavonoid biosynthesis, CHS uses P-coumaroyl-CoA as a substrate to synthesize chalcones and direct the metabolic stream for flavonoid metabolism. Studies have shown that cold stress can induce flavonoid structural gene expression, including CHS, CHI, and FLS, leading to the accumulation of flavonoids and thus improving plant adaptability to a low-temperature environment [49,50]. Furthermore, the down-regulation of the CHS gene decreases rutin content in tobacco leaves, and the salt-resistance ability of RNAi plants is weakened. Whereas CHS overexpression can significantly increase flavonoid accumulation and improve the clearance efficiency of ROS [51].
Metabolomics studies reveal that moderate salt and alkali stress causes the accumulation of prunin, naringin, and kaempferol derivatives in sorghum [52]. The concentrations of flavonoid metabolites such as epicatechin, epicatechin gallate, and epigallocatechin-3-gallate increase under drought conditions [53]. Exogenous catechins can significantly reduce the waterlogging damage of tomato leaves and roots by enhancing the free radical scavenging system and fully reducing the concentration of hydrogen peroxide and superoxide [54].
This study documented a notable reduction in dihydrokaempferol and kaempferol levels alongside an increase in dihydroquercetin. Consistent with this, two F3′H genes were also up-regulated one day following flooding stress (Fig. 5). Previous studies have also demonstrated that F3′H overexpression can significantly increase eriodictoyl and dihydroquercetin content, thereby enhancing Nicotiana benthamiana resistance to UV-B and drought stress [55]. Accordingly, the flavonoid metabolism pathway was also involved in the resistance of mulberries to flooding stress.
4.3 Metabolites Change in Glycolytic Pathway and TCA Cycle
Flooding induces hypoxic stress in plants, triggering a rapid shift from aerobic respiration to anaerobic metabolism, known as glycolysis. Since anaerobic metabolism is much less efficient in producing ATP than aerobic respiration, flooding stress often leads to energy shortages in plants. Transient flooding may not significantly impact plant growth but can trigger the expression of anaerobic response genes and alter related metabolites in plants, including but not limited to starch, sucrose, and amino acids. Following flooding stress, starch, sugar, and phenolics in Medicago truncatula leaves are significantly up-regulated [56]. The flooding stress experiments conducted on soybean seedlings demonstrated that the submergence leads to the accumulation of citrate, succinate, fumarate, alanine, and gamma-aminobutyric acid. These metabolites are closely associated with the TCA and amino acid metabolism [57].
It was found that sucrose accumulated significantly in mulberry leaves one-day post-flooding. Furthermore, the expression of genes coding for sucrose synthase, which is involved in the starch and sucrose metabolism pathway, was also induced by flooding stress. This result is consistent with previous studies on soybean and mulberry [30,58]. D-Mannose 6-phosphate, a precursor of lactose synthesis, also exhibited significant accumulation one day following flooding stress. Genes encoding enzymes involved in mannose metabolism were also up-regulated post-flooding. Similar results have been previously identified in waterlogged Medicago plants [56]. The accumulation levels of serine, phenylalanine, N-Acetyl-L-valine, and proline, which are derivatives of 3-phosphoglycerate, phosphoenolpyruvate, pyruvate, and 2-oxoglutarate, respectively, exhibited varying degrees of increase under flooding stress. Similar results have also been found in previous studies [59]. Contrary to previous studies [30,60], asparagine, a derivative of oxaloacetate, content was also up-regulated after flooding stress.
On the contrary, three metabolites involved in the citric acid cycle, specifically citric acid, fumaric acid, and 2-oxoglutarate, were significantly down-regulated after flooding stress. According to the above results, the glycolytic pathway in mulberry was also strengthened following flooding stress, while the aerobic citric acid cycle was significantly inhibited. This conclusion further confirms the results obtained in the transcriptomic analysis [4].
5 Conclusions and Perspectives
Analyzing the impact of flooding stress on plant growth and metabolism aids in understanding plant adaptation mechanisms to abiotic stress and helps in expanding cultivation ranges and selecting stress-resistant varieties. In the present study, a comprehensive analysis was conducted to examine the alterations in metabolites within mulberry leaves following exposure to flooding stress. Additionally, an analysis integrating the transcriptome and metabolome was conducted. Overall, the results of this study revealed that flooding stress has a significant impact on the composition of metabolites in mulberry leaves. In particular, many metabolites contained in metabolic pathways of organic acids and derivatives, phenylpropanoids and polyketides, and organic oxygen compounds were significantly altered. At the same time, many genes involved in these pathways were significantly induced by flooding stress. These DAMs and DEGs may play an important role in the response of mulberry to flooding stress. While this study focused on transcriptomic and metabolomic analyses of mulberry leaves post-flooding, the impact on differentially expressed genes and metabolites in roots and stems remains unexplored. After integrating the above data, a more scientific and credible adaptation mechanism for the flooding of mulberry should be proposed.
Acknowledgement: We express our profound gratitude to the personnel affiliated with the Ganning Base of Chongqing Three Gorges Academy of Agricultural Sciences, Chongqing, China.
Funding Statement: The funding for this research was provided by the General Program of Chongqing Natural Science Foundation (No. cstc2020jcyj-msxmX0073), Scientific and Technological Research Program of Chongqing Municipal Education Commission (Nos. KJQN202001209, KJZD-K202301206), Chongqing Graduate Research Innovation Project (CYS22698).
Author Contributions: The study was conceived and designed by J. Hu and L. Gan. The experiment, sampling, data analysis, and interpretation were conducted by W. Chen, Y. Duan, Y. Ru, W. Chen, P. Xiang and C. Huang. The manuscript was drafted by J. Hu. Funding was obtained by J. Hu and L. Gan. The study was supervised by J. Chen and L. Zhang, who also revised the manuscript. All authors have reviewed and approved the published version of the manuscript.
Availability of Data and Materials: The data utilized in this study have been incorporated within the tables and pictures. Furthermore, all of the raw reads from the transcriptome have been duly submitted to the NCBI SRA, accompanied by the corresponding accession number PRJNA901038.
Ethics Approval: None.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Supplementary Materials: The supplementary material is available online at https://doi.org/10.32604/phyton.2024.046521.
References
1. Jia W, Ma M, Chen J, Wu S. Plant morphological, Physiological and anatomical adaption to flooding stress and the underlying molecular mechanisms. Int J Mol Sci. 2021;22(3):1088. [Google Scholar] [PubMed]
2. Tanoue M, Hirabayashi Y, Ikeuchi H. Global-scale river flood vulnerability in the last 50 years. Sci Rep. 2016;6(1):36021. [Google Scholar] [PubMed]
3. Yuan Z, Ni X, Arif M, Dong Z, Zhang L, Tan X, et al. Transcriptomic analysis of the photosynthetic, respiration, and aerenchyma adaptation strategies in bermudagrass (Cynodon dactylon) under different submergence stress. Int J Mol Sci. 2021;22(15):7905. [Google Scholar] [PubMed]
4. Hu J, Duan Y, Yang J, Gan L, Chen W, Yang J, et al. Transcriptome analysis reveals genes associated with flooding tolerance in mulberry plants. Life. 2023;13(5):1087. [Google Scholar] [PubMed]
5. Bailey-Serres J, Voesenek L. Flooding stress: acclimations and genetic diversity. Annu Rev Plant Biol. 2008;59(1):313–39. [Google Scholar] [PubMed]
6. Lin Y, Li W, Zhang Y, Xia C, Liu Y, Wang C, et al. Identification of genes/proteins related to submergence tolerance by transcriptome and proteome analyses in soybean. Sci Rep. 2019;9(1):14688. [Google Scholar] [PubMed]
7. Ren CG, Kong CC, Yan K, Zhang H, Luo YM, Xie ZH. Elucidation of the molecular responses to waterlogging in Sesbania cannabina roots by transcriptome profiling. Sci Rep. 2017;7(1):9256. [Google Scholar] [PubMed]
8. Xu X, Chen M, Ji J, Xu Q, Qi X, Chen X. Comparative RNA-seq based transcriptome profiling of waterlogging response in cucumber hypocotyls reveals novel insights into the de novo adventitious root primordia initiation. BMC Plant Biol. 2017;17(1):129. [Google Scholar] [PubMed]
9. Zhang Y, Kong X, Dai J, Luo Z, Li Z, Lu H, et al. Global gene expression in cotton (Gossypium hirsutum L.) leaves to waterlogging stress. PLoS One. 2017;12(9):e0185075. [Google Scholar] [PubMed]
10. Savchenko T, Rolletschek H, Heinzel N, Tikhonov K. Waterlogging tolerance rendered by oxylipin-mediated metabolic reprogramming in Arabidopsis. J Exp Bot. 2019;70(10):2919–32. [Google Scholar] [PubMed]
11. Voesenek LA, Bailey-Serres J. Flood adaptive traits and processes: an overview. New Phytol. 2015;206(1):57–73. [Google Scholar] [PubMed]
12. Razzaq A, Sadia B, Raza A, Khalid Hameed M, Saleem F. Metabolomics: a way forward for crop improvement. Metabolites. 2019;9(12):303. [Google Scholar] [PubMed]
13. Dawid C, Hille K. Functional metabolomics—a useful tool to characterize stress-induced metabolome alterations opening new avenues towards tailoring food crop quality. Agronomy. 2018;8(8):138. [Google Scholar]
14. Piasecka A, Kachlicki P, Stobiecki M. Analytical methods for detection of plant metabolomes changes in response to biotic and abiotic stresses. Int J Mol Sci. 2019;20(2):379. [Google Scholar] [PubMed]
15. Durazzo A, Lucarini M, Souto EB, Cicala C, Caiazzo E, Izzo AA, et al. Polyphenols: a concise overview on the chemistry, occurrence, and human health. Phytother Res. 2019;33(9):2221–43. [Google Scholar] [PubMed]
16. Zhang A, Sun H, Wang P, Han Y, Wang X. Modern analytical techniques in metabolomics analysis. Analyst. 2012;137(2):293–300. [Google Scholar] [PubMed]
17. Xu J, Chen Z, Wang F, Jia W, Xu Z. Combined transcriptomic and metabolomic analyses uncover rearranged gene expression and metabolite metabolism in tobacco during cold acclimation. Sci Rep. 2020;10(1):5242. [Google Scholar] [PubMed]
18. Wang T, Zou Q, Guo Q, Yang F, Wu L, Zhang W. Widely targeted metabolomics analysis reveals the effect of flooding stress on the synthesis of flavonoids in Chrysanthemum morifolium. Molecules. 2019;24(20):3695. [Google Scholar] [PubMed]
19. Locke AM, BardingJr GA, Sathnur S, Larive CK, Bailey-Serres J. Rice SUB1A constrains remodelling of the transcriptome and metabolome during submergence to facilitate post-submergence recovery. Plant, Cell Environ. 2018;41(4):721–36. [Google Scholar] [PubMed]
20. Vashisht D, Akman M, Girke T, Mustroph A, Reinen E, Hartman S, et al. Transcriptomes of eight Arabidopsis thaliana accessions reveal core conserved, genotype-and organ-specific responses to flooding stress. Plant Physiol. 2016;172(2):668–89. [Google Scholar] [PubMed]
21. Minami A, Yano K, Gamuyao R, Nagai K, Kuroha T, Ayano M, et al. Time-course transcriptomics analysis reveals key responses of submerged deepwater rice to flooding. Plant Physiol. 2018;176(4):3081–102. [Google Scholar] [PubMed]
22. Komatsu S, Yamamoto R, Nanjo Y, Mikami Y, Yunokawa H, Sakata K. A comprehensive analysis of the soybean genes and proteins expressed under flooding stress using transcriptome and proteome techniques. J Proteome Res. 2009;8(10):4766–78. [Google Scholar] [PubMed]
23. Qi B, Yang Y, Yin Y, Xu M, Li H. De novo sequencing, assembly, and analysis of the Taxodium ‘Zhongshansa’ roots and shoots transcriptome in response to short-term waterlogging. BMC Plant Biol. 2014;14(1):1–12. [Google Scholar]
24. Zhang Q, Tang S, Li J, Fan C, Luo K. Integrative transcriptomic and metabolomic analyses provide insight into the long-term submergence response mechanisms of young Salix variegata stems. Planta. 2021;253(5):672. [Google Scholar]
25. Al-Khayri JM, Jain SM, Johnson DV. Advances in plant breeding strategies: fruits. Cham: Springer; 2018. [Google Scholar]
26. Liu Y, Willison JM. Prospects for cultivating white mulberry (Morus alba) in the drawdown zone of the three gorges reservoir, China. Environ Sci Pollut Res. 2013;20(10):7142–51. [Google Scholar]
27. Lu N, Luo Z, Ke Y, Dai L, Duan H, Hou R, et al. Growth, physiological, biochemical, and ionic responses of Morus alba L. seedlings to various salinity levels. Forests. 2017;8(12):488. [Google Scholar]
28. Rao L, Li S, Cui X. Leaf morphology and chlorophyll fluorescence characteristics of mulberry seedlings under waterlogging stress. Sci Rep. 2021;11(1):1–11. [Google Scholar]
29. Zhang Q, Ackah M, Wang M, Amoako FK, Shi Y, Wang L, et al. The impact of boron nutrient supply in mulberry (Morus alba) response to metabolomics, enzyme activities, and physiological parameters. Plant Physiol Biochem. 2023;200:107649. [Google Scholar] [PubMed]
30. Ackah M, Shi Y, Wu M, Wang L, Guo P, Guo L, et al. Metabolomics response to drought stress in Morus alba L. Variety Yu-711. Plants. 2021;10(8):1–35. [Google Scholar]
31. Jin X, Ackah M, Wang L, Amoako FK, Shi Y, Essoh LG, et al. Magnesium nutrient application induces metabolomics and physiological responses in mulberry (Morus alba) plants. Int J Mol Sci. 2023;24(11):9650. [Google Scholar] [PubMed]
32. Hodges DM, DeLong JM, Forney CF, Prange RK. Improving the thiobarbituric acid-reactive-substances assay for estimating lipid peroxidation in plant tissues containing anthocyanin and other interfering compounds. Planta. 1999;207(4):604–11. [Google Scholar]
33. Liang Q, Wu Y, Wang K, Bai Z, Liu Q, Pan Y, et al. Chrysanthemum WRKY gene DgWRKY5 enhances tolerance to salt stress in transgenic chrysanthemum. Sci Rep. 2017;7(1):4799. [Google Scholar] [PubMed]
34. Bian X, Zhao Y, Xiao S, Yang H, Han Y, Zhang L. Metabolome and transcriptome analysis reveals the molecular profiles underlying the ginseng response to rusty root symptoms. BMC Plant Biol. 2021;21(1):1–16. [Google Scholar]
35. Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13(8):1194–202. [Google Scholar] [PubMed]
36. Pei L, Liu J, Zhou Y, Jiang Y, Li H. Transcriptomic and metabolomic profiling reveals the protective role of anthocyanins in alleviating low phosphate stress in maize. Physiol Mol Biol Plants. 2021;27(5):889–905. [Google Scholar] [PubMed]
37. Zhang Q, Li B, Chen Q, Su Y, Wang R, Liu Z, et al. Non-targeted metabolomic analysis of the variations in the metabolites of two genotypes of Glycyrrhiza uralensis Fisch. under drought stress. Ind Crops Prod. 2022;176(5):114402. [Google Scholar]
38. Jiao Y, Zhang J, Pan C. Integrated physiological, proteomic, and metabolomic analyses of pecan cultivar ‘Pawnee’ adaptation to salt stress. Sci Rep. 2022;12(1):1841. [Google Scholar] [PubMed]
39. Zhang Q, Tang S, Li J, Fan C, Xing L, Luo K. Integrative transcriptomic and metabolomic analyses provide insight into the long-term submergence response mechanisms of young Salix variegata stems. Planta. 2021;253(5):1–18. [Google Scholar]
40. Ma W, Kim JK, Jia C, Yin F, Kim HJ, Akram W, et al. Comparative transcriptome and metabolic profiling analysis of buckwheat (Fagopyrum Tataricum (L.) Gaertn.) under salinity stress. Metabolites. 2019;9(10):225. [Google Scholar] [PubMed]
41. Zhang Q, Wang S, Qin B, Hy Sun X, Yuan, Wang Q, et al. Analysis of the transcriptome and metabolome reveals phenylpropanoid mechanism in common bean (Phaseolus vulgaris) responding to salt stress at sprout stage. Food Energy Secur. 2023;12(5):e481. [Google Scholar]
42. Ren G, Yang P, Cui J, Gao Y, Yin C, Bai Y, et al. Multiomics analyses of two sorghum cultivars reveal the molecular mechanism of salt tolerance. Front Plant Sci. 2022;13:886805. [Google Scholar] [PubMed]
43. Yan M, Xue C, Xiong Y, Meng X, Li B, Shen R, et al. Proteomic dissection of the similar and different responses of wheat to drought, salinity and submergence during seed germination. J Proteomics. 2020;220(2):103756. [Google Scholar] [PubMed]
44. Deluc L, Barrieu F, Marchive C, Lauvergeat V, Decendit A, Richard T, et al. Characterization of a grapevine R2R3-MYB transcription factor that regulates the phenylpropanoid pathway. Plant Physiol. 2006;140(2):499–511. [Google Scholar] [PubMed]
45. Zhang Q, Yang W, Liu J, Liu H, Lv Z, Zhang C, et al. Postharvest UV-C irradiation increased the flavonoids and anthocyanins accumulation, phenylpropanoid pathway gene expression, and antioxidant activity in sweet cherries (Prunus avium L.). Postharvest Biol Technol. 2021;175(Suppl 2):111490. [Google Scholar]
46. Parvin K, Hasanuzzaman M, Mohsin S, Nahar K, Fujita M. Coumarin improves tomato plant tolerance to salinity by enhancing antioxidant defence, glyoxalase system and ion homeostasis. Plant Biol. 2021;23(S1):181–92. [Google Scholar] [PubMed]
47. Döll S, Kuhlmann M, Rutten T, Mette MF, Scharfenberg S, Petridis A, et al. Accumulation of the coumarin scopolin under abiotic stress conditions is mediated by the Arabidopsis thaliana THO/TREX complex. Plant J. 2018;93(3):431–44. [Google Scholar]
48. Zhuang WB, Li YH, Shu XC, Pu YT, Wang XJ, Wang T, et al. The classification, molecular structure and biological biosynthesis of flavonoids, and their roles in biotic and abiotic stresses. Molecules. 2023;28(8):3599. [Google Scholar] [PubMed]
49. An JP, Wang XF, Zhang XW, Xu HF, Bi SQ, You CX, et al. An apple MYB transcription factor regulates cold tolerance and anthocyanin accumulation and undergoes MIEL1-mediated degradation. Plant Biotechnol J. 2020;18(2):337–53. [Google Scholar] [PubMed]
50. Zhang Q, Zhai J, Shao L, Lin W, Peng C. Accumulation of anthocyanins: an adaptation strategy of Mikania micrantha to low temperature in winter. Front Plant Sci. 2019;10:1049. [Google Scholar] [PubMed]
51. Dong NQ, Lin HX. Contribution of phenylpropanoid metabolism to plant development and plant—environment interactions. J Integr Plant Biol. 2021;63(1):180–209. [Google Scholar] [PubMed]
52. Ma S, Lv L, Meng C, Zhang C, Li Y. Integrative analysis of the metabolome and transcriptome of Sorghum bicolor reveals dynamic changes in flavonoids accumulation under saline–alkali stress. J Agr Food Chem. 2020;68(50):14781–9. [Google Scholar]
53. Hernández I, Alegre L, van Breusegem F, Munné-Bosch S. How relevant are flavonoids as antioxidants in plants? Trends Plant Sci. 2009;14(3):125–32. [Google Scholar]
54. Yiu JC, Tseng MJ, Liu CW. Exogenous catechin increases antioxidant enzyme activity and promotes flooding tolerance in tomato (Solanum lycopersicum L.). Plant Soil. 2011;344(1–2):213–25. [Google Scholar]
55. Baba SA, Ashraf N. Functional characterization of flavonoid 3′-hydroxylase, CsF3′H, from Crocus sativus L: insights into substrate specificity and role in abiotic stress. Arch Biochem Biophys. 2019;667:70–8. [Google Scholar] [PubMed]
56. Lothier J, Diab H, Cukier C, Limami AM, Tcherkez G. Metabolic responses to waterlogging differ between roots and shoots and reflect phloem transport alteration in Medicago truncatula. Plants. 2020;9(10):1373. [Google Scholar] [PubMed]
57. Nakamura T, Yamamoto R, Hiraga S, Nakayama N, Okazaki K, Takahashi H, et al. Evaluation of metabolite alteration under flooding stress in soybeans. Jpn Agr Res Q. 2012;46(3):237–48. [Google Scholar]
58. Coutinho ID, Henning LMM, Döpp SA, Nepomuceno A, Moraes LAC, Marcolino-Gomes J, et al. Flooded soybean metabolomic analysis reveals important primary and secondary metabolites involved in the hypoxia stress response and tolerance. Environ Exp Bot. 2018;153:176–87. [Google Scholar]
59. António C, Päpke C, Rocha M, Diab H, Limami AM, Obata T, et al. Regulation of primary metabolism in response to low oxygen availability as revealed by carbon and nitrogen isotope redistribution. Plant Physiol. 2016;170(1):43–56. [Google Scholar]
60. Souza SC, Mazzafera P, Sodek L. Flooding of the root system in soybean: biochemical and molecular aspects of N metabolism in the nodule during stress and recovery. Amino Acids. 2016;48(5):1285–95. [Google Scholar] [PubMed]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools