
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Comparative Transcriptome Analyses Reveal Genes Related to Spine Development in Cucumber (Cucumis sativus)
1 Department of Plant Science and Technology, Shanghai Vocational College of Agriculture and Forestry, Shanghai, 201699, China
2 School of Agriculture and Biology, Shanghai Jiaotong University, Shanghai, 200240, China
* Corresponding Authors: Weiwei She. Email: ; Weiwei Zhang. Email:
# These authors contributed equally to this work
(This article belongs to the Special Issue: Identification of Genetic/Epigenetic Components Responding to Biotic and Abiotic Stresses in Crops)
Phyton-International Journal of Experimental Botany 2023, 92(4), 1059-1070. https://doi.org/10.32604/phyton.2023.026486
Received 08 September 2022; Accepted 29 October 2022; Issue published 06 January 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Fruit spine is an important quality trait of cucumber. To better understand the molecular basis of cucumber spine development and function, RNA-Seq was performed to identify differentially expressed genes (DEGs) in fruit spines of different development stages, namely, 8 days before anthesis (SpBA8), anthesis (SpA) and 8 days after anthesis (SpAA8). Stage-wise comparisons obtained 2,259 (SpBA8 vs. SpA), 4,551 (SpA vs. SpAA8), and 5,290 (SpBA8 vs. SpAA8) DEGs. All the DEGs were classified into eight expression clusters by trend analysis. Among these DEGs, in addition to the Mict, Tril, CsTTG1, CsMYB6, NS, and Tu genes that have been reported to regulate fruit spine formation, we found that the CsHDG11, CsSCL8, CsSPL8, CsZFP6 and CsZFP8 may also be involved in spine development in cucumber. Our study provides a theoretical basis for further research on molecular mechanisms of spine development in cucumber.Keywords
Supplementary Material
Supplementary Material FilePlant trichomes, existed in almost all overground parts of terrestrial plants, are specialized epidermal appendages [1]. Depending on the morphology, trichomes can be divided into unicellular or multicellular, branched or unbranched, and glandular or non-glandular. Trichomes participate in a multitude of defence mechanisms to protect plants against biotic and abiotic stress such as drought, heat, insect predation and UV light [2].
Trichomes in Arabidopsis are large unicellular structures and the regulatory mechanisms of trichome development have been well studied. The GLABRA1 (GL1), encoding an R2R3 MYB transcription factor, plays a critical role in trichome initiation [3]. TRANSPARENT TESTA GLABRA1 (TTG1), a WD40 repeat protein, is essential for trichome formation [4]. Two bHLH regulators, GLABRA3 (GL3) and ENHANCER OF GLABRA3 (EGL3), formed a MYB–bHLH–WD repeat complex with GL1 and TTG1 [5]. This regulatory complex can activate the expression of GLABRA2 (GL2) and TRANSPARENT TESTA GLABRA2 (TTG2) to promote trichome initiation [6].
Cucumber (Cucumis sativus) is a major vegetable crop worldwide. The tubercules and trichomes (called spines) on its fruit are important traits that directly affect the appearance quality of fruit. Spines are composed of a base and a stalk [7]. Tubercules are formed by several layers of cells that lie near the spine base [8,9]. To date, several genes related to spines development in cucumber have been reported. The Cucumber GLABROUS3 (CsGL3), also called Trichome-less (Tril), encodes a homeodomain-leucine zipper (HDZip) family protein that plays a crucial role in the initiation of trichomes [10,11]. The Cucumber GLABROUS1 (CsGL1), also called Micro-trichome (Mict) and TINY BRANCHED HAIR (TBH), controls the development of trichomes in cucumber [12,13]. CsTTG1, a WD-repeat homolog protein, can directly interact with Mict to regulate the density of trichomes [14]. Tender spines (Ts) is involved in soft spine development in cucumber [15]. Tuberculate (Tu) encodes a C2H2 zinc finger transcription factor, which is required for the tubercule initiation. Tu binds directly to the CsTS1 promoter and promotes fruit tubercule formation in cucumber [9]. Yang et al. found that Tu not only affects the development of tubercule, but also regulates spine base size development by forming a complex with Mict and CsTTG1 [16].
Although extensive studies have been performed on cucumber spine, the initial differentiation and morphogenesis mechanism of spine are still unclear. To fully comprehend the transcriptional regulatory mechanism of cucumber spine development, we performed transcriptome analysis of spines in three stages and identified candidate genes responsible for the development of spine. Our study provides new insights into the regulatory molecular mechanisms of spine development in cucumber.
The North China type cucumber inbred line WZ1 was used as plant material in the current study. First, the seeds of WZ1 were cultivated in plug trays under 16/8 h day/night conditions until two true leaves emerged. The seedings were then planted in a greenhouse of Shanghai Jiao Tong University under natural light conditions and were cultivated with standard horticultural conditions of watering and fertilizer management. The spines were collected from WZ1 cucumber fruits at approximately 8 days before anthesis (SpBA8), anthesis (SpA) and 8 days after anthesis (SpAA8) using fine tweezers. Among them, spines were isolated from cucumber fruit at SpBA8 under a dissecting microscope and spines from at least 10 fruits were pooled as one biological sample.
2.2 Scanning Electron Microscopy (SEM) Analysis
The cucumber fruits at approximately 8 days before anthesis, anthesis and 8 days after anthesis were fixed in a formaldehyde/acetic acid/ethanol (FAA) at 4°C approximately 24 h. The samples then dehydrated through an ethanol series (30%, 50%, 60%, 70%, 85%, 90%, 95%, and 100%) for 3 min at each step, critical-point dried with liquid CO2 and coated with gold palladium using Leica EM SCD050. Images were taken with an ALTO 1000 SEM (Gatan, USA).
2.3 RNA Extraction Library and Illumina Sequencing
We used OminiPlant RNA Kit (DNase I) (CWBIO, China) extracting the total RNA of fruit spines at DBA8, anthesis, and DAA8. Three biological replications were carried out for each developmental period. The quality and concentration of RNA was assessed. RNA concentration was measured using a Nanodrop 2000c Spectrophotometer (Thermo Scientific, USA) and the quality of the RNA was assessed with agarose gel electrophoresis. RNA-Seq sequencing was performed individually using Illumina HiSeq 2000 (Personal Biotechnology, China).
Firstly, the raw reads were processed to obtain high quality clean reads through removing the adaptor sequences, ambiguous and low-quality reads. The clean reads from nine libraries were then mapped to the cucumber (Chinese Long) V2 reference genome using HISAT2 and the transcript abundance of each gene was normalized by fragments per kilobase of exon per million fragments mapped (FPKM) [17]. The R package DESeq was used to perform differentially expressed genes (DEGs) of three groups (SpBA8 vs. SpA, SpBA8 vs. SpAA8, SpA vs. SpAA8) on the basis of absolute log2 fold change > 1 and adjusted p-value ≤ 0.05. The trend analysis was performed by MeV (4.9). To further investigate these DEGs, GO and KEGG enrichment analyses were performed using the topGO package and KEGG database [18,19].
2.5 Quantitative Real-Time PCR (qRT-PCR)
First-strand cDNA was synthesized using a HiFiScript cDNA Synthesis Kit (CWBIO, China). qRT-PCR was implemented with the UltraSYBR Mixture (CWBIO, China) with a CFX96 Touch™ Real-Time PCR System. The cucumber CsACTIN2 (Csa6G484600) gene was selected as an internal control. Relative gene expression level of mRNA was analysed using the 2–ΔΔCT method [20]. All the primer sequences used for qRT-PCR were given in Table S1.
To characterize the spine phenotype, we conducted a scanning electron microscopy (SEM) assay. When the fruit length was about 1 cm long (approximately 8 d before anthesis), the spine consisted of a sharp stalk on the top and an inconspicuous base (Fig. 1A). Subsequently, the spine base expanded rapidly and developed to the maximum at anthesis (Fig. 1B), after which the shape and size of spine remained relatively constant (Fig. 1C).

Figure 1: The morphologic observation of fruit spines at approximately 8 days before anthesis (SpBA8) (A), anthesis (SpA) (B) and 8 days after anthesis (SpAA8) (C). (A1, B1, C1) The phenotype of fruit. (A2, B2, C2) SEM images of the spine. Bars, 0.5 cm (A1, B1); 2 cm (C1)
3.2 Sequencing and de novo Assembly
To understand the mechanism of fruit spine development in cucumber, nine transcriptome libraries were constructed from samples of SpBA8, SpA and SpAA8 groups. The data processing results were generalized in Table S2. After removing low-quality and primer/adapter containing reads, 51.39 Gb clean reads were generated with an average of 5.71 Gb per sample. The Q30 contents were 97.80%~98.03%. These data showed that the sequencing results were accurate. To evaluate the quality of the RNA-Seq data, the reads were mapped to cucumber (Chinese Long) V2 reference genome. Among them, 67.4%~95.4% clean reads in each sample were successfully mapped to the reference genome. The relatively low mapping rate in stage SpBA8 may be caused by the poor quality of RNA due to the smaller spines and the difficulty of sampling in that period.
To further validate the reliability of the transcriptome data, ten DEGs were selected and quantified through qRT-PCR. The results indicated that the expression patterns of the ten selected genes were consistent with those generated by RNA-Seq analysis (Fig. S1). Therefore, the qRT-PCR results confirmed that the transcriptome data were reliable.
3.3 Identification of Differentially Expressed Genes (DEGs) and Trend Analysis
To identify differentially expressed genes (DEGs) among spines at three different stages, the FPKM values of each gene was calculated (Fig. 2A). We identified a total of 6,831 DEGs, of which 2,259 DEGs were found in SpBA8 vs. SpA (1,005 upregulated, 1,254 downregulated) (Fig. 2A1, Table S3). 4,551 DEGs were found in SpA vs. SpAA8 (2,684 upregulated, 1,867 downregulated) (Fig. 2A2, Table S4). 5,290 DEGs were found in SpBA8 vs. SpAA8 (3,063 upregulated, 2,227 downregulated) (Fig. 2A3, Table S5). The results show that there is much difference during the three stages of fruit spine development. We than analyzed the trend and clustering of the DEGs in the samples from the three comparison groups. 6,831 genes were divided into eight clusters, namely, cluster 1–8 (Fig. 2B, Table S6). Clusters 1, 4 and 8 had the largest numbers of DEGs (1,471, 1,375 and 1,632, respectively).
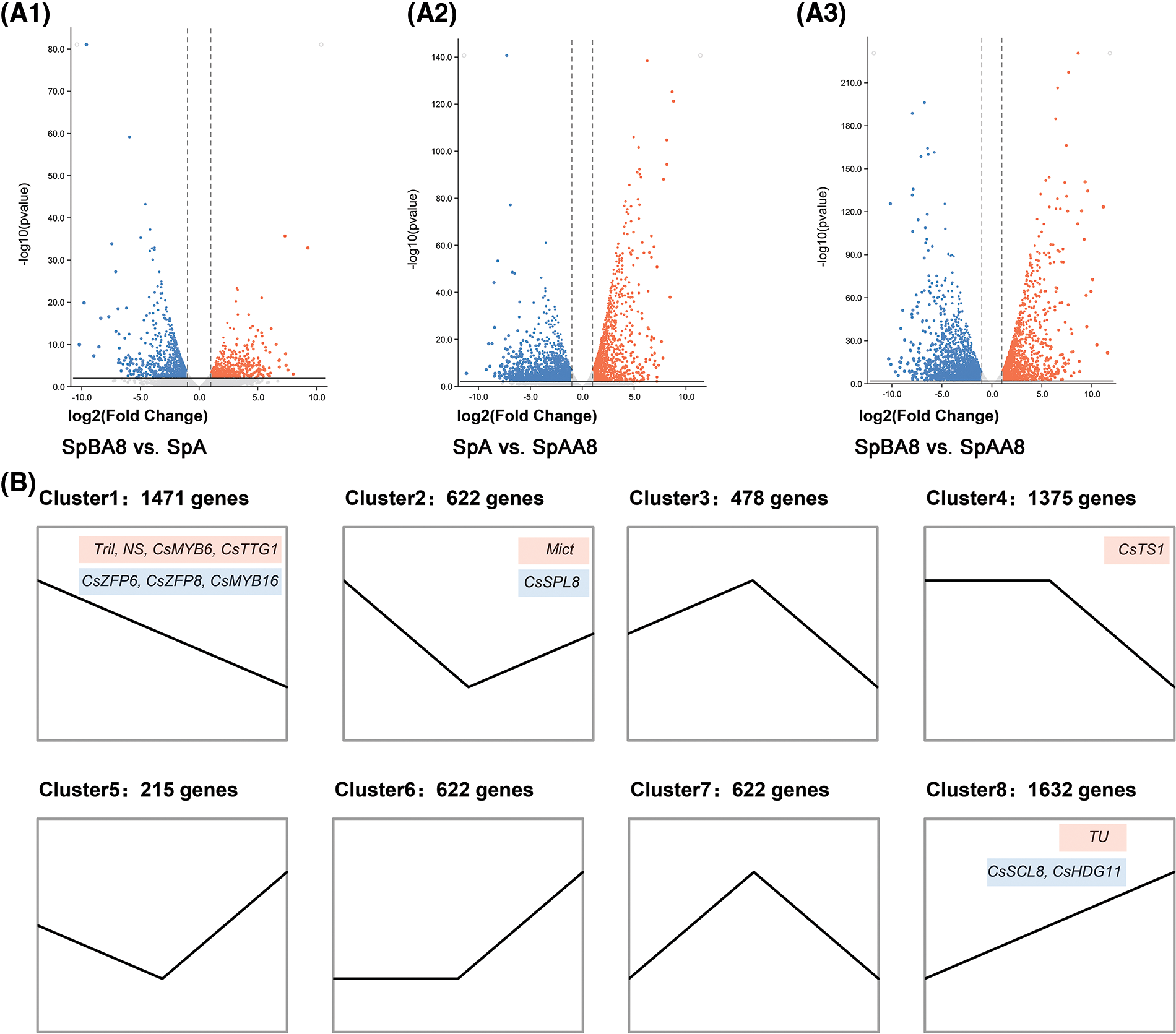
Figure 2: Volcano plot (A) and cluster analysis (B) of DEGs. (A1) SpBA8 vs. SpA. (A2) SpA vs. SpAA8. (A3) SpBA8 vs. SpAA8. The red dots indicate upregulated genes and the blue dots indicate downregulated genes. (B) Cluster analysis. The number of genes belonging to each pattern is indicated above the cluster. The genes in the red boxes represent the reported genes involved in spine development, while the genes in the blue boxes represent the genes that may regulate the formation of spine
Clusters 1, 2 and 4 included genes showing higher expression in SpBA8 stage, suggesting that the DEGs in the three clusters are mainly involved in the initiation of spines. Two important transcription factors for the initiation of spines, Tril and Mict, belong to clusters 1 and 2, respectively. We also found several genes in these clusters whose Arabidopsis orthologs are involved in the development of trichome, including two zinc finger proteins (ZFP6 and ZFP8), an R2R3 transcription factor (MYB16), and a squamosa promoter binding protein (SPL8) [21–23].
In clusters 3 and 7, the DEGs are highly expressed in SpA stage than in the other two stages. Clusters 5, 6 and 8 included genes showing higher expression in SpAA8 stage than in the other two stages. Notably, in cluster 8, the expression pattern of DEGs increases with the development of fruit spines. In this cluster, Tu gene regulates the development of spine and also determines the formation of tubercule. HOMEODOMAIN GLABROUS 11 (HDG11) and SCARECROW-LIKE 8 (SCL8) participate in trichome patterning in Arabidopsis [24,25], and their homologs in cucumber have similar expression patterns to Tu, suggesting that they may have similar function.
GO is a useful tool for the functional categorization of genes [26]. The DEGs in the fruit spine of SpBA8 vs. SpA, SpBA8 vs. SpAA8 and SpA vs. SpAA8 were annotated (Fig. 3, Table S7). GO terms included biological process (BP), cellular component (CC) and molecular function (MF), and the top 10 terms of the three categories are presented in descending order based on their p-value. The 310 GO terms were found to be significantly enriched with the DEGs of SpBA8 vs. SpA. The largest two subcategories in each of the BP, the cellular component CC, and the MF categories were ‘oxidation-reduction process’ and ‘anion transport’, the ‘membrane’ and ‘intrinsic component of membrane’, and the ‘oxidoreductase activity’ and ‘cofactor binding’, respectively (Fig. 3A). The DEGs in SpBA8 vs. SpAA8 and SpA vs. SpAA8 stage could be classified into 310 and 353 functional categories, respectively. The largest subcategories in these two stages were similar to the subcategories of SpBA8 vs. SpA (Figs. 3B and 3C).
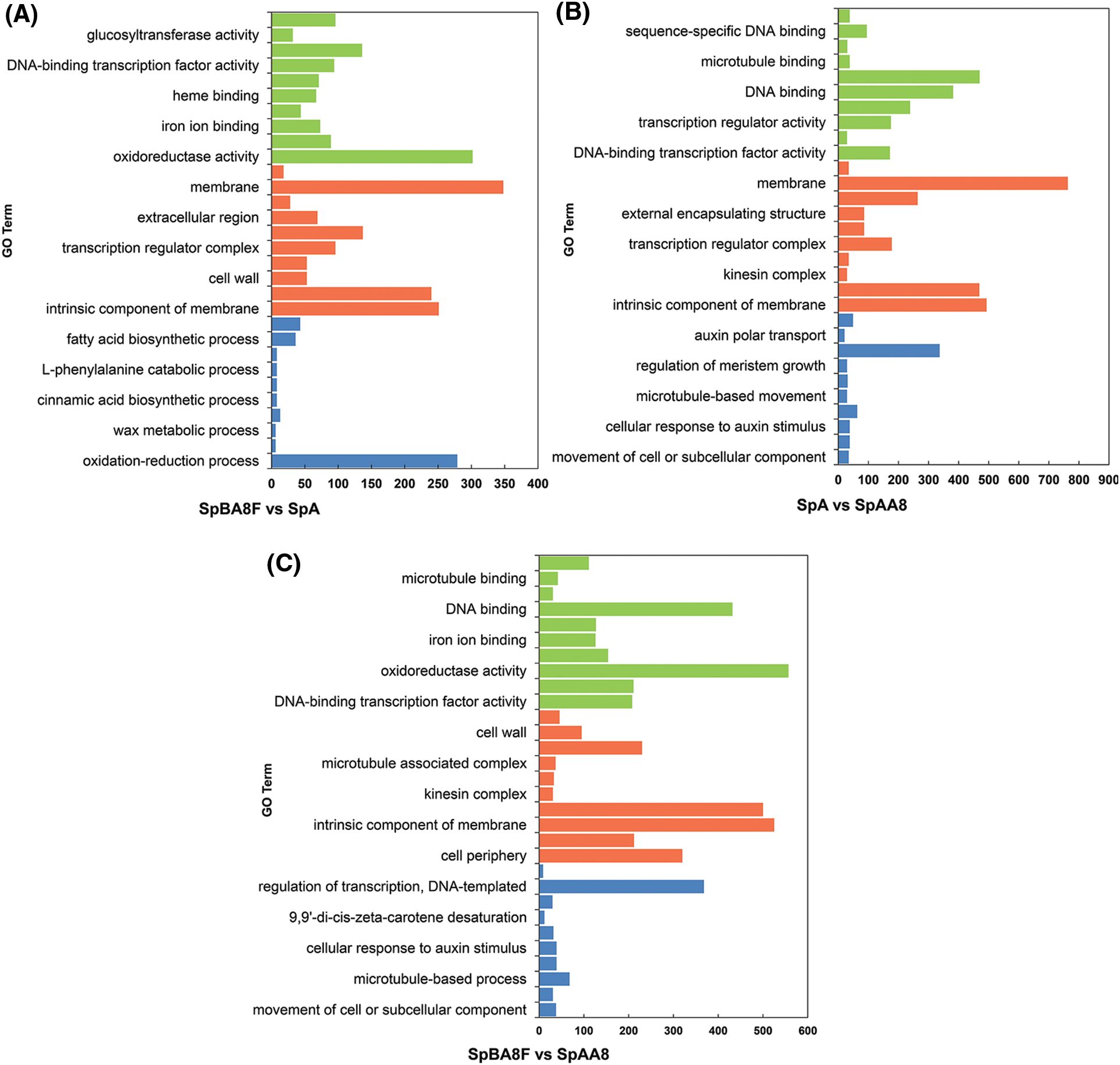
Figure 3: GO classification of DEGs. x-axis shows the number of DEGs in each category and y-axis shows the specific GO term. (A) SpBA8 vs. SpA. (B) SpA vs. SpAA8. (C) SpBA8 vs. SpAA8. The x-axis presents the number of DEGs in a category. The y-axis shows the specific GO term. The blue, orange and green histograms represent the biologic process, cellular component and molecular function, respectively
The pathway enrichment analysis of DEGs was annotated, and the top 20 KEGG pathways of the three comparison groups were displayed in descending order based on their Richness factor (Fig. S2, Table S8). The similar pathways of DEGs were observed in three groups, including ‘plant hormone signal transduction’, ‘phenylpropanoid biosynthesis’, ‘starch and sucrose metabolism’, ‘cysteine and methionine metabolism’ and ‘glutathione metabolism’.
3.5 Genes Related to Spine Development
Fruit spine is an important appearance quality of cucumbers, and extensive genes regulating fruit spine formation have been identified. To study the roles of these genes in different stages of fruit spine development, the expression levels of ten genes at different stages were analyzed (Fig. 4, Table S9). CsMYB6, Mict, Tril, NS, CsTTG1, these five genes were highly expressed in the SpBA8 stage spines. CsTS1, Ts and Tu were highly expressed in the SpA stage spines. Significantly, Tu gene was the only one highly expressed in spines at SpAA8 stage. However, the expression levels of CsHEC2 and CsTRY were lower in the three stages. The differential expression of these genes at three stages suggested their different functions during fruit spine development.

Figure 4: Expression patterns of the genes related to spine development. Genes highly expressed in spines are colored red and genes slightly expressed in spines are colored blue
To date, phytohormones have been shown to play important role in regulating the development of trichomes [27]. In this study, the DEGs involved in signal transduction and response contain almost all main types of phytohormones (Table S10), including auxin, cytokinin (CTK), abscisic acid (ABA), gibberellin (GA), brassinolide (BR), jasmonic acid (JA), ethylene (ET) and salicylic acid (SA). Specifically, auxin and CTK-related processes contain the largest number of genes, 76 and 32, respectively, indicating their important function in the development of cucumber spine. The tomato Aux/IAA gene SlIAA15 (the homolog in cucumber, Csa1G397130) regulates trichome formation and axillary shoot development [28]. The auxin response factor genes SlARF3 (the homolog in cucumber, Csa6G518210) and SlARF4 (the homolog in cucumber, Csa6G106780) are involved in the formation of trichome and epidermal cells [29,30]. Previous studies have also reported that meristem genes may play a crucial role in the development of spine [7]. We found several DEGs regulating the activity of vegetative and reproductive meristems in Arabidopsis, including CTK-related genes cytokinin oxidase 1 (CKX1), CKX3, CKX5, CKX6 and the GA-related gene gibberellin 2-oxidase 2 (GA2OX2) [31–34].
Cucumber is one of the most economically important vegetable crops in the world. Plants can be protected by trichomes from high temperature, excessive transpiration, UV light, pathogen infection and herbivore attack [2]. In cucumber, spines are still of important commercial quality. Although several key genes regulating spine development have been discovered, the initial differentiation and morphogenesis mechanism of spines are still unclear.
In this study, we sampled cucumber spines at three important development stages and measured the transcriptomics. Through quality assessment of clean datas, combined with qRT-PCR validation, we concluded that the sequencing data could be used to mine for fruit spine-related genes. Finally, 6831 genes were differentially expressed during spine development.
Transcription factors play an important role in plant trichome development. C2H2 zinc finger protein (C2H2-ZFP) was found to be involved in trichome formation in Arabidopsis [23]. The first-reported C2H2-ZFP that involved trichome initiation is GLABROUS INFLORESCENCE STEMS (GIS), which acts upstream of the complex (GL1-GL3-TTG1) to regulate inflorescence trichome initiation [35]. Subsequently, GIS2, GIS3, ZFP5 and ZFP6 were found to participate in regulating the density of trichome [36–38]. The HD-ZIP transcription factor is also critical in trichome development. GL2 is the first member of the HD-ZIP family identified to be involved in trichome development. In addition to GL2, a mutation of Arabidopsis HDG11 resulted in a decrease in the number of trichomes [24]. The Tril and Mict genes in cucumber play an important role in early spine development [11,39]. CsHDG11, whose expression increases consistently with spine development, is also likely to be involved in spine development [24]. R2R3MYB family members MYB106 and MYB16 can affect epidermal cell differentiation and trichome morphogenesis [22,40]. In cucumber, we found that CsMYB6 homologs from Arabidopsis MYB16 were highly expressed in the SpBA8 stage spines, and they play a key role in cucumber fruit spine formation [41].
In addition to transcription factors, the development of epidermal trichome is also regulated by a variety of plant hormones. C2H2-ZFPs GIS, GIS2, GIS3, ZFP5, ZFP6 and ZFP8 regulate epidermal hair initiation on floral organs by responding to GA and CTK signaling [23]. In cucumber, the physical interaction of Tu and CsHEC2 activates the expression of CTK hydroxylase-like1 gene (CsCHL1) [42]. Arabidopsis SPL8 acts as a positive regulator in GA-mediated trichomes density development [21]. We found CsSPL8 was significantly up-regulated in stage SpBA8, which may be involved in the GA-mediated spine development in cucumber.
In tomato, interfering with the Aux/IAA proteins SlIAA15 and SlARF3 both resulted in a decrease in the number of trichomes, suggesting that the auxin is also involved in the regulation of tomato trichomes [28,29]. In cucumber, NS encodes an auxin transport protein, and auxin levels in the spines were significantly lower and the density of fruit spines was significantly increased in the ns mutant [43]. We found a strong enrichment of categories involved in auxin-activated signaling pathway, but the expression of the NS gene did not change significantly in the three stages. However, another gene, CsTS1, can increase auxin levels in fruit tubercules, suggesting that it may respond to auxin signaling [8]. Lv et al. [44] analyzed the transcriptome of spines from cucumber fruits with a length of 0.2, 0.35, and 0.6 cm (approximately SpBA8). The number of DEGs related to the auxin response was the largest at different fruit spine developmental stages. This observation is consistent with the findings of this study, indicating that auxin may played a dominant role in the formation of cucumber spine.
The phytohormone ethylene has an important effect of promoting root hair growth. Ethylene induces root hair formation through activating the ethylene activated transcription factors EIN3/EIL1 and down-regulation of GL2 expression [45]. In cucumber, Mict can directly bind to the promoters of cucumber 1-Aminocyclopropane-1-Carboxylate Synthase (CsACS) genes and regulate their expression, which regulates the development of fruit spines by affecting ethylene accumulation [39]. JA treatment increases trichome density and number of Arabidopsis, whereas salicylic acid treatment has the opposite result [46]. In Artemisia annua, HOMEODOMAIN PROTEIN 1 (AaHD1) positively controls trichome initiation via the JA signalling pathway [47]. HOMEODOMAIN PROTEIN8 (SlHD8) is a downstream regulator of JA signaling that promotes trichome elongation in tomato [48]. The DEGs related to diverse phytohormone signaling pathways changed in this study, including auxin, CTK, ABA, GA, ET, JA, SA, and BR, suggesting that these phytohormones may play an important role in the spine development of cucumber.
Thus, we proposed a working model to explain the possible mechanisms of spine and tubercule development in cucumber (Fig. 5). During spine development, the Tril gene play an important role in spine initiation [10,11,49]. CsTTG1 directly interacts with Tu and Mict functions in spine differentiation [7,16]. Mict regulates spine development by promoting ethylene biosynthesis [39]. Besides, NS regulates the fruit spine density by promoting auxin biosynthesis. Genetic analyses showed that Tril is epistatic to Mict, and Mict is epistatic to NS and Tu [43]. During tubercule development, Tu binds directly to the CsTS1 promoter and promotes fruit tubercule formation through the auxin pathway [8]. CsHEC2 interacts with Tu and CsTTG1 to regulate fruit wart initiation by promoting CTK biosynthesis [42]. The genes CsZFP6, CsZFP8, CsHDG11, CsMYB6, CsSCL8 and CsSPL8, identified in this study, may also be involved in spine development, but their functions still need further validation.
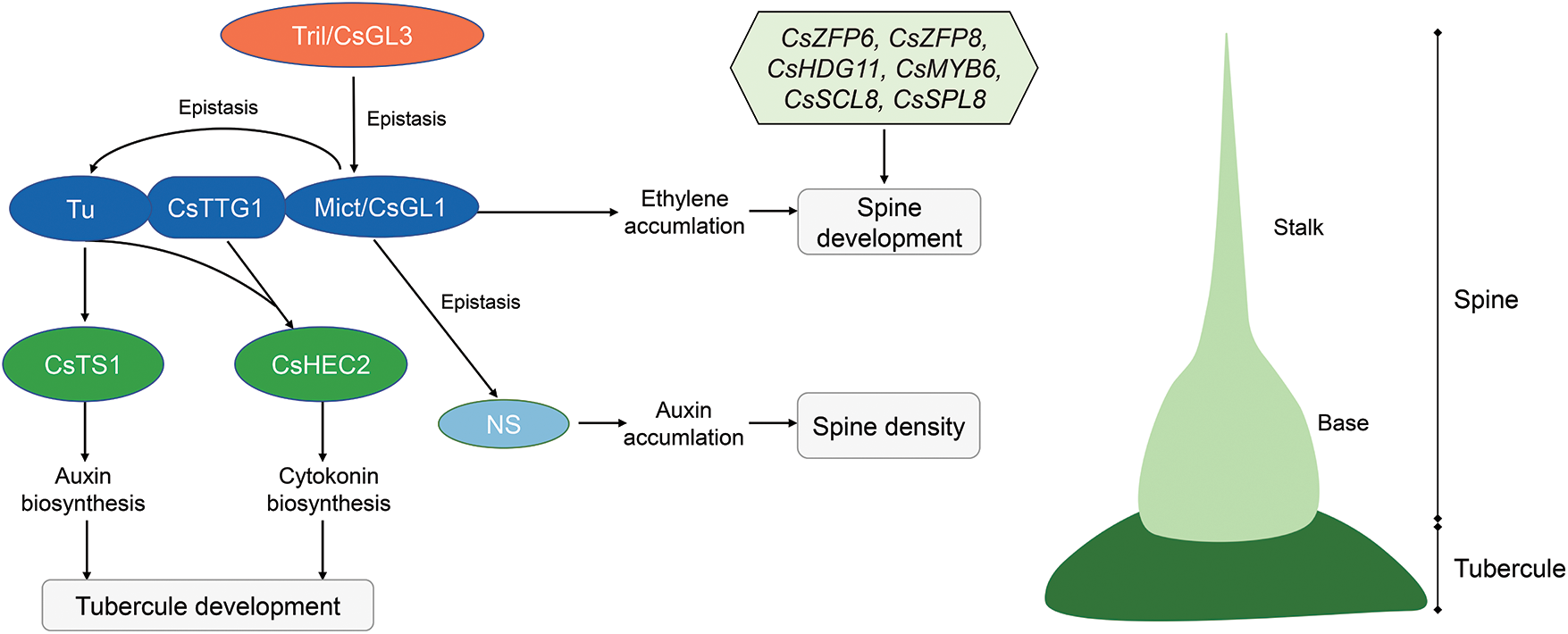
Figure 5: Working model of spine and tubercule development in cucumber
In this study, nine transcriptome libraries were constructed from samples of SpBA8, SpA and SpAA8 groups. Based on the comparative transcriptome analysis, 6831 DEGs were identified. A significant number of genes are involved in cucumber spine development. These results provide new insights into the regulatory molecular mechanisms of spine development and also contribute to breeding spineless cucumber varieties.
Authorship: The authors confirm contribution to the paper as follows: study conception and design: Yue Chen, Huan Wang and Weiwei She; data collection: Huan Wang, Taibai Xu and Peng Zhou; analysis and interpretation of results: Yue Chen; resources, Junsong Pan; draft manuscript preparation: Yue Chen and Huan Wang; writing–review & editing, Weiwei Zhang; funding acquisition, Weiwei Zhang. All authors reviewed the results and approved the final version of the manuscript.
Data Availability Statement: The RNA-Seq data associated with this study has been deposited in the NCBI database with BioProject Accession PRJNA852610.
Funding Statement: This work was sponsored by the Natural Science Foundation of Shanghai (20ZR1439600, 19ZR1436500), the Agricultural Field Project of Shanghai “Action Plan for Scientific and Technological Innovation” (20392001300), the Young and Middle-Aged Leading Talent Project of Shanghai Vocational College of Agriculture and Forestry (A2-0273-20-01-16) and the Project of Shanghai Vocational College of Agriculture and Forestry (KY2-0000-20-01).
Conflicts of Interest: The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Pattanaik, S., Patra, B., Singh, S. K., Yuan, L. (2014). An overview of the gene regulatory network controlling trichome development in the model plant, Arabidopsis. Frontiers in Plant Science, 5, 259. [Google Scholar]
2. Balkunde, R., Pesch, M., Hülskamp, M. (2010). Trichome patterning in Arabidopsis thaliana: From genetic to molecular models. Current Topics in Developmental Biology, 91, 299–321. [Google Scholar]
3. Marks, M. D., Feldmann, K. A. (1989). Trichome development in Arabidopsis thaliana. I. T-DNA tagging of the GLABROUS1 gene. The Plant Cell, 1(11), 1043–1050. [Google Scholar]
4. Walker, A. R., Davison, P. A., Bolognesi-Winfield, A. C., James, C. M., Srinivasan, N. et al. (1999). The TRANSPARENT TESTA GLABRA1 locus, which regulates trichome differentiation and anthocyanin biosynthesis in Arabidopsis, encodes a WD40 repeat protein. The Plant Cell, 11(7), 1337–1349. [Google Scholar]
5. Payne, C. T., Zhang, F., Lloyd, A. M. (2000). GL3 encodes a bHLH protein that regulates trichome development in Arabidopsis through interaction with GL1 and TTG1. Genetics, 156(3), 1349–1362. [Google Scholar]
6. Ishida, T., Hattori, S., Sano, R., Inoue, K., Shirano, Y. et al. (2007). Arabidopsis TRANSPARENT TESTA GLABRA2 is directly regulated by R2R3 MYB transcription factors and is involved in regulation of GLABRA2 transcription in epidermal differentiation. The Plant Cell, 19(8), 2531–2543. [Google Scholar]
7. Chen, C., Liu, M., Jiang, L., Liu, X., Zhao, J. et al. (2014). Transcriptome profiling reveals roles of meristem regulators and polarity genes during fruit trichome development in cucumber (Cucumis sativus L.). Journal of Experimental Botany, 65(17), 4943–4958. [Google Scholar]
8. Yang, S., Wen, C., Liu, B., Cai, Y., Xue, S. et al. (2019). A CsTu -TS1 regulatory module promotes fruit tubercule formation in cucumber. Plant Biotechnology Journal, 17(1), 289–301. [Google Scholar]
9. Yang, X., Zhang, W., He, H., Nie, J., Bie, B. et al. (2014). Tuberculate fruit gene Tu encodes a C2H2 zinc finger protein that is required for the warty fruit phenotype in cucumber (Cucumis sativus L.). The Plant Journal, 78(6), 1034–1046. [Google Scholar]
10. Wang, Y., Nie, J., Chen, H., Guo, C., Pan, J. et al. (2016). Identification and mapping of Tril, a homeodomain-leucine zipper gene involved in multicellular trichome initiation in Cucumis sativus. Theoretical and Applied Genetics, 129(2), 305–316. [Google Scholar]
11. Du, H., Wang, G., Pan, J., Chen, Y., Xiao, T. et al. (2020). The HD-ZIP IV transcription factor Tril regulates fruit spine density through gene dosage effects in cucumber. Journal of Experimental Botany, 71(20), 6297–6310. [Google Scholar]
12. Li, Q., Cao, C., Zhang, C., Zheng, S., Wang, Z. et al. (2015). The identification of Cucumis sativus Glabrous 1 (CsGL1) required for the formation of trichomes uncovers a novel function for the homeodomain-leucine zipper I gene. Journal of Experimental Botany, 66(9), 2515–2526. [Google Scholar]
13. Zhao, J. L., Pan, J. S., Guan, Y., Zhang, W. W., Bie, B. B. et al. (2015). Micro-trichome as a class I homeodomain-leucine zipper gene regulates multicellular trichome development in Cucumis sativus. Journal of Integrative Plant Biology, 57(11), 925–935. [Google Scholar]
14. Chen, C., Yin, S., Liu, X., Liu, B., Yang, S. et al. (2016). CsTTG1 encodes a WD-repeat protein that regulates fruit wart formation in Cucumis sativus through interaction with the homeodomain-leucine zipper I protein Mict. Plant Physiology, 171(2), 1156–1168. [Google Scholar]
15. Guo, C., Yang, X., Wang, Y., Nie, J., Yang, Y. et al. (2017). Identification and mapping of ts (tender spinesa gene involved in soft spine development in Cucumis sativus. Theoretical and Applied Genetics, 131(1), 1–12. [Google Scholar]
16. Yang, S., Wang, Y., Zhu, H., Zhang, M., Wang, D. et al. (2022). A novel HD-Zip I/C2H2-ZFP/WD-repeat complex regulates the size of spine base in cucumber. New Phytologist, 233(6), 2643–2658. DOI 10.1111/nph.17967. [Google Scholar] [CrossRef]
17. Siren, J., Valimaki, N., Makinen, V. (2014). Indexing graphs for path queries with applications in genome research. IEEE/ACM Transactions on Computational Biology and Bioinformatic, 11(2), 375–388. DOI 10.1109/TCBB.2013.2297101. [Google Scholar] [CrossRef]
18. Alexa, A., Rahnenführer, J. (2009). Gene set enrichment analysis with topGO. Bioconductor, 27, 1–26. [Google Scholar]
19. Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., Tanabe, M. (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Research, 44(D1), 457–462. DOI 10.1093/nar/gkv1070. [Google Scholar] [CrossRef]
20. Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods, 25(4), 402–408. DOI 10.1006/meth.2001.1262. [Google Scholar] [CrossRef]
21. Zhang, Y., Schwarz, S., Saedler, H., Huijser, P. (2006). SPL8, a local regulator in a subset of gibberellin-mediated developmental processes in Arabidopsis. Plant Molecular Biology, 63(3), 429–439. [Google Scholar]
22. Oshima, Y., Mitsuda, N. (2013). The MIXTA-like transcription factor MYB16 is a major regulator of cuticle formation in vegetative organs. Plant Signaling Behavior, 8(11), e26826. [Google Scholar]
23. Yan, A., Wu, M., Zhao, Y., Zhang, A., Liu, B. et al. (2014). Involvement of C2H2 zinc finger proteins in the regulation of epidermal cell fate determination in Arabidopsis. Journal of Integrative Plant Biology, 56(12), 1112–1117. [Google Scholar]
24. Khosla, A., Paper, J. M., Boehler, A. P., Bradley, A. M., Neumann, T. R. et al. (2014). HD-Zip proteins GL2 and HDG11 have redundant functions in Arabidopsis trichomes, and GL2 activates a positive feedback loop via MYB23. The Plant Cell, 26(5), 2184–2200. [Google Scholar]
25. Pu, L., Brady, S. (2010). Systems biology update: Cell type-specific transcriptional regulatory networks. Plant Physiology, 152(2), 411–419. [Google Scholar]
26. Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H. et al. (2000). Gene ontology: Tool for the unification of biology. Nature Genetics, 25(1), 25–29. [Google Scholar]
27. Yang, C., Ye, Z. (2013). Trichomes as models for studying plant cell differentiation. Cellular and Molecular Life Sciences, 70(11), 1937–1948. [Google Scholar]
28. Deng, W., Yang, Y., Ren, Z., Audran-Delalande, C., Mila, I. et al. (2012). The tomato SlIAA15 is involved in trichome formation and axillary shoot development. New Phytologist, 194(2), 379–390. [Google Scholar]
29. Zhang, X., Yan, F., Tang, Y., Yuan, Y., Deng, W. et al. (2015). Auxin response gene SlARF3 plays multiple roles in tomato development and is involved in the formation of epidermal cells and trichomes. Plant and Cell Physiology, 56(11), 2110–2124. [Google Scholar]
30. Yuan, Y., Xu, X., Luo, Y., Gong, Z., Hu, X. et al. (2021). R2R3 MYB-dependent auxin signalling regulates trichome formation, and increased trichome density confers spider mite tolerance on tomato. Plant Biotechnology Journal, 19(1), 138–152. [Google Scholar]
31. Holst, K., Schmülling, T., Werner, T. (2011). Enhanced cytokinin degradation in leaf primordia of transgenic Arabidopsis plants reduces leaf size and shoot organ primordia formation. Journal of Plant Physiology, 168(12), 1328–1334. [Google Scholar]
32. Bartrina, I., Otto, E., Strnad, M., Werner, T., Schmülling, T. (2011). Cytokinin regulates the activity of reproductive meristems, flower organ size, ovule formation, and thus seed yield in Arabidopsis thaliana. The Plant Cell, 23(1), 69–80. [Google Scholar]
33. Werner, S., Bartrina, I., Novák, O., Strnad, M., Werner, T. et al. (2021). The cytokinin status of the epidermis regulates aspects of vegetative and reproductive development in Arabidopsis thaliana. Frontiers in Plant Science, 12, 613488. [Google Scholar]
34. Li, H., Torres-Garcia, J., Latrasse, D., Benhamed, M., Schilderink, S. et al. (2017). Plant-specific Histone Deacetylases HDT1/2 regulate GIBBERELLIN 2-OXIDASE2 expression to control Arabidopsis root meristem cell number. The Plant Cell, 29(9), 2183–2196. [Google Scholar]
35. Gan, Y., Kumimoto, R., Liu, C., Ratcliffe, O., Yu, H. et al. (2006). GLABROUS INFLORESCENCE STEMS modulates the regulation by gibberellins of epidermal differentiation and shoot maturation in Arabidopsis. The Plant Cell, 18(6), 1383–1395. [Google Scholar]
36. Zhou, Z., An, L., Sun, L., Zhu, S., Xi, W. et al. (2011). Zinc Finger Protein5 is required for the control of trichome initiation by acting upstream of zinc finger protein8 in Arabidopsis. Plant Physiology, 157(2), 673–682. [Google Scholar]
37. Zhou, Z., Sun, L., Zhao, Y., An, L., Yan, A. et al. (2013). Zinc Finger Protein6 (ZFP6) regulates trichome initiation by integrating gibberellin and cytokinin signaling in Arabidopsis thaliana. New Phytologist, 198(3), 699–708. [Google Scholar]
38. Sun, L., Zhang, A., Zhou, Z., Zhao, Y., Yan, A. et al. (2015). GLABROUS INFLORESCENCE STEMS3 (GIS3) regulates trichome initiation and development in Arabidopsis. New Phytologist, 206(1), 220–230. [Google Scholar]
39. Zhang, Y., Shen, J., Bartholomew, E. S., Dong, M., Chen, S. et al. (2021). TINY BRANCHED HAIR functions in multicellular trichome development through an ethylene pathway in Cucumis sativus L. The Plant Journal, 106(3), 753–765. [Google Scholar]
40. Jakoby, M. J., Falkenhan, D., Mader, M. T., Brininstool, G., Wischnitzki, E. et al. (2008). Transcriptional profiling of mature Arabidopsis trichomes reveals that NOECK encodes the MIXTA-like transcriptional regulator MYB106. Plant Physiology, 148(3), 1583–1602. [Google Scholar]
41. Yang, S., Cai, Y., Liu, X., Dong, M., Zhang, Y. et al. (2018). A CsMYB6-CsTRY module regulates fruit trichome initiation in cucumber. Journal of Experimental Botany, 69(8), 1887–1902. [Google Scholar]
42. Wang, Z., Wang, L., Han, L., Cheng, Z., Liu, X. et al. (2021). HECATE2 acts with GLABROUS3 and Tu to boost cytokinin biosynthesis and regulate cucumber fruit wart formation. Plant Physiology, 187(3), 1619–1635. [Google Scholar]
43. Liu, X., Yang, X., Xie, Q., Miao, H., Bo, K. et al. (2022). NS encodes an auxin transporter that regulates the ’numerous spines’ trait in cucumber (Cucumis sativus) fruit. The Plant Journal, 110(2), 325–336. [Google Scholar]
44. Lv, D., Wang, G., Zhang, Q., Yu, Y., Qin, P. C. et al. (2022). Comparative transcriptome analysis of hard and tender fruit spines of cucumber to identify genes involved in the morphological development of fruit spines. Frontiers in Plant Science, 13, 797433. [Google Scholar]
45. Qiu, Y., Tao, R., Feng, Y., Xiao, Z., Zhang, D. et al. (2021). EIN3 and RSL4 interfere with an MYB-bHLH-WD40 complex to mediate ethylene-induced ectopic root hair formation in Arabidopsis. PNAS, 118(51), e2110004118. [Google Scholar]
46. Traw, M. B., Bergelson, J. (2003). Interactive effects of jasmonic acid, salicylic acid, and gibberellin on induction of trichomes in Arabidopsis. Plant physiology, 133(3), 1367–1375. [Google Scholar]
47. Yan, T., Chen, M., Shen, Q., Li, L., Fu, X. et al. (2016). HOMEODOMAIN PROTEIN 1 is required for jasmonate-mediated glandular trichome initiation in Artemisia annua. New Phytologist, 213(3), 1145–1155. [Google Scholar]
48. Hua, B., Chang, J., Xu, Z., Han, X., Xu, M. et al. (2021). HOMEODOMAIN PROTEIN 8 mediates jasmonate-triggered trichome elongation in tomato. New Phytologist, 230(3), 1063–1077. [Google Scholar]
49. Zhang, H., Wang, L., Zheng, S., Liu, Z., Wu, X. et al. (2016). A fragment substitution in the promoter of CsHDZIV11/CsGL3 is responsible for fruit spine density in cucumber (Cucumis sativus L.). Theoretical and Applied Genetics, 129(7), 1289–1301. [Google Scholar]
Appendix:
Figure S1: qRT-PCR verification of ten differentially expressed genes (DEGs) from RNA-Seq. The relative expression level of each gene is expressed as the fold change among fruit spines at three stages in the RNA-Seq (blue bar) and qRT-PCR (red bar). Error bars represent the SD
Figure S2: The bubble chart of KEGG pathway enrichment
Table S1: Primers used for qRT-PCR
Table S2: Summary of sequenced RNA libraries
Table S3: List of identified DEGs identified in SpBA8 vs. SpA comparison
Table S4: List of identified DEGs identified in prickleless SpA vs. SpAA8 comparison
Table S5: List of identified DEGs identified in SpBA8 vs. SpAA8 comparison
Table S6: Clusters of DEGs
Table S7: GO enrichment analysis of DEGs identified in the pairwise comparisons
Table S8: KEGG enrichment analysis of DEGs identified in the pairwise comparisons
Table S9: List of DEGs involved in spine development of cucumber
Table S10: List of DEGs related to phytohormones
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools