
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Exploring Splicing Variants and Novel Genes in Sacred Lotus Based on RNA-seq Data
State Key Laboratory of Biocatalysis and Enzyme Engineering, School of Life Sciences, Hubei University, Wuhan, 430026, China
* Corresponding Author: Pingfang Yang. Email:
Phyton-International Journal of Experimental Botany 2023, 92(6), 1665-1679. https://doi.org/10.32604/phyton.2023.029482
Received 21 February 2023; Accepted 06 March 2023; Issue published 11 April 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Sacred lotus (Nelumbo nucifera) is a typical aquatic plant, belonging to basal eudicot plant, which is ideal for genome and genetic evolutionary study. Understanding lotus gene diversity is important for the study of molecular genetics and breeding. In this research, public RNA-seq data and the annotated reference genome were used to identify the genes in lotus. A total of 26,819 consensus and 1,081 novel genes were identified. Meanwhile, a comprehensive analysis of gene alternative splicing events was conducted, and a total of 19,983 “internal” alternative splicing (AS) events and 14,070 “complete” AS events were detected in 5,878 and 5,881 multi-exon expression genes, respectively. Observations made from the AS events show the predominance of intron retention (IR) subtype of AS events representing 33%. IR is followed by alternative acceptor (AltA), alternative donor (AltD) and exon skipping (ES), highlighting the universality of the intron definition model in plants. In addition, functional annotations of the gene with AS indicated its relationship to a number of biological processes such as cellular process and metabolic process, showing the key role for alternative splicing in influencing the growth and development of lotus. The results contribute to a better understanding of the current gene diversity in lotus, and provide an abundant resource for future functional genome analysis in lotus.Keywords
Supplementary Material
Supplementary Material FileAlternative splicing (AS) is a unique and versatile means of genetic regulation that determines the maintenance and elimination of a portion of coding sequence in the pre-mRNA. This gives rise to transcript isoforms resulting to diverse proteins that differ in chemical and biological activity [1]. In principle, the four basic and most common main subgroups of AS have been reported, including exon skipping (ES), alternative donor (AltD), alternative acceptor (AltA) as well as intron retention (IR) [2]. However, other types of AS have also been observed in Arabidopsis and rice [3]. In addition, there are “complex events” where different subtypes of AS can occur in a combinatorial way or one exon can be subjected to multiple AS types [4]. As a major mechanism for the improvement of transcriptome and proteome diversity, AS events have been found in numerous eukaryotes. In mammals, the AS plays a vital role in development, such as stem cell high self-renewal and multi-directional differentiation [5]. However, the distribution of AS events varies greatly between different species, and ES is the most prevalent type of AS in animals whereas IR is the most predominant in plants [3,4].
With the advent of the second generation sequencing technologies, transcriptome sequencing (RNA-seq) provides precise measurement levels for transcripts and their isoforms. These technologies offer a great opportunity for identifying novel genes and surveying the AS events in many species [6]. For instance, the analysis of deep developmental transcriptome in Amphimedon uncovered 101,062 previously unannotated protein encoding genes, greatly increasing the total number of genes by 25% [7]. Recently, the de novo transcriptome analysis of sugarcane identified a total of 164,803 genes from 275,018 transcripts, and a few of differentially expressed transcripts associated with the progress of leaf abscission during maturation were revealed through the comparative transcriptome analysis of the leaf abscission sugarcane plants (LASP) and leaf packaging sugarcane plants (LPSP) [8]. In addition, the transcriptome survey revealed approximately 61% of intron-containing genes with AS and ~150,000 splice junctions under normal conditions in Arabidopsis thaliana [9]. Transcriptome data analysis from fourteen maize tissues showed a 40% increase in the ratio of multi-exonic genes undergoing AS events. The role of AS in plants was previously unclear until recently when scientists showed its role in tissue identity and genotypic variation in Zea mays [10].
Lotus is an important food crop in China and other Asian countries due to its high nutrient content in its rhizomes and seeds [11]. In addition, the entire plant can also be used as medicine for the treatment of various diseases [12]. In 2013, the complete lotus genome was sequenced covering 86.5% of the 929 Mb genome [13]. However, the detected gene number is lower compared to Arabidopsis and rice having 92.32% and 95% genes out of the estimated genome size, respectively [14,15]. In addition, the distribution of AS events in lotus is not clearly understood. In 2013, Robert Vanburen et al. found 174 AS events distributed in approximately 161 genes, of which 62.6% were retained introns while the “complex events” contained more than one basic event detected through 454 pyrosequencing of a cDNA library [16]. However, around 17,000 AS events have been identified where 64% of the expressed genes are based on the RNA-seq data analyzed from four different cultivars, and alternative 5′ first exon was the main type of AS events accounting for 41.2% of the detected AS events [17]. The sample numbers used in previous studies are small, hence a more comprehensive investigation of AS events in lotus should be conducted.
To generate a better understanding of the genetic diversity and the complexity of alternative splicing events in lotus, 41 public RNA-seq data were analyzed from 13 tissues including root, rhizome, leaf, and flower. A total of 26,819 encoding genes, including 1,081 novel genes, were identified. Among them, abundant alternative splicing events were detected, with the IR being the most frequent event. In addition, Gene Ontology (GO) analysis was also conducted on the genes generating AS isoforms. In summary, the discovery of novel genes and the comprehensive survey of AS events in this research provide a resource for further understanding of gene diversity and a good foundation for the genetics research and breeding of lotus.
A total of 41 public transcriptome sequencing datasets from lotus were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/) [18–24]. Clean reads were mapped to the reference genome and the transcripts in each sample were assembled and quantified using the new tool hierarchical indexing for spliced alignment of transcripts (HISAT2 v2.1.0) and StringTie (v1.3.6), respectively, after removing low-quality sequences (Q < 20), adapter sequences, and the reads containing ploy-N [25]. The aligned transcripts from multiple RNA-seq data sets and the novel splice variants were assembled using TACO tool (v0.7.3) to reconstruct a consensus transcriptome [26]. Default parameters were used in the analysis tools. Then the longest potential candidate open reading frames (ORFs) within transcript sequences were identified through TransDecoder v2.0.1 (available at http://transdecoder.Github.io), and the weighted consensus gene structures were generated through the EVidenceModeler (EVM, v1.1.1) software, combining pre-existing ab intio predicted genes and generated transcript alignments [27].
2.2 Identification and Visualization of AS Isoforms
Both “internal” AS events with alternative splicing of internal exons and “complete” AS events with transcription start and end sites were retrieved from the GTF files generated using the TACO tool and astalavista-4.0 tool (version 2.2.1) having the key parameters as “-e [ASI]” and “-d 0”, respectively. The AS events were classified into five types, including ES, IR, AltD, AltA and other complex event by the AStalavista server (http://genome.crg.es/astalavista/) [28–30]. The AS events can be systematically browsed using IGV Browse [28]. In addition, the correlation analysis was performed between genes with AS event (AS gene) or genes with no AS event (no AS gene) and their gene CDS length, exon number, and expression quantity, respectively.
2.3 Functional Ontology of Genes
The consensus and novel genes were searched against Non Redundant (NR, http://www.ncbi.nlm.nih.gov/), Gene Ontology (GO, http://geneontology.org/), Clusters of Orthologous Groups of proteins (COGs, http://www.ncbi.nlm.nih.gov/COG/), EuKaryotic Orthologous Groups (KOG, ftp://ftp.ncbi.nih.gov/pub/COG/KOG) databases, respectively.
The functionalities of genes with AS were identified using BLASTX against Non Redundant (NR, http://www.ncbi.nlm.nih.gov/) and the Gene ontology (GO, http://geneontology.org/). Information was retrieved by mapping NR to GO and analyzing it using agriGO (http://bioinfo.cau.edu.cn/agriGO/analysis.php) [31], with default parameters, to obtain the GO plot and the corresponding values for the biological process, molecular function, and cellular component.
3.1 Summary of All RNA-Seq Data Set
RNA-seq data sets collected from a variety of cultivated species and organizations were downloaded from the NCBI database to capture a comprehensive and representative transcriptome of lotus. In this research, 41 public RNA-seq samples were analyzed, including apical buds, root, leaf, petiole, rhizome, and different stage rhizome internode from different cultivars and ecotypes. These cultivars included many ‘BG’ (temperate lotus), ‘WR1’ (tropical lotus), “China Antique”, wild flower lotus (WFL) plants and cultivated rhizome lotus (CRL) plants. A total of 833,789,625-base single reads and 581,210,638-base paired reads from 41 lotus RNA-seq data sets were mapped to the reference genome of lotus after filtering and removing low quality sequences and adapters [13]. A total of 800,013,401 and 529,377,632 high-quality clean reads were generated from the two sequences, respectively. The mapping rates of the 41 sample are above 80%, while the mean mapping ratio is 93.47%, and six samples had a mapping rate of lower than 90%, where the least recorded rate is 82.18% (Fig. S1). Approximately 63,869 novel transcripts have been assembled from 41 samples transcriptome sequencing. Using TransDecoder, a total of 57,074 transcripts (89.36%) were predicted as having a completed ORF box for protein translations with at least 100 amino acids.
3.2 Features of Consensus and Novel Genes
A total of 26,819 consensus genes (EVM genes) are predicted using EVM tool with parameters having a segment-size of 100,000 bp and an overlap-size of 10,000 bp, of which 19,869 comprise the expression genes. The average CDS length of these genes is 1,144 bp with sizes ranging from 100 to 20,973 bp, and around 86.67% gene CDS length is less than 2,000 bp while the longest CDS length is 20,973 bp (Fig. 1). The 26,819 genes were compared to the pre-existing predicted genes (NNU genes) in the reference genome. 24,475 EVM genes (91.26%) have a one-to-one relationship with NNU genes while 654 EVM genes contain one EVM gene corresponding many NNU genes. In addition, 260 and 41 EVM genes can correspond to one or more than one NNU genes, respectively. However, there are still 529 NNU genes having no transcripts in the assembled transcriptome, indicating the inaccuracy of the pre-existing predicated NNU genes (Table 1).
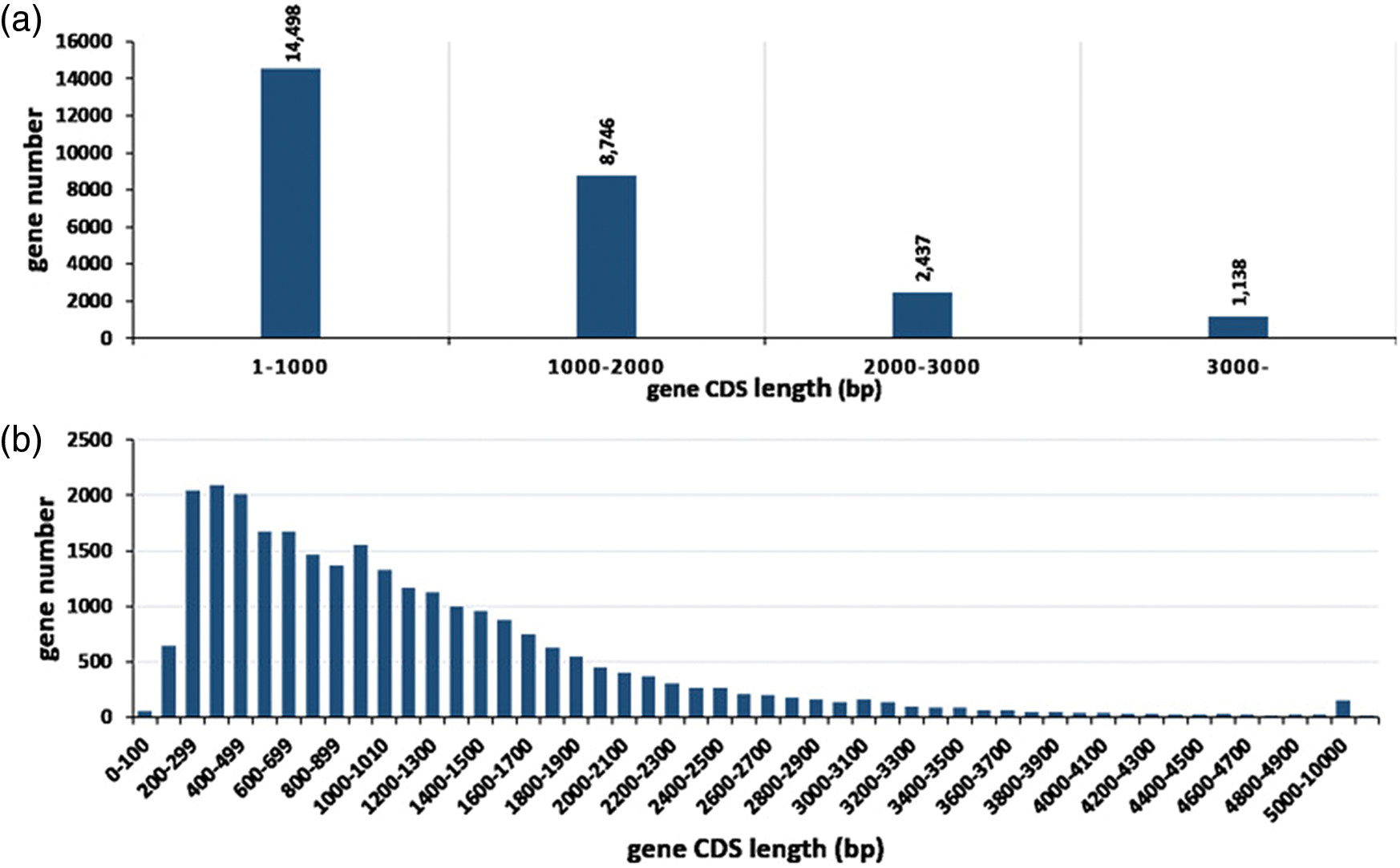
Figure 1: Distribution of gene coding sequence (CDS) length of consensus genes. (a) General information of CDS distribution. (b) Shows a detailed distribution of smaller CDS

To improve the genomic annotation information, 1,081 novel genes were annotated on NR, COGs, KOG and GO databases, respectively. 1,063 novel genes out of the total novel genes have homologous genes with other species in the NR database. And 1,032 (97.08%) could be identified with an annotated CDS region against the draft lotus genome with a cut off E-value of 1e-10 though, 21 novel genes aligned to Vitis vinifera, Cucumis sativus, Elaeis guineensis and other species related to aquatic features of lotus (Fig. 2a). In addition, the functional distribution of novel genes was estimated using COG and KOG classification, and about 363 and 718 novel genes have been classified 21 and 24 terms, respectively. In COG classification, most novel genes are distributed in the general function prediction terms, though the novel gene involving in the four terms of RNA processing and modification, cell motility, extracellular structures and nuclear structures is absent. In contrast, the KOG functional classification of consensus genes is more comprehensive. A total of 718 novel genes are classified into 24 terms and most genes are categorized under posttranslational modification, protein turnover, chaperones and intracellular trafficking, secretion, and vesicular transport excluding the term for the general function prediction. This highlights the role of most novel genes involvement in the protein synthesis and post-processing (Figs. 2b and 2c).
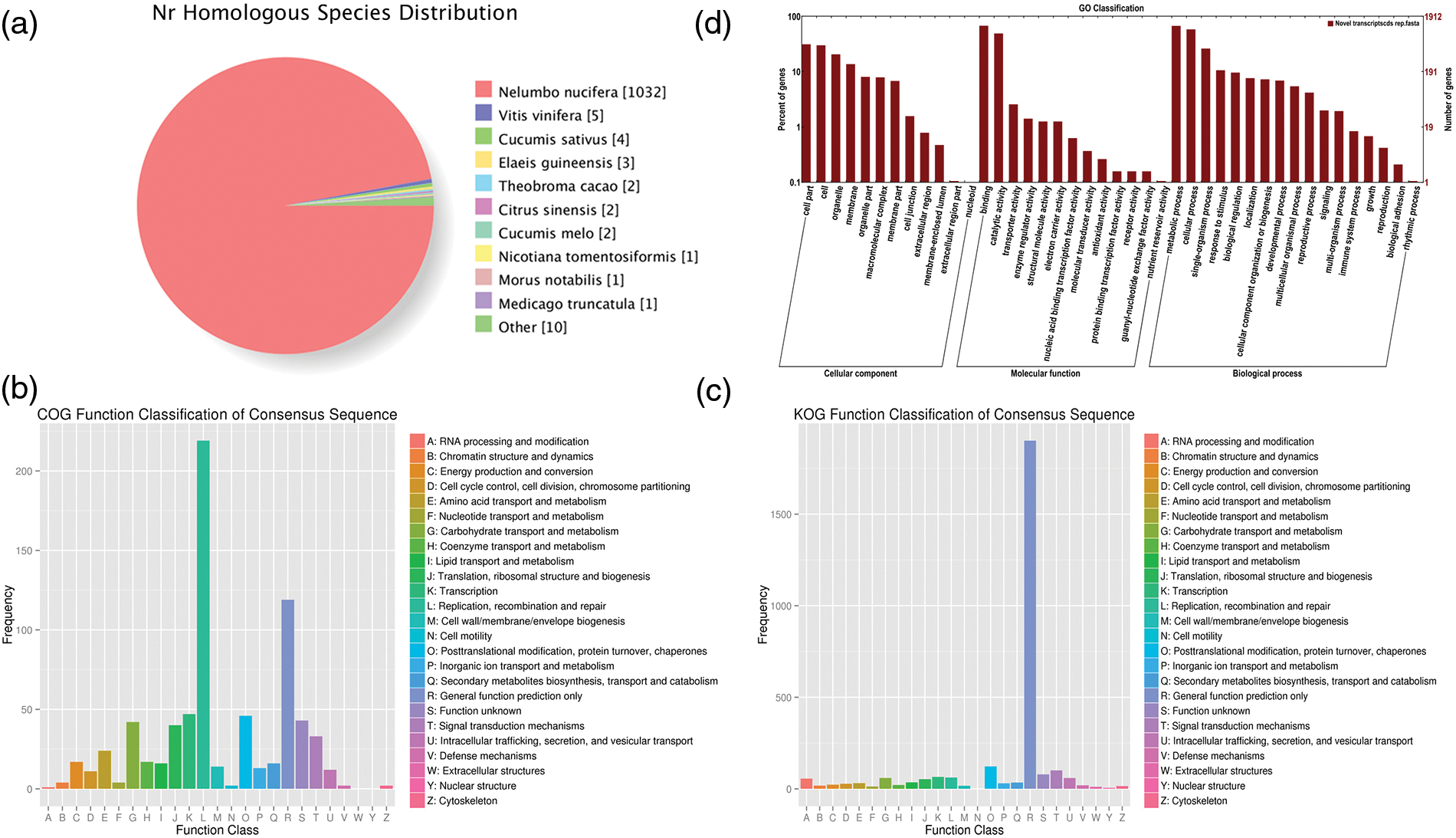
Figure 2: The annotation of 1,081 novel genes in NR (a), COG (b), KOG (c) and GO (d). The number in brackets in figure a is the number of genes having homologous gene within the species
Further classification of all the novel genes was carried out according to their GO categories, i.e., biological processes, molecular functions and cellular component (http://www.geneontology.org/). Among 1,078 novel genes having a BLASTX hit against NR/COG/KOG/Pfam/NT database with an identity of 80%, 618 had more than one GO annotation. A total of 3,147 GO terms were extracted and classified further. Cellular Component classification showed 48% cellular component proteins, 29% organelle and organelle part proteins, 15% membrane proteins separately, and diverse distributions in other subcellular components. Molecular function analyses indicated 47% potential proteins encoded by the specific genes having catalytic or enzyme activity, including oxidoreductase activity, kinase activity, ATPase activity and transmembrane transporter activity, respectively. Approximately 41% proteins had the molecular binding function including ion binding, DNA binding and RNA binding. In addition, 1,952 GO identifiers involving in a variety of biological processes are present in metabolic process, cellular process, single-organism process, biological regulation, response to stimulus, localization, etc. (Fig. 2d).
3.3 Detection and Classification of AS Events
In this study, tentative consensus transcripts were aligned to the reference genome, which could improve the original genome structure and annotation information. To increase the accuracy of the outputs and decrease of the ratio of false-positive potential AS isoforms, complete internal AS events were detected and classified according to the four types of AS events in the lotus genome based on the comparatively comprehensive putative map. A total of 14,070 complete and 19,983 internal AS events were obtained from 30% of all consensus expression genes corresponding to those reported earlier in rice (48%), Arabidopsis (61%), and maize (40%) [2–4]. In addition, the frequency of AS events in lotus is also comparable with that in Lotus japonicus (~30%) and Medicago truncatula (~28%) [32]. Among them, 445 complete AS also contain the internal AS events.
AS events were retrieved from the GTF files produced by the TACO tool used the online software ASTALAVISTA [26,28] to obtain the AS events patterns of lotus. The IR was observed to be the most predominant subtype of AS events accounting for 32.72%, followed by AltA (3,603, 17.64%), AltD (2,790, 13.63%) and ES (1,998, 9.78%). In addition, the second predominant type of AS with 5,352 “complex AS events” had more than one basic type of AS events in sacred lotus [3,33]. From these figures above, the IR and “complex AS events” are the most prevalent AS types while the ES is minor event in sacred lotus. Moreover, the use of AltA is higher than the use of AltD corresponding to initial study (Figs. 3a and 3b) [34]. These results are consistent with the previous study conducted by Robert VanBuren et al considering the IR as the most common AS type accounting for 62.6% of the 174 AS events based on expressed sequence tags (ESTs) analysis in 2013. Recently, research shows alternative 5′ first exon as the most dominant type of AS events was in contrast to IR in lotus accounting for 5.01% of the AS events. This contradiction could be caused by the space and time expression specificity of AS types and the various sequencing depth of four different cultivars by Yang et al. compared with the previous study [16,17].
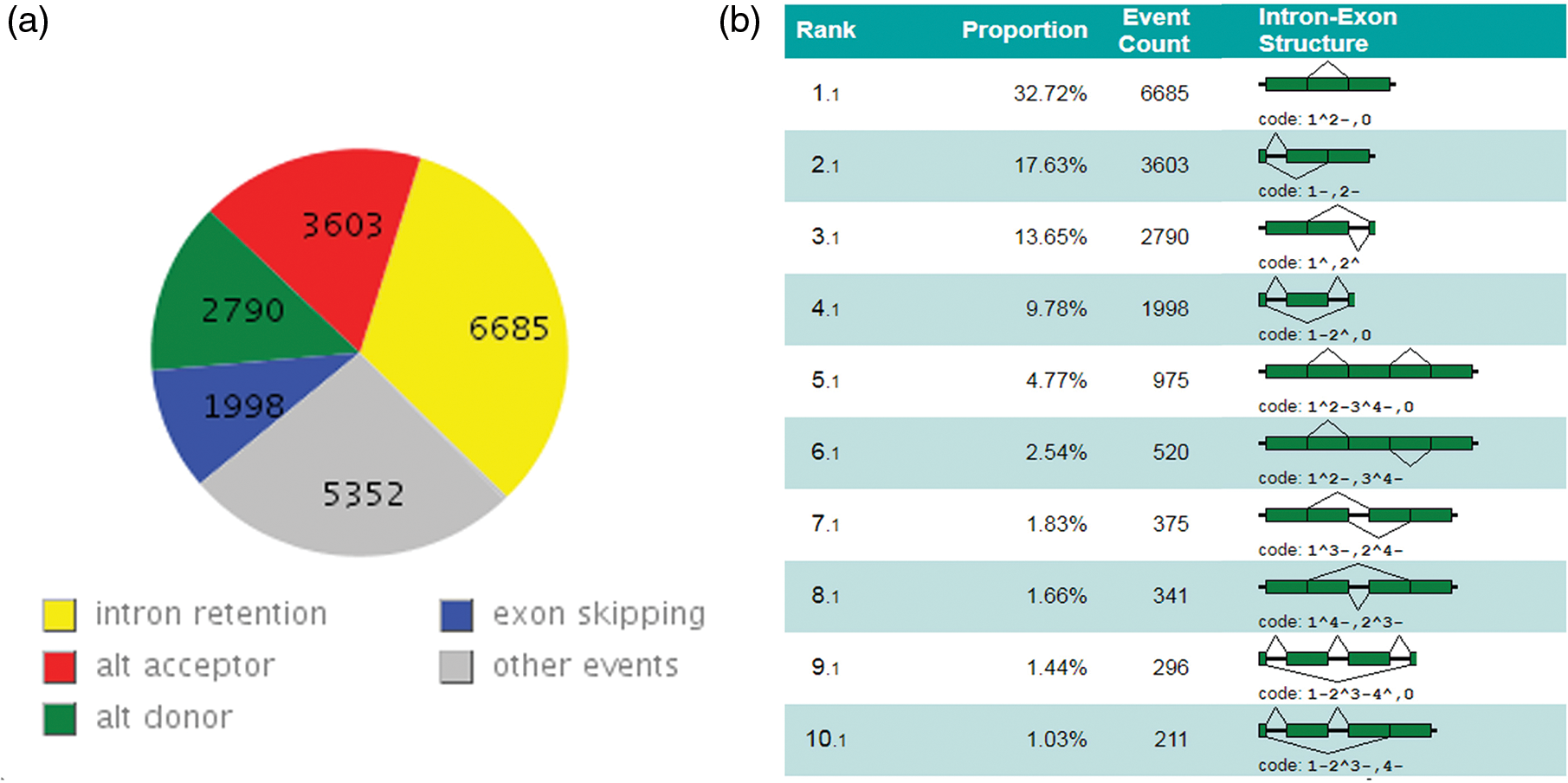
Figure 3: Landscape of AS events in sacred lotus. (a) The number before the comma represents the type of AS event, and the number after the comma represents the proportion of the AS event in all the AS events. (b) Detailed information of different types of alternative splicing is shown in figure. 1.1 Intron retention. 2.1 Alternative acceptor. 3.1 Alternative donor. 4.1 Exon skipping. From 5.1 to 10.1 and other trans forms (not shown) categorized as complex events. In the figure, exons and introns are represented by dark green boxes and solid lines, respectively, and broken lines indicate splicing options
The abundance of IR (~33%) is higher than other types of AS events in sacred lotus, while ES has the least AS occurrence as evidenced in other plant studies. Researches conducted on Soybean, Medicago, Rice, Common Bean, Poplar, Tomato, Grape, Brachypodium distachyon, Sorghum bicolor, and even in Volvox carteri shown the predominance of IR AS subtype. Arabidopsis had the largest quantity of IR event category (65.3%) based on an extensive transcripts profile analysis [9,35–41]. The wide distribution of IR type in plants supports the intron definition model, where the introns area is identified directly by the splicing machinery spliceosomes in pre-messenger RNA splicing plants, corresponding to the exon-definitive model always seen in animals. In this case, the frequency of ES events is much higher than IR events [42]. IR evidence in plants indicates the transcript generation form IR is a product of incomplete splicing which is saved as a transcript having the translation potential in the cytoplasm of the cell, resulting in different fates of a mRNA through the insertion in the coding region of a pre-mature stop codon inside the transcript frame [40,43]. The results obtained in this study increased the landscape of the AS events by incorporating a much higher number of different-length transcripts. Intron retention is a common AS form among four basic AS events in plant. Despite the great difference in the proportion of AS genes and the ratio of the four foremost prevalent types in sacred lotus using expressed sequence tag mapping and deep high-throughput transcriptome sequencing, respectively [16,17].
3.4 Features of Exons Number, Gene Length and Gene Expression Level of Genes with AS
The relationship between AS event number per gene and the genetic characteristics is analyzed statistically with Perl algorithm scripts, such as the numbers of exon in the most-exon transcripts of AS genes, the lengths of coding sequence of AS genes and the expression level of AS genes. This calculation can help to obtain an overview of the distribution of AS event frequency in all EVM expression genes and its relationships with other individual genomic characteristics, following the alignment of the transcripts to the reference genomic sequences. In our study, a total of 5,881 “complete” AS genes and 5,878 “internal” AS genes were observed, respectively, accounting for 30% and 29.58% of all expression genes. Further research in our study indicates the association of the two types AS genes’ frequency with of most-exon numbers, the length of CDS and the coefficient association level for gene expression. The results obtained in this study are consistent with previous findings in maize, teosinte, soybean, which show a positive correlation with the total AS events to the exon number and gene length [44,45].
EVM expression genes were classified into four groups: high AS gene (more than eleven AS events per gene), middle AS gene (six to ten AS events per gene), low AS gene (one to five AS events per gene), and non-AS gene (no AS events in the gene) to confirm the associations. A number of 5,482 low and 324 middle AS genes were identified, respectively, out of the 5,881 “complete” AS genes, accounting for about 98.72% of total EVM expression AS genes. The highest frequency transcripts generated through alternative splicing had 43 genes among the 75 high AS genes. In contrast, the frequencies of middle AS genes and high AS genes involving “internal” AS events are higher than “complete” AS genes while the transcript number of most “internal” AS genes are 110, constituting to more than “complete” AS genes (44). The average frequency of the “internal” AS genes (3) is slightly higher than “complete” AS genes in sacred lotus (Fig. S2).
The gene internal features were compared among high AS gene, middle AS gene and low AS gene involving two AS models. As shown in Fig. 4, AS events per gene between middle AS gene and low AS gene show a slight positive correlation with the three individual gene internal features. This is consistent with the analysis above, though there is an inverse correlation between AS frequency, exon number and gene length observed in the high AS gene due to the limited number of AS genes detected. Regard less of the type of AS frequency among EVM expression genes, the relationship between AS frequency and coefficient of variation of gene expression FPKM value, indicates the tissue expression specificity in AS genes similar to other plants. Recently, more splice variants identified per gene have been generated within fruit tissues although the AS frequencies are similar among early fruit growths, flowers and seedlings [40,44].
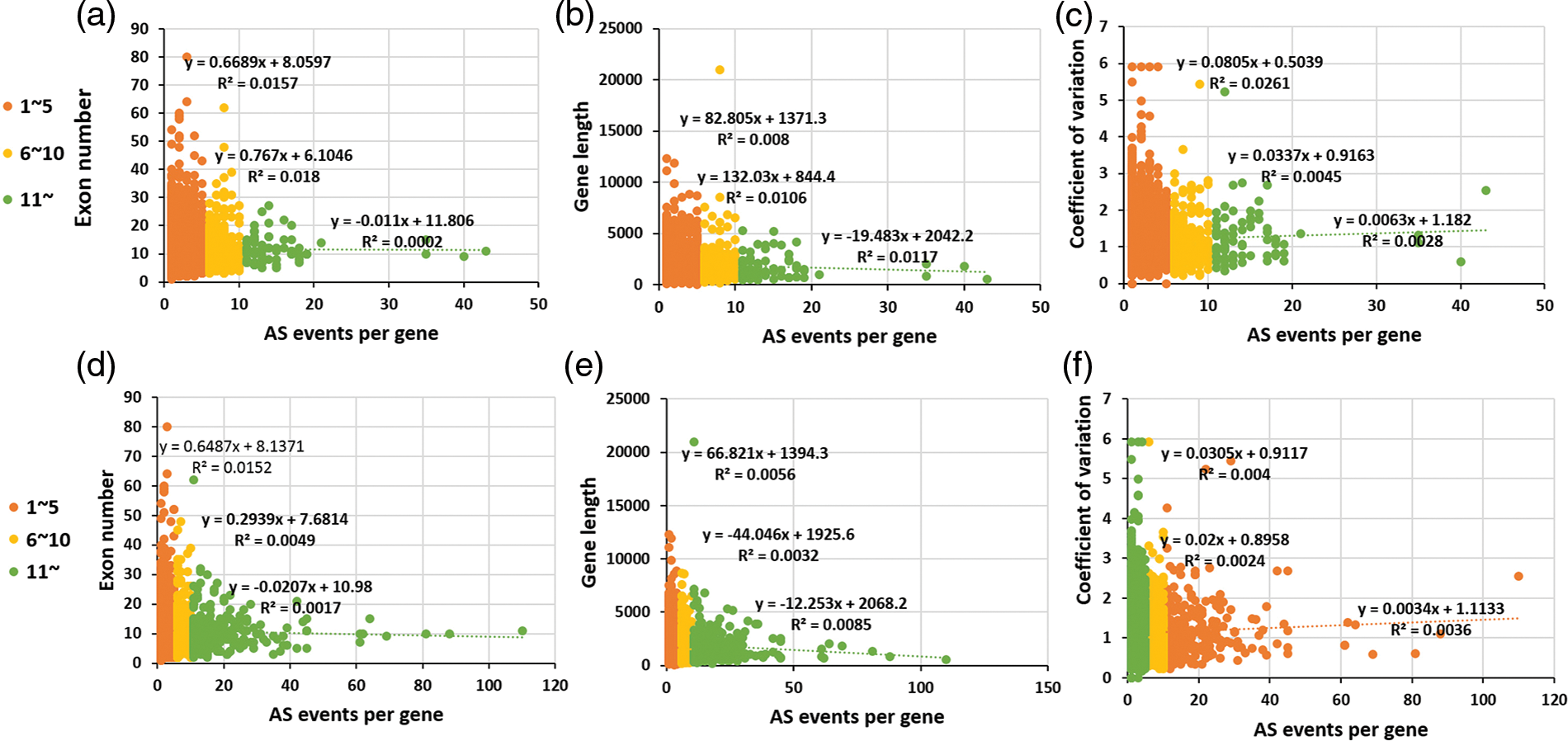
Figure 4: The comparison of the gene internal features among high AS gene, middle AS gene and low AS gene involved in the two AS models. The upper figures (a–c) shown the “complete” AS genes, while the below ones (d–f) shown “internal” AS gene by alternative splicing
3.5 Functional Ontology of AS Genes
AS changes the diversity of protein-parts by increasing the abundance of mRNAs generated from the genome with a significant functional effect. In recent years, tremendous efforts have been directed towards functional characterization of the majority of known plant alternative splicing events to comprehend the mechanistic overview of the biological functional impact of the AS events. As shown in O Kelemen’ review, AS events emerged as a crucial central element with molecular versatility and widespread usage that is involved in most biological functions regulating gene expression [46].
The AS and no AS genes were classified on the basis of their GO categories, i.e., molecular functions, cellular component, and biological processes to further improve the annotation information of AS genes in lotus. Compared to the no-AS genes, the distribution of AS genes function is higher on the cellular process and metabolic process, catalytic activity and binding properties in biological processes and molecular functions, respectively (Fig. 5).
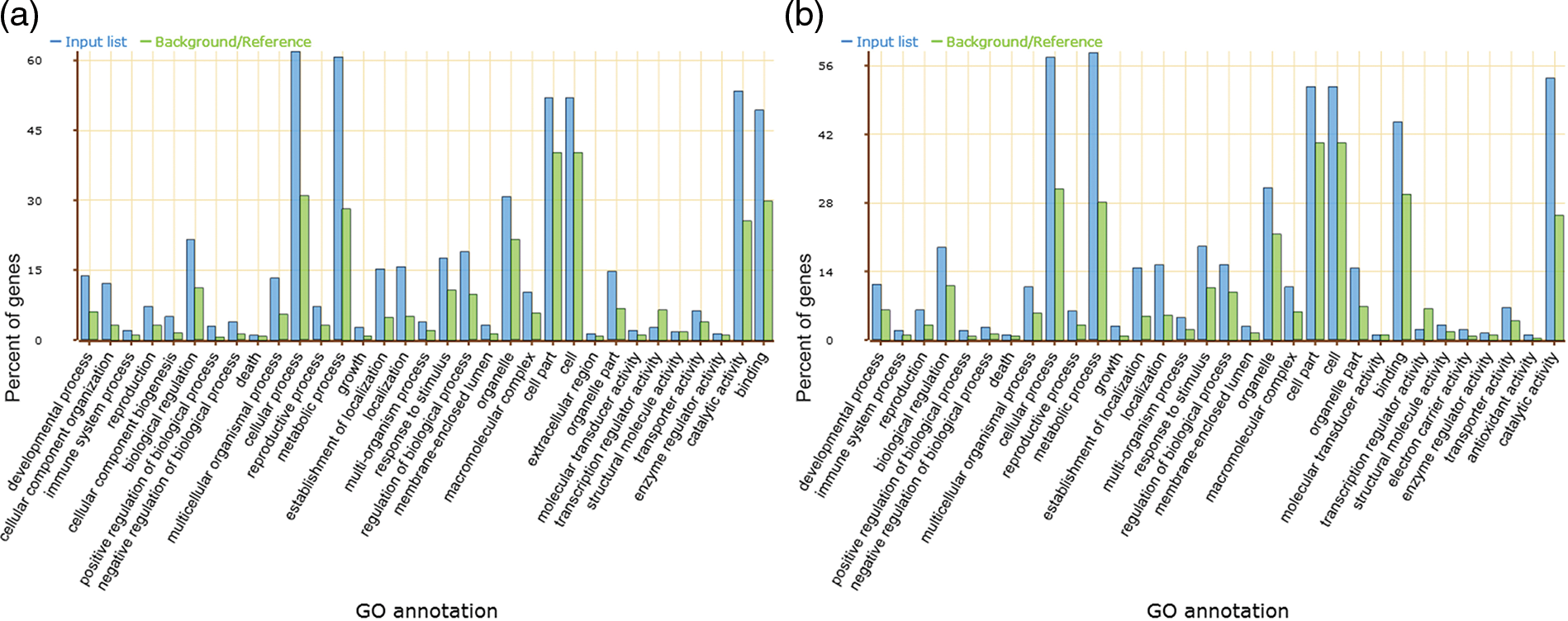
Figure 5: Function ontology diversity. Biological process, cellular component and molecular function for AS genes (a) and no AS genes (b), respectively
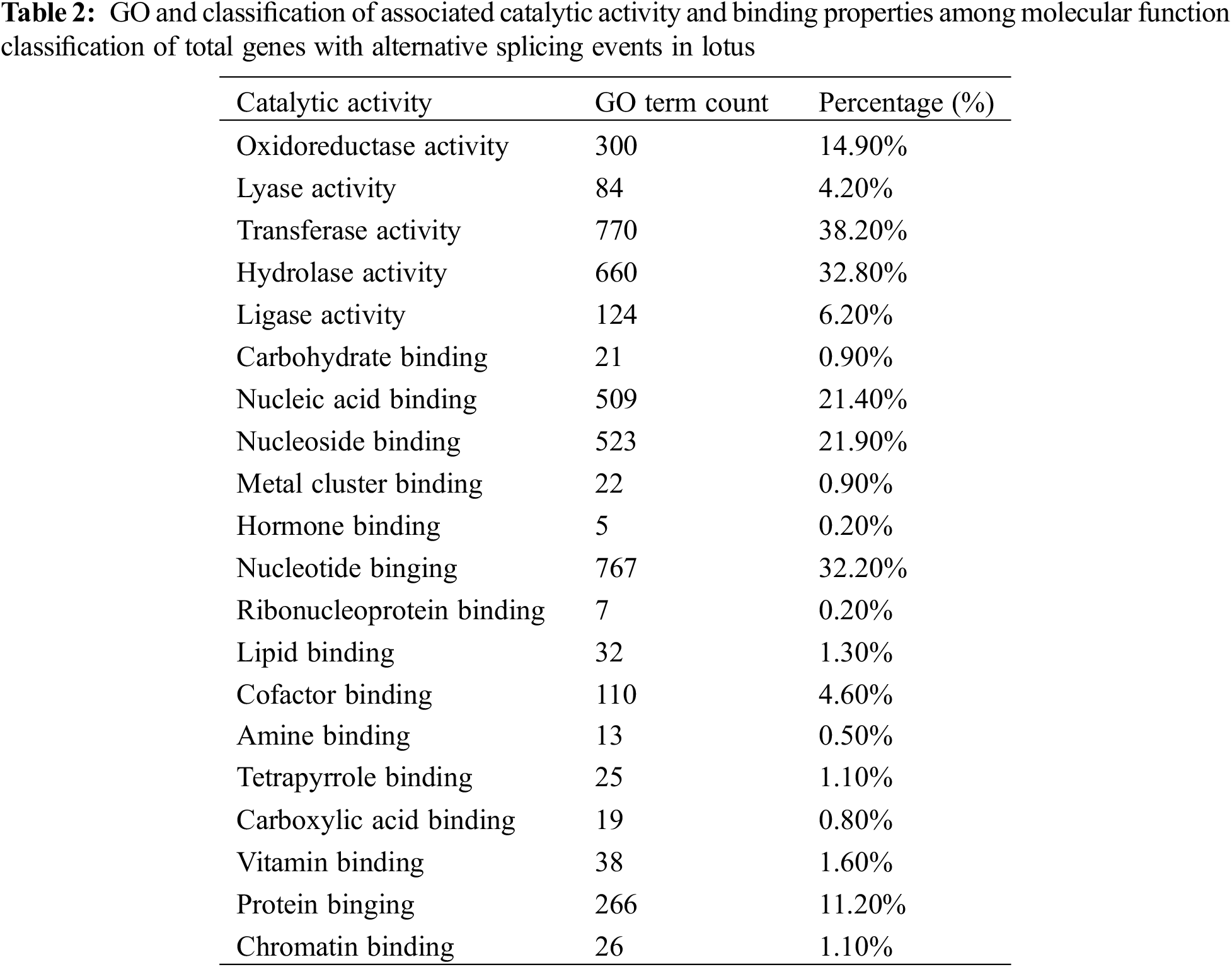
Gene ontology classification for molecular function ontology for 3,657 AS genes accounted for 17.5% of the total 20,853 annotated genes, indicating that approximately 45.2% and 41.7% were associated with catalytic activity and binding properties, respectively. Majority of the functionally annotated genes in the detailed dissection of each classified group in the catalytic activity ontology were related to transferase activity (38.2%), hydrolase activity (32.8%) and oxidoreductase activity (14.9%). 14.1% of the genes were associated with ligase activity, lyase activity in these sub-groups (Table 2).
As shown in recent studies, AS plays a key role in determining species- and tissue-specific differentiation models while the transcripts generated through splicing can participate in a variety of signaling pathways and respond to transcription factors and chromatin structure [47]. Recently, results from several functional analyses have shown the effect of AS on plant biological and biochemical processes such as histone modification [48], DNA-binding preference of transcription factor [49], mRNA processing [50]. The effect was evidenced in plants through high-throughput sequencing for transcriptome, which alternative splicing had been proposed to play a fundamental role in growth, development, and stress responses of plant, such as internal circadian clock, cell fate, plant defense, and tolerance/sensitivity to stress [51].
In sacred lotus, nucleotide binding proteins are the most prevalent proteins affected by alternative splicing (32.20%) though they play a significant role in in disease resistance and activating of downstream signal response, such as the genes generated from the nucleotide binding site-leucine-rich repeats sequences by AS in rice [52]. The other types of sub-binding specialty among annotated AS genes comprise the nucleoside binding (21.90%), nucleic acid binding (21.40%), protein binding (11.20%), cofactor binding (4.60%), vitamin binding (1.60%) and lipid binding (1.30%), and the other types of binding proteins (Table 2).
GO analysis for the category of biological process indicated the prevalent composition association to cellular process and metabolic process (Fig. 5). The cellular process was anatomized and about 80.3% of the genes were classified into the cellular metabolic process, while the primary metabolic process accounted for 80.2% of the metabolic process. In addition, a large number of sub-distributions in other processes have been observed in sacred lotus including the macromolecule metabolic process, the biosynthetic process, the catabolic process, the nitrogen compound metabolic process, the regulation of metabolic process, the secondary metabolic process, the organophosphate metabolic process and the negative regulation of metabolic process.
A total of 4,067 GO terms were classified under the cellular component part and about 46.30% cell part proteins, 27.40% organelle proteins, and variable classification in other subcellular components were observed. These genes are involved in a variety of biological processes including transport, biosynthesis, metabolic, and stress responses.
The results from gene functional ontology inspection of the AS gene list and non-AS gene list in this research proposes the involvement of AS in supporting metabolism, physiology, development, resistance of biotic and abiotic stresses, and immune system. These processes regulate lotus biological characteristics and phenotypes as evidenced in other plants. Previously, the involvement of AS events was demonstrated in the floral promoter FCA where the biological pathways promote the floral transitions process in fca mutants of Arabidopsis [53]. Similarly, alternative spliced transcripts of Rj2 gene in soybean have the ability to restrict the nodulation of specific rhizobial strains [54]. In 2011, Seo et al. proposed two splice variants of INDERMINATE DOMAIN 14 (IDD14) transcription factor gene in Arabidopsis produced a self-restrained regulatory loop regulating starch metabolism in response to cold stress by producing a competitive inhibitor [55]. In addition, alternative splicing plays a vital role in linking the circadian clock to temperature response in Arabidopsis through the splice variants CCA1β and the CCA1β production of the self-regulated CIRCADIAN CLOCK-ASSOCIATED1 (CCA1), which is part of the key clock components associated with low temperature acclimation in Arabidopsis [56]. In lotus, we observed total GO classifications enriched in cellular process (GO:0009987) and metabolic process (GO:0008152) in the total AS genes (Fig. 5), providing a strong evidence on its regulating the biosynthetic pathway and improving the gene protein abundance for variable subsets through alternative splicing. However, a detailed understanding of functions of all AS genes still remain uncovered in sacred lotus. Lotus is a model aquatic plant for the ancient dicotyledonous system, and a thorough experimental investigation are required to verify the functionally annotated AS events identified in this study and previous studies to provide substantial evidence about the importance of AS biological function.
In this study, we successfully detected 26,819 consensus intron-containing genes and 1,081 novel genes in the lotus genome. The functional annotation information for all the genes were substantially improved through the analysis of numerous high-sequencing transcriptome data in combination with a well-designed bio informatics approach. To identify the post-transcriptional modifications, particularly the alternative splicing, the genome-wide investigation of AS events in lotus was conducted based on total mapping consensus transcript sequences to the reference genome scaffolds. Distribution results for the four main AS types and AS-gene functional annotations greatly increased the gene diversity contributing to in-depth laboratory experiments and large-scale transcriptomics or genomics studies in sacred lotus.
Acknowledgement: The authors are grateful to the colleagues who have generated the transcriptomic data.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: The authors confirm contribution to the paper as follows: PY designed this project. XZ and ZY downloaded and analyzed the data. XZ wrote the manuscript. PY revised the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: All the data are accessible within the text.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Black, D. L. (2003). Mechanisms of alternative pre-messenger RNA splicing. Annual Review of Biochemistry, 72(1), 291–336. https://doi.org/10.1146/annurev.biochem.72.121801.161720 [Google Scholar] [PubMed] [CrossRef]
2. Busch, A., Hertel, K. J. (2015). Splicing predictions reliably classify different types of alternative splicing. RNA, 21(5), 813–823. https://doi.org/10.1261/rna.048769.114 [Google Scholar] [PubMed] [CrossRef]
3. Liu, L., Tang, Z., Liu, F., Mao, F., Gu, Y. J. et al. (2021). Normal, novel or none: Versatile regulation from alternative splicing. Plant Signaling Behavior, 16(7), 1917170. https://doi.org/10.1080/15592324.2021.1917170 [Google Scholar] [PubMed] [CrossRef]
4. Zhang, H., Jia, J., Zhai, J. (2022). Plant intron-splicing efficiency database (PISEExploring splicing of ∼1,650,000 introns in Arabidopsis, maize, rice, and soybean from ∼57,000 public RNA-seq libraries. Science China Life Science, 1–10. https://doi.org/10.1007/s11427-022-2193-3 [Google Scholar] [PubMed] [CrossRef]
5. Keren, H., Lev-Maor, G., Ast, G. (2010). Alternative splicing and evolution: Diversification, exon definition and function. Nature Reviews Genetics, 11(5), 345–355. https://doi.org/10.1038/nrg2776 [Google Scholar] [PubMed] [CrossRef]
6. Wang, Z., Gerstein, M. (2009). RNA-Seq: A revolutionary tool for transcriptomics. Nature Reviews Genetics, 10, 57–63. [Google Scholar] [PubMed]
7. Fernandezvalverde, S. L., Calcino, A. D., Degnan, B. M. (2015). Deep developmental transcriptome sequencing uncovers numerous new genes and enhances gene annotation in the sponge Amphimedon queenslandica. BMC Genomics, 16, 1–11. [Google Scholar]
8. Li, M., Liang, Z., Zeng, Y., Jing, Y., Wu, K. C. et al. (2016). De novo analysis of transcriptome reveals genes associated with leaf abscission in sugarcane (Saccharum officinarum L.). BMC Genomics, 17, 195. [Google Scholar] [PubMed]
9. Marquez, Y., Brown, J. W., Simpson, C., Barta, A., Kalyna, M. (2012). Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Research, 22, 1184–1195. [Google Scholar] [PubMed]
10. Thatcher, S. R., Zhou, W., Leonard, A., Wang, B. B., Beatty, M. et al. (2014). Genome-wide analysis of alternative splicing in Zea mays: Landscape and genetic regulation. Plant Cell, 26(9), 3472–3487. [Google Scholar] [PubMed]
11. Shenmiller, J. (2002). Sacred lotus, the long-living fruits of China Antique. Seed Science Research, 12, 131–143. [Google Scholar]
12. Mukherjee, P. K., Mukherjee, D., Maji, A. K., Rai, S., Heinrich, M. (2009). The sacred lotus (Nelumbo nucifera)—Phytochemical and therapeutic profile. Journal of Pharmacy & Pharmacology, 61, 407–422. [Google Scholar]
13. Ming, R., VanBuren, R., Liu, Y., Yang, M., Han, Y. P. et al. (2013). Genome of the long-living sacred lotus (Nelumbo nucifera Gaertn.). Genome Biology, 14, R41. [Google Scholar] [PubMed]
14. Initiative, A. G. (2000). Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature, 408, 796. [Google Scholar]
15. International Rice Genome Sequencing Project (2005). The map-based sequence of rice genome. Nature, 436(7052), 793–800. [Google Scholar]
16. Vanburen, R., Walters, B., Ming, R., Min, X. J. (2013). Analysis of expressed sequence tags and alternative splicing genes in sacred lotus (Nelumbo nucifera Gaertn.). Plant Omics, 6, 311–317. [Google Scholar]
17. Yang, M., Xu, L., Liu, Y., Yang, P. (2015). RNA-Seq uncovers SNPs and alternative splicing events in Asian lotus (Nelumbo nucifera). PLoS One, 10, e0125702. [Google Scholar] [PubMed]
18. Cheng, L., Li, S., Yin, J., Li, L., Chen, X. (2013). Genome-wide analysis of differentially expressed genes relevant to rhizome formation in lotus root (Nelumbo nucifera Gaertn). PLoS One, 8, e67116. [Google Scholar] [PubMed]
19. Hu, J., Jin, J., Qian, Q., Huang, K., Ding, Y. (2016). Small RNA and degradome profiling reveals miRNA regulation in the seed germination of ancient eudicot Nelumbo nucifera. BMC Genomics, 17, 684. [Google Scholar] [PubMed]
20. Kim, M. J., Nelson, W., Soderlund, C. A., Gang, D. R. (2013). Next-generation sequencing-based transcriptional profiling of sacred lotus “China Antique”. Tropical Plant Biology, 6, 161–179. [Google Scholar]
21. Yang, M., Zhu, L., Pan, C., Xu, L., Liu, Y. L. et al. (2015). Transcriptomic analysis of the regulation of rhizome formation in temperate and tropical lotus (Nelumbo nucifera). Scientific Reports, 5, 13059. [Google Scholar] [PubMed]
22. Yang, M., Zhu, L., Xu, L., Pan, C., Liu, Y. (2014). Comparative transcriptomic analysis of the regulation of flowering in temperate and tropical lotus (Nelumbo nucifera) by RNA-Seq. Annals of Applied Biology, 165, 73–95. [Google Scholar]
23. Zhang, W., Tian, D., Huang, X., Xu, Y., Mo, H. B. et al. (2014). Characterization of flower-bud transcriptome and development of genic SSR markers in Asian lotus (Nelumbo nucifera Gaertn.). PLoS One, 9, e112223. [Google Scholar] [PubMed]
24. Zheng, X. F., You, Y. N., Zheng, X. W., Xie, K. Q., Zhou, M. Q. et al. (2014). Development and characterization of genic-SSR markers from different Asia lotus (Nelumbo nucifera) types by RNA-seq. Genetics & Molecular Research, 14, 11171–11184. [Google Scholar]
25. Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., Salzberg, S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nature Protocols, 11, 1650–1667. [Google Scholar] [PubMed]
26. Niknafs, Y. S., Pandian, B., Iyer, H. K., Chinnaiyan, A. M., Iyer, M. K. (2017). TACO produces robust multisample transcriptome assemblies from RNA-seq. Nature Methods, 14(1), 68–70. https://doi.org/10.1038/nmeth.4078 [Google Scholar] [PubMed] [CrossRef]
27. Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, E. J. et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biology, 9(1), R7. https://doi.org/10.1186/gb-2008-9-1-r7 [Google Scholar] [PubMed] [CrossRef]
28. Foissac, S., Sammeth, M. (2007). ASTALAVISTA: Dynamic and flexible analysis of alternative splicing events in custom gene datasets. Nucleic Acids Research, 35(Web Server), W297. https://doi.org/10.1093/nar/gkm311 [Google Scholar] [PubMed] [CrossRef]
29. Sammeth, M. (2009). Complete alternative splicing events are bubbles in splicing graphs. Journal of Computational Biology, 16(8), 1117–1140. https://doi.org/10.1089/cmb.2009.0108 [Google Scholar] [PubMed] [CrossRef]
30. Sammeth, M., Foissac, S., Guigó, R. (2008). A general definition and nomenclature for alternative splicing events. PLoS Computational Biology, 4(8), e1000147. https://doi.org/10.1371/journal.pcbi.1000147 [Google Scholar] [PubMed] [CrossRef]
31. Tian, T., Liu, Y., Yan, H., You, Q., Yi, X. et al. (2017). agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Research, 45(W1), W122–W129. [Google Scholar] [PubMed]
32. Wang, Z., Zhang, H., Gong, W. (2019). Genome-wide identification and comparative analysis of alternative splicing across four legume species. Planta, 249, 1133–1142. [Google Scholar] [PubMed]
33. Barbazuk, W. B., Fu, Y., Mcginnis, K. M. (2008). Genome-wide analyses of alternative splicing in plants: Opportunities and challenges. Genome Research, 18, 1381–1392. [Google Scholar] [PubMed]
34. Wang, B. B., Brendel, V. (2006). Genome-wide comparative analysis of alternative splicing in plants. Proceedings of the National Academy of Sciences of the United States of America, 103(18), 7175–7180. [Google Scholar] [PubMed]
35. Bao, H., Li, E., Mansfield, S. D., Cronk, Q. C., El-Kassaby, A. Y. et al. (2013). The developing xylem transcriptome and genome-wide analysis of alternative splicing in Populus trichocarpa (Black cottonwood) populations. BMC Genomics, 14, 359. [Google Scholar] [PubMed]
36. Braden, W., Gengkon, L., Gaurav, S., Jia, M. X. (2013). Genome-wide landscape of alternative splicing events in Brachypodium distachyon. DNA Research, 20, 163–171. [Google Scholar]
37. Chamala, S., Feng, G., Chavarro, C., Barbazuk, W. B. (2015). Genome-wide identification of evolutionarily conserved alternative splicing events in flowering plants. Frontiers in Bioengineering & Biotechnology, 3, 33. [Google Scholar]
38. Kianianmomeni, A., Cheng, S. O., Rätsch, G., Hallmann, A. (2014). Genome-wide analysis of alternative splicing in Volvox carteri. BMC Genomics, 15, 1–21. [Google Scholar]
39. Ner-Gaon, H., Leviatan, N., Rubin, E., Fluhr, R. (2007). Comparative cross-species alternative splicing in plants. Plant Physiology, 144, 1632–1641. [Google Scholar] [PubMed]
40. Panahi, B., Abbaszadeh, B., Taghizadeghan, M., Ebrahimie, E. (2014). Genome-wide survey of alternative splicing in Sorghum bicolor. Physiology & Molecular Biology of Plants, 20, 323–329. [Google Scholar]
41. Potenza, E., Racchi, M. L., Sterck, L., Coller, E., Asquini, E. et al. (2015). Exploration of alternative splicing events in ten different grapevine cultivars. BMC Genomics, 16(1), 1–9. https://doi.org/10.1186/s12864-015-1922-5 [Google Scholar] [PubMed] [CrossRef]
42. Mcguire, A. M., Pearson, M. D., Neafsey, D. E., Galagan, J. E. (2008). Cross-kingdom patterns of alternative splicing and splice recognition. Genome Biology, 9(3), 1–19. https://doi.org/10.1186/gb-2008-9-3-r50 [Google Scholar] [PubMed] [CrossRef]
43. Seoighe, C., Gehring, C. (2010). Heritability in the efficiency of nonsense-mediated mRNA decay in humans. PLoS One, 5(7), e11657. https://doi.org/10.1371/journal.pone.0011657 [Google Scholar] [PubMed] [CrossRef]
44. Huang, J., Gao, Y., Jia, H., Liu, L., Zhang, D. et al. (2015). Comparative transcriptomics uncovers alternative splicing changes and signatures of selection from maize improvement. BMC Genomics, 16(1), 363. https://doi.org/10.1186/s12864-015-1582-5 [Google Scholar] [PubMed] [CrossRef]
45. Shen, Y., Zhou, Z., Wang, Z., Li, W., Fang, C. et al. (2014). Global dissection of alternative splicing in paleopolyploid soybean. Plant Cell, 26(3), 996–1008. https://doi.org/10.1105/tpc.114.122739 [Google Scholar] [PubMed] [CrossRef]
46. Kelemen, O., Convertini, P., Zhang, Z., Wen, Y., Shen, M. L. et al. (2013). Function of alternative splicing. Gene, 514(1), 1–30. https://doi.org/10.1016/j.gene.2012.07.083 [Google Scholar] [PubMed] [CrossRef]
47. Kornblihtt, A. R., Schor, I. E., Alló, M., Dujardin, G., Petrillo, E. et al. (2013). Alternative splicing: A pivotal step between eukaryotic transcription and translation. Nature Reviews Molecular Cell Biology, 14(3), 153–165. https://doi.org/10.1038/nrm3525 [Google Scholar] [PubMed] [CrossRef]
48. de Almeida, S. F., Grosso, A. R., Koch, F., Fenouil, R., Carvalho, S. et al. (2011). Splicing enhances recruitment of methyltransferase HYPB/Setd2 and methylation of histone H3 Lys36. Nature Structural & Molecular Biology, 18(9), 977–983. https://doi.org/10.1038/nsmb.2123 [Google Scholar] [PubMed] [CrossRef]
49. Gabut, M., Samavarchi-Tehrani, P., Wang, X., Slobodeniuc, V., O’Hanlon, D. et al. (2011). An alternative splicing switch regulates embryonic stem cell pluripotency and reprogramming. Cell, 147(1), 132–146. https://doi.org/10.1016/j.cell.2011.08.023 [Google Scholar] [PubMed] [CrossRef]
50. Rauch, H. B., Patrick, T. L., Klusman, K. M., Battistuzzi, F. U., Mei, W. B. et al. (2014). Discovery and expression analysis of alternative splicing events conserved among plant SR proteins. Molecular Biology & Evolution, 31(3), 605–613. https://doi.org/10.1093/molbev/mst238 [Google Scholar] [PubMed] [CrossRef]
51. Sellmann, D., Becker, T., Knoch, F. (2013). Alternative splicing at the intersection of biological timing, development, and stress responses. The Plant Cell, 25(10), 3640–3656. https://doi.org/10.1105/tpc.113.113803 [Google Scholar] [PubMed] [CrossRef]
52. Gu, L., Guo, R. (2007). Genome-wide detection and analysis of alternative splicing for nucleotide binding site-leucine-rich repeats sequences in rice. Acta Genetica Sinica, 34, 247–257. [Google Scholar]
53. Macknight, R., Duroux, M., Laurie, R., Dijkwel, P., Simpson, G. et al. (2002). Functional significance of the alternative transcript processing of the Arabidopsis floral promoter FCA. Plant Cell, 14(4), 877–888. https://doi.org/10.1105/tpc.010456 [Google Scholar] [PubMed] [CrossRef]
54. Tang, F., Yang, S., Zhu, H. (2016). Functional analysis of alternative transcripts of the soybean Rj2 gene that restricts nodulation with specific rhizobial strains. Plant Biology, 18(3), 537–541. https://doi.org/10.1111/plb.12442 [Google Scholar] [PubMed] [CrossRef]
55. Seo, P. J., Mi, J. K., Ryu, J. Y., Jeong, E. Y., Park, C. M. (2011). Two splice variants of the IDD14 transcription factor competitively form nonfunctional heterodimers which may regulate starch metabolism. Nature Communication, 2(1), 303. https://doi.org/10.1038/ncomms1303 [Google Scholar] [PubMed] [CrossRef]
56. Park, M., Seo, P. J., Park, C. (2012). Alternative splicing as a way of linking the circadian clock to temperature response in Arabidopsis. Plant Signaling & Behavior, 7(9), 1194–1196. https://doi.org/10.4161/psb.21300 [Google Scholar] [PubMed] [CrossRef]
Supplementary Materials
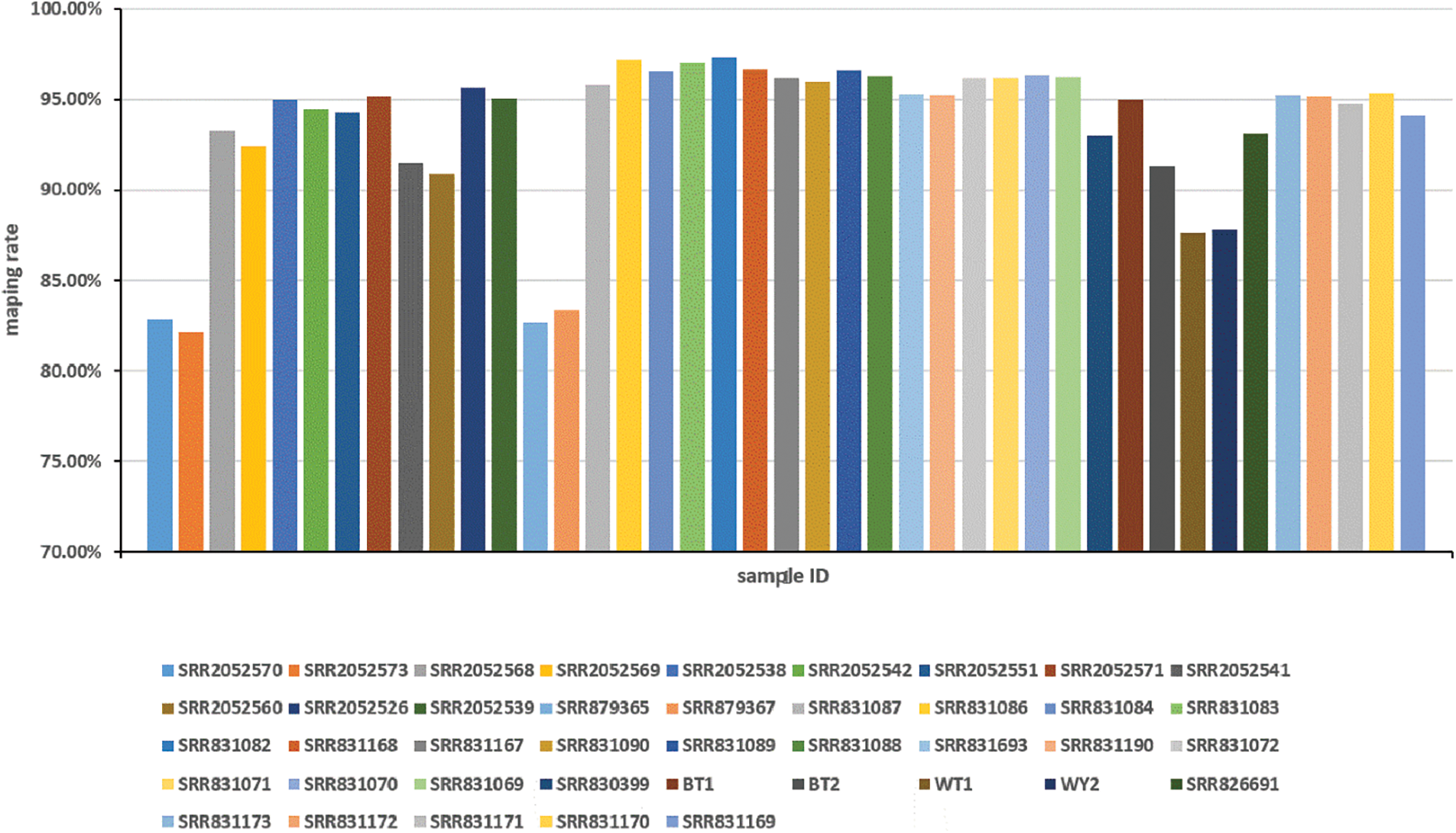
Figure S1: Overview of sample mapping rate distribution
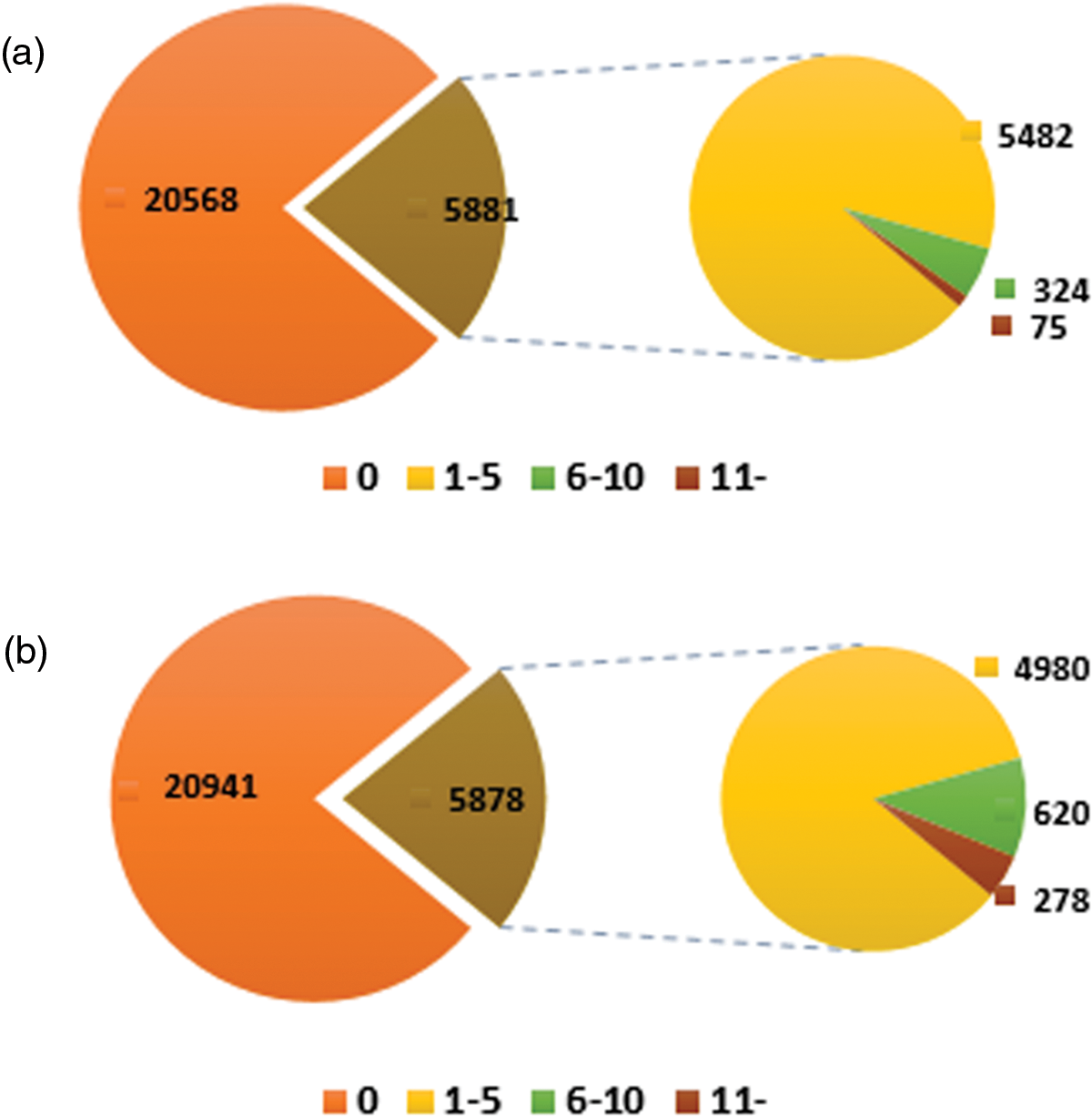
Figure S2: Overview of the frequency of AS genes. The picture above shows the distribution of genes involving “complete” AS events, while the figure below indicates the distribution of total genes having “internal” AS events. 0 no alternative splicing genes. 1–5 the frequency of AS genes between one to five. 6–11 the frequency of AS genes between six to ten. 11- the frequency of AS genes more than eleven. The number in the figure represents the number and type of AS genes
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools