| Phyton-International Journal of Experimental Botany |
DOI: 10.32604/phyton.2022.020390
ARTICLE
Comparative Analysis of the Complete Chloroplast Genome Sequences of Four Origin Plants of Lonicerae Flos (Lonicera; Caprifoliaceae)
1Hunan Academy of Forestry, Changsha, China
2Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China
3College of Pharmacy, Shaanxi Qinling Application Development and Engineering Center of Chinese Herbal Medicine, Shaanxi University of Chinese Medicine, Xi’an, China
*Corresponding Authors: Yongxin Li. Email: liyongxin@hnlky.cn; Gang Zhang. Email: jay_gumling2003@aliyun.com
Received: 20 November 2021; Accepted: 21 December 2021
Abstract: Lonicerae Flos (LF) derived from the dried flower buds or opening flowers of four Lonicera plants (Lonicera macranthoides, L. hypoglauca, L. confusa, and L. fulvotnetosa), is a popular traditional Chinese medicine. Because the four origin plants are very similar in morphology, it is difficult to control the quality of LF in actual production. Over the past decade, many reports have pointed out the differences among them, including the botanical characteristics and active ingredients. However, there is still a lack of rapid methods that can be applied to the identification of the four origins. In this study, comparative analysis of the four chloroplast genomes was performed, and they showed low diversity (Pi = 0.00267), three variation hotspots regions (rbcL-accD, rps12-ndhF and rps12-trnN-trnG) were identified as potentially molecular marker of highly informative. Meanwhile, the most obvious difference in SSR comparative analysis is reverse and complement repeats were only identified in L. confusa and L. hypoglauca, respectively. Lastly, the phylogenetic tree showed that L. confusa is more closely related to L. fulvotnetosa, while L. macranthoides is closer to L. hypoglauca. This study systematically revealed the differences among the four chloroplast genomes, and it provides valuable genetic information for identifying the origin of LF.
Keywords: Lonicerae Flos; Lonicera; chloroplast genome; origin plants; comparative genome
Lonicerae Flos (LF, Shanyinhua in Chinese) is one of the most commonly used traditional Chinese medicines [1], which has been officially listed in Chinese Pharmacopeia (Edition 2020) and described as “Used for carbuncle boils, throat arthralgia, erysipelas, toxic blood dysentery, wind-heat cold, febrile fever”. LF is the dried flower buds or opening flowers of four plants (Lonicera macranthoides, L. hypoglauca, L. confusa, and L. fulvotnetosa) [2], and they are widely cultivated in Southern China. Inflorescences and bracts are the main differences among the four origin plants in terms of botanical traits [3]. At present, L. macranthoides is the most cultivated one in the market, and L. confusa was the least cultivated and used [3,4]. Because they are so similar, how to distinguish the differences among them is one of the difficulties of current research [5,6]. In order to solve this problem, studies on genetic diversity and relationships might be a good choice.
The use of plant DNA analysis to identify plant species, genotypes, and relationships has gradually replaced earlier techniques based on other biochemical markers [7]. With the rapid development of DNA analysis technology, it is becoming more accurate, conventional and low-cost, and is commonly used in plant research [8,9]. At present, phylogenetic analysis of plants is mainly based on the structure and changes of chloroplast genome and nuclear genome. However, it is difficult to screen low copy genes in plants due to the complexity of nuclear genomes. Chloroplast (cp) genome is of great significance in revealing the origin and evolution of species, genetic diversity, genetic relationship and biodiversity because of their small molecular weight (115 to 165 kb), non-recombination, highly conserved, and uniparental inheritance characteristics [10–13]. It has been widely accepted as a powerful tool for distinguishing the difference among related similar species [14,15]. With the accumulation of cp genome of Lonicera, comparative analysis of the complete cp genome of four origin plants of LF is helpful for deepening and expanding our systematic understanding of it.
In most previous report, studies focused on inferring the phylogeny of Lonicera or Caprifoliaceae based on complete chloroplast genome [16–19]. In this study, we report three sequenced complete cp genomes from three different origin plants of LF (L. macranthoides, L. confusa, and L. fulvotnetosa), respectively, and genomic comparative analyses with the other published cp genome (L. hypoglauca) [20]. The comparative analysis focuses on features, structure, nucleotide diversity, simple sequence repeats (SSRs) and phylogenetic analysis. The aims of our study are: (1) to comprehensive understanding the complete cp genome features from four origin plants of LF, (2) to the systematic analysis of similarities and differences from the four origin plants, (3) to infer the phylogenetic relationship among the four and between the four and Lonicerae Japonicae Flos (LJF, Jinyinhua in Chinese, L. japonica,), and (4) to provide genetic resources for developing chloroplast markers to identify LF species and future research on Lonicera.
2.1 Plant Material and Genome Sequencing
Fresh leaves samples were collected from three Lonicera species (Lonicera macranthoides, L. confusa, and L. fulvotnetosa; Table 1). Voucher specimens were deposited at the Hunan Academy of Forestry. A Genomic DNA extraction and high-throughput sequencing were performed with an Illumina NovaSeq6000 by Suzhou GENEWIZ Biotech. Co., Ltd. (Suzhou, China).

2.2 Assembly and Annotation of Chloroplast Genome
NGS QC Tool Kit software package was used for data quality detection and filtering to remove low quality sequences, joint sequences and sequences containing uncertain bases to obtain high quality sequences (clean reads). The clean reads then assembled using Velvet 1.2.10 [21], SSPACE v3.0 [22] and GapFiller v2.1.2 [23] with the cp genome of L. japonica (GenBank: KJ170923) as the reference [24]. The assembled sequence was annotated using the Plann [25], transfer RNAs (tRNAs) were detected in the genome using the program tRNAscan-SE [26] with default parameter settings and rRNA were identified by using RNAmmer [27]. All gene annotations were verified by Geneious 11.1.4 software [28] and the circular genome maps were drawn with OGDRAW (Organellar Genome DRAW) [29]. Finally, three Lonicera species annotated chloroplast genomes were submitted to GenBank (Table 2).
2.3 Comparative Analysis of Chloroplast Genomes
This study, except for the L. macranthoides, L. confusa and L. fulvotnetosa complete chloroplast genomes sequenced here, L. hypoglauca chloroplast genome (NC_054350) [20] were used for comparative genomic analysis by mVISTA with LAGAN alignment program [30]. The major variations of gene contents or features in four Lonicera species chloroplast genome were manually identified with Geneious [28]. For accurate comparisons, gene annotations of NC_054350 was checked again with Plann, RNAmmer, and tRNAscan-SE [26]. DNA polymorphisms analysis including highly variable sites and nucleotide diversity (Pi) was performed using DnaSP (DNA Sequence Polymorphism) v6 [31], which the window length was set to 800 bp and the step size was set to 200 bp. The four chloroplast genome sequences were aligned by using Geneious.
2.4 Analysis of Simple Sequence Repeats
The repetitive simple sequence repeat (SSR) sequences in four chloroplast genomes of Lonicera were identified by MISA software (https://webblast.ipk-gatersleben.de/misa/) [32]. By using REPuter software [33] with a minimum repeat size of 20 bp, four types of repeat sequences (forward, reverse, complement and palindrome) were determined. The minimum repeat number thresholds for mononucleotide, dinucleotide, trinucleotide, tetranucleotide, pentanucleotide and hexanucleotide repeats were set to 10, 5, 4, 3, 3 and 3, respectively.
In the phylogenetic reconstruction of four LF species, 17 Lonicera species chloroplast genomes were used to conduct the phylogenetic tree. Except for the three LF species (L. macranthoides, L. confusa and L. fulvotnetosa) chloroplast genomes sequenced here, the other fourteen genomes were obtained from the NCBI (https://www.ncbi.nlm.nih.gov) included L. hypoglauca (NC_054350), L. japonica (KJ170923.1), L. maximowiczii (MN986996.1), L. insularis (MH028739.1), L. sachalinensis (MH028742.1), L. maackii (MH028741.1), L. nervosa (MK176510.1), L. ferdinandi (MK176512.1), L. vesicaria (MH028743.1), L. hispida (MK176511.1), L. stephanocarpa (MG738668.1), L. praeflorens (MH028740.1), L. tragophylla (MG738667.1), and L. calcarata (MN524650.1). MAFFT 7.487 (https://mafft.cbrc.jp/alignment/software/windows.html) [34] were used for multi-sequence alignment. Finally, by using IQTREE 1.6.12 software (http://www.iqtree.org) [35] with maximum likelihood method (GTR + I + G) and Figtree 1.4.4 software (http://tree.bio.ed.ac.uk/software/figtree/), the phylogenetic tree was built and edited.
3.1 Features of Chloroplast Genome
The chloroplast genome sizes of three Lonicera species (L. macranthoides, L. confusa and L. fulvotnetosa) were 155,515, 155,157 and 155,126 bp with 1198X, 1580X and 2154X depth, respectively. Each complete chloroplast genome sequences of three LF species had a typical quadripartite structure (Fig. 1), in which a large single-copy region (LSC) and a small single-copy region (SSC) were separated by two inverted repeats (IRs). L. macranthoides, L. confusa and L. fulvotnetosa chloroplast genome consisting of 89,303, 88,942 and 88,910 bp LSC region, 18,656, 18,661 and 18,662 bp SSC region and 23,778, 23,777 and 23,777 bp IRs regions (Table 2), respectively. 110 genes (75 PCGs, 31 tRNAs, 4 rRNAs), 106 genes (72 PCGs, 30 tRNAs, 4 rRNAs) and 111 genes (76 PCGs, 31 tRNAs, 4 rRNAs) were encoded by the three chloroplast genomes, respectively. In addition, The GC contents were identical in the three chloroplast genomes (Fig. 1; Table 2).
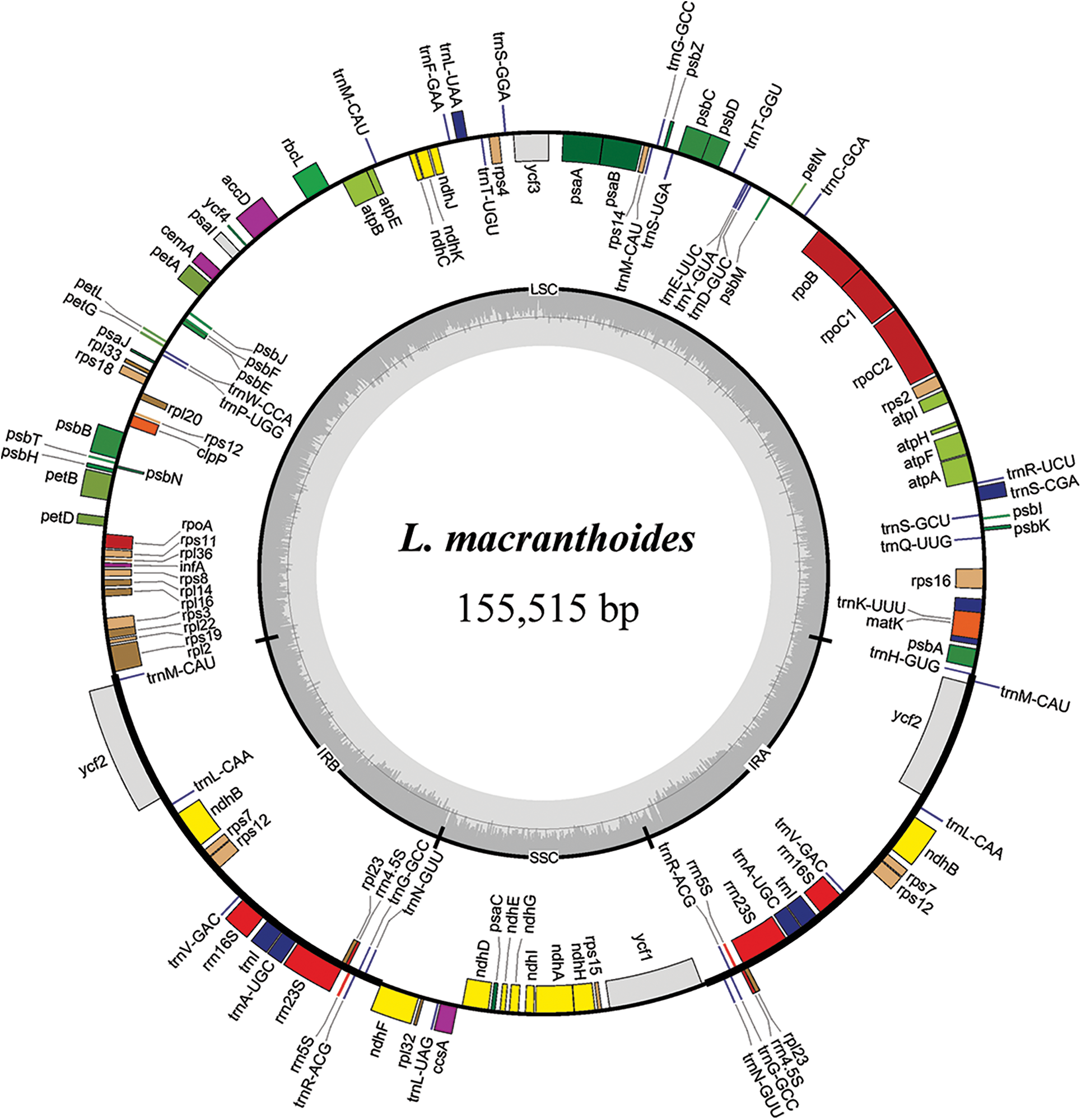
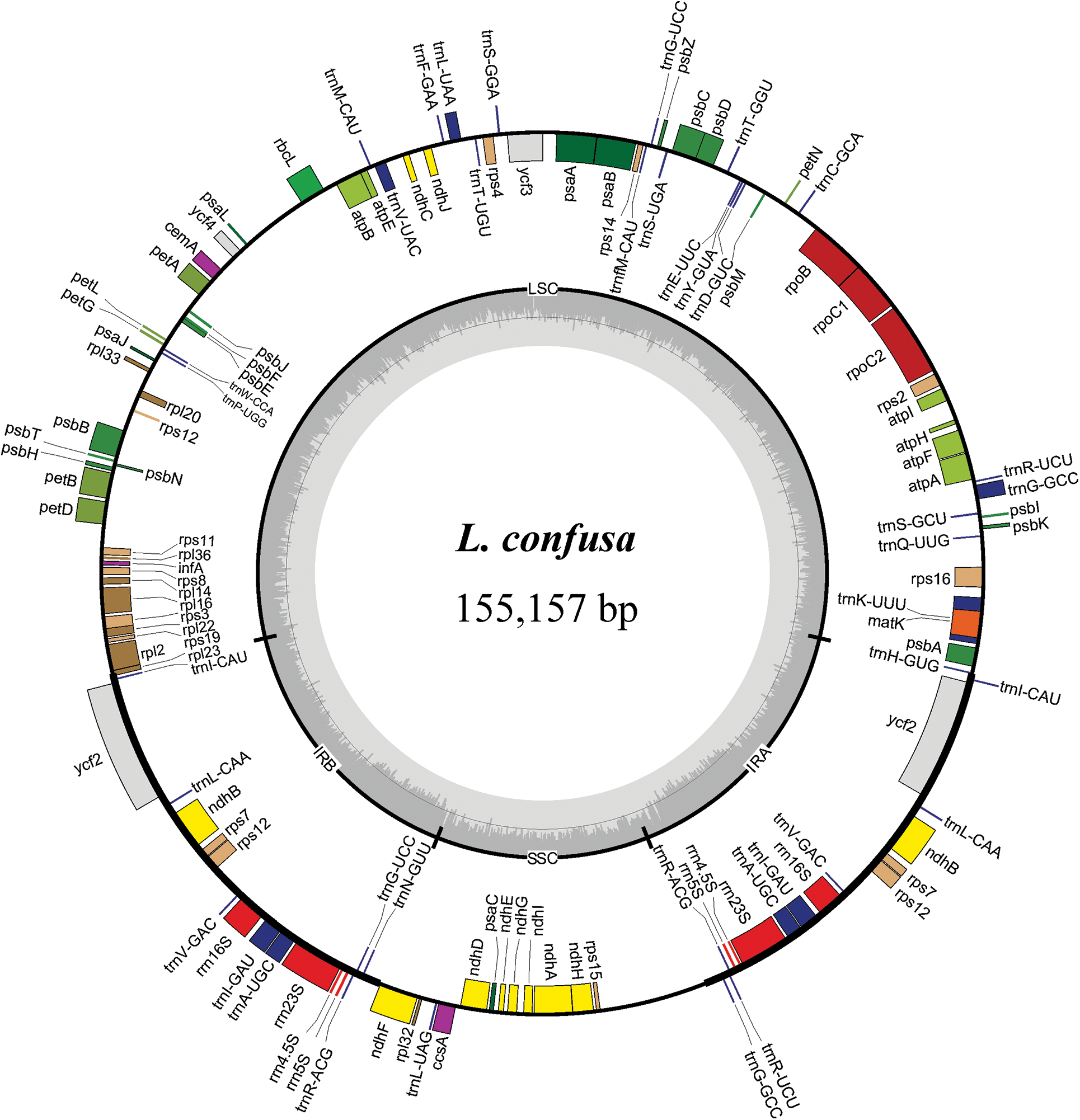
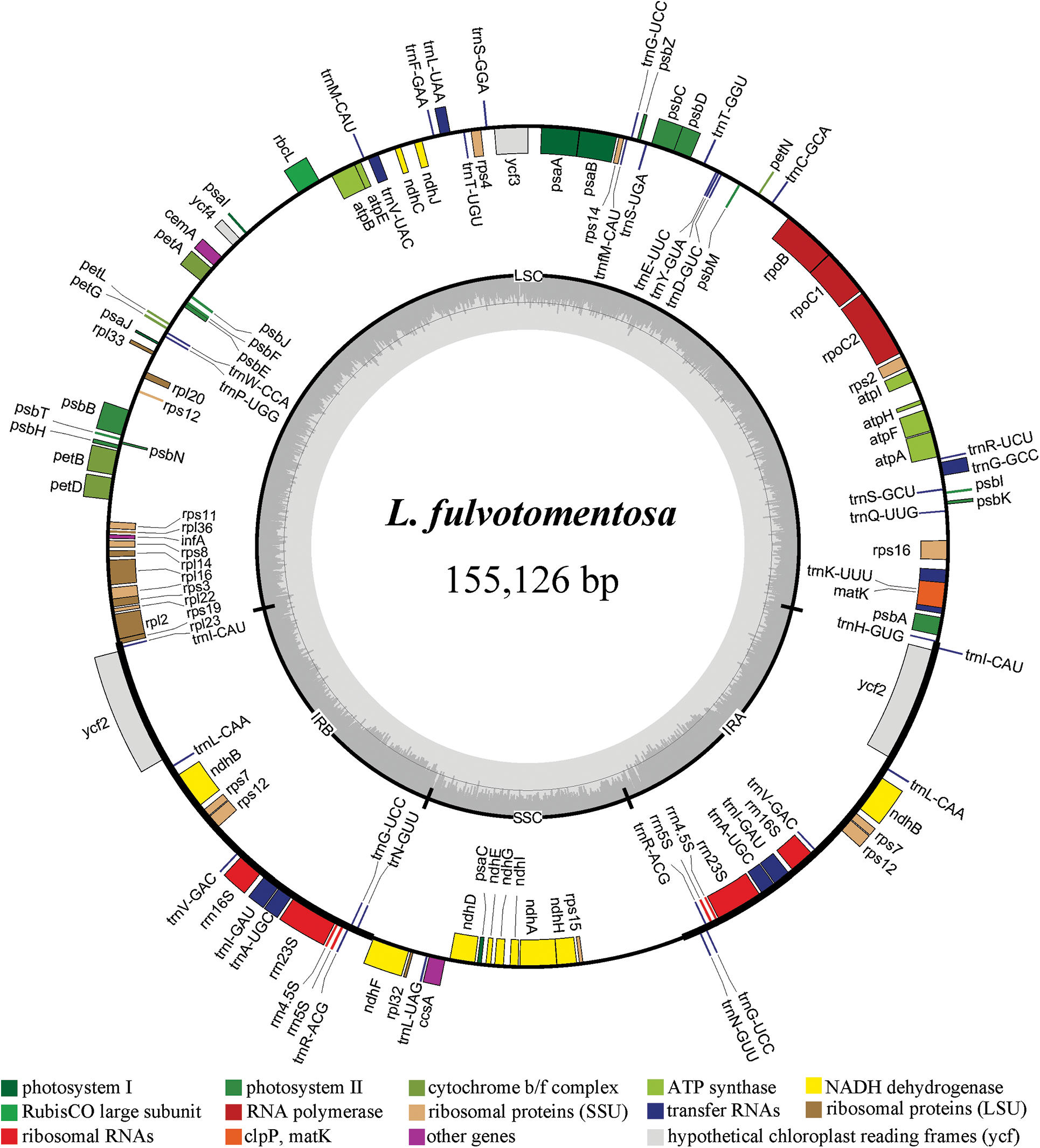
Figure 1: Chloroplast gene maps of L. macranthoides, L. confusa and L. fulvotnetosa. Different functional genes were shown in different colors. Transcribed clockwise or counter-clockwise were shown inside or outside the circle. LSC, large single-copy region; SSC, small single-copy region; IR, inverted repeat
Comparison of chloroplast genomes from four origin plants of LF, L. macranthoides chloroplast genome had the largest genome size, however L. hypoglauca had the smallest genome size. Interestingly enough, “L. macranthoides and L. hypoglauca”, “L. confusa and L. fulvotnetosa” shared the completely same number of genes. This is largely due to the simple and relatively conserved structure of chloroplast genome and the difference in chloroplast genome size may be caused by different homologous gene length for plants of the same genus.
3.2 Comparative Analyses of Lonicerae Flos Species
There were no large differences among the four origin plants of LF as a whole. Meanwhile, the chloroplast genomes of L. confusa and L. fulvotnetosa showing the least differences. It is worth noting that, when compared to the L. macranthoides, L. confusa, L. fulvotnetosa and L. hypoglauca have a large gap between rbcL and accD genes with five gaps located in conserved non-coding sequences (CNS) and two gaps located in accD (Fig. 2). In addition, L. confusa and L. fulvotnetosa have more variable sites compared to L. macranthoides and L. hypoglauca, L. hypoglauca has more variable sites than L. macranthoides.
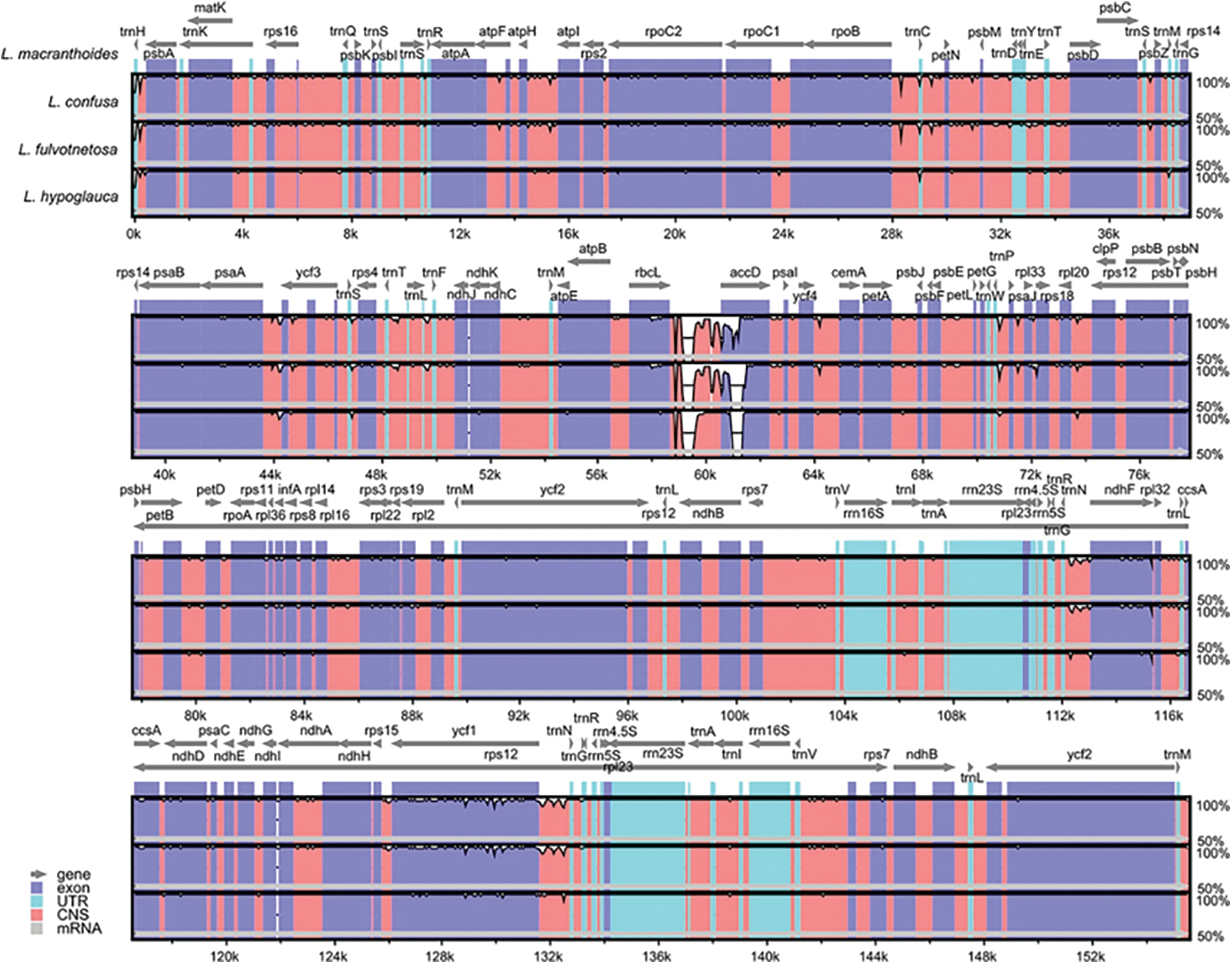
Figure 2: Comparisons of four Lonicerae Flos species chloroplast genomes. L. macranthoides chloroplast genome was used as reference sequence, the x-axis represents the aligned sequence of base and the y-axis represents the pairwise percent identity (50%–100%). Gray arrows, purple bars, sky blue, red bars and gray bars represent gene, exon, UTR, CNS and mRNA, respectively
By using DnaSP software, nucleotide diversity (Pi) was calculated to estimate the genetic distance among four LF species chloroplast genomes. The Pi value for four LF chloroplast genomes included (L. macranthoides, L. confusa, L. fulvotnetosa and L. hypoglauca) was 0.00267. By comparing the chloroplast genomes of four LF species, several variation hotspots were found (Fig. 3). There are three hotspots showed higher Pi values than other regions (Pi > 0.02), among these variation hotspots, rbcL-accD region showed the highest Pi (0.06875), followed by two regions (rps12-ndhF and rps12-trnN-trnG).
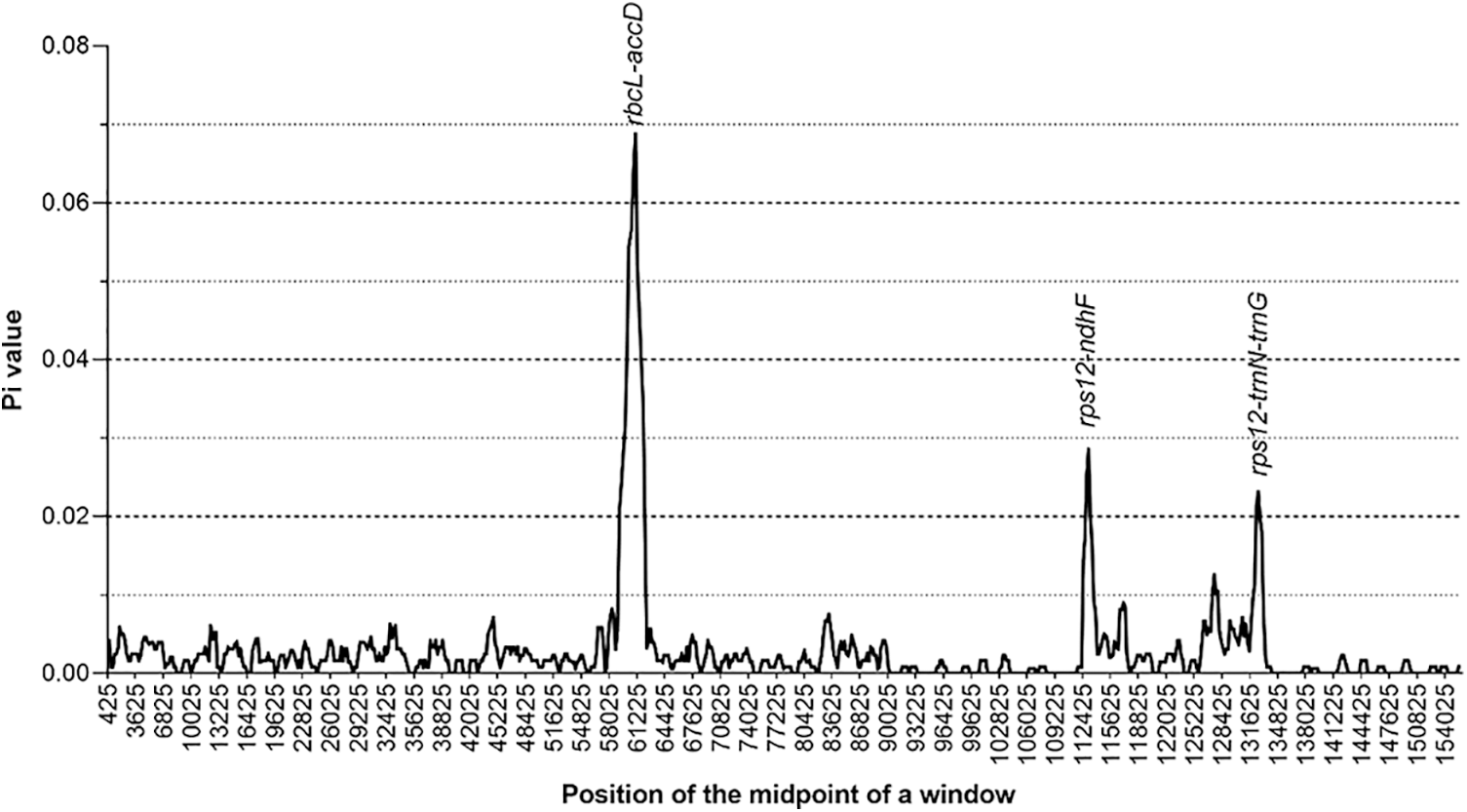
Figure 3: Nucleotide diversity of Lonicerae Flos chloroplast genomes. The x-axis represents the aligned sequence of base and the y-axis represents the Pi value. This graph shows each variation hotspot for Lonicerae Flos chloroplast genomes
There were some differences among these four chloroplast genomes. By using MISA software, 51, 54, 54 and 51 microsatellites were identified in L. macranthoides, L. confusa, L. fulvotnetosa and L. hypoglauca, respectively. For the 51 microsatellites identified from L. macranthoides, 32 mono-nucleotide, 7 di-nucleotide, 4 tri-nucleotide, 8 tetra-nucleotide repeats were identified. No penta-nucleotide or hexa-nucleotide was found (Fig. 4a). Among these microsatellites, 6, 13, and 32 microsatellites were located in the intron, exon and intergenic regions (Fig. 4b). Of the 54 microsatellites identified from the L. confusa and L. fulvotnetosa, 36 mono-nucleotide, 4 di-nucleotide, 2 tri-nucleotide, 8 tetra-nucleotide and 4 hexa-nucleotide repeats were identified. No penta-nucleotide was found (Fig. 4a). Among these microsatellites, 4, 7, and 43 microsatellites were located in the intron, exon and intergenic regions (Fig. 4b). Of the 51 microsatellites identified from the L. hypoglauca, 35 mono-nucleotide, 5 di-nucleotide, 3 tri-nucleotide, 7 tetra-nucleotide and 1 hexa-nucleotide repeats were identified. No penta-nucleotide was found (Fig. 4a). Among these microsatellites, 3, 10, and 38 microsatellites were located in the intron, exon and intergenic regions (Fig. 4b). Furthermore, there were forward (76.0%, 68.0%, 70.0% and 56.0%), palindromic (24.0%, 30.0%, 30.0% and 42.0%), reverse (0.0%, 2.0%, 0.0% and 0.0%) and complement (0.0%, 0.0%, 0.0% and 2.0%) in L. macranthoides, L. confusa, L. fulvotnetosa and L. hypoglauca, respectively (Fig. 4c). Reverse repeats and complement repeats were only identified in L. confusa and L. hypoglauca (Fig. 4c), respectively.
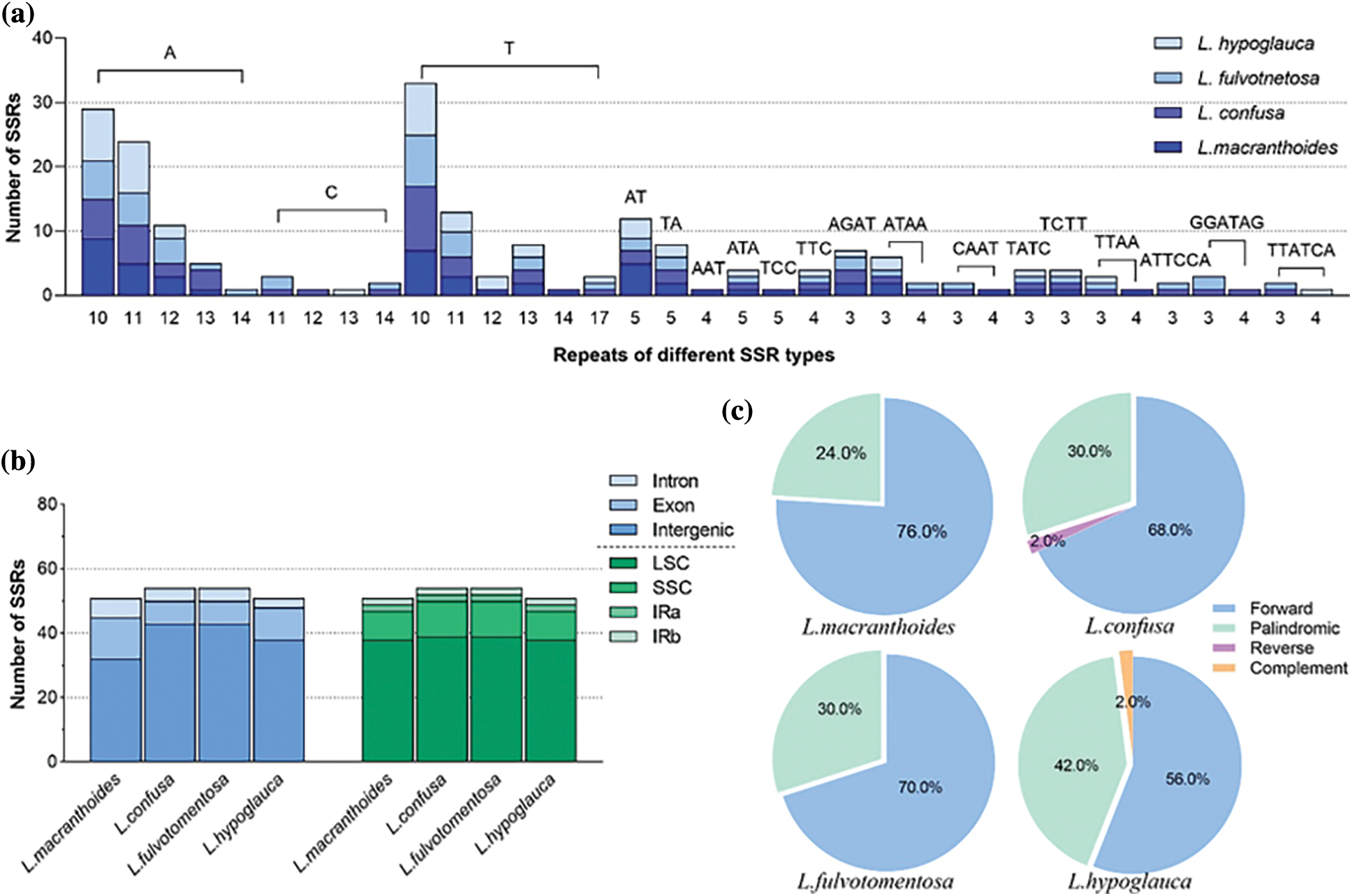
Figure 4: Statistics of Lonicerae Flos chloroplast genomes. (a) The number of repeat and repeated sequences; (b) Distribution of SSRs in the different regions; (c) The proportion of different SSR types
In this study, the phylogeny of Lonicera was reconstructed using four complete chloroplast genomes of LF species (L. macranthoides, L. confusa, L. fulvotnetosa and L. hypoglauca) and thirteen other species from the Lonicera genus. According to the phylogenetic tree (Fig. 5), two main clades can be identified, one of these includes four Lonaria species (96% bootstrap) and the remaining species, including the four species of interest, are located in the other clade (100% bootstrap). All four species of this study are included in the same clade (100% bootstrap) which is divided in two, with L. confusa, L. fulvotomentosa and L. japonica together in one clade, while L. macranthoide and L. hypoglauca along with L. maximowiczii are grouped in the other. In other words, the L. confusa is closer to L. fulvotnetosa and L. macranthoides is closer to L. hypoglauca. Compared to other species of Lonicera genus, the four origin plants of LF to be more closely related.
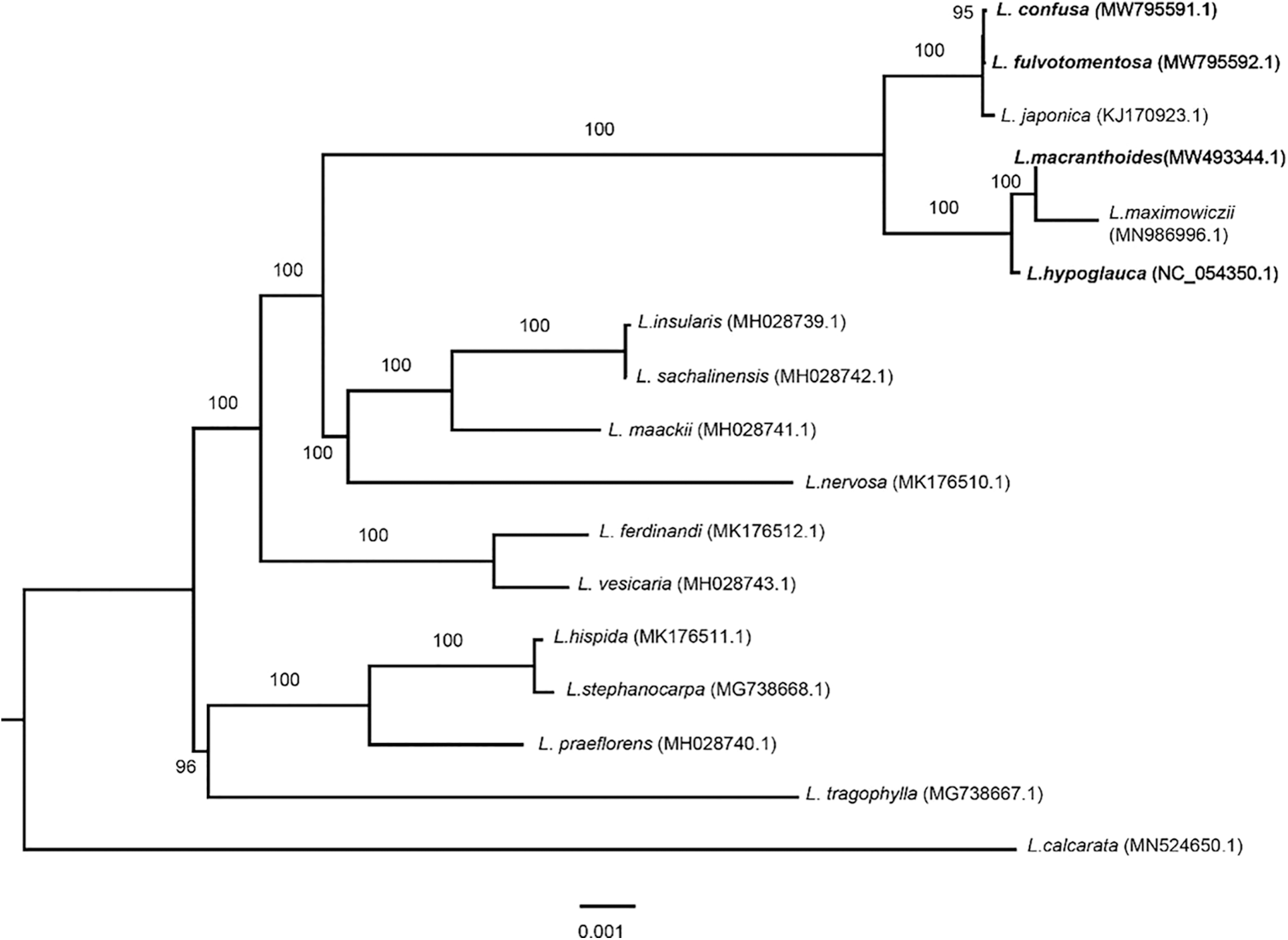
Figure 5: Phylogenetic analysis of seventeen Lonicera chloroplast genomes. The phylogenetic tree was built and edited by using IQTREE with maximum likelihood method and Figtree software. A total of 17 chloroplast sequences of Lonicera including L. macranthoides (MW493344.1), L. confusa (MW795591.1), L. fulvotnetosa (MW795592.1), L. hypoglauca (NC_054350), L. japonica (KJ170923.1), L. maximowiczii (MN986996.1), L. insularis (MH028739.1), L. sachalinensis (MH028742.1), L. maackii (MH028741.1), L. nervosa (MK176510.1), L. ferdinandi (MK176512.1), L. vesicaria (MH028743.1), L. hispida (MK176511.1), L. stephanocarpa (MG738668.1), L. praeflorens (MH028740.1), L. tragophylla (MG738667.1), and L. calcarata (MN524650.1)
Although chloroplast genomes of four origin plants of LF (L. macranthoides, L. confusa, L. fulvotnetosa and L. hypoglauca) have been reported [17,36,37], comparative analysis of the four chloroplast genomes for the first time in this study. In addition, the results of chloroplast genome assembly and annotation were different due to different plant varieties, sequencing platforms and assembly methods, the results of this study will be complementary. There is small difference in the feature of chloroplast genome among the four Lonicera plants, such as size of LSC, SSC and IR, the number of PCGs, tRNAs and rRNAs, GC content, etc. Within four origin plants of LF, the four chloroplast genomes showed low diversity (Pi = 0.00267), meanwhile, there are three variation hotspots regions which including rbcL-accD, rps12-ndhF and rps12-trnN-trnG were found. To find more differences, SSR analysis was performed. The results showed two obvious differences among these four chloroplast genomes. One is the percentage of microsatellites were located in the intron, exon and intergenic regions (Fig. 4b), and the other is the proportion of different SSR types. Reverse, complement repeats were only identified in L. confusa and L. hypoglauca (Fig. 4c), respectively. Additionally, the chloroplast genome sequences of 17 Lonicera species were constructed the genetic phylogenetic analysis based on maximum likelihood method. According to the phylogenetic tree, four origin plants of LF (L. macranthoides, L. confusa, L. fulvotnetosa and L. hypoglauca), L. japonica and L. maximowiczii have a closer relationship.
The differences among the four origin plants of LF were evident in the genetic structure and repetitive sequences. Although L. confusa and L. fulvotnetosa, L. macranthoides and L. hypoglauca have the same number of microsatellites and coding regions without rearrangement, L. confusa, L. fulvotnetosa and L. hypoglauca have a large gap between rbcL and accD genes with seven small gaps compared to the L. macranthoides. Then by analyzing the SSR type, reverse and complement repeats were only found in L. confusa and L. hypoglauca, respectively. In order to better control the quality of traditional Chinese medicine, we need to identify the origin plants of Chinese medicinal materials quickly and accurately. Unquestionably, these differences were found in this study will benefit of the development of molecular markers to identify the origin of LF. Meanwhile, these results will provide more genetic information for molecular assisted breeding of LF.
Traditional Chinese medicine Lonicerae Japonicae Flos (LJF, Jinyinhua in Chinese) is dried flower buds or the flower with opening of L. japonica. Because LJF and LF both have the same pharmacologic effects and extremely similar appearances, there are easily confused, abuse and other phenomena [38–40]. As can be seen from Fig. 5, LF chloroplast genomes were classified into two branches, L. japonica was clustered into a branch with L. confusa and L. fulvotnetosa, however, L. macranthoides and L. hypoglauca were clustered into a branch with L. maximowiczii. The phylogenetic analysis showed that L. japonica have more closer relationship with L. confusa and L. fulvotnetosa than L. macranthoides and L. hypoglauca. Most current studies showed that it is difficult to point out their similarities or differences in-depth [41–43]. Therefore, studies on genetic diversity, relationships, bioactive compounds and modern pharmacological effects should be highlighted.
Author Contributions: Sisi Liu and Yongxin Li conceived and designed the experiments. Sisi Liu and Lisi Zhou performed the experiment and analyzed the data. Zhongquan Qiao provided technical guidance. Sisi Liu drafted the manuscript. Jiaoli Huang, Huijie Zeng and Gang Zhang revised the manuscript. All authors reviewed the manuscript.
Funding Statement: This research was funded by the Science and Technology Project of Changsha City (No. kq2004038) and Program for Innovative Leading Talents for Science and Technology of Xianyang City.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Tang, Y. R., Zeng, T., Zafar, S., Yuan, H. W., Li, B. et al. (2018). Lonicerae flos: A review of chemical constituents and biological activities. Digital Chinese Medicine, 1(2), 173–188. DOI 10.1016/S2589-3777(19)30022-9. [Google Scholar] [CrossRef]
2. Chinese Pharmacopoeia Commission (2020). Chinese pharmacopoeia, vol. 1, pp. 32. Beijing: China Medical Science Press. [Google Scholar]
3. Li, Y., Li, W., Fu, C., Song, Y., Fu, Q. (2020). Lonicerae japonicae flos and Lonicerae flos: A systematic review of ethnopharmacology, phytochemistry and pharmacology. Phytochemistry Reviews, 19(1), 1–61. DOI 10.1007/s11101-019-09655-7. [Google Scholar] [CrossRef]
4. Liu, S., Qiao, Z., Wang, X., Zeng, H., Li, Y. et al. (2019). Analysis of codon usage patterns in “Lonicerae Flos” (Lonicera macranthoides Hand.-Mazz.) based on transcriptome data. Gene, 705, 127–132. DOI 10.1016/j.gene.2019.04.065. [Google Scholar] [CrossRef]
5. Ryuk, J. A., Lee, H. W., Ko, B. S. (2012). Discrimination of Lonicera japonica and Lonicera confusa using chemical analysis and genetic marker. The Korea Journal of Herbology, 27(6), 15–21. DOI 10.6116/kjh.2012.27.6.15. [Google Scholar] [CrossRef]
6. Nakaji, M., Tanaka, N., Sugawara, T. (2015). A molecular phylogenetic study of Lonicera L. (Caprifoliaceae) in Japan based on chloroplast DNA sequences. Acta Phytotaxonomica et Geobotanica, 66(3), 137–151. DOI 10.18942/apg.KJ00010115701. [Google Scholar] [CrossRef]
7. Li, B., Lin, F., Huang, P., Guo, W., Zheng, Y. (2017). Complete chloroplast genome sequence of Decaisnea insignis: Genome organization, genomic resources and comparative analysis. Scientific Reports, 7(1), 1–10. DOI 10.1038/s41598-017-10409-8. [Google Scholar] [CrossRef]
8. Kress, W. J. (2017). Plant DNA barcodes: Applications today and in the future. Journal of Systematics and Evolution, 55(4), 291–307. DOI 10.1111/jse.12254. [Google Scholar] [CrossRef]
9. Zhang, Y., Iaffaldano, B. J., Zhuang, X., Cardina, J., Cornish, K. (2017). Chloroplast genome resources and molecular markers differentiate rubber dandelion species from weedy relatives. BMC Plant Biology, 17(1), 1–14. DOI 10.1186/s12870-021-03391-x. [Google Scholar] [CrossRef]
10. Daniell, H., Lin, C. S., Yu, M., Chang, W. J. (2016). Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biology, 17(1), 1–29. DOI 10.1186/s13059-016-1004-2. [Google Scholar] [CrossRef]
11. Kirchhoff, H. (2019). Chloroplast ultrastructure in plants. New Phytologist, 223(2), 565–574. DOI 10.1111/nph.15730. [Google Scholar] [CrossRef]
12. Chan, K. X., Phua, S. Y., Crisp, P., McQuinn, R., Pogson, B. J. (2016). Learning the languages of the chloroplast: Retrograde signaling and beyond. Annual Review of Plant Biology, 67, 25–53. DOI 10.1146/annurev-arplant-043015-111854. [Google Scholar] [CrossRef]
13. Bobik, K., Burch-Smith, T. M. (2015). Chloroplast signaling within, between and beyond cells. Frontiers in Plant Science, 6, 781. DOI 10.3389/fpls.2015.00781. [Google Scholar] [CrossRef]
14. Li, C., Zheng, Y., Huang, P. (2020). Molecular markers from the chloroplast genome of rose provide a complementary tool for variety discrimination and profiling. Scientific Reports, 10(1), 1–15. DOI 10.1038/s41598-020-68092-1. [Google Scholar] [CrossRef]
15. Schroeder, H., Höltken, A., Fladung, M. (2011). Chloroplast SNP-marker as powerful tool for differentiation of Populus species in reliable poplar breeding and barcoding approaches. BMC Proceedings, 5(7), 1–2. DOI 10.1186/1753-6561-5-S7-P56. [Google Scholar] [CrossRef]
16. Fan, W. B., Wu, Y., Yang, J., Shahzad, K., Li, Z. H. (2018). Comparative chloroplast genomics of dipsacales species: Insights into sequence variation, adaptive evolution, and phylogenetic relationships. Frontiers in Plant Science, 9, 689. DOI 10.3389/fpls.2018.00689. [Google Scholar] [CrossRef]
17. Wang, H. X., Liu, H., Moore, M. J., Landrein, S., Liu, B. et al. (2020). Plastid phylogenomic insights into the evolution of the Caprifoliaceae s.l. (Dipsacales). Molecular Phylogenetics and Evolution, 142, 106641. DOI 10.1016/j.ympev.2019.106641. [Google Scholar] [CrossRef]
18. Wang, H. F., Landrein, S., Dong, W. P., Nie, Z. L., Kondo, K. et al. (2015). Molecular phylogeny and biogeographic diversification of Linnaeoideae (Caprifoliaceae s.l.) disjunctly distributed in Eurasia, North America and Mexico. PLoS One, 10(3), e0116485. DOI 10.1371/journal.pone.0116485. [Google Scholar] [CrossRef]
19. Smith, S. A., Donoghue, M. J. (2010). Combining historical biogeography with niche modeling in the Caprifolium clade of Lonicera (Caprifoliaceae, Dipsacales). Systematic Biology, 59(3), 322–341. DOI 10.1093/sysbio/syq011. [Google Scholar] [CrossRef]
20. Gu, L., Wu, Q., Yi, Y., Yu, Z. (2021). The complete chloroplast genome of Lonicera hypoglauca Miq (Caprifoliaceae: Dipsacales) from Guangxi, China. Mitochondrial DNA Part B, 6(2), 450–451. DOI 10.1080/23802359.2020.1870903. [Google Scholar] [CrossRef]
21. Zerbino, D. R., Birney, E. (2008). Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Research, 18(5), 821–829. DOI 10.1101/gr.074492.107. [Google Scholar] [CrossRef]
22. Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D., Pirovano, W. (2011). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics, 27(4), 578–579. DOI 10.1093/bioinformatics/btq683. [Google Scholar] [CrossRef]
23. Nadalin, F., Vezzi, F., Policriti, A. (2012). GapFiller: A de novo assembly approach to fill the gap within paired reads. BMC Bioinformatics, 13(14), 1–16. DOI 10.1186/1471-2105-13-S14-S8. [Google Scholar] [CrossRef]
24. He, L., Qian, J., Li, X., Sun, Z., Xu, X. et al. (2017). Complete chloroplast genome of medicinal plant Lonicera japonica: Genome rearrangement, intron gain and loss, and implications for phylogenetic studies. Molecules, 22(2), 249. DOI 10.3390/molecules22020249. [Google Scholar] [CrossRef]
25. Huang, D. I., Cronk, Q. C. (2015). Plann: A command-line application for annotating plastome sequences. Applications in Plant Sciences, 3(8), 1500026. DOI 10.3732/apps.1500026. [Google Scholar] [CrossRef]
26. Lowe, T. M., Chan, P. P. (2016). tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Research, 44(W1), W54–W57. DOI 10.1093/nar/gkw413. [Google Scholar] [CrossRef]
27. Lagesen, K., Hallin, P., Rødland, E. A., Stærfeldt, H. H., Rognes, T. et al. (2007). RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Research, 35(9), 3100–3108. DOI 10.1093/nar/gkm160. [Google Scholar] [CrossRef]
28. Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M. et al. (2012). Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics, 28(12), 1647–1649. DOI 10.1093/bioinformatics/bts199. [Google Scholar] [CrossRef]
29. Greiner, S., Lehwark, P., Bock, R. (2019). Organellar Genome DRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Research, 47, W59–W64. DOI 10.1093/nar/gkz238. [Google Scholar] [CrossRef]
30. Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., Dubchak, I. (2004). VISTA: Computational tools for comparative genomics. Nucleic Acids Research, 32(suppl_2), W273–W279. DOI 10.1093/nar/gkh458. [Google Scholar] [CrossRef]
31. Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P. et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Molecular Biology and Evolution, 34(12), 3299–3302. DOI 10.1093/molbev/msx248. [Google Scholar] [CrossRef]
32. Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: A web server for microsatellite prediction. Bioinformatics, 33(16), 2583–2585. DOI 10.1093/bioinformatics/btx198. [Google Scholar] [CrossRef]
33. Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J. et al. (2001). REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Research, 29(22), 4633–4642. DOI 10.1093/nar/29.22.4633. [Google Scholar] [CrossRef]
34. Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molecular Biology and Evolution, 30(4), 772–780. DOI 10.1093/molbev/mst010. [Google Scholar] [CrossRef]
35. Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D. et al. (2020). IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Molecular Biology and Evolution, 37(5), 1530–1534. DOI 10.1093/molbev/msaa015. [Google Scholar] [CrossRef]
36. Hu, H., Liu, J., An, J., Wang, M., Wang, Q. (2018). Characterization of the complete chloroplast genome of Lonicera macranthoides. Mitochondrial DNA Part B, 3(2), 1000–1001. DOI 10.1080/23802359.2018.1507643. [Google Scholar] [CrossRef]
37. Yu, Z., Yi, Y., Gu, L. (2021). The complete chloroplast genome of Lonicera fulvotomentosa Hsu et SC Cheng and its phylogenetic analysis. Mitochondrial DNA Part B, 6(3), 842–843. DOI 10.1080/23802359.2021.1884027. [Google Scholar] [CrossRef]
38. Li, Y., Cai, W., Weng, X., Li, Q., Wang, Y. et al. (2015). Lonicerae Japonicae Flos and Lonicerae Flos: A systematic pharmacology review. Evidence-Based Complementary and Alternative Medicine, 2015, 905063. DOI 10.1155/2015/905063. [Google Scholar] [CrossRef]
39. Chen, C. Y., Qi, L. W., Li, H. J., Li, P., Yi, L. et al. (2007). Simultaneous determination of iridoids, phenolic acids, flavonoids, and saponins in Flos Lonicerae and Flos Lonicerae Japonicae by HPLC-DAD-ELSD coupled with principal component analysis. Journal of Separation Science, 30(18), 3181–3192. DOI 10.1002/(ISSN)1615-9314. [Google Scholar] [CrossRef]
40. Shi, Z. L., Liu, Z. J., Liu, C. S., Wu, M. Q., Su, H. B. et al. (2016). Spectrum-effect relationships between chemical fingerprints and antibacterial effects of Lonicerae Japonicae Flos and Lonicerae Flos base on UPLC and microcalorimetry. Frontiers in Pharmacology, 7, 12. DOI 10.3389/fphar.2016.00012. [Google Scholar] [CrossRef]
41. Fu, S. S., Zhang, T. T., Lö, J. L., Guo, J. J., Yuan, H. L. et al. (2011). Comparison of microcalorimetric fingerprint profiles of Lonicerae japonicae Flos and Lonicerae Flos. Acta Pharmaceutica Sinica, 46(10), 1251–1256. [Google Scholar]
42. Yang, X., Liu, Y., Hou, A., Yang, Y., Tian, X. et al. (2017). Systematic review for geo-authentic Lonicerae Japonicae Flos. Frontiers of Medicine, 11(2), 203–213. DOI 10.1007/s11684-017-0504-0. [Google Scholar] [CrossRef]
43. Zeng, A. Q., Hua, H., Chen, C. R., Liu, L., Zhang, M. et al. (2020). Comparative study on anti-inflammatory effect of Lonicerae Japonicae Flos and Lonicerae Flos. China Journal of Chinese Materia Medica, 45(16), 3938–3944. DOI 10.19540/j.cnki.cjcmm.20200520.401. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |