
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Trilobatin Acts as a Marker Metabolite Involved in Flavonoid Accumulation Regulated by CsWRKY28-MYC2 with Trypsin Activation in Cucumber (Cucumis sativus)
1 College of Food and Bioengineering, Henan University of Science and Technology, Luoyang, 471023, China
2 Chinese Academy of Agricultural Sciences Cotton Research Institute, Anyang, 455000, China
3 The National and Local Joint Engineering Laboratory of High Efficiency and Superior-Quality Cultivation and Fruit Deep Processing Technology of Characteristic Fruit Trees in South Xinjiang, Tarim University, Alar, 843300, China
4 National Demonstration Center for Experimental Food Processing and Safety Education, Luoyang, 471000, China
* Corresponding Author: Xin Li. Email:
# These authors contributed equally to this work
(This article belongs to the Special Issue: Influence of Biotic and Abiotic Stresses Signals on Plants and their Performance at Different Environments)
Phyton-International Journal of Experimental Botany 2024, 93(11), 2837-2856. https://doi.org/10.32604/phyton.2024.057932
Received 31 August 2024; Accepted 18 October 2024; Issue published 30 November 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
During post-harvest storage of Cucumis sativus fruit, the application of trypsin treatment could increase flavonoid compound levels and reduce oxidative damage. To investigate the mechanism of trypsin-induced flavonoid biosynthesis in C. sativus, we conducted a combined analysis of transcriptomics and widely targeted metabolomics. One hundred and seventy-five significantly different metabolites were obtained from metabolomics data. The kyoto encyclopedia of genes and genomes (KEGG) functional enrichment results indicated that these metabolites were mainly involved in the phenylpropanoid biosynthesis pathway. By combining the results of the weighted gene co-expression network analysis (WGCNA) with the 130 upregulated phenylpropanoid metabolites, 22 significantly upregulated phenylpropanoid metabolites were identified. Trilobatin was identified as the most prominent metabolite through cluster analysis and variable importance in projection (VIP) analysis. High performance liquid chromatography (HPLC) experiments confirmed that trilobatin was the key metabolite induced by trypsin. The transcriptomic results showed that 1068 genes in the brown module of WGCNA were highly positively correlated with flavonoid biosynthesis. The gene set enrichment analysis (GSEA) identified leading edges in 4 key KEGG pathways. Finally, combined with WGCNA and GSEA analysis results, 35 core genes were obtained. The co-expression network of transcriptomics and metabolomics suggested that CsWRKY28 and CsMYC2 regulated the biosynthesis of trilobatin. The quantitative real-time polymerase chain reaction (RT-qPCR) and dual luciferase experiments confirmed the activation effect of CsWRKY28 on CsMYC2 and downstream target genes. This study revealed the key transcription factors involved in the trypsin-controlled biosynthesis of trilobatin in C. sativus and provided a new theoretical basis for elucidating the molecular mechanism of trypsin preservation.Keywords
Supplementary Material
Supplementary Material FilePhenylpropanoid compounds were commonly found in plants and were synthesized through a series of enzymatic reactions, including catalysis by phenylalanine deaminase (PAL), cinnamate-4 hydroxylase (C4H), and 4-coumaryl CoA ligase (4CL). They were transformed into different phenylpropanoid metabolites through downstream biosynthesis pathways, including flavonoids, coumarins, terpenes, lignin, anthocyanins and others [1,2]. Flavonoids are a type of plant secondary metabolite that are widely present in nature and have various physiological activities and protective effects [3].
Flavonoids could act as natural antioxidants for plants, neutralizing hydrogen peroxide and radicals produced during photosynthesis and other biochemical reactions, thus preventing excessive accumulation and oxidative damage to cells [2]. Additionally, they also enhanced gene expression levels and antioxidant enzyme activities, promoted the content and clearance of antioxidant compounds, and played an important role in maintaining the post-harvest quality of fruits and vegetables while delaying aging [4]. Research on lychee and banana fruits has shown that proanthocyanidins help maintain fruit quality by promoting antioxidant activity and enhancing endogenous flavonoid biosynthesis [5–7]. Flavonoid biosynthesis and regulatory mechanisms have been extensively studied in plants due to the bioactive functions of these compounds.
According to reports, several transcription factors (TFs) were involved in the regulation of key genes in the phenylpropanoid pathway, thereby affecting the levels of anthocyanins, lignins, and flavonoids [8]. For instance, VaMYBF1 could bind to the VaCHS promoter regulating flavonol biosynthesis in Vitis adenoclada grapes [9]. In cotton, GhWRKY41 promoted the accumulation of lignin and flavonoids by activating the expression of GhC4H and Gh4CL [10]. In strawberries, FaWRKY71 mediated resistance to active oxygen species (ROS) by accelerating anthocyanin accumulation, thereby reducing the content of MDA (Malondialdehyde) [11]. Our laboratory also discovered that the expression of HuWRKY40 in pitaya was closely related to flavonoid biosynthesis [12].
Previous studies conducted in our laboratory have demonstrated that trypsin could act as a biological preservative, significantly enhancing the antioxidant capacity of fruits and vegetables, thereby extending their storage period [13]. Trypsin not only cleared superoxide anions and regulated endogenous ROS levels in pitaya peel, but also induced the biosynthesis of lignin, chlorogenic acid [14,15], and flavonoids in the phenylpropanoid pathway [16,12]. This affected the flavonoids’ ability to scavenge radicals such as DPPH (2,2-Diphenyl-1-picrylhydrazyl) and ABTS (2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)) [17,18], enhancing the fruit’s resistance. Trypsin was found to activate the expression of CsMYC2, promoting flavonoid biosynthesis in C. sativus [19]. However, the promotion of flavonoid biosynthesis by trypsin required more professional and in-depth research to fully reveal its mechanism of action.
C. sativus was a nutritious vegetable with high water content, but it was prone to quality deterioration and decay after harvest [20,21]. In this study, C. sativus was used as the experimental material. The key flavonoid compounds promoted by trypsin were screened using widely targeted metabolomics. Key pathway genes and transcription factors influenced by trypsin were selected from the transcriptome data. Co-expression network analysis was used to identify the connections between key metabolites and their biosynthesis-related genes. The function of CsWRKY28 was validated, and its impact on the expression of downstream target genes was verified through RT-qPCR and dual-luciferase assays. The results of this study provided new theoretical evidence for the mechanism by which trypsin enhances flavonoid biosynthesis, thus providing a more comprehensive understanding of the molecular mechanisms of trypsin in flavonoid biosynthesis in C. sativus.
2.1 C. sativus Treatment Methods
The C. sativus (Jinyou 38) was harvested from Miaoshui Base, Ruyang County, Luoyang City, Henan Province, China. The fruit with bright green color, no obvious mechanical damage, and uniform length were selected for grouping research. Experiments were conducted following the treatment method proposed by Zhang et al. [12], took photos and recorded data regularly.
2.2 Weighted Gene Co-Expression Network Analysis (WGCNA)
Unsupervised co-expression modules were analyzed using WGCNA. This method utilized the PickSoftThreshold function from the WGCNA package to determine the weights of each indicator, ensuring conformity to a scale-free network distribution [22]. For each identified co-expression module of biological molecules, representative feature genes were computed (using the WGCNA function “module feature genes”), and the correlation between module feature genes and stress parameters was calculated (adjusting the corresponding p-values using the R function p adjust (FDR) with Benjamin-Hochberg correction).
Differential expression analysis of the transcriptome was performed using the DESeq2 software [23], with parameters set as follows: p adjust < 0.05 & |log2FC| >= 1.5, to identify differentially expressed genes. Enrichment analysis of KEGG pathways was performed on the selected differentially expressed genes using an R script. Additionally, the gene set enrichment analysis (GSEA) method was employed. This method, based on the Signal2Noise algorithm, comprehensively detects genes with significant biological significance at the overall level, without the need to specify a threshold for the degree of differential expression [24].
2.4 Widely Targeted Metabolomics Analyses
Metabolites with VIP > 1.0 (variable importance in the projection) and p < 0.05 were considered as differentially expressed metabolites (DEMs) [14]. Additionally, supervised models such as OPLS-DA/PLS-DA were employed. Paired differences were examined and predicted through 7-fold cross-validation. Variable importance in projection (VIP) analysis based on the first principal component was utilized to identify metabolites contributing significantly to classification.
2.5 Correlation Network Analyses
Differential genes and metabolites selected from the transcriptomics and metabolomics profiles were used to compute correlations using Spearman. A co-expression network diagram of genes and metabolites was constructed using Cytoscape software (https://cytoscape.org/index.html, accessed on 17 October 2024, ver3.9.1). The cytoHubba plugin was employed to rank nodes based on closeness using 11 topological analysis methods [25].
2.6 RNA Isolation and First-Strand cDNA Synthesis
Total RNA was extracted from C. sativus peel samples using Triquick reagent. The purity of the extracted RNA was assessed using a microvolume spectrophotometer. Subsequently, cDNA synthesis was performed using the TransScript® One-Step gDNA Removal and cDNA Synthesis SuperMix Kit.
2.7 Construction of CsWRKY28 Vector
Fragments of CsWRKY28 (279 bp) obtained through PCR amplification and purification were ligated with the pTRV2 vector. The full-length CsWRKY28 (1431 bp) was also ligated with the P3300 vector. Both constructs were separately transformed into Agrobacterium GV3101 for subsequent experiments (primer sequences used are provided in Table S1). C. sativus samples were divided into four groups: the blank control group (CK group), trypsin-treated group (Try group), trypsin combined with CsWRKY28 silencing treatment group (TV-WRKY28), and trypsin combined with CsWRKY28 overexpression treatment group (TO-WRKY28). The infection protocol followed the method described by Li et al. [26], whereby the peel was immersed in a bacterial suspension with an OD600 of 0.6. After soaking in bacterial solution, samples were cultured in darkness for one day, followed by trypsin treatment, and then incubated at 25°C for 18 days. Distilled water treatment was used as the control group (CK), while the group treated with only trypsin was designated as the Try group. The group soaked in bacterial solution containing the recombinant pTRV2 plasmid, followed by trypsin treatment, was referred to as the TV-WRKY28 group, and the group soaked in bacterial solution containing the recombinant P3300 plasmid, followed by trypsin treatment, was designated as the TO-WRKY28 group.
The promoter sequence of the target gene was analyzed using the PlantCARE database website (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) (accessed on 17 October 2024) [27]. Subsequently, the specific promoter sequence of the target gene was cloned and ligated with the Luc plasmid (primer sequences used are provided in Table S1). The construct was then transformed into Agrobacterium GV3101 containing helper plasmids for subsequent infection of tobacco [28].
2.9 RT-qPCR and Quality of Fruits
The method of 2−ΔΔCt was used to calculate the expression levels of differential genes, with experiments repeated three times [29]. All primers are listed in Table S1. The loss rate was periodically measured according to the method introduced previously. Hardness testing was performed using a TA. XT EXPRESS food texture analyzer. The maximum force required to penetrate to a depth of 10 mm at a rate of 1 mm/s with a P/5 probe was measured. pH detection was conducted using a PHS-3E pH meter, measuring the pH value of C. sativus juice sequentially. Each sample was measured three times, and the average value was recorded [14,30].
2.10 Metabolites Determination by HPLC
The dried sample of C. sativus was ground and filtered by 70% absolute ethanol, bathed in water at 65°C for 2.5 h, and placed overnight at −20°C. The supernatant was centrifuged and filtered through a 0.2 μM filter membrane, and the filtrate was analyzed by high-performance liquid chromatography (HPLC). The Agilent 1200 series HPLC system (Agilent Technologies, Santa Clara, CA, USA) was used in the current work. Chromatographic column: reverse liquid chromatographic column (Agilent ZORBAX SB-C18, 4.6 mm × 250 mm, 5 μm). The column temperature was kept at 30°C. A gradient mobile phase consisted of 0.1% formic acid in water (A) and methanol (B) and separation was achieved using the following gradient program: 1–5 min, 5% B; 5–8 min, 5%–30% B; 8–15 min, 30%–40% B; 15–20 min, 40%–60% B; 20–30 min, 60%–70% B; 30–33 min, 70%–5% B; 33–38 min, 5% B. The inject volume was 20 μL. Flow rate: 0.8 mL/min; Detection wave length: 280 nm. The loading concentration of the standard products of trilobatin (Shanghai Yuanye Bio-Technology Co., Ltd., Shanghai, China), phloretin and phloretin (Shanghai Aladdin Bio-Chem Technology Co., Ltd., Shanghai, China) were all 1 mg/mL, respectively [19].
Data analysis and graph plotting were performed using Origin 2022 software. The t-test between samples was conducted using SPSS software to calculate the differences between samples. Significance and high significance differences were denoted by *p < 0.05 and **p < 0.01, respectively [31].
3.1 Screening of Important Metabolic Pathways and Key Metabolites
Differential metabolite analysis was conducted on C. sativus metabolites after trypsin treatment, and a total of 704 metabolites were identified (Fig. 1A), among which 175 metabolites showed significant differences (Table S2). KEGG enrichment analysis was conducted on the 175 significantly different metabolites. It was found that among the top 20 pathways enriched by these significantly different metabolites, the phenylpropanoid biosynthesis pathway was the most prominent (Fig. 1B). Therefore, we further focused on the phenylpropanoid biosynthesis pathway and selected 216 compounds related to it from the 704 metabolites in the metabolomic data, including 130 upregulated metabolites.
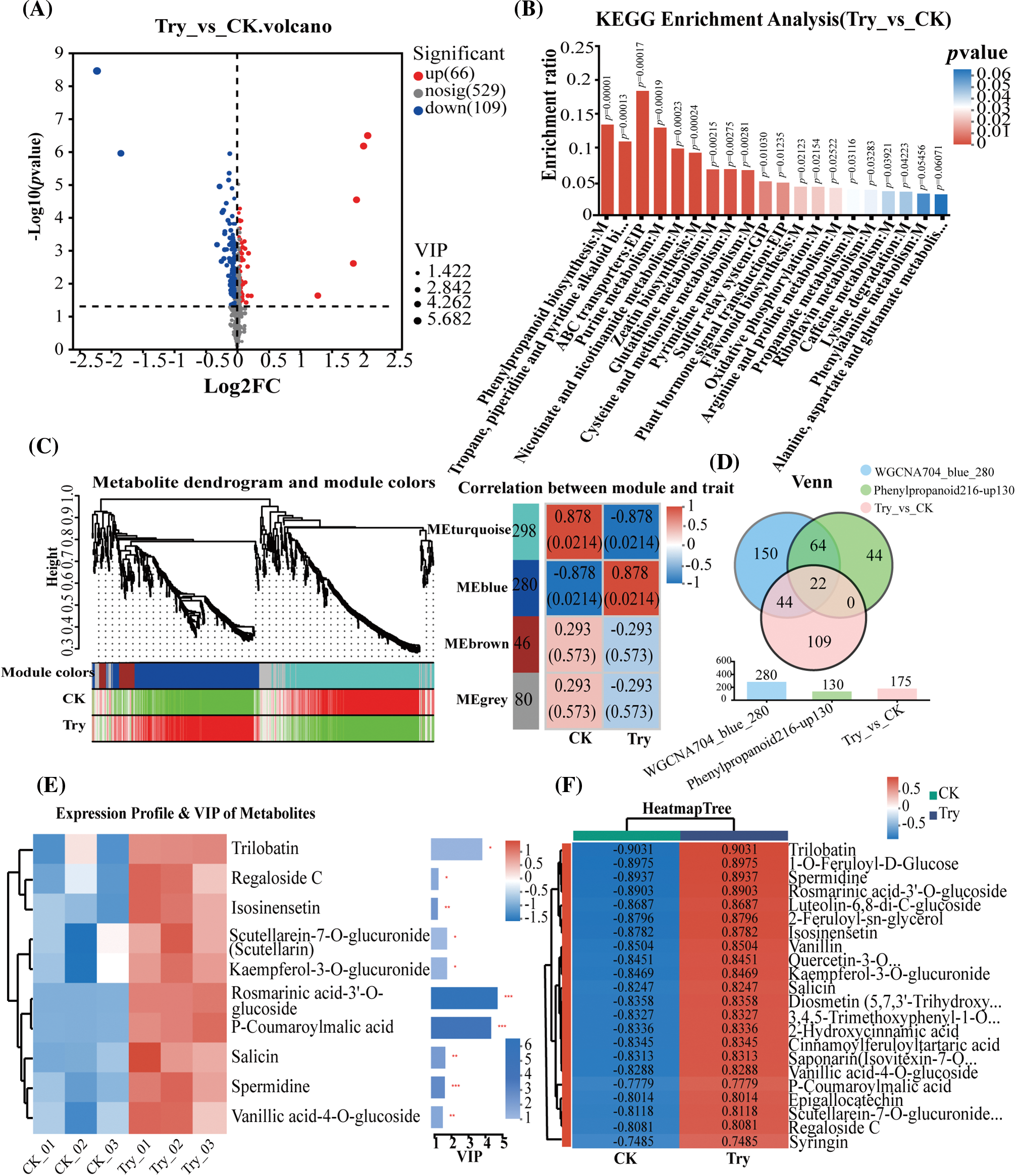
Figure 1: Metabolomics analysis of key metabolites regulated by trypsin. (A) Volcano plot of significant differential metabolites: red represented significantly upregulated metabolites, and blue represented significantly downregulated metabolites. (B) KEGG pathway enrichment plot of 175 metabolites. (C) Weighted gene co-expression network analysis of metabolites. On the left was the clustering analysis of metabolites and phenotypes, with the upper part showing the hierarchical clustering tree of metabolites, the middle part indicating the modules to which the metabolites belong, and the lower part representing the heat map of the correlation between metabolites within the module and phenotypes. On the right was the analysis of the correlation between modules and phenotypes, where the left column of numbers indicates the number of metabolites in the module, and each group of data on the right represented the correlation coefficient and significance p-value (in parentheses) between the module and phenotype. A larger absolute value indicates a stronger correlation, with red indicating positive correlation and blue indicating negative correlation. (D) Venn diagram of metabolites in three groups. (E) VIP analysis. (F) Clustering analysis of metabolites
Additionally, Weighted Gene Co-Expression Network Analysis (WGCNA) could partition metabolites into modules based on their expression trends, providing a more comprehensive assessment of the correlation between phenotypes and metabolites. Therefore, we employed WGCNA again to reanalyze 704 metabolites, identifying a total of 4 color modules. Among these modules, only the blue module, consisting of 280 metabolites, exhibited a highly positive correlation with trypsin (Fig. 1C), while the remaining three modules showed a negative correlation with trypsin. Consequently, we selected the 280 metabolites showing significant positive correlation with trypsin for subsequent analysis.
Venn analysis was performed on the 175 significantly different metabolites, the 280 metabolites significantly positively correlated with trypsin, and the 130 upregulated phenylpropanoid metabolites, confirmed 22 core metabolites significantly upregulated by trypsin and associated with phenylpropanoid (Fig. 1D; Table S3). After VIP analysis, it was found that significantly increased compounds included trilobatin, rosmarinic acid-3′-O-glucoside, and P-coumaroylmalic acid, among other phenylpropanoid compounds (Fig. 1E). Subsequently, cluster analysis was conducted on the 22 differentially upregulated metabolites, with trilobatin exhibiting the highest expression level (Fig. 1F). To further elucidate the mechanisms regulated by relevant genes, transcriptomic data will be used for in-depth analysis.
3.2 Transcriptome Analysis for Screening Key Genes
Based on the gene expression profile induced by trypsin treatment, an analysis of differential expression levels was completed. According to the transcriptomic PCA analysis, the samples were divided into two independent groups (Fig. 2A), with significant differences observed between the two groups (PC1 = 55.58%). Samples marked with the same color within each group were closely clustered together (PC2 = 12.63%). Subsequently, to elucidate the regulatory mechanism of trypsin on C. sativus preservation, we would analyze genes regulated by trypsin from multiple perspectives and focus on identifying functional genes that regulate flavonoid compounds, in combination with the results of metabolomic analysis.
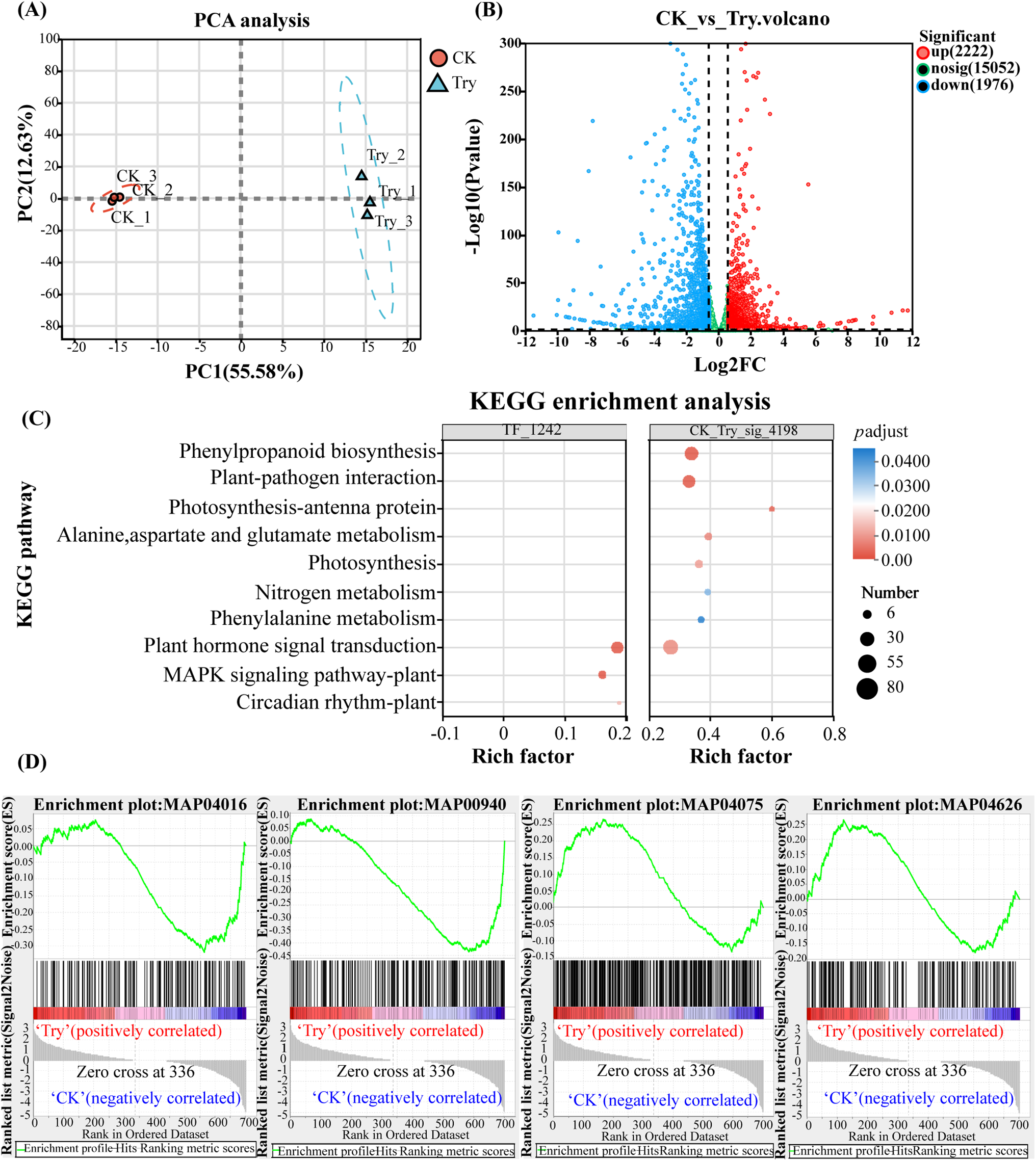
Figure 2: Transcriptomic analysis of differentially expressed genes regulated by trypsin. (A) PCA analysis of samples. (B) Volcano plot of expression differences. (C) KEGG pathway enrichment analysis of multiple gene sets, with transcription factor enrichment results on the left and differential gene expression enrichment results on the right. (D) GSEA analysis of genes related to 4 pathways. The upper curve represented the trend of cumulative ES values, with the highest point being the ES value of the gene set; the middle vertical line represented the position of the prior gene set in the sorted gene list, with black lines marking the position of each gene in the prior gene set in the sorted gene list; the lower heatmap and gray area graph represented the distribution of genes in the sorted gene list
Significant changes in gene expression were observed in C. sativus after trypsin treatment (Fig. 2B), with a total of 4198 differentially expressed genes identified (p < 0.05, FC > 1.5). KEGG pathway enrichment analysis (Table S4) revealed that the most enriched pathways among the differentially expressed genes were “phenylpropanoid biosynthesis” (map00940) pathway (68/4198) and “plant-pathogen interaction” (map04626) pathway (58/4198). In our previous work, it was found that trypsin treatment significantly altered gene expression in C. sativus during storage, with transcription factors playing important regulatory roles in this process [32]. Therefore, we identified 1242 transcription factors from the transcriptomic data, of which 270 were significantly upregulated and 255 were significantly downregulated. KEGG pathway enrichment analysis (Table S5) of these 1242 transcription factors showed that the most enriched pathways were “MAPK signaling pathway-plant” (map04016) pathway (26/1242) and “plant hormone signal transduction” (map04075) pathway (55/1242). The analysis identified four major pathways involved in the regulation of genes and transcription factors significantly influenced by trypsin (Fig. 2C). However, relying solely on the significance of differential gene expression as a basis to assume that these genes played a key role was not comprehensive enough, as it may overlook genes with important biological significance but non-significant expression differences.
Therefore, we targeted these four pathways and re-screened all genes enriched in these pathways in C. sativus. Subsequently, we performed GSEA analysis on the genes of these four pathways to identify their leading subsets (Fig. 2D). A total of 197 genes were obtained, with 46 genes enriched in the leading edge of map00940, 36 genes enriched in the leading edge of map04016, 76 genes enriched in the leading edge of map04075, and 41 genes enriched in the leading edge of map04626.
Combining the results of our previous metabolomics analysis, the “phenylpropanoid biosynthesis” pathway was significantly enriched in both omics results. Moreover, it was found during the metabolomics analysis that flavonoid compounds were likely to play a crucial role in the phenylpropanoid biosynthesis pathway. Therefore, we selected 280 genes related to flavonoids from the transcriptome data, among which 56 genes were significantly upregulated. To prevent the omission of important genes related to flavonoids, we further identified all genes through WGCNA analysis, and after associating the modules with flavonoid phenotypic features, we obtained a heatmap of gene-phenotype correlation (Fig. 3A), which included a total of 5106 genes and were divided into 7 modules. Among these modules, the “yellow”, “brown”, and “turquoise” modules showed a positive correlation between their expression patterns and the dynamic changes of flavonoid compounds. Moreover, the brown module, consisting of 1068 genes, exhibited a highly positive correlation with flavonoid biosynthesis, with 607 genes showing upregulated expression.
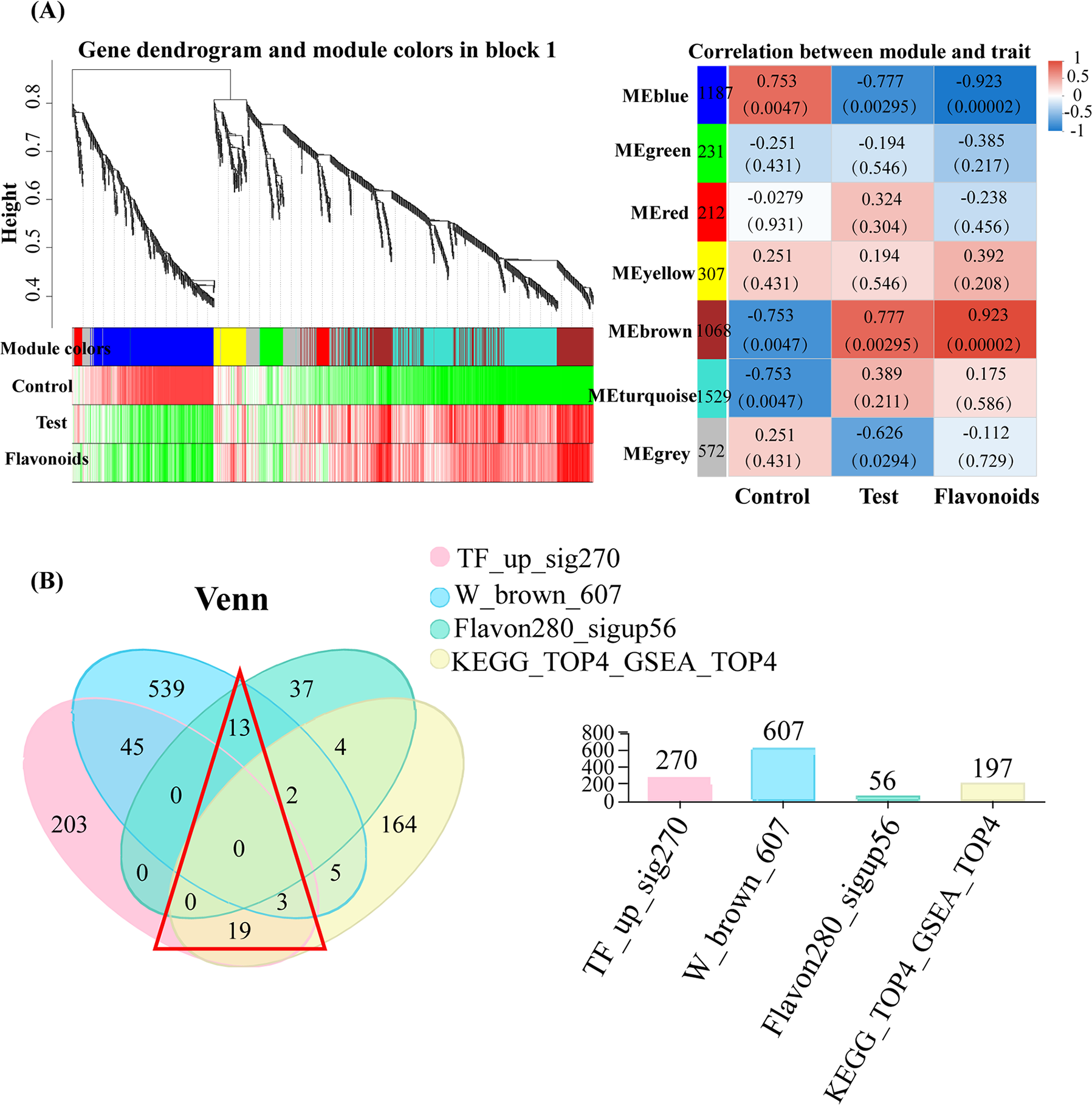
Figure 3: Screening of key genes. (A) Weighted co-expression analysis plot. On the left was the clustering analysis of metabolites and phenotypes, with the top being the hierarchical clustering tree of metabolites, the middle being the modules to which metabolites belong, and the bottom representing the heat map of the correlation between metabolites and phenotypes within the modules. On the right was the analysis of the correlation between modules and phenotypes, where each group of data on the right represented the correlation coefficient and significance p-value (in parentheses) between the module and phenotype, with a larger absolute value indicating a stronger correlation, red indicating positive correlation, and blue indicated negative correlation. (B) Venn diagram analysis of multiple gene sets, with the red triangular area representing the core genes
Finally, by combining the 270 significantly upregulated transcription factors, GAEA analyzed a total of 197 genes in the leading edge of 4 key pathways, 56 flavonoid-related significantly upregulated genes were selected from the transcriptome data, and 607 genes upregulated in the WGCNA brown module were analyzed using Venn plots (Fig. 3B). At last, a total of 35 genes highly related to flavonoids, with high pathway enrichment in KEGG, playing a key role, and related to upregulated transcription factors were selected.
3.3 Correlation Network of Genes and Metabolites
The 35 key genes selected above were co-expressed with the 130 metabolites related to phenylpropanoid and upregulated to construct a co-expression network and analyzed the correlation between the genes and metabolites. The selected list of key genes and metabolites was input into Cytoscape software, and an interaction diagram between co-expressed network genes was created based on their weights (Fig. 4A). The size of the node shape represented the number of edges connected to the node, and the width of the edges represented the weight of the connections between the two nodes. The key metabolite trilobatin was used as the central node, and the transcription factors CsWRKY28 (CsaV3_3G033350) and CsMYC2 (CsaV3_2G010120) related to it were selected (Fig. 4B). Then, CsWRKY28 was used as the central node to screen for genes and metabolites related to it, obtaining the top 10 genes and metabolites ranked by the Closeness algorithm (Table S6). The results indicated that the CsMYC2 transcription factor may also have been regulated by CsWRKY28 and further regulated the biosynthesis of quercetin by affecting CsUGT91C1 (CsaV3_3G048090) and CsUGT71K1 (CsaV3_4G032530) (Fig. 4C).
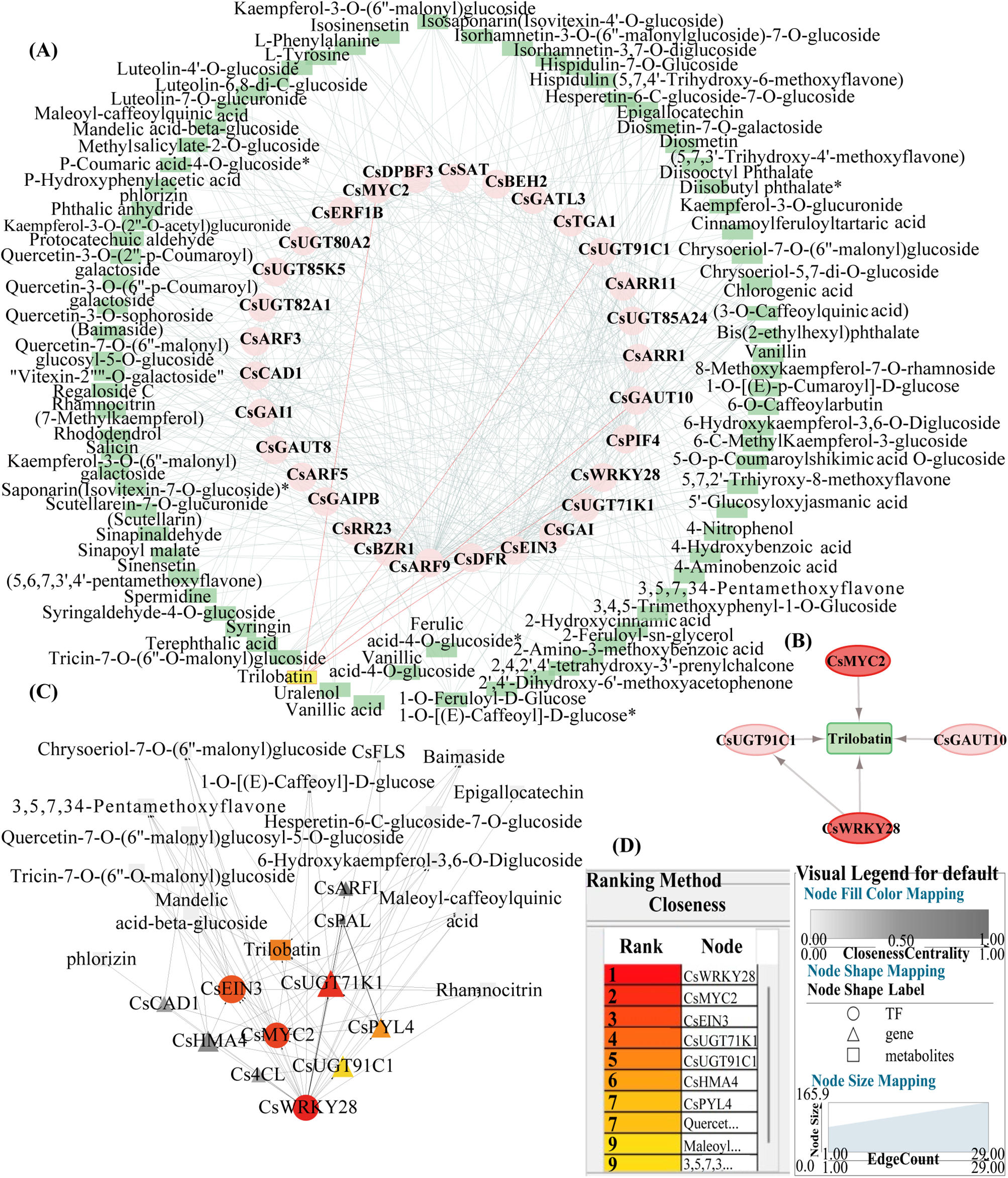
Figure 4: Expression correlation network analysis. (A) Co-expression network diagram, where pink represented genes, green represented metabolites, and the target metabolite quercetin was highlighted in yellow. The pink lines indicated the correlated genes. (B) Trilobatin biosynthesis network interaction diagram, with the central green representing the trilobatin metabolite and surrounding genes. (C) CsWRKY28 regulation network interaction diagram, with circles representing transcription factors, triangles representing genes, and rectangles representing metabolites. (D) The left panel showed the top 10 genes and metabolites related to CsWRKY28 obtained according to the Closeness algorithm in (C), and the right panel showed the parameters of (C)
3.4 Validation of the Function of CsWRKY28
To validate the function of CsWRKY28, RNA was extracted from C. sativus and reverse transcribed into C. sativus cDNA. Using this as a template, a silencing fragment (279 bp) for RNAi interference and the full-length open reading frame (1431 bp) of the target gene CsWRKY28 were obtained through PCR. Subsequently, the silencing fragment was ligated to the pTRV2 vector and the full-length fragment was ligated to the p3300-35S-Tone vector through double enzyme digestion. The structures of the recombinant plasmids for overexpression and RNAi silencing were shown in Fig. 5A. The successfully constructed recombinant plasmids were then separately transformed into Agrobacterium tumefaciens strain pTRV1 for cultivation.
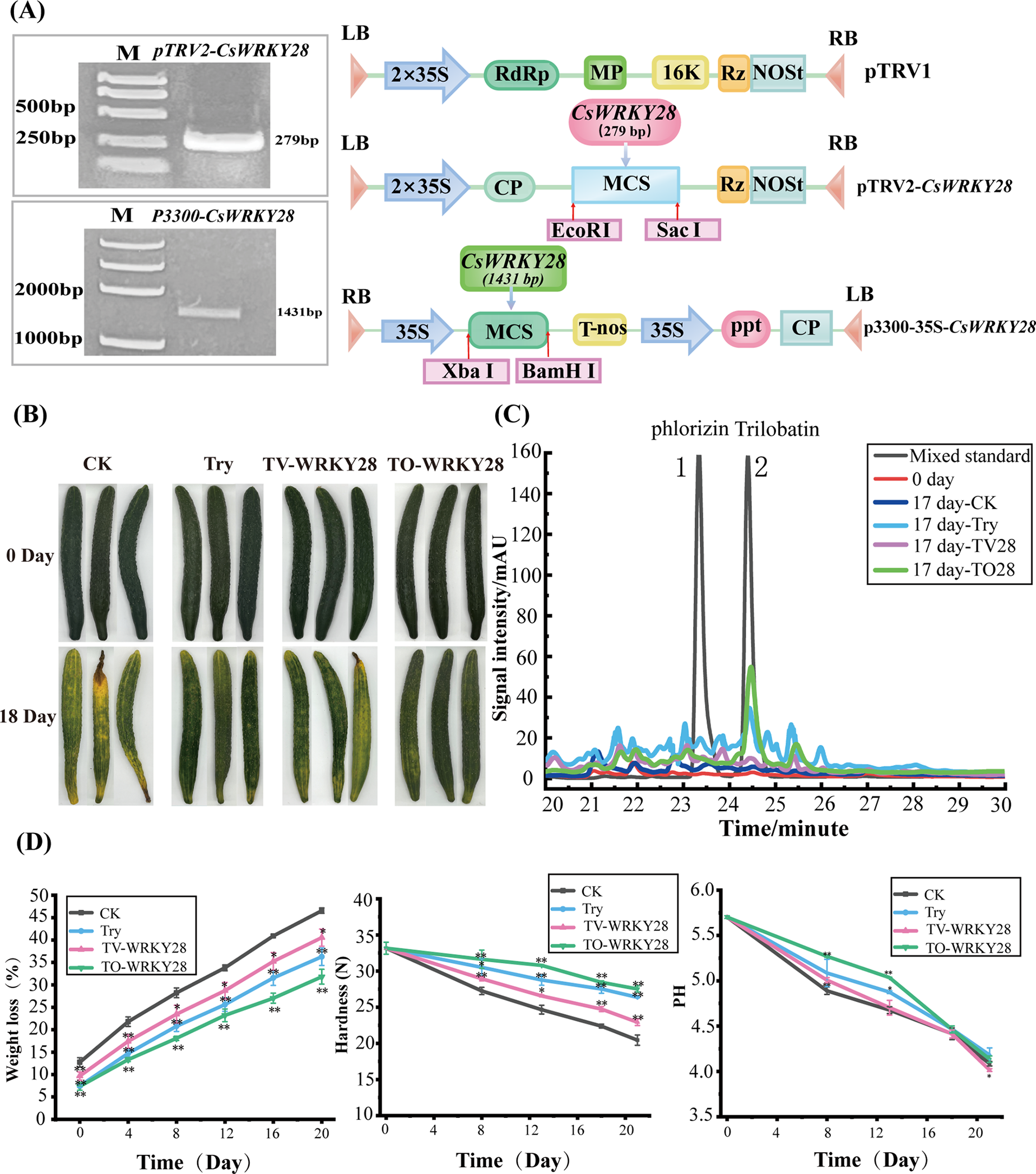
Figure 5: Depicted the construction and functional validation of the CsWRKY28 vector. (A) Schematic diagram of CsWRKY28 vector construction. The left side displays the electrophoresis image of the target gene cloning and the right side illustrates the connection mode between the target gene and vector. (B) The phenotypic results of C. sativus in different treatment groups. CK represented the blank control group, Try represented the trypsin treatment group, TV-WRKY28 represented the trypsin combined with WRKY28 silencing treatment group, and TO-WRKY28 represented the trypsin combined with WRKY28 overexpression treatment group. (C) HPLC determination of trilobatin content in C. sativus of different groups. (D) The results of C. sativus weight loss rate, hardness, and pH measurements
After being subjected to different treatments and stored for 18 days, the overall phenotypic changes of C. sativus were shown in (Fig. 5B). C. sativus from the CK group and TV-WRKY28 group gradually turned yellow and exhibited wrinkles. In contrast, C. sativus from the try group and TO-WRKY28 group appeared comparatively fresher, with the color maintaining stability. Over time, the C. sativus’ dehydration rate increased gradually. After 18 days of storage, the C. sativus dehydration rate was lowest in the TO-WRKY28 group (36.59%), followed by the trypsin-treated group (38.14%), TV-WRKY28 silenced group (42.79%), and the control group (43.72%). This trend was also reflected in the hardness measurements of the four C. sativus groups and the pH values after juicing (Fig. 5D), further indicating that the decay of C. sativus was delayed after treatment.
After trypsin treatment, the content of trilobatin in C. sativus was gradually increased and accumulated, and in the process affecting trilobatin biosynthesis, a key role may have been played by the transcription factor CsWRKY28. To verify this speculation, the trilobatin content in C. sativus samples after 17 days of storage in four groups was analyzed by HPLC. As shown in Fig. 5C, compared to fresh C. sativus at 0 h, the trilobatin content was found to have increased in all four groups after 17 days of storage, with the highest trilobatin content observed in the CsWRKY28 overexpression group, followed by the trypsin-treated group, CsWRKY28 silenced group, and CK group.
3.5 Verification of Downstream Target Genes of CsWRKY28
An analysis was performed on five central transcription factors of the WRKY family (Fig. 6A) using RT-qPCR, and it was demonstrated that their expression levels were consistent between RT-qPCR and RNA-seq data. A high degree of fit was shown by linear regression analysis between the data from the two methods, with an R2 value greater than 0.99 (Fig. 6B), confirming the reliability of the transcriptome data.
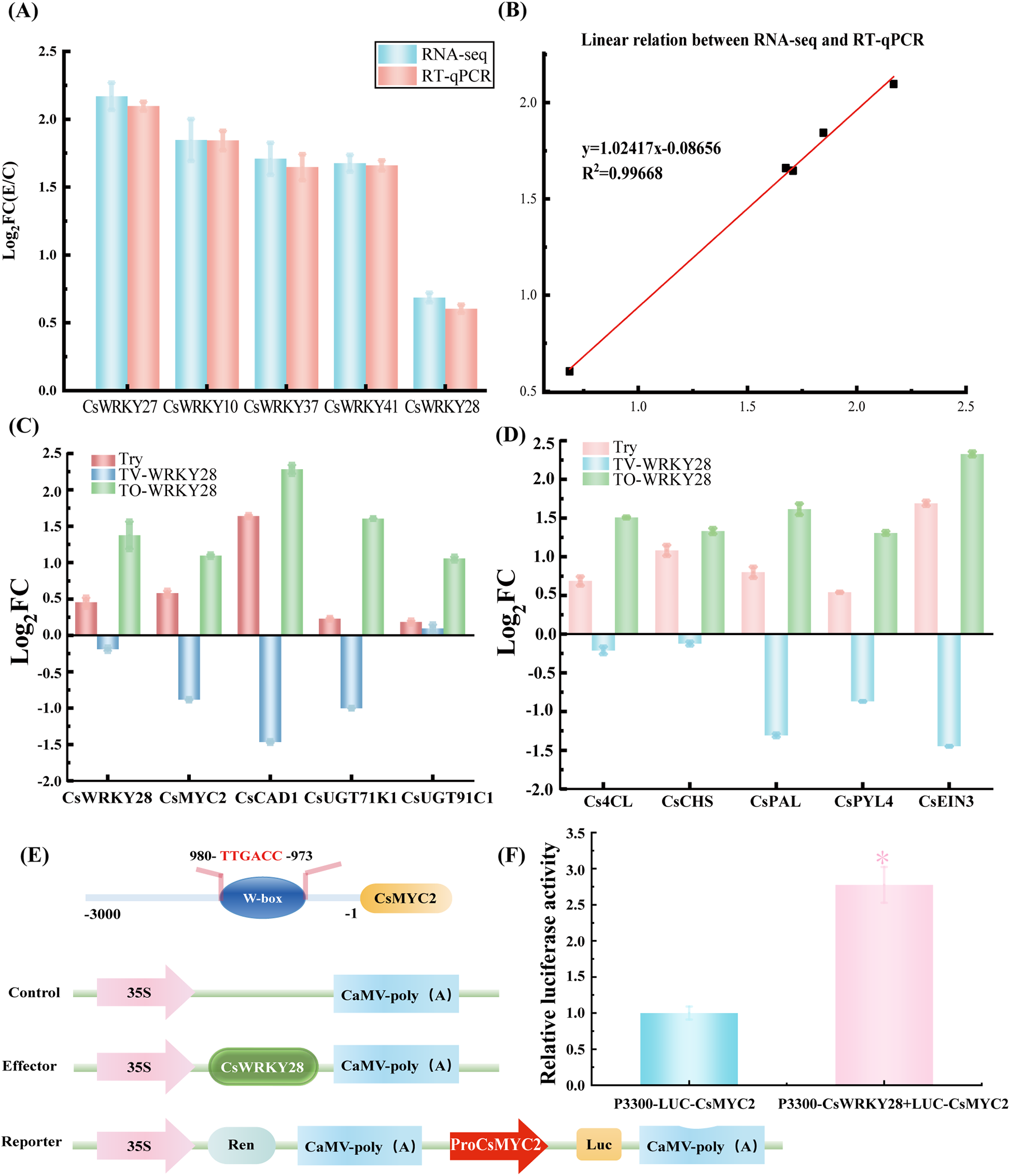
Figure 6: Validation of downstream target genes regulated by CsWRKY28. (A) Expression patterns of five genes determined by transcriptomics and RT-qPCR. (B) Linear relationship of selected gene expression rates. (C and D) Validation of expression levels of CsWRKY28 downstream target genes. (E) Schematic diagram of dual-luciferase vector construction. The upper part showed the schematic diagram of the W-box structure in the MYC2 promoter sequence, and the lower part displayed the vector construction. (F) Interaction assay between CsWRKY28 and CsMYC2 promoter
Predicted downstream target genes of CsWRKY28, identified from the co-expression network analysis of genes and metabolites along with the key genes involved in the phenylpropanoid metabolic pathway, were analyzed using RT-qPCR to examine changes in gene expression levels in C. sativus after different treatments (Fig. 6C,D). After 18 days of storage, with the gene expression changes in the control group C. sativus set as 0 for comparison, the expression levels of genes such as CsMYC2, CsUGT91C1, Cs4CL, and CsCHS were significantly upregulated in the trypsin-treated group, whereas in the CsWRKY28-silenced group, the expression of these genes was generally down-regulated. Conversely, the expression levels of these genes were further enhanced in the CsWRKY28-overexpression group, confirming the impact of the transcription factor CsWRKY28 on the biosynthesis pathway of C. sativus flavonoids.
CsMYC2 was one of the transcription factors regulating the biosynthesis of saponins and might have been one of the important downstream transcription factors regulated by CsWRKY28. In order to verify the activation of CsMYC2 by CsWRKY28, the promoter sequence of CsMYC2 was analyzed. According to the results from the PlantCARE database network tool, the W-box element capable of directly binding with WRKY TFs was found. Subsequently, the CsMYC2 promoter sequence containing the W-box region was inserted into the LUC0800 vector as a reporter. It was then transferred into GV3101 (pSoup) containing the helper plasmid. CsWRKY28 was inserted into the p3300-35S-Tons vector as an effector and transferred into Agrobacterium tumefaciens, as shown in Fig. 6E. The LUC/REN ratio was obtained through fluorescence detection (Fig. 6F). The results indicated that CsWRKY28 could bind to the promoter of CsMYC2, leading to an increased expression level of CsMYC2 upon overexpression of CsWRKY28 (Fig. 6C). This further verified the promoting effect of CsWRKY28 on CsMYC2 expression.
C. sativus, as a high-moisture vegetable, was losing water, leading to cell structure damage, soft texture, causing internal oxidative damage to fruits, and making them more susceptible to infections such as Fusarium and Verticillium wilt, and bacterial soft rot, resulting in significant economic losses [33]. Flavonoids, as a class of metabolites with biological activity in plants, had significant effects in scavenging free radicals and resisting oxidative damage [34]. In this study, the mechanism by which pancreatin activated the CsWRKY28 transcription factor in C. sativus to regulate the phenylpropanoid biosynthesis pathway was revealed, thereby promoting flavonoid biosynthesis.
KEGG enrichment analysis of all significantly different genes and metabolites using metabolomics and transcriptomics data revealed that the phenylpropanoid biosynthesis pathway was consistently enriched in the first place. This strongly suggested that trypsin may have been primarily enhancing C. sativus fruit resistance to aging and decay by regulating the phenylpropanoid biosynthesis pathway. Numerous studies have shown that phenylpropanoid compounds play important roles in regulating plant growth, maintaining redox homeostasis, and responding to biotic and abiotic stress [8]. Activation of the phenylpropanoid pathway could enhance the host’s resistance to invading pathogens [30]. The involvement of the phenylpropanoid pathway in regulating C. sativus disease resistance was identified by Wang et al. through metabolomics analysis [35]. Additionally, the phenylpropanoid secondary metabolism pathway could also be influenced by the biosynthesis of flavonoids and lignins, thereby enhancing plant resilience [36–38]. Phenylpropanoid compounds have been extensively studied due to their antibacterial properties and their role in signal transduction during defense responses [39,40].
In our metabolomics study, it was found that the biosynthesis of various flavonoid compounds in C. sativus could also be promoted by pancreatin, and 22 core metabolites were identified. Particularly, trilobatin was identified as the most crucial metabolite, with a significant increase of 32-fold. Trilobatin was an important compound in apples (Malus spp.) and had also been detected in sweet tea (Lithocarpus polystachyus rehd.) and crabapples [41–43]. The presence of trilobatin was detected in C. sativus peel by our laboratory, which not only expanded our understanding of trilobatin distribution but also provided important information for further exploration of the biological activity and potential applications of trilobatin in C. sativus. Therefore, in-depth studies on the metabolic pathways and regulatory mechanisms of trilobatin could provide a new perspective for elucidating the resistance and disease resistance of C. sativus.
Trilobatin was a natural dihydroxy flavonoid compound that was synthesized through a branch of the phenylpropanoid and flavonoid pathways, involving the participation of various enzymes and the coordinated regulation of multiple transcription factors [44–46]. It was demonstrated by studies in mango and cotton that WRKY transcription factors played a positive role in the accumulation of flavonoid compounds [47,10]. Our previous studies indicated that trilobatin was regulated by CsUGT91C1 and CsMYC2, with the W-box motif being capable of binding to WRKY transcription factors identified in the promoter sequence of CsMYC2, a relationship that was confirmed by dual-luciferase assays conducted in our laboratory. The results of this study further revealed that CsUGT91C1 and CsMYC2 were activated by the upstream transcription factor CsWRKY28, with CsMYC2 acting as an intermediate regulatory factor, thereby influencing the expression of downstream CsUGT91C1. These research findings provided important references for elucidating the regulatory mechanisms of trilobatin biosynthesis in C. sativus.
The functions of trilobatin in plants primarily include promoting growth, enhancing stress resistance, and improving nutritional components, and it is believed to regulate plant metabolic pathways. Research has indicated that it may also influence the synthesis of certain hormones [48]. In Wang et al.’s experiment [43], trilobatin, as a plant-derived sweetener, was produced in high concentrations in the young leaves of transgenic plants that overexpressed PGT2, resulting in a significantly sweeter taste compared to the control group. In our study, we observed that C. sativus in the CsWRKY28 overexpression group exhibited better overall conditions compared to other groups, with significantly delayed water loss, hardness, and pH changes. Additionally, we found a notable increase in trilobatin synthesis in the overexpression group compared to the others. Therefore, we propose that trilobatin enhances the stress resistance of C. sativus, maintaining their overall quality.
Through Dual-luciferase assays, CsWRKY28 was proven to regulate CsMYC2. Using RT-qPCR, the expression changes of the predicted downstream target genes of CsWRKY28 were analyzed, such as CsUGT91C1, Cs4CL, and CsCHS, under different treatments in C. sativus. The results indicated that the expression of these genes was generally downregulated in C. sativus with silenced CsWRKY28, while the expression levels were further elevated in the CsWRKY28 overexpression group, consistent with CsWRKY28 expression. This also confirmed the influence of the transcription factor CsWRKY28 on the flavonoid compound synthesis pathway in C. sativus. In our previous study [19], silencing of the CsMYC2 gene also significantly downregulated multiple enzyme-coding genes in the downstream phenylpropanoid pathway, particularly CsPAL, Cs4CL, and CsUGT91C1. It is evident that both CsWRKY28 and CsMYC2 can affect key enzymes in the flavonoid synthesis pathway. We concluded that trypsin treatment upregulates CsWRKY28, which then binds to CsMYC2 to regulate key downstream genes in the flavonoid pathway, such as CsUGT91C1, thereby achieving a preservation effect for C. sativus.
Furthermore, it was found that CsWRKY28 was enriched in the MAPK pathway, and it was likely to have been activated through this pathway. Based on this finding, a hypothesis was proposed: trypsin may have activated the expression of CsWRKY28 through the MAPK cascade pathway and activated the expression of downstream target genes CsMYC2, CsUGT91C1, and genes related to flavonoid biosynthesis, thereby promoting the biosynthesis of trilobatin (Fig. 7). Interestingly, the conclusions of Wang et al. were consistent with our findings, as they proposed a novel disease defense mechanism in cotton where the WRKY-MAPK pathway promoted GhMYC2-mediated flavonoid biosynthesis to defend against pathogen infection [49]. In summary, the biosynthetic pathway of trilobatin in C. sativus was elucidated in this study, demonstrating that trilobatin could serve as an important feature of flavonoid metabolism in trypsin-treated C. sativus. These findings not only contributed to a better understanding of trypsin-mediated resistance regulation mechanisms in C. sativus but also provided a theoretical basis for improving C. sativus quality and extending its shelf life.
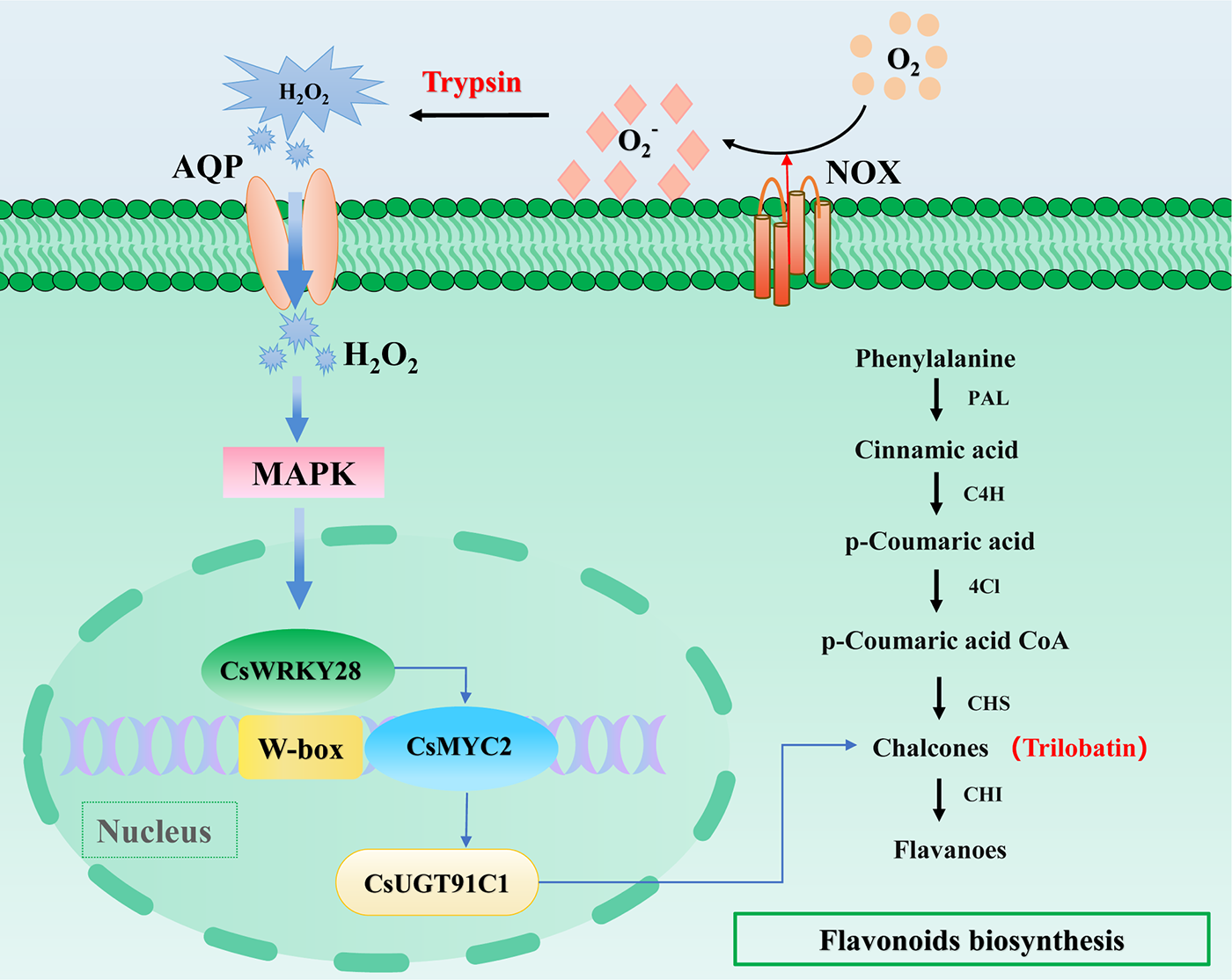
Figure 7: Process diagram of trypsin regulating CsWRKY28 to promote flavonoid biosynthesis
Trypsin protease treatment effectively delayed C. sativus water loss played a positive role in maintaining fruit firmness and normal morphology, induced the biosynthesis of phenylpropanoid compounds, enhanced fruit resistance, and further extended the storage time of C. sativus. Metabolomics analysis revealed that trypsin protease induced differential expression of metabolites in the phenylpropanoid metabolic pathway, with a significant increase in the content of flavonoids, including quercetin, among the regulated metabolites. Transcriptomics analysis identified the key transcription factor CsWRKY28 and downstream target gene CsMYC2, which were potentially regulated, and their regulatory relationship was experimentally validated through in vitro RNA interference, transient overexpression of target genes, dual-luciferase assay, and fluorescence semi-quantification, demonstrating the regulatory role of CsWRKY28 in promoting the expression of genes involved in flavonoid biosynthesis. HPLC results confirmed the increase in the content of quercetin in C. sativus. As a novel bio-preservative, trypsin protease activated the expression of CsWRKY28 and its downstream genes, improved endogenous flavonoid biosynthesis, enhanced fruit resistance, and thereby delayed C. sativus decay. Trypsin protease contributed to quality control during C. sativus storage, providing further theoretical support for bio-preservation technology.
This study preliminarily demonstrated that CsWRKY28 was a key transcription factor influenced by chymotrypsin in the biosynthesis of flavonoids via the phenylpropanoid pathway, and it could activate the expression of downstream target genes such as CsMYC2, thereby enhancing the biosynthesis of trilobatin.
Acknowledgement: We wish to thank professor Jianye Chen of South China Agricultural University for providing pTRV1 and pTRV2 VIGS vectors and Agrobacterium strain (GV3101) for this work. We are grateful for the free online platform of Majorbio I-Sanger Cloud Platform (www.i-sanger.com) (accessed on 17 October 2024).
Funding Statement: This work was supported by the National Key Research and Development Program of China (2017YFC1600802) and the National and Local Joint Engineering Laboratory of High Efficiency and Superior-Quality Cultivation and Fruit Deep Processing Technology of Characteristic Fruit Trees in South Xinjiang of China (No. FE202303).
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Enyan Chen, Xin Li; data collection: Jingyu Jia; analysis and interpretation of results: Jiaju Sun, Jie Wang; draft manuscript preparation: Xinxin Chen. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: All data and materials used in this research are publicly available. Raw sequence data from this study have been submitted to the NCBI sequence read archive under the BioProject accession (PRJNA 728562).
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Supplementary Materials: The supplementary material is available online at https://doi.org/10.32604/phyton.2024.057932.
References
1. Dong NQ, Lin HX. Contribution of phenylpropanoid metabolism to plant development and plant-environment interactions. J Integr Plant Biol. 2021;63(1):180–209. doi:10.1111/jipb.v63.1. [Google Scholar] [PubMed] [CrossRef]
2. Zhu Z, Chen R, Zhang L. Simple phenylpropanoids: recent advances in biological activities, biosynthetic pathways, and microbial production. Nat Prod Rep. 2024;41(1):6–24. doi:10.1039/D3NP00012E. [Google Scholar] [PubMed] [CrossRef]
3. Aktar S, Bai P, Wang L, Xun H, Zhang R, Wu L, et al. Identification of a BAHD acyltransferase gene involved in plant growth and secondary metabolism in tea plants. Plants. 2022;11(19):2483. doi:10.3390/plants11192483. [Google Scholar] [PubMed] [CrossRef]
4. Tao H, Zhao Y, Li L, He Y, Zhang X, Zhu Y, et al. Comparative metabolomics of flavonoids in twenty vegetables reveal their nutritional diversity and potential health benefits. Food Res Int. 2023;164:112384. doi:10.1016/j.foodres.2022.112384. [Google Scholar] [PubMed] [CrossRef]
5. Rajput R, Naik J, Stracke R, Pandey A. Interplay between R2R3 MYB-type activators and repressors regulates proanthocyanidin biosynthesis in banana (Musa acuminata). New Phytol. 2022;236(3):1108–27. doi:10.1111/nph.v236.3. [Google Scholar] [CrossRef]
6. Xie C, Wang K, Liu X, Liu G, Hu Z, Zhao L. Characterization and bioactivity of A-type procyanidins from litchi fruitlets at different degrees of development. Food Chem. 2023;405:134855. doi:10.1016/j.foodchem.2022.134855. [Google Scholar] [PubMed] [CrossRef]
7. Zou S, Zhuo M, Abbas F, Hu G, Wang H, Huang X. Transcription factor LcNAC002 coregulates chlorophyll degradation and anthocyanin biosynthesis in litchi. Plant Physiol. 2023;192(3):1913–27. doi:10.1093/plphys/kiad118. [Google Scholar] [PubMed] [CrossRef]
8. Pratyusha DS, Sarada DV. MYB transcription factors—Master regulators of phenylpropanoid biosynthesis and diverse developmental and stress responses. Plant Cell Rep. 2022;41(12):2245–60. doi:10.1007/s00299-022-02927-1. [Google Scholar] [PubMed] [CrossRef]
9. Cheng G, Wu D, Guo R, Li H, Wei R, Zhang J, et al. Chromosome-scale genomics, metabolomics, and transcriptomics provide insight into the synthesis and regulation of phenols in Vitis adenoclada grapes. Front Plant Sci. 2023;14:1124046. doi:10.3389/fpls.2023.1124046. [Google Scholar] [PubMed] [CrossRef]
10. Xiao S, Ming Y, Hu Q, Ye Z, Si H, Liu S, et al. GhWRKY41 forms a positive feedback regulation loop and increases cotton defence response against Verticillium dahliae by regulating phenylpropanoid metabolism. Plant Biotechnol J. 2023;21(5):961–78. doi:10.1111/pbi.14008. [Google Scholar] [PubMed] [CrossRef]
11. Yue M, Jiang L, Zhang N, Zhang L, Liu Y, Wang Y, et al. Importance of FaWRKY71 in strawberry (Fragaria × ananassa) fruit ripening. Int J Mol Sci. 2022;23(20):12483. doi:10.3390/ijms232012483. [Google Scholar] [PubMed] [CrossRef]
12. Zhang Y, Li B, Zhang M, Jia J, Sun S, Chen X, et al. Transcriptome analyses and virus-induced gene silencing identify HuWRKY40 acting as a hub transcription factor in the preservation of Hylocereus undatus by trypsin. J Food Biochem. 2022;46(12):e14437. [Google Scholar] [PubMed]
13. Li X, Zhang Y, Wu Y, Li B, Sun J, Gu S, et al. Lipid metabolism regulated by superoxide scavenger trypsin in Hylocereus undatus through multi-omics analyses. J Food Biochem. 2022;46(7):e14144. [Google Scholar] [PubMed]
14. Li X, Liu X, Yin Y, Yu H, Zhang M, Jing H, et al. Transcriptomic analysis reveals key genes related to antioxidant mechanisms of Hylocereus undatus quality improvement by trypsin during storage. Food Funct. 2019;10(12):8116–28. doi:10.1039/C9FO00809H. [Google Scholar] [PubMed] [CrossRef]
15. Li X, Zhang Y, Zhao S, Li B, Cai L, Pang X. Omics analyses indicate the routes of lignin related metabolites regulated by trypsin during storage of pitaya (Hylocereus undatus). Genomics. 2021;113(6):3681–95. doi:10.1016/j.ygeno.2021.08.005. [Google Scholar] [PubMed] [CrossRef]
16. Pang X, Zhao S, Zhang M, Cai L, Zhang Y, Li X. Catechin gallate acts as a key metabolite induced by trypsin in Hylocereus undatus during storage indicated by omics. Plant Physiol Biochem. 2021;158:497–507. doi:10.1016/j.plaphy.2020.11.036. [Google Scholar] [PubMed] [CrossRef]
17. Li X, Liu X, Pang X, Yin Y, Yu H, Yuan Y, et al. Transcriptomic analysis reveals hub genes and subnetworks related to ROS metabolism in Hylocereus undatus through novel superoxide scavenger trypsin treatment during storage. BMC Genom. 2020;21:1–13. [Google Scholar]
18. Pang X, Li X, Liu X, Cai L, Li B, Li X. Transcriptomic analysis reveals Cu/Zn SODs acting as hub genes of SODs in Hylocereus undatus induced by trypsin during storage. Antioxidants. 2020;9(2):162. doi:10.3390/antiox9020162. [Google Scholar] [PubMed] [CrossRef]
19. Wang J, Jia J, Sun J, Pang X, Li B, Yuan J, et al. Trypsin preservation: csugt91c1 regulates trilobatin biosynthesis in Cucumis sativus during storage. Plant Growth Regul. 2023;100(3):633–46. doi:10.1007/s10725-023-00962-w. [Google Scholar] [CrossRef]
20. Feng S, Zhang J, Mu Z, Wang Y, Wen C, Wu T, et al. Recent progress on the molecular breeding of Cucumis sativus L. in China. Theor Appl Genet. 2020;133:1777–90. doi:10.1007/s00122-019-03484-0. [Google Scholar] [PubMed] [CrossRef]
21. Gebretsadik K, Qiu X, Dong S, Miao H, Bo K. Molecular research progress and improvement approach of fruit quality traits in cucumber. Theor Appl Genet. 2021;134:3535–52. doi:10.1007/s00122-021-03895-y. [Google Scholar] [PubMed] [CrossRef]
22. Sheng C, Song S, Zhou W, Dossou SSK, Zhou R, Zhang Y, et al. Integrating transcriptome and phytohormones analysis provided insights into plant height development in sesame. Plant Physiol Biochem. 2023;198:107695. doi:10.1016/j.plaphy.2023.107695. [Google Scholar] [PubMed] [CrossRef]
23. Zhang N, Yu H, Yu H, Cai Y, Huang L, Xu C, et al. A core regulatory pathway controlling rice tiller angle mediated by the LAZY1-dependent asymmetric distribution of auxin. Plant Cell. 2018;30(7):1461–75. doi:10.1105/tpc.18.00063. [Google Scholar] [PubMed] [CrossRef]
24. Li C, Zhang J, Wu R, Liu Y, Hu X, Yan Y, et al. A novel strategy for rapidly and accurately screening biomarkers based on ultraperformance liquid chromatography-mass spectrometry metabolomics data. Anal Chim Acta. 2019;1063:47–56. doi:10.1016/j.aca.2019.03.012. [Google Scholar] [PubMed] [CrossRef]
25. Kong L, Chen J, Ji X, Qin Q, Yang H, Liu D, et al. Alcoholic fatty liver disease inhibited the co-expression of Fmo5 and PPARα to activate the NF-κB signaling pathway, thereby reducing liver injury via inducing gut microbiota disturbance. J Exp Clin Cancer Res. 2021;40:1–18. doi:10.1186/s13046-020-01782-w. [Google Scholar] [PubMed] [CrossRef]
26. Li G, Li Y, Yao X, Lu L. Establishment of a virus-induced gene-silencing (VIGS) system in tea plant and its use in the functional analysis of CsTCS1. Int J Mol Sci. 2022;24(1):392. doi:10.3390/ijms24010392. [Google Scholar] [PubMed] [CrossRef]
27. Tang Y, Yang X, Li H, Shuai Y, Chen W, Ma D, et al. Uncovering the role of wheat magnesium transporter family genes in abiotic responses. Front Plant Sci. 2023;14:1078299. doi:10.3389/fpls.2023.1078299. [Google Scholar] [PubMed] [CrossRef]
28. Feng C, Zhang X, Du B, Xiao Y, Wang Y, Sun Y, et al. MicroRNA156ab regulates apple plant growth and drought tolerance by targeting transcription factor MsSPL13. Plant Physiol. 2023;192(3):1836–57. doi:10.1093/plphys/kiad099. [Google Scholar] [PubMed] [CrossRef]
29. Feng Y, Lin Y, Li C, Tian P, Trapp S, Yu X. Integration of RT-qPCR analysis and grey situation decision-making model for evaluating the effects of plant growth regulators on the gene expression in rice seedlings under thiocyanate exposure. Sci Total Environ. 2021;783:146805. doi:10.1016/j.scitotenv.2021.146805. [Google Scholar] [PubMed] [CrossRef]
30. Liu X, Cui X, Ji D, Zhang Z, Li B, Xu Y, et al. Luteolin-induced activation of the phenylpropanoid metabolic pathway contributes to quality maintenance and disease resistance of sweet cherry. Food Chem. 2021;342:128309. doi:10.1016/j.foodchem.2020.128309. [Google Scholar] [PubMed] [CrossRef]
31. Liu J, Gao Y, Gong F, Hou F, Zhang Z, Cheng X, et al. The transcriptome and metabolome reveal stress responses in sulfur-fumigated Cucumber (Cucumis sativus L.). Front Plant Sci. 2021;12:778956. doi:10.3389/fpls.2021.778956. [Google Scholar] [PubMed] [CrossRef]
32. Wang Y, Cao R, Yang L, Duan X, Zhang C, Yu X, et al. Transcriptome analyses revealed the wax and phenylpropanoid biosynthesis pathways related to disease resistance in rootstock-grafted cucumber. Plants. 2023;12(16):2963. doi:10.3390/plants12162963. [Google Scholar] [PubMed] [CrossRef]
33. He Y, Wei M, Yan Y, Yu C, Cheng S, Sun Y, et al. Research advances in genetic mechanisms of major cucumber diseases resistance. Front Plant Sci. 2022;13:862486. doi:10.3389/fpls.2022.862486. [Google Scholar] [PubMed] [CrossRef]
34. Ferreyra MLF, Serra P, Casati P. Recent advances on the roles of flavonoids as plant protective molecules after UV and high light exposure. Physiol Plant. 2021;173(3):736–49. doi:10.1111/ppl.v173.3. [Google Scholar] [CrossRef]
35. Wang J, Tian P, Sun J, Li B, Jia J, Yuan J, et al. CsMYC2 is involved in the regulation of phenylpropanoid biosynthesis induced by trypsin in cucumber (Cucumis sativus) during storage. Plant Physiol Biochem. 2023;196:65–74. doi:10.1016/j.plaphy.2023.01.041. [Google Scholar] [PubMed] [CrossRef]
36. Chun HJ, Baek D, Cho HM, Lee SH, Jin BJ, Yun DJ, et al. Lignin biosynthesis genes play critical roles in the adaptation of Arabidopsis plants to high-salt stress. Plant Signal Behav. 2019;14(8):1625697. doi:10.1080/15592324.2019.1625697. [Google Scholar] [PubMed] [CrossRef]
37. Hou Y, Wang Y, Liu X, Ahmad N, Wang N, Jin L, et al. A cinnamate 4-HYDROXYLASE1 from Safflower promotes flavonoids accumulation and stimulates antioxidant defense system in Arabidopsis. Int J Mol Sci. 2023;24(6):5393. doi:10.3390/ijms24065393. [Google Scholar] [PubMed] [CrossRef]
38. Ma D, Reichelt M, Yoshida K, Gershenzon J, Constabel CP. Two R2R3-MYB proteins are broad repressors of flavonoid and phenylpropanoid metabolism in poplar. Plant J. 2018;96(5):949–65. doi:10.1111/tpj.2018.96.issue-5. [Google Scholar] [CrossRef]
39. Chen O, Deng L, Ruan C, Yi L, Zeng K. Pichia galeiformis induces resistance in postharvest citrus by activating the phenylpropanoid biosynthesis pathway. J Agric Food Chem. 2021;69(8):2619–31. doi:10.1021/acs.jafc.0c06283. [Google Scholar] [PubMed] [CrossRef]
40. Yadav V, Wang Z, Wei C, Amo A, Ahmed B, Yang X, et al. Phenylpropanoid pathway engineering: an emerging approach towards plant defense. Pathogens. 2020;9(4):312. doi:10.3390/pathogens9040312. [Google Scholar] [PubMed] [CrossRef]
41. Shi Y, Zhang Y, Luo H, Xu F, Gao J, Shi J, et al. Trilobatin, a natural food additive, exerts anti-type 2 diabetes effect mediated by Nrf2/ARE and IRS-1/GLUT2 signaling pathways. Front Pharmacol. 2022;13:828473. doi:10.3389/fphar.2022.828473. [Google Scholar] [PubMed] [CrossRef]
42. Wang Y, Hu S, Xiao F, Dong Z, Ye J, Zheng X, et al. Dihydrochalcones in sweet tea: biosynthesis, distribution and neuroprotection function. Molecules. 2022;27(24):8794. doi:10.3390/molecules27248794. [Google Scholar] [PubMed] [CrossRef]
43. Wang Y, Yauk Y, Zhao Q, Hamiaux C, Xiao Z, Gunaseelan K, et al. Biosynthesis of the dihydrochalcone sweetener trilobatin requires Phloretin Glycosyltransferase2. Plant Physiol. 2020;184(2):738–52. doi:10.1104/pp.20.00807. [Google Scholar] [PubMed] [CrossRef]
44. Ibdah M, Martens S, Gang DR. Biosynthetic pathway and metabolic engineering of plant dihydrochalcones. J Agric Food Chem. 2017;66(10):2273–80. [Google Scholar] [PubMed]
45. Zhang Y, Zhang W, Wang H, Shu C, Chen L, Cao J, et al. The combination treatment of chlorogenic acid and sodium alginate coating could accelerate the wound healing of pear fruit by promoting the metabolic pathway of phenylpropane. Food Chem. 2023;414:135689. doi:10.1016/j.foodchem.2023.135689. [Google Scholar] [PubMed] [CrossRef]
46. Zhuang W, Li Y, Shu X, Pu Y, Wang X, Wang T, et al. The classification, molecular structure and biological biosynthesis of flavonoids, and their roles in biotic and abiotic stresses. Molecules. 2023;28(8):3599. doi:10.3390/molecules28083599. [Google Scholar] [PubMed] [CrossRef]
47. Qian M, Wu H, Yang C, Zhu W, Shi B, Zheng B, et al. RNA-Seq reveals the key pathways and genes involved in the light-regulated flavonoids biosynthesis in mango (Mangifera indica L.) peel. Front Plant Sci. 2023;13:1119384. doi:10.3389/fpls.2022.1119384. [Google Scholar] [PubMed] [CrossRef]
48. Gaucher M, de Bernonville TD, Lohou D, Guyot S, Guillemette T, Brisset MN, et al. Histolocalization and physico-chemical characterization of dihydrochalcones: insight into the role of apple major flavonoids. Phytochemistry. 2013;90:78–89. doi:10.1016/j.phytochem.2013.02.009. [Google Scholar] [PubMed] [CrossRef]
49. Wang L, Guo D, Zhao G, Wang J, Zhang S, Wang C, et al. Group IIc WRKY transcription factors regulate cotton resistance to Fusarium oxysporum by promoting GhMKK2-mediated flavonoid biosynthesis. New Phytol. 2022;236(1):249–65. doi:10.1111/nph.v236.1. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools