
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Nucleotide Sequence Assessment of Four ORFs of Citrus Tristeza Virus: Evidence of Recombination
1
Department of Agricultural Biotechnology, College of Agricultural and Food Science, King Faisal University, Al-Ahsa, 31982,
Saudi Arabia
2
Virus and Phytoplasma Research Department, Plant Pathology Research Institute, Agricultural Research Center (ARC), Giza,
12619, Egypt
* Corresponding Author: Adel A. Rezk. Email:
(This article belongs to the Special Issue: Plant–Environment Interactions)
Phyton-International Journal of Experimental Botany 2023, 92(3), 691-705. https://doi.org/10.32604/phyton.2022.024208
Received 26 May 2022; Accepted 19 August 2022; Issue published 29 November 2022
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Citrus Tristeza Virus (CTV), usually occurs in nature as a mixture of genotypes. Six naturally infected citrus (Citrus sinensis) trees grafted on sour orange rootstock were collected from three citrus growing governorates in Egypt (Sharqia, Qalyubia and Garbia). In this study, RT-PCR, Single-Strand Conformation Polymorphism (SSCP) and nucleotide sequence analysis were used for four independent CTV genomic regions (p65, p18, p20, and p23) to detect and assess the sequence and genetic variabilities among CTV Egyptian isolates. RTPCR products (650 bp) for the CTV p23 gene obtained from the selected isolates were used for the SSCP analysis and DNA sequencing. SSCP patterns of p23 gene for individual isolates yielded different complex haplotype patterns. Nucleotide sequence analysis of p23 region amplified from six isolates under study revealed that p23 shared high nucleotide identity 98.7% with T36 isolate from USA, Florida. Phylogenetic analysis of p23 gene indicated a close evolutionary relationship between all examined isolates and Qaha isolate (T36 isolate group), suggesting that they may have originated from closely related ancestors. Nucleotide sequence analysis of the three genes located on CTV 3′-coterminal overhang, p18, p20 and p65, amplified from isolate A3, Sharqia governorate, revealed that the p18, p65, and p20 genes were related to the T3-KB isolate from South Africa with 99%–100% sequence homology. Phylogenetic relationship analysis for p65, p18 and p20 ORFs clustered the current A3 isolate with T3 genotype group. The recombination analysis identified three of six isolates from Sharqia, and Garbia as potential recombinant for p23 gene. The isolates T36 and T3 were identified as major donors for recombination events in isolate A3. Our results concluded that p23 ORF likely to be as a hotspot region for recombination and originated through recombination event. The current study indicated that recombination is an important factor for the origin of CTV strains in Egypt.Keywords
Citrus Tristeza Virus (CTV) is widespread in its distribution and is the most destructive and economically important virus of commercial citrus worldwide. CTV, a member of the genus Closterovirus within the family Closteroviridae, is phloem limited and is transmitted by aphids in a semi persistent manner. CTV was first reported in Qanater region, Qalyubia governourate in Egypt in 1957 [1]. CTV virions are flexuous thread-like particles about 2,000 × 11 nm long, with a genomic RNA (gRNA) molecule and two coat proteins (CP and CPm) coating 95% and 5% of the particle length, respectively [2]. The CTV genome is a 19.3-kb single stranded, positive sense RNA genome that contains 12 open reading frames (ORFs) with untranslated regions (UTRs) of 108 and 290 nucleotides at the 5′- and 3′-termini, respectively [3]. The CTV genome has two conserved genes, the replication gene block (ORF 1a and 1b), which are unique of this group of viruses. The 10 other ORFs, expressed through subgenomic RNAs, encode the major and minor capsid proteins (p25 and p27), two genes expressing a heat shock protein homolog (p65) and p61, both required for virion assembly [4]. The three genes identified as host range genes (p33, p13, and p18) appear to have a role in stem pitting development. Certain deletion combinations of these three genes have been shown to improve stem pitting symptoms, while others were shown to reduce stem pitting symptoms [5]. p33 protein is necessary for effective transmission by the aphid vector (Toxoptera citricida) [6], three suppressors of RNA silencing (p25, p20, and p23) [7,8]. The p23 protein is an RNA binding protein and thought to play a regulatory role in the expression of other CTV genes and could be used as a disease severity indicator [9]. The p23 protein is an important pathogenicity factor and it is involved in symptom production in Citrus and related species [10,11]. The p20 protein is the major component of CTV amorphous inclusion bodies [12]. CTV is unique among the Closteroviridae in that it has a diversity of strains and defined isolates that create a wide range of phenotypic combinations in its hosts [13]. The majority of viral isolates consist of a population of various strains that co-infect a host and interact to create a variety of symptoms [13,14]. Seedling yellows, stem pitting, and quick decline are three prominent host syndromes caused by CTV, the last two of which are important problems for the citrus industry [15]. CTV and Candidatus Liberibacter asiaticus have recently been linked to citrus decline in India, with greater incidences of 70.58% and 27.45%, respectively [16]. Because of CTV’s genetic diversity, determining the virus strains present in an area is critical for determining control strategies. The most precise method for CTV differentiation and genetic diversity assessment is nucleotide sequence analysis. Complete genome analysis has revealed wide variation among CTV isolates worldwide [17,18] and classified them into six CTV genotype groups, designated as T36, T30, VT, B165, HA16-5 and Resistant Breaking (RB) [19,20]. Several genomic loci have been tested and described to determine the genetic variation within the CTV isolates [21–24]. The CTV genotype strains based on ORF1a region were classified into six groups namely T3, T30, T36, T68, VT and RB [13]. New strains have recently been proposed, including NC (aka HA16-5), S1, L1, and M1 [25,26]. Based on CP and P18 gene sequencing, severe CTV isolates close to the T36 genotype and moderate CTV isolates similar to the T30 genotype have recently been found in eastern Mexico and Georgia state, USA, respectively [27,28]. Phylogenetic study based on four genomic ORFs (ORF1a, p25, p23, and p18) has recently allowed CTV isolates to be classified into nine broad groups: RB, T36, T30, T3, T68, VT or VT-B, HA16-5, B1, and B2 [22].
Recombination is an important evolutionary factor for the evolution and divergence of many positive sense RNA viruses. It contributes to the diversification of plant viruses and their adaptation to new hosts, which frequently results in the emergence of new variants and viral strains that overcome plant viral resistance. The identification of genomic regions that are hotspots for repeated recombination is important in understanding the supposed role of recombination in the development and emergence of new viral strains/variants [29,30]. Recombination of viral genomes is especially significant in long-term persistent infections, where many viral variants co-replicate in a single host, resulting in a large number of genotypic variants, some of which may have novel host-colonizing and pathogenicity properties. As a result, the persistent infection of numerous viral genotypes within a host organism is enough to cause large-scale generation of viral genetic variants that could evolve into new and emerging viruses [31–33]. To date, new strains or genome variation and recombinant isolates of CTV continue to be reported in many countries [25,34,35].
In Egypt, however, there is scanty information on the molecular-based characterization of CTV. In 2006, the coat protein gene (CP) of three Egyptian CTV isolates from two locations (Inshas and El Qanater) revealed no significant differences in sequence composition. Each isolate had remarkably similar haplotypes that clustered closely with the severe Florida strain T3 [21]. Egyptian CTV isolates from the governorates of Qalyubia and Ismailia had their p20 and p23 genes analyzed, and the isolates shared significant nucleotide identity with VT severe strains [24]. The complete nucleotide sequence ofa CTV Egyptian isolate (Qaha) is available in Genbank (accession no. AY340974), but no further information about its origin was given. As a result, in order to acquire a better knowledge of the genetic features of CTV Egyptian isolates, in this study, we used the SSCP and nucleotide sequence analysis for four CTV genomic region to assess the sequence and genetic diversity among CTV Egyptian isolates. The recombination analyses were also done to better understand its population structure of variants and evolution.
Naturally infected citrus (Citrus sinensis (Washington and Valencia)) trees grafted on sour orange rootstock with putative CTV decline inducing symptoms (decline, scion overgrowth, and honey-combing) were collected from three different governorates in citrus-growing regions in Egypt. The six CTV isolates from Egypt included in this study (Table 1) were chosen to represent CTVs that are currently spreading under natural conditions. Isolates were maintained on Mexican lime seedlings (C. aurantifolia (Christm.) Swing) as indicator plant in the greenhouse at 28°C. The isolates were tested against polyclonal and Monoclonal (MCA13) antibodies specific for CTV by DAS-ELISA and indirect IDAS ELISA [36].
The p23, p20, p18 and p65 genes were isolated from the phloem of the tested isolates. Total RNA was isolated using Total RNA Mini Kit for plant (Geneaid Biotech, Ltd., Taiwan) according to the manufacturing manual protocol. The one-step RT-PCR was performed using Verso TM one-step RT- PCR kit (Thermo Scientific, USA) according to the instruction manual. The reaction was performed in 25 μL total volume of amplification mixture using 12 ng of RNA and 20 μM of each PCR specific primer (Table 2). The Amplification proceeded in the thermocycler (Uno) at 50°C for 15 min and 95°C for 15 min, and through 35 cycles of 94°C for 30 s and 54°C (for amplification of p65, p20 and p23 gene) and 42°C (for amplification of P18 gene) for 45 s and 72°C for 1 min, with a final step at 72°C for 10 min. The RT-PCR products were directly analyzed on a 1% prestained agarose electrophoresis gel using EZView stain and the gel electrophoresis as described by Sambrook et al. [37] and then visualized by UV illumination.
2.3 Molecular Cloning and SSCP Analysis
The amplified products for the four genes were cloned in a pCR2.1-TOPO cloning vector (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instruction manual using ampicillin as resistance antibiotic. The amplified products (1–2 μL) obtained from the RT-PCR and clones of selected colonies were used for the Single-Strand Conformation Polymorphism (SSCP) analysis. SSCP analysis was performed as antecedent represented by Rubio et al. [38] and Kong et al. [39]. Samples were resolved by electrophoresis in 8% polyacrylamide gels at 200 volts, 4°C for 3 h.
2.4 Nucleotide Sequencing and Phylogram Analysis
The clones for each gene were sequenced in both directions using ABI PRISM DNA 377sequncer (Perkinelmer, USA). A multiple sequence alignment was done along with 14 reference sequences from the GenBank that represented each the internationally recognized CTV genotype (Table 3), using DNAMAN 8.0 software (Lynnon Biosoft, Quebec, Canada). The Expasy Translate Tool (http://us.expasy.org/tools/dna.html) was used to translate amino acids (aa). The Maximum likelihood phylogenetic tree (ML) at 1000 bootstrap were constructed using DNAMAN software in order to compare our four amplified regions as individual ORFs. The tree was visualised with the TreeView (Win32) Version 1.6.6 application.
2.5 Detection of Recombination Hotspot
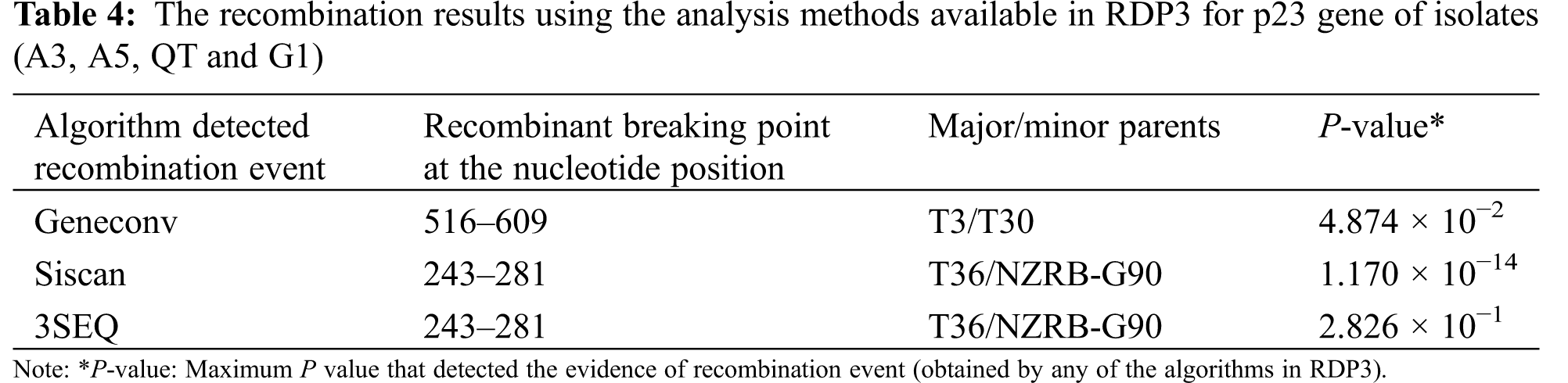
A multiple sequence alignment was together with fourteen reference CTV Isolates obtained from the Genbank using DNAMAN 8.0 software. To determine the genomic regions that were affected by the possible recombination events, the four isolated genes (p23, p20, p18 and p65), at the 3′end of CTV genome, were screened with six totally different algorithms (RDP, GENECONV, MaxChi, 3Seq, Chimeara and SiScan) implemented within the RDP3 (Recombination Detection Program version 3) Alpha 44 program for the analysis according to RDP3 guide. Any event that supported all the five methods with P-values ≤ 0.05 was the criteria used for positive recombination breakpoints identification. The breakpoint position and therefore the supposed parental sequences were also determined.
All isolates of CTV displayed CTV decline inducing symptoms. Fig. 1 indicated the scion overgrowth and honey-combing. Mild chlorotic flecks, vein clearing, leaf cupping with only minor stunting with twig discoloration symptom were induced on Mexican lime indicator plant as shown in Fig. 2. No stem pitting symptom was observed either on Field tree or Mexican lime indicator plant. The isolates were reacted positively with polyclonal (PAbs) and monoclonal antibodies (MCA-13) using DAS-ELISA (Table 1). The infected samples showed OD values, which ranged from 0.900 to 0.446 using PAbs and 0.748–0.303 using MCA-13. Healthy control showed OD value of 0.084 and 0.133 using PAbs and MCA-13, respectively, as illustrated in Table 1.

Figure 1: Tristeza disease caused by Citrus Tristeza Virus (CTV) in Navel orange field tree grafted on sour orange rootstock. A: Scion overgrowth symptom, B: Honeycombing of the internal bark of Sour orange rootstock of infected Navel orange field tree

Figure 2: Tristeza disease symptoms on Mexican lime indicator plant. A: stunting reaction of Mexican lime (Citrus auranrifolia) indicator plant (the left seedlings are healthy plants), B: leaf-cupping symptom of infected Mexican lime, C&D: mild vein clearing and leaf-flecking symptoms in Mexican lime leaf. E: leaf deformation, F: xylem discoloration of Mexican lime twig
3.2 RT-PCR Amplification and SSCP Analysis
The current Egyptian isolates yielded an expected DNA fragments 650 bp corresponding to p23 gene as shown in Fig. 3B. The obtained RT-PCR products were used for the SSCP analysis. SSCP patterns of p23 gene for each isolate displayed a complex pattern (Fig. 4A). The SSCP pattern was the same in isolates A3, A5, and QT. Isolates Q2 and Q3 had similar SSCP profile, whereas isolates G1 had different SSCP patterns (Fig. 4A). The amplified products for the p23 gene for isolates (A3, A5, Q2, Q3, QT and G1) showing identical and different SSCP patterns were cloned. SSCP analysis of all P23 clones from the isolate A3 revealed a complicated SSCP profile similar to the corresponding RT-PCR product (Fig. 4B). Hence, only one clone for each isolate was chosen for sequence analysis.
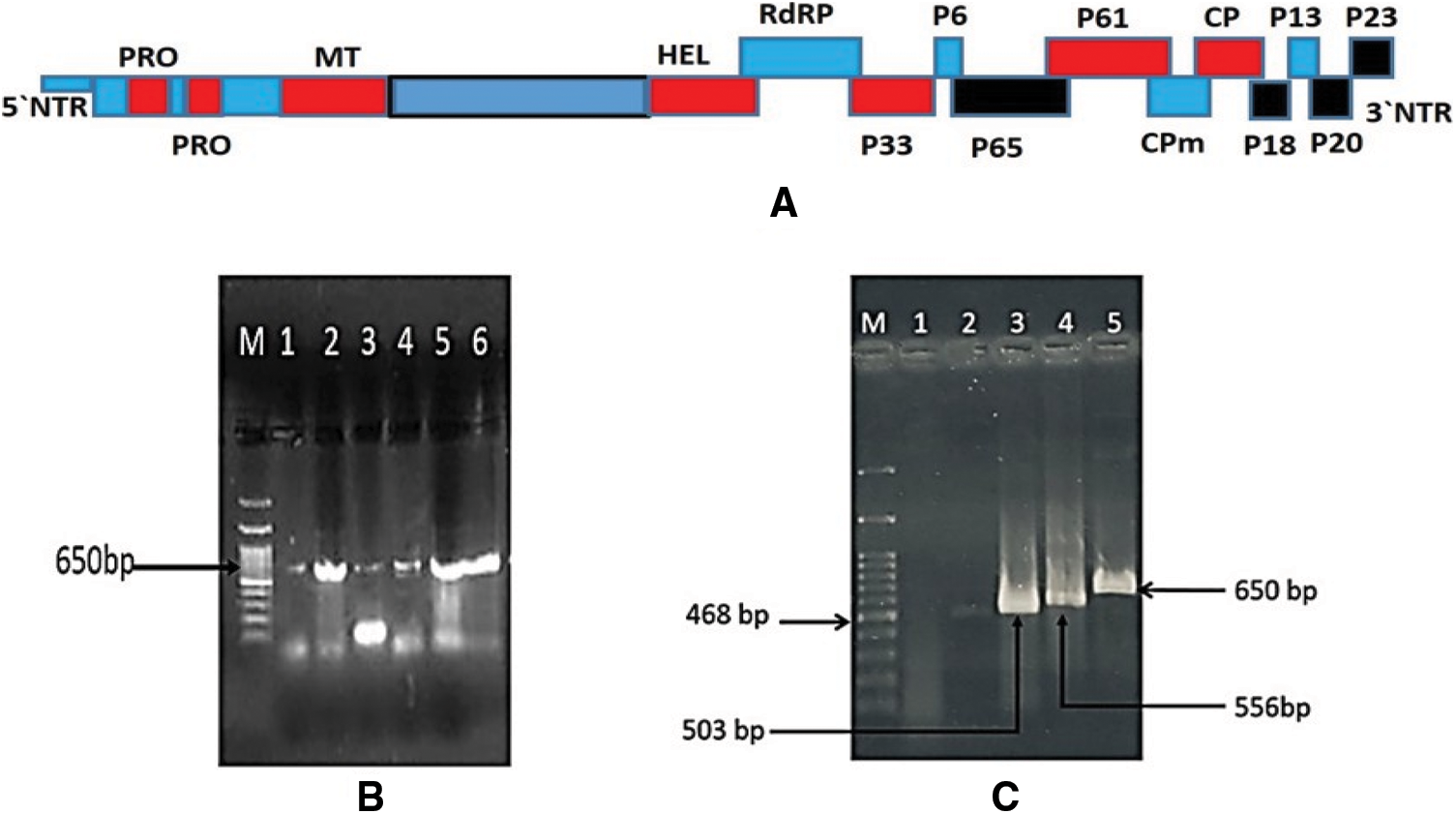
Figure 3: A: Schematic representation of the CTV genome and its corresponding putative protein products encoded by them. Black boxes represent location of CTV ORFs examined in this study; B: 1% agarose gel electrophoresis analysis of the RT-PCR products of p23 gene amplified from infected trees with CTV isolates. Lane 1 corresponding to isolate G1; Lan 2: isolate QT; Lane 3: isolate Q2; Lane 4: isolate Q3; Lane 5: isolate A3; Lane 6: isolate A5 and Lane M: 100 bp DNA ladder. C: 1% agarose gel electrophoresis analysis of the RT-PCR amplified products from four CTV ORFs isolated from Mexican lime seedling infected with A3 CTV isolate and corresponding to Lane1: healthy Mexican lime seedling Plant. Lane 2: p65 (468 bp); Lane 3: p18 (503 bp) Lane 4: p20 (556 bp) and Lane 5: p23 (650 bp)
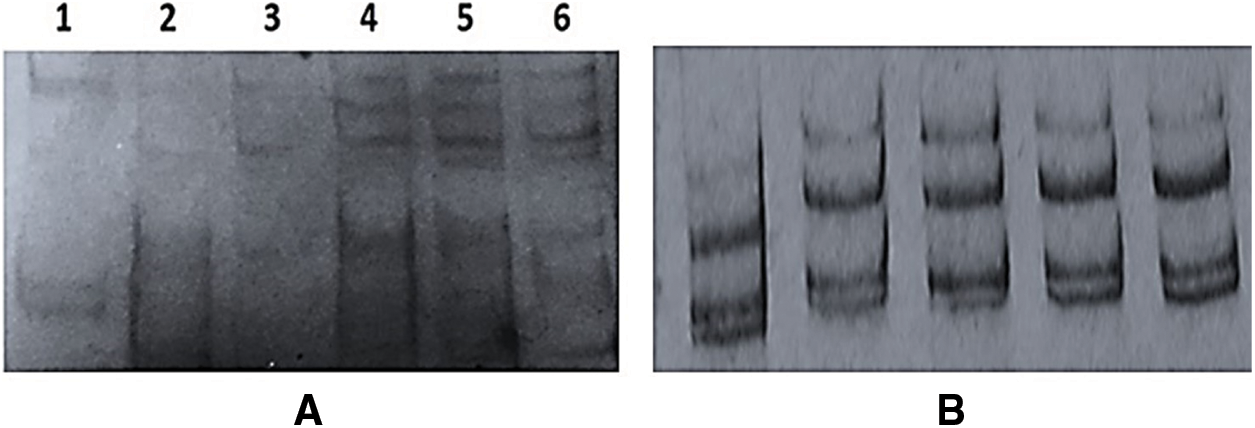
Figure 4: Single-Strand Conformation Polymorphism (SSCP) patterns of CTV-p23 gene from A, RT-PCR products of P23 gene for the present Egyptian CTV isolates; Lane 1: isolate G1; Lan 2: isolate Q2; Lane 3: isolate Q3; Lane 4: isolate QT; Lane 5: isolate A3 and Lane 6: isolate A5; and B, PCR amplification from p23 clones of isolate A3
The CTV isolate A3 from the Sharqia Governorate was chosen for further sequence analysis across the CTV genomic regions. The genomic regions p18, p65 and p20 at 3′ prime of A3 CTV isolate (Fig. 3A) were also amplified. The products size for the four regions of CTV isolate A3 were 556 bp for p20 gene, 468 bp for p65, 650 bp for p23 and 503 bp for p18 gene (Fig. 3C). No amplification was obtained from uninfected plants grown in the greenhouse. The amplified products for the p20, p18 and p65 regions for isolate A3 were cloned and subjected to nucleotide sequencing. The SSCP pattern for cDNA clones of p20 and p18 genes each showed the same SSCP pattern between cDNA clones (data not shown). All examined clones were subjected for sequence analysis.
The sequence alignments for p23 gene of the examined isolates showed that the p23 gene with the same SSCP pattern differed by 1 to 2 nt, except for one isolate (G1 isolate) with eight nucleotide changes and showed nucleotide distance matrix 0.12–0.019 between all tested isolates. Multiple sequence alignments of current CTV isolates (A3, A5, Q2, Q3, QT and G1) showed 98.1%–100% sequences homology between each other (data not shown). The sequence comparison for p23 gene revealed that the p23 gene have high sequence homology 97%–98.7% with Qaha and T36 isolates from Egypt and USA, Florida, respectively. Whereas, The P23 gene of current isolates showed low nucleotide identity 85.8%–89.8% with NZRB-G90 resistant breaking (RB) isolate, 88.4%–90.5% with T3 genotype isolates (T3 and T3-KB). As well as 88.4%–90.0% with severe stem pitting isolate (VT and VT-ISR) and 86.5%–90.5% with Indian HA16-5, B165 and T68-1 isolates (data not shown). All of the examined isolates had 95.8%–100% amino acid (aa) sequence similarity for the p23 gene. While they were 93.0%–97.6% similar to the T36 strain, they were 81.7%–86.1% aa identity to VT and T3 strains. Because of 98.1%–100% sequence identity among all present Egyptian isolates, one isolate A3 was chosen for further sequence analysis of the CTV isolate across the CTV genomic regions.
Positive clones of RT- PCR products (2 to 5) were obtained from the genes coding for p18 (503 nt) and p20 (556 nt), and one clone from p65 (468 nt) at 3` half of CTV genome of isolate A3. The sequence alignments for the clones of p20 and p18 genes showed that the sequence of the p20 and p18 genes were almost conserved in all clones differing by 1–2 nt with average distance matrix 0.007. For p65 (HSP70 analogue) gene shared 97%–99.6% nucleotide identity with T3 isolate from Florida and T3-KB isolate from South Africa as illustrated in Table 3, but show low nt identity 88%–88.2% with Florida T30 and SY568 isolates (Table 3). Analyses of the p18 gene of isolate A3 revealed nucleotide identity of 99.4% with T3-KB from South Africa and 97.4% with T3 isolate from Florida, respectively. While the P18 gene showed sequence homology of 92.3%–93.8% with all used GenBank reference isolates with the lowest identity of 89.9% with Hawaii isolate HA16-5 (Table 3). Sequence comparison of p20 gene showed nucleotide identity 99.6% with T3-KB from South Africa and 98% identity with T3 isolate from Florida. It revealed sequence homology 95.9%–96.4% with SY568, T318A, B165, T68-1 and 94.1%–94.6% with VT isolates from Israel and VT from Florida (EU937519.1). Whereas distant from other CTV isolates, it shared 90.7% identity with NZRB-G90 resistant breaking isolate and all the T36 genotype CTV isolates (Qaha, and T36) from Egypt, Florida and USA, respectively (Table 3). The nucleotide sequences of the present study have been submitted in the GenBank database under accession numbers ON035624 to ON035637.
The rooted maximum likelihood Phylogenetic tree (ML) at 1000 bootstrap were constructed using DNAMAN 8.0 software. The bootstrap values for all tested genes were over 900. The phylogenetic tree for the p23 gene sequence of the current isolates (A3, A5, Q2, Q3,QT and G1) clustered in T36 genotype group and all tested isolates were closely related to T36 genotype isolates (Qaha and T36) from Egypt and USA (Fig. 5A). The phylogenetic analysis of the p23 gene indicated a close evolutionary relationship between the current isolates and Qaha isolate from Egypt (T36 isolate group) which might have been originated from closely related ancestors. The rooted phylogenetic tree for p23 deduced amino acids revealed that the present isolates were closely related to T36 protein (Fig. 5B). Whereas, phylogenetic analysis for p65, p20 and p18 ORFs of the isolate A3 revealed that it was closely related to decline inducing T3 genotype isolates (T3-KB and T3) and they considered the common ancestor as illustrated in Figs. 6A–6C.
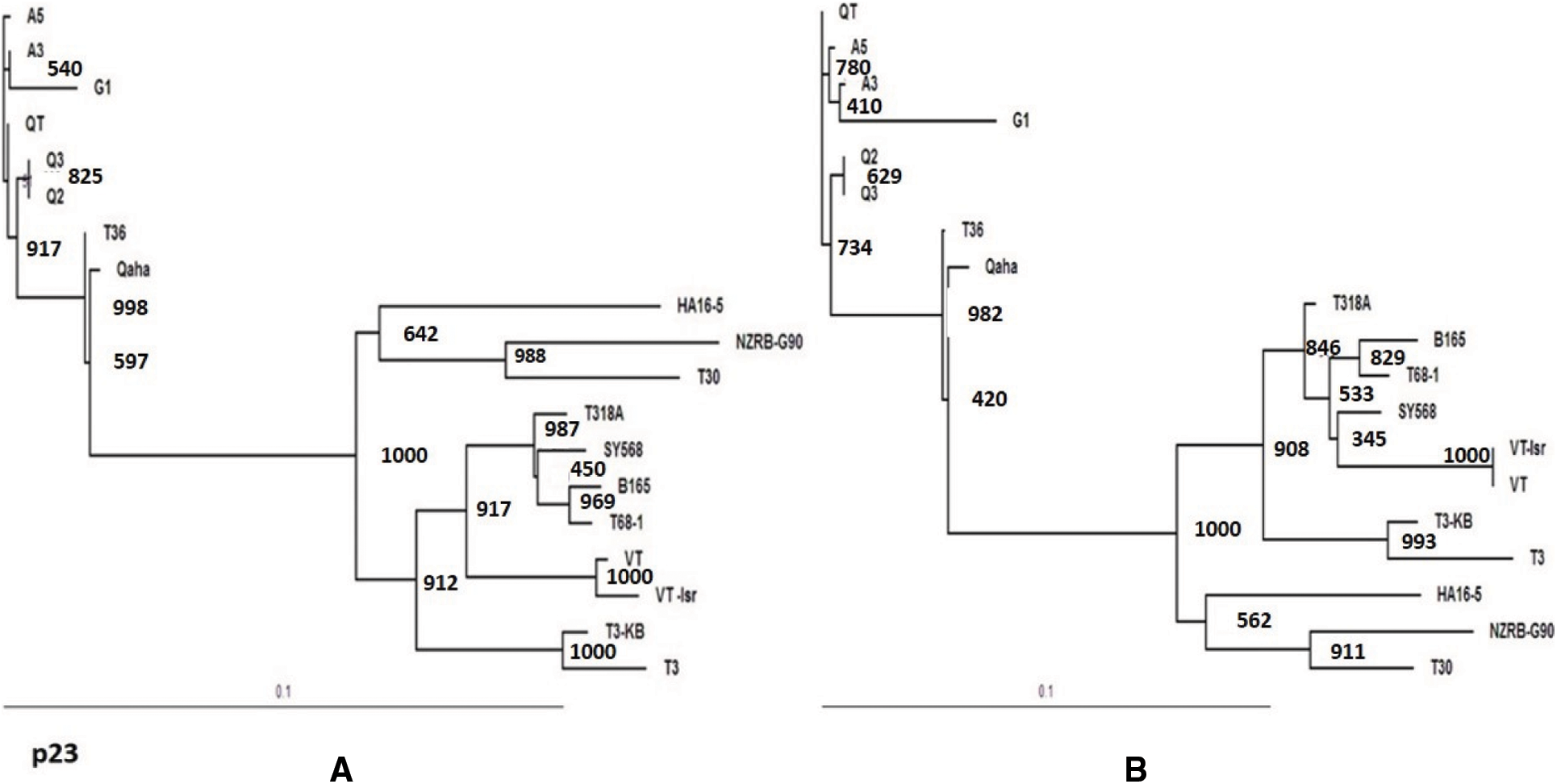
Figure 5: Phylogenetic relationships of the present CTV isolates (A3, A5, Q2, A3, QT and G1) with other reference GenBank isolates based on the nucleotide sequences of the p23 gene (A) and the deduce amino acids (B)
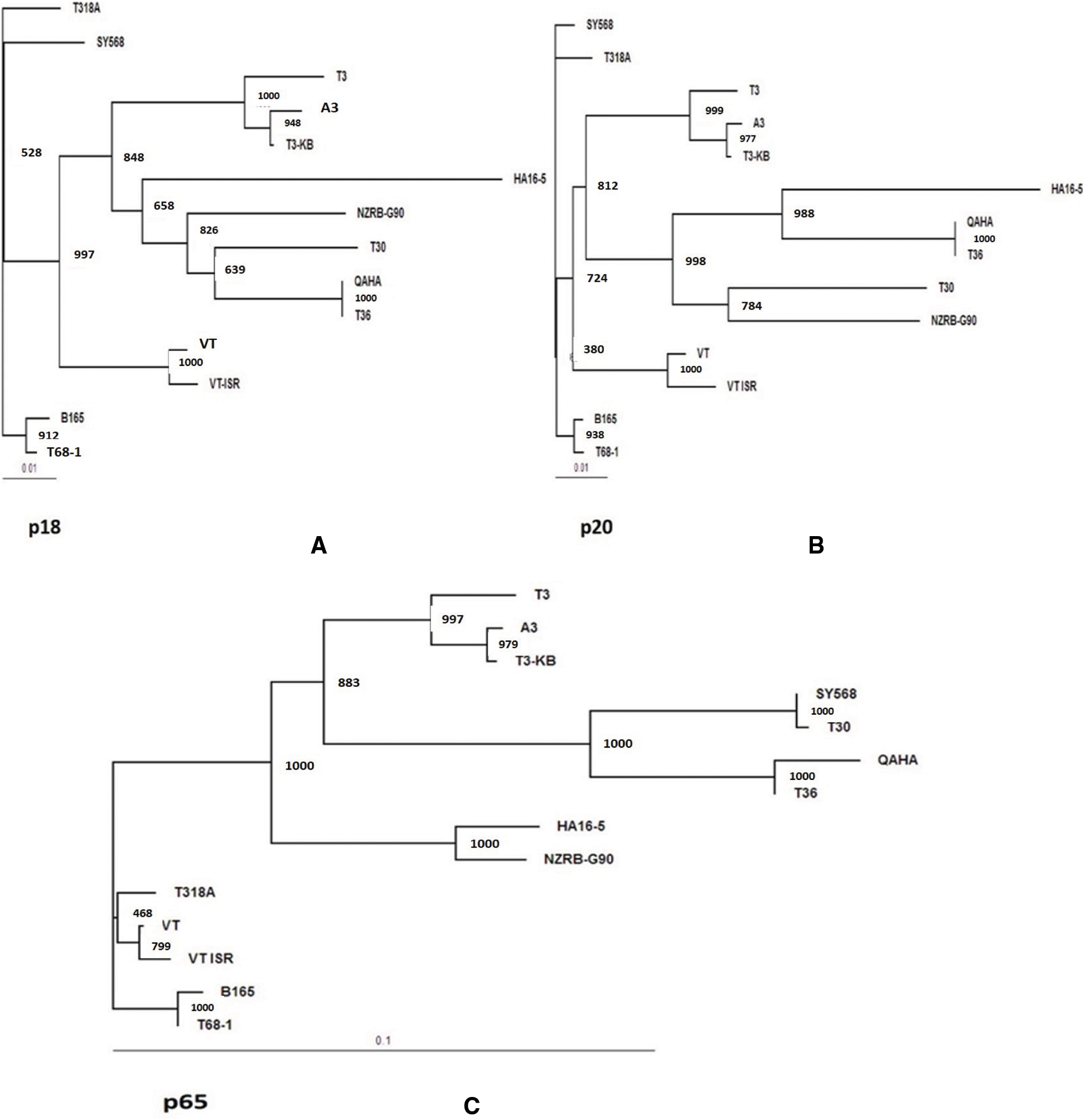
Figure 6: Phylogenetic tree for the present CTV isolate A3 with other reference CTV isolates based on the nucleotide sequences for three genes. A: p18 gene, B: p20 gene and C: p65 gene, at 3` end region of the viral genome. Maximum likelihood Phylogenetic tree (ML) was at 1000 bootstrap repetition
The detection of recombination events in present isolates was carried out in this investigation using RDP3 v.3.44 program. Three algorithms (GENECONV, 3Seq and SiScan) of RDP3 software identified four of six isolates A3, A5, QT and G1 with accession numbers; ON035635, ON035630, ON035629 and ON035628 as potential recombinant for the p23 gene (Table 4). Two recombination events were detected at nucleotide positions of 243–281 and 516–609. The isolates T36 and T3 were found to be major parents for recombination events of the three identified isolates. In addition, the isolates T36 and T3 were detected as the predominant major parents with percentage 99.4% and 89.9%, respectively. The mild isolate T30 from Florida, and the (RB) resistant breaking NZRB-G90 isolate from New-Zealand were detected as minor parents for recombination of detected isolates (Table 4). The predominant breakpoint position was at nucleotide 243 and 281 as illustrated in Table 4. No evidence of recombination in the p65, p18, or p20 ORFs of isolate A3.
CTV has expanded widely in various parts of the Mediterranean basin in the recent decade, with citrus trees infected with severe strains of the virus being documented in several countries [40,41]. CTV infection causes various disease symptoms. CTV isolate usually falls into one or more of the general biological symptom categories as previously identified by Hilf et al. [32]. The current Egyptian CTV isolates (A3, A5, Q2, Q3, QT, and G1), which were selected for this study, were described as decline inducing (DI) isolates according to the biological symptoms. The present isolates reacted positively with polyclonal and monoclonal antibodies (MCA-13). The MCA-13 reacts primarily positively with most severe strains [42]. For all tested CTV isolates, SSCP analysis of cDNA from the p23 CTV gene was performed to define the population of CTV variants. SSCP patterns of RT-PCR amplicons from p23 gene for the present isolates revealed a complicated pattern. The obtained complex SSCP pattern of p23 region of each isolate indicates the presence of CTV-variants within a single isolate. Nonetheless, SSCP profile from p23 clones of isolate A3 revealed a complicated SSCP profile similar to the corresponding RT-PCR product indicating that the RT-PCR SSCP profile could be resulted from the same predominant RNA strand and did not result from amplification of different virus populations from the same plant. Sharqia and Qalyubia governorate isolates A3, A5, and QT showed haplotype matching, whereas Qalyubia and Garbia governorate isolates Q2, Q3, and G1 displayed unique haplotype conformations. Despite this, the tested isolates have a high level of nucleotide sequence similarity (98.1% to 100%) among themselves. However, SSCP is sensitive enough to detect single nucleotide differences in rather large DNA fragments [43]. The nucleotide sequence comparison of p23 region amplified from six isolates under study with the GenBank reference isolates suggested that all of the sequences had a high similarity to the CTV T36 sequence. The T36 isolate from USA, Florida considered biologically as intermediate decline inducing (DI) strain of CTV [34,44,45]. The phylogenetic analysis revealed that the p23 gene sequences for the current isolates (A3, A5, Q2, Q3, QT and G1) belonged to the CTV T36 genotype and closely related to Florida T36 isolate and Qaha isolate from Egypt and they considered the common ancestor.
Because all of the examined Egyptian isolates have a high percentage of p23 sequence identity, A3 isolate from Sharqia Governorate was chosen for further sequencing investigation of the CTV isolate across the CTV genomic regions. The genomic regions p18, p65 and p20 at 3′ end of CTV isolate A3 were amplified and cloned. The nucleotide sequence of three ORFs (p65, p20 and p18) from the A3 isolate were assessed. The sequence comparison and the phylogeny analysis of the three tested genes (p65, p18 and p20) with GenBank CTV reference strains was mentioned previously by Harper [13], Zablocki et al. [46], Biswas et al. [34] and Ghosh et al. [22]. They revealed that the present A3 isolate from Sharqia had high sequence identity with T3-strains and was closely related to the South Africa decline inducing CTV isolate T3-KB and the T3 isolate from Florida that considered the common ancestor. Meanwhile, the tested genes (p65, p18 and p20) of A3 isolate are distant from the Egyptian Qaha and Florida T36 isolates. The sequence comparisons of p65 and p20 gene of A3 isolate from Sharqia detected that p65 is distant from mild Florida T30 and seedling yellow SY568 isolates. While the P20 gene revealed that the A3 isolate is related to VT, SY568, T318A, B165 and T68-1, severe isolates with sequence homology ranged from 94.1% to 96.4%. Comparisons of sequences of four ORFs from the 3-terminal region of Egyptian CTV isolate (A3), provided additional evidence that more than one genotype may exist within an isolate [47]. The T3-KB isolate from South Africa is severe quick decline inducing of citrus species grafted on sour orange rootstock and it induces stem-pitting in sweet orange [15]. The stem pitting symptom was not detected on the tested isolates, either in the field or in the greenhouse indicator plants. According to the present result, the greenhouse and field symptoms could be due to the p23 gene (ORF11), which is closest to the T36 genotype. The T36 strain was biologically classified as an intermediate decline inducing (DI) strain of CTV [44]. Our findings support the theory that the CTV p23 gene is responsible for pathogenicity and disease severity [4,34,48]. In previous investigations by Amin et al. [21], SSCP and nucleotide sequencing investigation based on coat protein (p25) gene of three Egyptian CTV isolates from two places in Egypt (El-Kanater and Anshas) clustered the isolates very close to the severe strain T3 from Florida that causes rapid decline and stem pitting. Youssef et al. [24] identified additional strains, including a VT haplotype (severe stem pitting isolate) for samples collected from Qalyubia and Ismailia in Egypt using SSCP and nucleotide sequencing based on the p20 and p23 regions of the CTV genome. So, the incidence of severe isolates and their consequences for future CTV epidemics must be considered.
Many positive RNA viruses use recombination as one of their primary strategies of development and divergence. The identification of genomic regions that are hotspots of repeated recombination is critical in understanding the supposed role of recombination in the development and emergence of new viral strains/variants [29,30,49]. The recombination analysis identified four of six isolates (A3, A5. QT and G1) from Sharqia, Qalubyia and Garbia, respectively, as potential recombinant for the p23 gene. The T36 and T3 genotypes were found to be the major donors for recombination events. The mild isolate T30 and the (RB) resistant breaking NZRB-G90 isolate were detected as minor parents. Our data suggested that the CTV genome of isolates A3, A5. QT and G1 originated through recombination events by exchanging the genetic material between Severe and mild divergent sequences. This result supports the finding that the majority of the sequences associated with severe isolates had additional sequences associated with mild and/or atypical isolates. The existence of the severe and/or unusual sequence types is thought to be associated with the severe phenotype [49]. The absence of recombination events in the other CTV genomic regions of isolate A3 (p18, p20, and p65) indicates that the p23 region sequence may be a preferred region for potential recombination events and the prevalent isolate genotype was T3 genotype.
This result came in line with results of Sambade et al. [50,51]. On the other hand, it is inconsistent with Biswas et al. [34] reported that most CTV isolates did not display recombination events in the CP, p20, or p23 regions, while the majority of CTV isolates preferred recombination in the HSP70, p33, and p6 regions. The current study indicated that recombination is an important factor for the origin of CTV strains in Egypt. Obvious recombination patterns within the genomes of the viruses can reveal a great deal about their biology and evolution [30,13]. In addition, this could be a useful tool for eradicating and cross protection programs which significant to predict the mild and severe isolates.
The sequence comparison with 14 CTV GenBank reference isolates, representing each the internationally recognized CTV genotypes showed that the amplified p23 gene was closely related to Florida T36 isolate and Qaha isolate from Egypt. The p65, p20 and p18 genes amplified from isolate A3 was closely related to T3-KB isolate from South Africa. The present study revealed that the SSCP analysis is a powerful and accurate technique to estimate the CTV genetic diversity. Recombination analysis of the p18, p20, p65 and p23 genomic regions of the isolate A3 was performed individually. The results indicate that the p23 region sequence may be a favorable region for potential recombination events. The current study confirm that more than a single introduction of CTV could have occurred in Egypt over the years.
Acknowledgement: Authors extend their appreciation to the Deanship of Scientific Research, King Faisal University, Saudi Arabia, for supporting this research for work through Project No. GRANT494.
Authorship: The authors confirm contribution to the paper as follows: study conception and design: A.A.R. and H.A.A.; data collection: A.A.R. and H.A.A.; analysis and interpretation of results: A.A.R. and H.A.A.; draft manuscript preparation: A.A.R. and H.A.A. All authors reviewed the results and approved the final version of the manuscript.
Funding Statement: Authors extend their appreciation to Deanship of Scientific Research, King Faisal University, Saudi Arabia, for supporting this research (GRANT494).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Nour-Eldin, F., Bishay, F. (1958). Presence of the tristeza virus disease in Egypt. FAO Plant Protection Bulletin, 6, 153–154. [Google Scholar]
2. Bar-Joseph, M., Dawson, W. O. (2008). Citrus tristeza virus. In: Mahy, B. W. J., van Regenmortel, M. H. V. (Eds.Encyclopedia of virology. Third Edition, pp. 520–525. USA: Academic Press. [Google Scholar]
3. Fuchs, M., Bar-Joseph, M., Candresse, T., Maree, H. J., Martelli, G. P. et al. (2020). ICTV virus taxonomy profile: Closteroviridae. Journal of General Virology, 101(4), 364–365. DOI 10.1099/jgv.0.001397. [Google Scholar] [CrossRef]
4. Satyanayanana, T., Gowda, S., Ayllon, M. A., Dawson, W. O. (2004). Closterovirus bipolar virion: Evidence for initiation of assembly by minor coat protein and its restriction to the genomic RNA 5 region. PNAS, 101(3), 799–804. DOI 10.1073/pnas.0307747100. [Google Scholar] [CrossRef]
5. Tatineni, S., Dawson, W. O. (2012). Enhancement or attenuation of disease by deletion of genes from Citrus tristeza virus. Journal of Virology, 86(15), 7850–7857. DOI 10.1128/JVI.00916-12. [Google Scholar] [CrossRef]
6. Shilts, T., El-Mohtar, C., Dawson, W. O., Killiny, N. (2020). Citrus tristeza virus P33 protein is required for efficient transmission by the Aphid Aphis (Toxoptera) citricidus (Kirkaldy). Viruses, 12(10), 1131. DOI 10.3390/v12101131. [Google Scholar] [CrossRef]
7. Lu, R., Folimonov, A., Shintaku, M., Li, W. X., Falk, B. W. et al. (2004). Three distinct suppressors of RNA silencing encoded by a 20-kb viral RNA genome. PNAS, 101(44), 15742–15747. DOI 10.1073/pnas.0404940101. [Google Scholar] [CrossRef]
8. Ruiz-Ruiz, S., Soler, N., Sánchez-Navarro, J., Fagoaga, C., López, C. et al. (2013). Citrus tristeza virus p23: Determinants for nucleolar localization and their influence on suppression of RNA silencing and pathogenesis. Molecular Plant-Microbe Interactions, 26(3), 306–318. DOI 10.1094/MPMI-08-12-0201-R. [Google Scholar] [CrossRef]
9. Lόpez, C., Navas-Castillo, J., Gowda, S., Moreno, P., Flores, R. (2000). The 23-kDa protein coded by the 3′-terminal gene of Citrus tristeza virus is an RNA-binding protein. Virology, 269, 462–470. [Google Scholar]
10. Fagoaga, C., López, C., Moreno, P., Navarro, L., Flores, R. et al. (2009). The P23 protein from Citrus tristeza virus is a pathogenicity determinant in transgenic citrus hosts. Tree and Forestry Science and Biotechnology, 3(2), 23–29. [Google Scholar]
11. Amin, H. A., Falk, B. W. (2009). Citrus tristeza virus p23 gene correlated with the pathogenicity in non-citrus hosts. Egyptian Journal of Phytopathology, 37(1), 83–98. [Google Scholar]
12. Gowda, S., Satyanarayana, T., Davis, C. L., Navas-Castillo, J., Albiach-Martí, M. R. et al. (2000). The p20 gene product of Citrus tristeza closterovirus accumulates in the amorphous inclusion bodies. Virology, 274(2), 246–254. [Google Scholar]
13. Harper, S. J. (2013). Citrus tristeza virus: Evolution of complex and varied genotypic groups. Frontiers in Microbiology, 4, 1–18. DOI 10.3389/fmicb.2013.00093. [Google Scholar] [CrossRef]
14. Hernández-Rodríguez, L., Benítez-Galeano, M. J., Bertalmío, A., Rubio, L., Rivas, F. et al. (2019). Diversity of Uruguayan Citrus tristeza virus populations segregated after single aphid transmission. Tropical Plant Pathology, 44(4), 352–362. DOI 10.1007/s40858-019-00288-x. [Google Scholar] [CrossRef]
15. Cook, G., van Vuuren, S. P., Breytenbach, J. H. J., Steyn, C., Burger, J. T. et al. (2016). Characterization of Citrus tristeza virus single-variant sources in grapefruit in greenhouse and field trials. Plant Disease, 100(11), 2251–2256. DOI 10.1094/PDIS-03-16-0391-RE. [Google Scholar] [CrossRef]
16. Ghosh, D. K., Kokane, A. D., Kokane, S. B., Tenzin, J., Gubyad, M. G. et al. (2021). Detection and molecular characterization of ‘Candidatus liberibacter asiaticus’ and Citrus tristeza virus associated with citrus decline in Bhutan. Phytopathology, 111(5), 870–881. DOI 10.1094/PHYTO-07-20-0266-R. [Google Scholar] [CrossRef]
17. Biswas, K. K. (2010). Molecular characterization of Citrus tristeza virus isolates from the Northeastern Himalayan region of India. Archives of Virology, 155, 959–963. [Google Scholar]
18. Biswas, K. K., Tarafdar, A., Sharma, S. K. (2012). Complete genome sequence of mandarin decline Citrus tristeza virus of the Northeastern Himalayan hill region of India: Comparative analyses determine recombinant. Archives of Virology, 157(3), 579–583. DOI 10.1007/s00705-011-1165-y. [Google Scholar] [CrossRef]
19. Melzer, M. J., Borth, W. B., Sether, D. M., Ferreira, S., Gonsalves, D. et al. (2010). Genetic diversity and evidence for recent modular recombination in Hawaiian Citrus tristeza virus. Virus Genes, 40, 111–118. [Google Scholar]
20. Licciardello, G., Scuderi, G., Ferraro, R., Giampetruzzi, A., Russo, M. et al. (2015). Deep sequencing and analysis of small RNAs in sweet orange grafted on sour orange infected with two Citrus tristeza virus isolates prevalent in Sicily. Archives of Virology, 160(10), 2583–2589. DOI 10.1007/s00705-015-2516-x. [Google Scholar] [CrossRef]
21. Amin, H. A., Fonseca, F., Santos, C., Nolasco, G. (2006). Typing of Egyptian Citrus tristeza virus (CTV) isolates based on the capsid protein gene. Phytopathologia Mediterranea, 45(1), 10–14. [Google Scholar]
22. Ghosh, D. K., Kokane, A., Kokane, S., Mukherjee, K., Tenzin, J. et al. (2022). A comprehensive analysis of Citrus tristeza variants of bhutan and across the world. Frontiers in Microbiology, 13, 797463. DOI 10.3389/fmicb.2022.797463. [Google Scholar] [CrossRef]
23. Warghane, A., Kokane, A., Kokane, S., Motghare, M., Surwase, D. et al. (2020). Molecular detection and coat protein gene based characterization of Citrus tristeza virus prevalent in Sikkim state of India. Indian Phytopathology, 73(1), 135–143. DOI 10.1007/s42360-019-00180-3. [Google Scholar] [CrossRef]
24. Youssef, S., Shalaby, A. A. (2016). Single-strand conformation polymorphism (SSCP) and nucleotide sequence analysis of Citrus tristeza virus in Egypt. International Journal of Advanced Research in Biological Sciences, 3(5), 84–92. [Google Scholar]
25. Yokomi, R., Selvarajan, R., Maheshwari, Y., Chiumenti, M., Saponari, M. et al. (2018). Molecular and biological characterization of a novel mild strain of Citrus tristeza virus in California. Archives of Virology, 163(7), 1795–1804. DOI 10.1007/s00705-018-3799-5. [Google Scholar] [CrossRef]
26. Wang, J., Zhou, T., Shen, P., Zhang, S., Cao, M. et al. (2020). Complete genome sequences of two novel genotypes of Citrus tristeza virus infecting Poncirus trifoliata in China. Journal of Plant Pathology, 102(3), 903–907. DOI 10.1007/s42161-020-00535-0. [Google Scholar] [CrossRef]
27. Rivas-Valencia, P., Domínguez-Monge, S., Santillán-Mendoza, R., Loeza-Kuk, E., Pérez-Hernández, O. et al. (2020). Severe Citrus tristeza virus isolates from eastern mexico are related to the T36 genotype group. American Journal of Plant Sciences, 11(10), 1521–1532. DOI 10.4236/ajps.2020.1110110. [Google Scholar] [CrossRef]
28. Ali, M. E., Bennett, A., Stackhouse, T., Waliullah, S., Oliver, J. E. (2021). First report of Citrus tristeza virus infecting citrus trees in Georgia, USA. Plant Disease, 105(7), 2024. DOI 10.1094/PDIS-02-21-0365-PDN. [Google Scholar] [CrossRef]
29. Martin, D. P., Murrell, B., Golden, M., Khoosal, A., Muhire, B. (2015). RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evolution, 1(1), vev003. DOI 10.1093/ve/vev003. [Google Scholar] [CrossRef]
30. Monjane, A. L., Martin, D. P., Lakay, F., Muhire, B. M., Pande, D. et al. (2014). Extensive recombination-induced disruption of genetic interactions is highly deleterious but can be partially reversed by small numbers of secondary-recombination events. Journal of Virology, 88(14), 7843–7851. [Google Scholar]
31. Weng, Z., Barthelson, R., Gowda, S., Hilf, M. E., Dawson, W. O. et al. (2007). Persistent infection and promiscuous recombination of multiple genotypes of an RNA virus within a single host generate extensive diversity. PLoS One, 2(9), e917. DOI 10.1371/journal.pone.0000917. [Google Scholar] [CrossRef]
32. Hilf, M. E., Mavrodieva, V. A., Garnsey, S. M. (2005). Genetic marker analysis of a global collection of isolates of Citrus tristeza virus: Characterization and distribution of CTV genotypes and association with symptoms. Phytopathology, 95, 909–917. [Google Scholar]
33. Vives, M. C., Rubio, L., Sambade, A., Mirkov, T. E., Moreno, P. et al. (2005). Evidence of multiple recombination events between two RNA sequence variants within a Citrus tristeza virus isolate. Virology, 331(2), 232–237. DOI 10.1016/j.virol.2004.10.037. [Google Scholar] [CrossRef]
34. Biswas, K. K., Palchoudhury, S., Sharma, S. K., Saha, B., Godara, S. et al. (2018). Analyses of 3′ half genome of Citrus tristeza virus reveal existence of distinct virus genotypes in citrus growing regions of India. Virus Disease, 29(3), 308–315. DOI 10.1007/s13337-018-0456-2. [Google Scholar] [CrossRef]
35. Bester, R., Cook, G., Maree, H. J. (2021). Citrus tristeza virus genotype detection using high-throughput sequencing. Viruses, 13(2), 168. DOI 10.3390/v13020168. [Google Scholar] [CrossRef]
36. Bar-Joseph, M., Malkinson, M. (1980). Lien egg yolk as a source of antiviral antibodies with enzyme-linked immunosorbent assay (ELISAA comparison of two plant viruses. Journal of Virological Methods, 1, 179–183. [Google Scholar]
37. Sambrook, J., Fritsch, E. F., Maniatis, T. (1989). Molecular cloning: Laboratory manual. 2nd edition, pp. 246–254. New York, USA: Cold Spring Harbor laboratory Press. [Google Scholar]
38. Rubio, L., Ayllόn, M. A., Guerri, J., Pappu, H. R., Niblett, C. L. et al. (1996). Differentiation of citrus tristeza virus (CTV) isolates by single-strand conformation polymorphism analysis of the coat protein gene. Annals of Applied Biology, 129(3), 479–489. [Google Scholar]
39. Kong, P., Rubio, L., Polek, M., Falk, B. W. (2000). Population structure and genetic diversity of California citrus tristeza virus (CTV) field isolates. Virus Genes, 21, 139–145. [Google Scholar]
40. Saponari, M., Abou Kubaa, R., Loconsole, G., Percoco, A., Savino, V. (2009). Low genetic complexity in the Citrus tristeza virus population spreading in Apulia. Journal of Plant Pathology, 91(4), 87–88. [Google Scholar]
41. Malandraki, I., Marouli, E., Varveri, C. (2011). New isolates of Citrus tristeza virus naturally occurring in old lemon and mandarin trees in Greece. New Disease Reports, 23(1), 2. DOI 10.5197/j.2044-0588.2011.023.002. [Google Scholar] [CrossRef]
42. Vela, C., Cambra, M., Cortés, E., Moreno, P., Miguet, T. G. et al. (1986). Production and characterization of monoclonal antibodies specific for Citrus tristeza virus and their use for diagnosis. Journal of General Virology, 67, 91–96. [Google Scholar]
43. Rubio, L., Ayllόn, M. A., Kong, P., Fernandez, A., Polek, M. L. et al. (2001). Genetic variation of Citrus tristeza virus isolates from California and Spain: Evidence for mixed infections and recombination. Journal of Virology, 75, 8054–8062. [Google Scholar]
44. Singh, J. K., Tarafdar, A., Sharma, S. K., Biswas, K. K. (2013). Evidence of recombinant Citrus tristeza virus isolate occurring in acid lime cv. pant lemon orchard in Uttarakhand Terai Region of Northern Himalaya in India. Indian Journal of Virology, 24(1), 35–41. DOI 10.1007/s13337-012-0118-8. [Google Scholar] [CrossRef]
45. Anonymous (2012). Virus taxonomy: Classification and nomenclature of viruses. In: King, A. M. Q., Adams, M. J., Carstens, E. B., Lefkowitz, E. J. (Eds.Ninth report of the International Committee on Taxonomy of Viruses. pp. 1121–1234. Netherlands, Amsterdam: Elsevier. [Google Scholar]
46. Zablocki, O., Pietersen, G. (2014). Characterization of a novel Citrus tristeza virus genotype within three cross-protecting source GFMS12 sub-isolates in South Africa by means of Illumina sequencing. Archives of Virology, 159(8), 2133–2139. DOI 10.1007/s00705-014-2041-3. [Google Scholar] [CrossRef]
47. Roy, A., Manjunath, K. L., Brlansky, R. H. (2005). Assessment of sequence diversity in the 5-terminal region of Citrus tristeza virus from India. Virus Research, 113, 132–142. [Google Scholar]
48. Ghorbel, R., López, C., Fagoaga, C., Moreno, P., Navarro, L. et al. (2001). Transgenic citrus plants expressing the citrus tristeza virus p23 protein exhibit viral-like symptoms. Molecular Plant Pathology, 2, 27–36. [Google Scholar]
49. Roy, A., Brlansky, R. H. (2010). Genome analysis of an orange stem pitting Citrus tristeza virus isolate reveals a novel recombinant genotype. Virus Research, 151, 118–130. [Google Scholar]
50. Sambade, A., López, C., Rubio, L., Flores, R., Guerri, J. et al. (2003). Polymorphism of a specific region in gene p23 of Citrus tristeza virus allows discrimination between mild and severe isolates. Archives of Virology, 148, 2325–2340. [Google Scholar]
51. Martín, S., Sambade, A., Rubio, L., Vives, M. C., Moya, P. et al. (2009). Contribution of recombination and selection to molecular evolution of Citrus tristeza virus. Journal of General Virology, 90, 1527–1538. DOI 10.1099/vir.0.008193-0. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools