| Phyton-International Journal of Experimental Botany |
DOI: 10.32604/phyton.2022.019630
ARTICLE
Comparative Transcriptome Analysis Reveals Different Mechanisms of Adaptation to Environment among Three Species of Saussurea DC.
1School of Ecol-Environmental Engineering, Qinghai University, Xining, 810016, China
2State Key Laboratory of Plateau Ecology and Agriculture, Qinghai University, Xining, 810016, China
3Key Laboratory of Landscape Plants of Qinghai Province, Qinghai University, Xining, 810016, China
*Corresponding Author: Tao He. Email: hetaoxn@yahoo.com
Received: 03 October 2021; Accepted: 16 December 2021
Abstract: Saussurea medusa, Saussurea hypsipeta and Saussurea obvallata are typical alpine snowline plants growing in the Qinghai-Tibet plateau. They are characterized by a specialized morphology. S. medusa and S. hypsipeta have very dense trichomes on whole plant, whereas S. obvallata has transparent bracts covered inflorescences. The different forms reflect their adaptation to cold environments. To investigate the different mechanisms of adaptation of these species to cold temperatures, transcriptome sequencing was performed in three species of Saussurea DC. A total of 116394 137237 and 113879 Unigenes were identified from S. medusa, S. hypsipeta and S. obvallata, respectively. Of these, 55909 (48.03%), 65519 (47.74%) and 51889 (45.56%) Unigenes were matched in public databases. GO analysis identified that most of annotated Unigenes in the three species of plants were related to cellular, metabolic, and single−organism processes, and binding and catalytic activities. The differential expression of 37 genes related to environmental adaptation were discovered by pairwise comparisons. Of these, two candidate genes (Interaptin-like and CSLB3) related to trichome development were identified only in S. medusa and S. hypsipeta, which was consistent with their distinct morphology. Our data can provide a valuable resource for the further studies on the adaptive mechanisms of molecular and functional ecology in Saussurea DC.
Keywords: Saussurea DC.; transcriptome; adaptive mechanisms
Saussurea medusa Maxim., Saussurea hypsipeta Diels. and Saussurea obvallata (DC.) Edgew. are perennial herbaceous plants in the family Asteraceae, distributed mainly in the alpine zone of the Qinghai-Tibet Plateau at altitudes from 3800 to 5000 m [1,2]. Many of the high-altitude Saussurea are known for their spectacular growth forms. S. medusa and S. hypsipeta, which are called “woolly” plants, have dense layers of woolly trichomes on their stems, leaves, bracts, and inflorescences [2]. S. obvallata is called called “glasshouse” plant, which encloses their inflorescences in large translucent bracts [3]. These spectacular growth forms are thought to represent an evolutionary response to cold and windy environments [4,5]. However, how are the morphological differences among three the species of Saussurea DC. produced? What are the molecular mechanisms behind these adaptations?
Transcriptomes are the whole RNA transcripts in cells or tissues. They reflect expressed genes at different life stages, tissue types, physiological state and environmental conditions. Transcriptome-based techniques provide useful means of studying gene expression and gene structure, and revealing the molecular mechanism involved in a specific biological process [6]. Recently, this technique has been widely used in the study of molecular mechanisms of plant responses to drought [7], waterlogging [8], low temperature [9], salt [10], high light [11], and irradiation stresses [12]. By comparative transcriptome analysis, new genes are found and molecular mechanisms are revealed in these plants.
Most studies of Saussurea plants growing in the Qinghai-Tibet plateau have focused on their adaptive shapes and anatomies as well as physiological functions [1–3]. Little is known about molecular mechanisms behind these plant adaptations [13]. The present work was performed to examine the comparative transcriptome analysis by transcriptome sequencing and gene function annotation on three species of Saussurea plants growing in the Daban Mountain in the northeastern Qinghai-Tibet Plateau. The studies of molecular mechanisms in alpine plant species will contribute to further understand the adaptation of plants to alpine environments since their responses are associated with cold climates; it is probably not useful to know them in the context of a warming climate.
S. medusa, S. hypsipeta and S. obvallata were collected at flowering stag from Qilian Mountains (37°5′-59′N, 100°55′-102°41′E, 4,200 m a.s.l.) in the northeastern Qinghai-Tibet plateau, China. Growth form of these species is depicted in Fig. 1. This field site features typical plateau continental climate with an annual average (a) temperature of –2°C, (b) rainfall of 482 mm, (c) solar radiation of 6.46 × 109 Jm−2, and (d) barometric pressure of 684.2 hPa. Three plants were collected from each species. The roots, stems, leaves, and flowers obtained from one individual were pooled into a single sample. These materials were flash-frozen in liquid nitrogen.

Figure 1: Saussurea plants in the high-elevation scree or rock fields. (A) S. medusa; (B) S. hypsipeta; (C) S. obvallata
2.2 RNA Extraction and Transcriptome Sequencing
Trizol® (Invitrogen-Thermo Fisher Scientific, Carlsbad, CA, USA) was used to extract total RNA on the three species. All RNA was treated with DNase I. The NanoPhotometer® spectrophotometer (IMPLEN, CA, USA) was used to test RNA purity; the Assay Kit in Qubit® 2.0 Flurometer (Life Technologies, CA, USA) was used to determine RNA concentrations; the RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA) was used to assess RNA integrity, and RNA Integrated Number (RIN) values ≥8.
We used NEBNext® Ultra™ RNA Library Prep Kit from Illumina® (NEB, USA) to construct the sequences library and the Agilent Bioanalyzer 2100 system to evaluate library quality. We sequenced library preparation to obtain raw reads by Illumina HiSeqTM 2500; clean reads were obtained by removing reads containing adapter, reads containing poly-N and low-quality reads from raw reads. At the same time, Q20, Q30, GC-content and sequence duplication level of the clean data were calculated. All the downstream analyses were based on clean data with high quality. Transcriptome was obtained by splicing clean reads with Trinity [14]. The longest transcript in each gene was taken as the Unigene for subsequent analysis.
2.3 Unigene Functional Annotation
Unigenes of S. medusa, S. hypsipeta and S. obvallata were searched in the public databases (Nr, Nt, Pfam, KOG/COG, Swiss-Prot, KEGG, GO), and gene function was annotated according to gene similarity. The E-value in Nr is less than or equal to 1E-5 (Nt, ≤1E-5; Pfam, ≤0.01, KOG/COG, ≤1E-3; Swiss-Prot, ≤1E-5; KEGG, ≤1E-10; GO, ≤1E-6).
2.4 Orthology Genes Screening and Analysis
Orthology genes were searched for full-length CDS sequences by OrthoMCL, and one-to-one orthology genes were screened out [15]. Nonsynonymous substitution rate (Ka), synonymous substitution rate (Ks) and Ka/Ks of orthology genes were calculated by PALM [16]. GO gene enrichment analysis was carried out for the orthology genes (Ka/Ks>1) that were positively selected by GOseq [17]. Pathway significant enrichment analysis was carried out on the orthology genes positively selected by the hypergeometric test, with KEGG Pathway as the unit [18].
2.5 Analysis of Environmental Adaptation Related Unigenes
By using the classification results of Unigene in the GO database and the functional annotations in the seven public databases, the environmental adaptation-related Unigenes of the three species were analysed by interspecies reduction.
3.1 Transcriptome Data Assembly of the Three Species
S. medusa, S. obvallata and S. hypsipeta obtained 150217, 162036 and 192813 transcripts, respectively (Table 1). The average length of the transcript of S. medusa was 748 bp and N50 was 1292 bp. The average length of the transcript and N50 of S. obvallata and S. hypsipeta were 760 bp, 1270 bp and 698 bp, 1156 bp, respectively. The longest transcript in each gene was taken as Unigene for subsequent analysis. S. medusa obtained 116394 Unigenes with an average length of 623 bp and N50 of 966 bp. S. obvallata showed 113879 Unigenes with an average length of 635 bp and N50 of 1000 bp. S. hypsipeta obtained 13723 Unigenes with an average length of 581 bp and N50 of 857 bp. The results showed that the sequencing quality was sufficient for future analysis needs.
3.2 Functional Annotation and Classification of the Three Species’ Unigene
3.2.1 Unigene Annotation in the Public Database
S. medusa, S. hypaipeta and S. obvalata had 7620, 7245 and 9366 Unigenes, respectively, that can be matched in all databases (Table 2).
3.2.2 GO Functional Classification of Unigene
The Unigenes of S. medusa, S. obvallata and S. hypsipeta were divided into 55, 56 and 56 types of function respectively, and the 55 types of function were the same (Fig. 2). The Unigene of S. obvallata and S. hypsipeta found a gene related to cell aggregation. Cell agglutination is associated to the resistance of organisms to environmental UV stress [19].
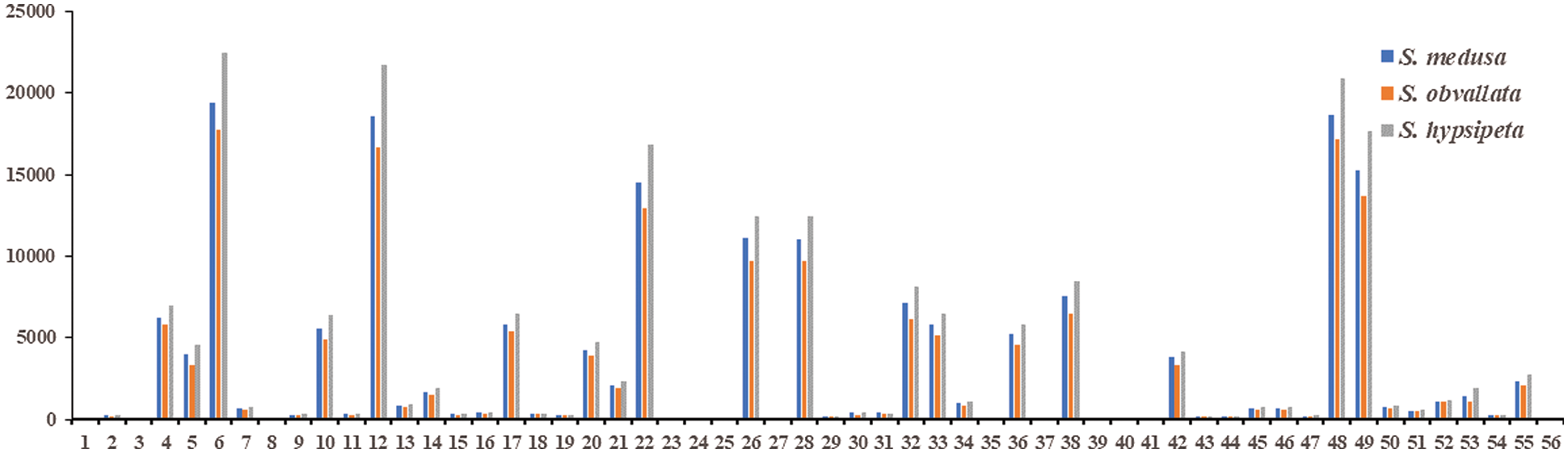
Figure 2: The number of GO function annotated for Unigene in S. medusa, S. hypsipeta and S. obvallata
Notes: 1: behavior; 2: biological adhesion; 3: biological phase; 4: biological regulation; 5: cellular component organization or biogenesis; 6: cellular process; 7: developmental process; 8: growth; 9: immune system process; 10: localization; 11: locomotion; 12: metabolic process; 13: multicellular organismal process; 14: multi−organism process; 15: negative regulation of biological process; 16: positive regulation of biological process; 17: regulation of biological process; 18: reproduction; 19: reproductive process; 20: response to stimulus; 21: signaling; 22: single-organism process; 23: rhythmic process; 24: cell killing; 25: cell aggregation; 26: cell; 27: cell junction; 28: cell part; 29: extracellular matrix; 30: extracellular region; 31: extracellular region part; 32: macromolecular complex; 33: membrane; 34: membrane-enclosed lumen; 35: synapse part; 36: membrane part; 37: synapse; 38: organelle; 39: extracellular matrix component; 40: nucleoid; 41: symplast; 42: organelle part; 43: other organism; 44: other organism part; 45: virion; 46: virion part; 47: antioxidant activity; 48: binding; 49: catalytic activity; 50: molecular function regulator; 51: molecular transducer activity; 52: nucleic acid binding transcription factor activity; 53: structural molecule activity; 54: transcription factor activity and protein binding; 55: transporter activity; 56: metallochaperone activity; 1~25, 26~46 and 47~56 correspond to secondary functional classification of Biological Process, Cellular Component and Molecular Function, respectively.
3.2.3 KOG Functional Classification of Unigene
The KOG annotation results of Unigene of S. medusa, S. hypsipeta and S. obvallata were similar. There were 21053 Unigenes of S. medusa, 17117 Unigenes of S. hypsipeta and 24003 Unigenes of S. obvallata annotated on 25 KOG classifications. Firstly, the general function prediction only accounted for most of the Unigenes, which were 16.41%, 18.05% and 16.46% in S. medusa, S. hypsipeta and S. obvallata, respectively. Next, there were posttranslational modification, proteins turnover and chaperones. The proportion of S. medusa, S. hypsipeta and S. obvallata in this category was 14.32%, 14.41% and 11.64%, respectively. Signal transduction mechanisms were 8.74%, 8.54% and 7.65%, in S. medusa, S. hypsipeta and S. obvallata, respectively (Fig. 3). They accounted for the least Unigene in the classification of Cell motility, Extracellular structures and Nuclear structures.
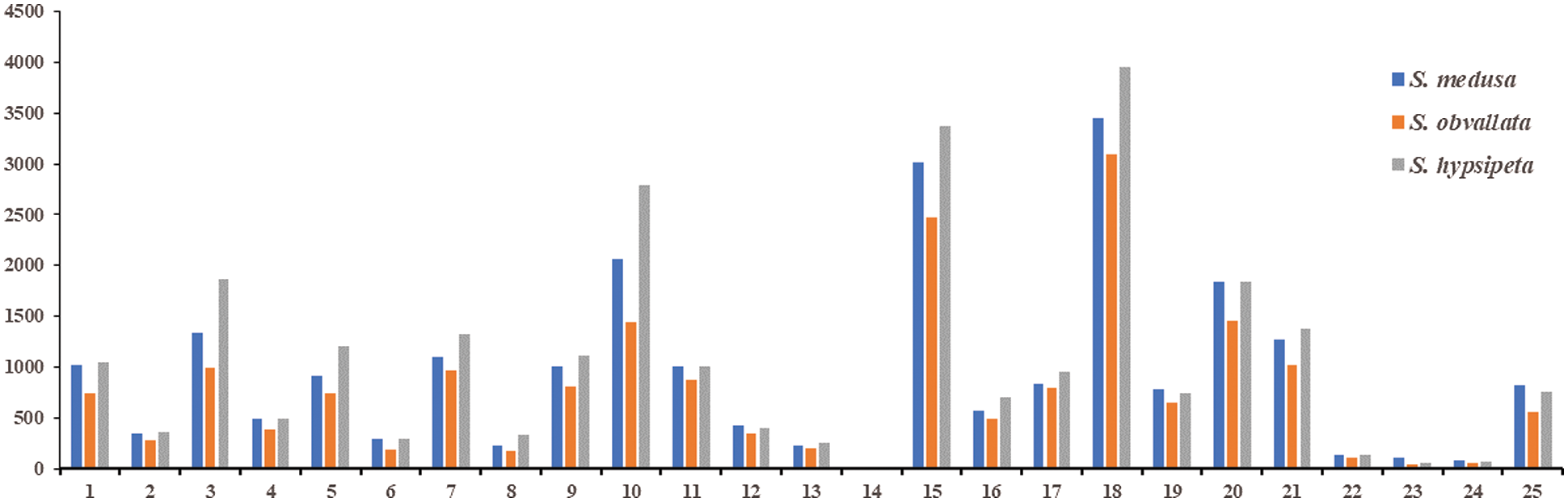
Figure 3: The number of KOG function annotated for Unigene in S. medusa, S. hypsipeta and S. obvallata
Note: 1: RNA processing and modification; 2: chromatin structure and dynamics; 3: energy production and conversion; 4: cell cycle control or cell division or chromosome partitioning; 5: amino acid transport and metabolism; 6: nucleotide transport and metabolism; 7: carbohydrate transport and metabolism; 8: coenzyme transport and metabolism; 9: lipid transport and metabolism; 10: translation or ribosomal structure or biogenesis; 11: transcription; 12: replication and recombination and repair; 13: cell wall or membrane or envelope biogenesis; 14: cell motility; 15: posttranslational or modification or protein turnover, chaperones; 16: inorganic ion transport and metabolism; 17: secondary metabolites biosynthesis or transport and catabolism; 18: general function prediction only; 19: function unknown; 20: signal transduction mechanisms; 21: intracellular trafficking or secretion or vesicular transport; 22: defense mechanisms; 23: extracellular structures; 24: nuclear structure; 25: cytoskeleton.
3.2.4 KEGG Functional Classification of Unigene
The Unigene annotated by the three species belonged to Cellular Processes, Environmental Information Processing, Genetic Information Processing, Metabolism and Organismal Systems, including 19 secondary pathways (Fig. 4). Thereinto, 7 types of pathway Unigene accounted for a large proportion (>50%), and they included Cellular Processes, Folding and sorting and degradation, Translation, Amino acid metabolism, Carbohydrate metabolism, Energy metabolism, and Overview, respectively. The KEGG classification results indicated that the Unigene functions were mainly related to protein translation and material metabolism.
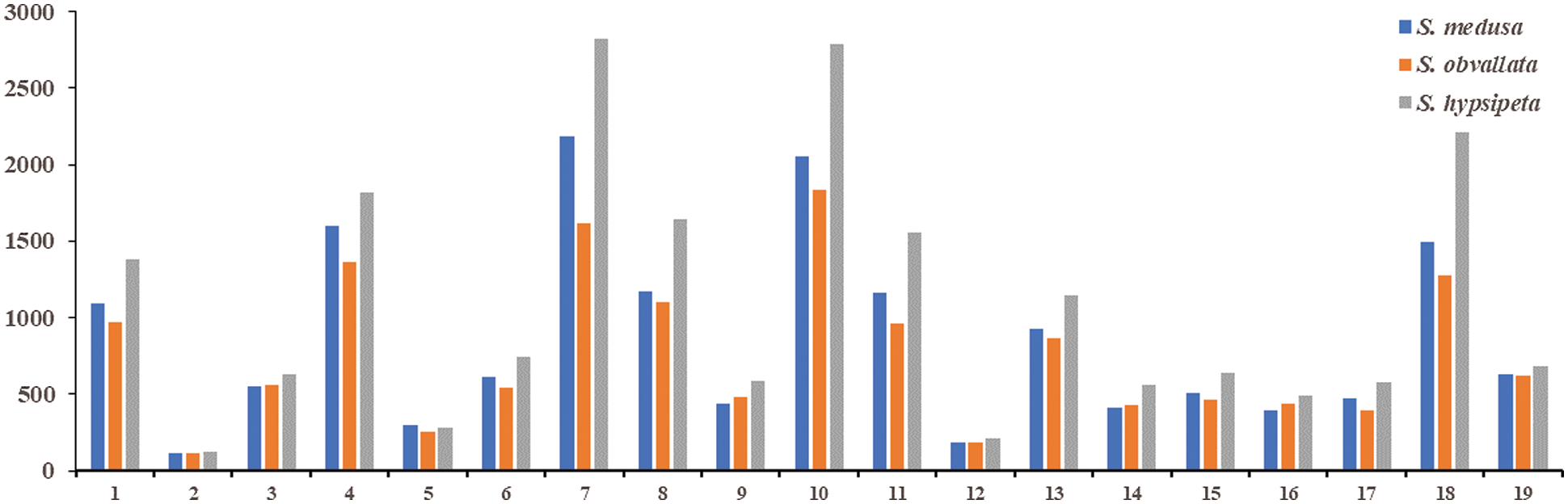
Figure 4: The number of KEGG function annotated for Unigene in S. medusa, S. hypsipeta and S. obvallata
Notes: 1: transport and catabolism; 2: membrane transport; 3: signal transduction; 4: folding and sorting and degradation; 5: replication and repair; 6: transcription; 7: translation; 8: amino acid metabolism; 9: biosynthesis of other secondary metabolites; 10: carbohydrate metabolism; 11: energy metabolism; 12: glycan biosynthesis and metabolism; 13: lipid metabolism; 14: metabolism of cofactors and vitamins; 15: metabolism of other amino acids; 16: metabolism of terpenoids and polyketides; 17: nucleotide metabolism; 18: overview; 19: environmental adaptation; 1, 2~3, 4~7, 8~18 and 19 correspond to secondary functional classification of cellular processes, environmental information processing, genetic information processing, metabolism, and Organismal systems, respectively.
3.3 Orthology Genes Screening of the Three Species
10811 sets of orthologous genes were obtained through OrthoMCL in the three species. There were 463 sets of orthologous genes which Ka/Ks>1, indicating that these genes were rapidly divergent (Fig. 5).
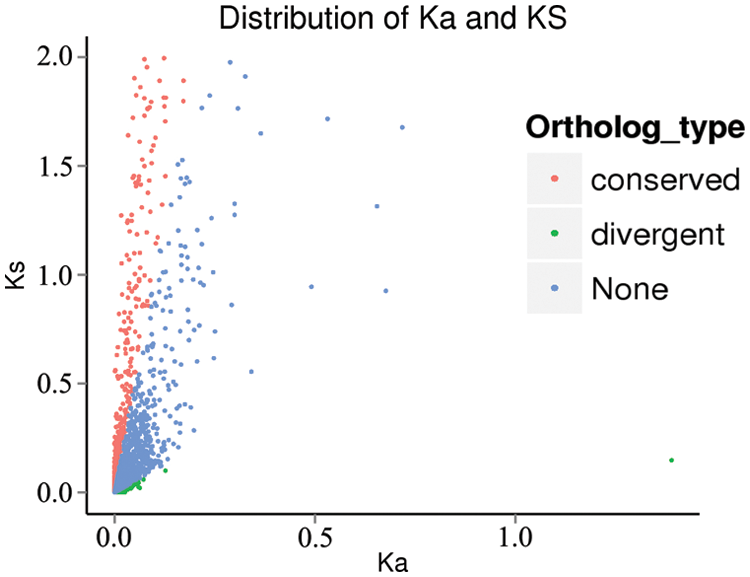
Figure 5: Distribution of Ka and Ks
The GO functional enrichment classification was carried out for the orthologous genes of positive selection in the three species. We did not find functional enrichment genes, but 78 genes were related to plant resistance. The resistance genes included peroxidase, ubiquitin, zinc-finger protein and acyl-CoA-binding protein, which play an important role in plant environmental stress. The KEGG gene enrichment analysis revealed that Ribosome had the largest number of genes enriched in this pathway, with a total of 14 genes, revealing that the genes related to Ribosome synthesis in these three species are subject to strong positive selection (Fig. 6).
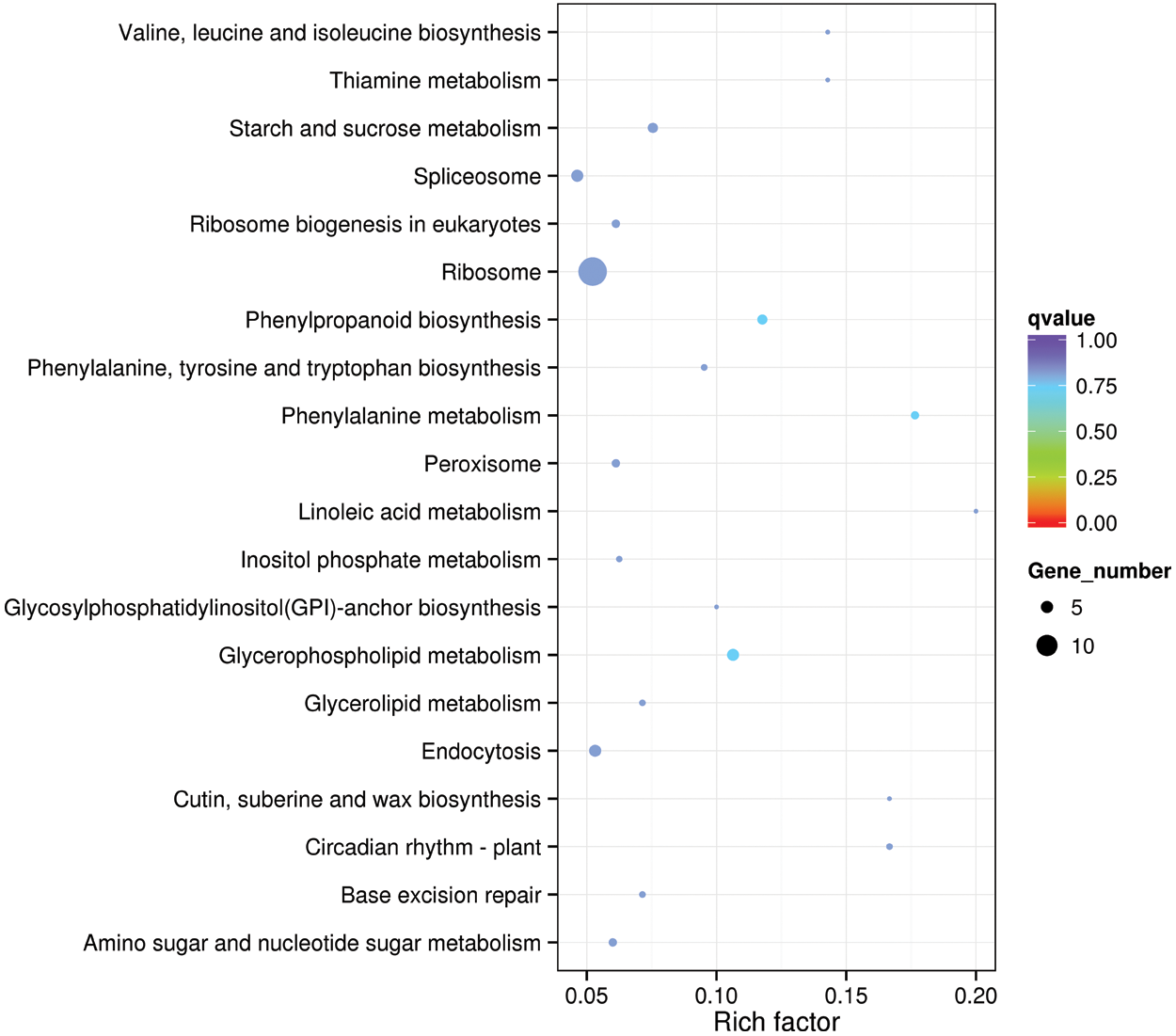
Figure 6: Statistic of KEGG enrichment
3.4 Analysis of Environmental Adaptation Related Unigenes
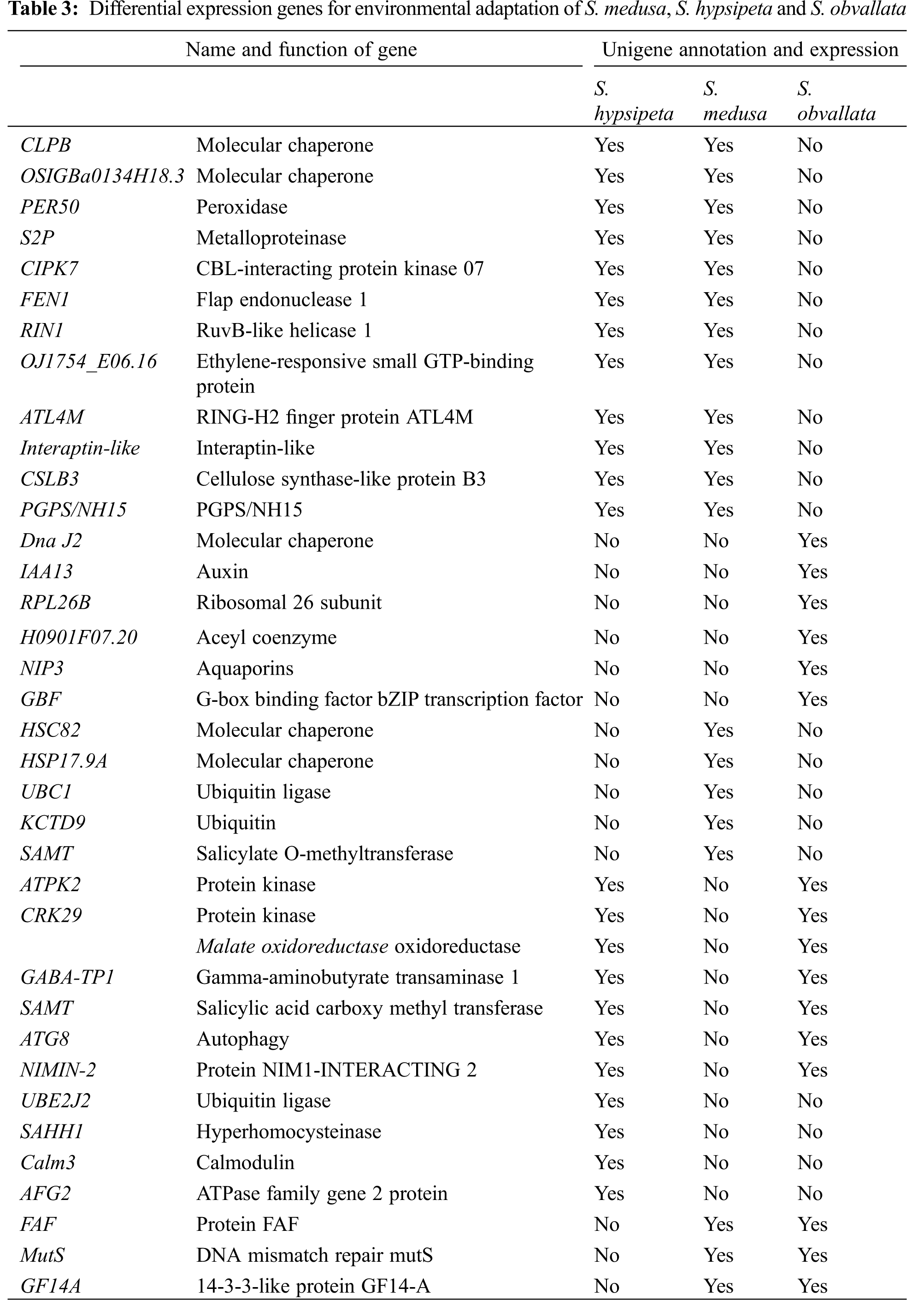
Based on the classification of the three species Unigenes in GO database, and analysis of Unigene about environmental adaptation, the results showed that the genes related to environmental adaptation in the three species were mainly molecular chaperone, ubiquitin, calmodulin, enzyme and ribosomes (Table 3). HSC82, CLPB and HSP17.9A can enhance the effect of plants on high temperature and salt stresses [20–22]. Dna J2 and osigba0134h18.3 were annotated by GO, which are involved in the adaptation of plants to high temperature stress. PER50 is one of the enzymes with which plants respond to oxidative stress [23], while S2P corresponds to salt stress [24]. ATPK2 is involved in plant adaptation to cold and high salt stresses [25], while CRK29 and CIPK7 are in connection with plant immune responses and adaptation to cold stress [26,27]. FEN1 is one of the key enzymes in DNA replication and repair in eukaryotes [28]. RIN1 annotated by GO is involved in plant defensive responses to fungi. Malate oxidoreductase can regulate plant growth and respiration [29]. Gene expression is regulated by SAHH1 through gene methylation modification [30]. GABA-TP1 participates in plant adaptation to temperature stress [31]. SAMT is related to plants’ defense responses to biologic stimulation [32,33]. Calm3 mediates the regulation of enzymes and ion channels through calcium ions [34]. IAA13 regulates growth and development of plants through ARFs [35]. H0901F07.20 annotated by GO involves in the binding of lipoyl coenzyme A. ATG8 is related to the formation of autophagosomes in plants [36]. NIP3 is correlated with the transport of the heavy metal arsenic in plants [37]. OJ1754_E06.16 is associated with the processes of protein secretion and signal transmission. FAF takes part in plant ABA activation signaling pathway and phosphorylation regulation. MutS participates in DNA mismatch repair. GF14A is connected with plant adaptation to drought stress [38]. NIMIN-2 is related to the acquisition of systemic resistance in plants [39], while ATL4M is related to abiotic stress responses [40]. Interaptin-like links the intracellular system to the cytoskeleton, which regulates the morphogenesis of the multicellular trichomes [41,42]. CSLB3 is related to the formation of non-cellulosic polysaccharide skeletons in plant cell walls. GBF is one of the transcription factors in which cells respond to signals [43]. AFG2 is associated with the maturation of 60S ribosomal subunit [44]. PGPS/NH15 annotated by GO participates in the regulation of oxidation-reduction enzyme activities.
Our sequencing results showed that Unigene N50 of the three species were 857~1000 bp, the base quality value Q30 was over 92%, and the GC content was over 43%. The amount of sequencing data and the number of Unigene spliced were relatively large, which showed that the sequencing quality was good, and the sequence information generated was sufficient and effective [45]. The results of Go, KOG and KEGG of three species Unigenes were very similar, indicating that their genetic relationship was very similar.
The temperature in the collecting area was between 0–30°C, UV radiation up to 7000 μW·cm−2, and the oxygen partial pressure was 13.25 kPa for the three species. Stresses of low oxygen, strong radiation and large temperature differences made that makes the plants growing in this area formed special stress resistance mechanisms in the long-term to achieve collaborative resilience, such as trichomes, ABA signaling pathway, molecular chaperones and ubiquitin pathway. In our study, there was a gene expression related to cell aggregation in both S. hypsipeta and S. obvallata. Some research shows that Gloeocapsa sp. cells with low metabolic activity aggregate to form a lamellar biofilm to protect organisms against ultraviolet radiation [19]. Then, we compared leaf structure among the three species; S. medusa had the longest, thickest and white trichomes on the leaf epidermis. This structure can effectively resist and reflect ultraviolet radiation. S. hypsipeta and S. obvallata may resist the ultraviolet radiation stress through cell aggregation, but the specific mechanism needs to be verified in future experiments. Interaptin protein was the binding protein between the components of the inner membrane and the actin of the cytoskeleton, Microtubules and actin promote the development of trichomes [41,42]. CSLB3 participates in the formation of the noncellulose polysaccharide skeleton in plant cell wall. In this study, we found that the Interaptin-like and CSLB3 genes were both expressed in S. hypsipeta and S. medusa. It is speculated that the expression of these two genes can promote the occurrence of trichomes and improve the resistance of these two plant species to radiation and temperature stresses. This finding was also consistent with the structural characteristics of the three species. S. medusa and S. hypsipeta are densely covered with trichomes. The ground tissues of S. obvallata showed no trichomes, and the inflorescences were enclosed in translucent bracts. At the same time, we found that the three plant species expressed different molecular chaperones, ubiquitin, protease and other genes to allow the adaptation of plants to the environmental stresses, and their mechanisms need further study.
Funding Statement: This work was financially supported by the National Natural Science Foundation of China (31960222, 31360095).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Tsukaya, H., Fujikawa, K., Wu, S. (2002). Thermal insulation and accumulation of heat in the downy inflorescences of Saussurea medusa (Asteraceae) at high elevation in Yunnan, China. Journal Plant Research, 115(4), 263–268. DOI 10.1007/s10265-002-0030-1. [Google Scholar] [CrossRef]
2. Yang, Y., KÖrner, C., Sun, H. (2008). The ecological significance of pubescence in Saussurea medusa, a high-elevation himalayan woolly plant. Arctic Antarctic and Alpine Research, 40(1), 250–255. DOI 10.1657/1523-0430(07-009)[YANG]2.0.CO;2. [Google Scholar] [CrossRef]
3. Semwal, P., Pauw, A., Palni L.M., S., Thapliyal, A. (2019). Bumblebees (Bombus rufofasciatus Smith) pollinate the enclosed inflorescences of the endangered Brahma’s lotus (Saussurea obvallata: Asteraceae) of the Indian Himalaya. South African Journal of Botany, 121, 435–441. DOI 10.1016/j.sajb.2018.12.015. [Google Scholar] [CrossRef]
4. Tsukaya, H., Tsuge, T. (2001). Morphological adaptation of inflorescence in plants that develops at low temperature in early spring: The convergent evolution of “downy plants”. Plant Biology, 3, 536–543. DOI 10.1055/s-2001-17727. [Google Scholar] [CrossRef]
5. KÖrner, C. (2003). Alpine plant life. Berlin, Berlin Heidelberg: Springer-Verlag. [Google Scholar]
6. Garber, M., Grabherr, M. G., Guttman, M., Trapnell, C. (2011). Computational methods for transcriptome annotation and quantification using RNA-seq. Nature Methods, 8(6), 469–477. DOI 10.1038/nmeth.1613. [Google Scholar] [CrossRef]
7. Kumar, J., Gunapati, S., Kianian, S. F., Singh, S. P. (2018). Comparative analysis of transcriptome in two wheat genotypes with contrasting levels of drought tolerance. Protoplasma, 255(5), 1487–1504. DOI 10.1007/s00709-018-1237-x. [Google Scholar] [CrossRef]
8. Zhao, N., Li, C., Yan, Y., Cao, W., Song, A. et al. (2018). Comparative transcriptome analysis of waterlogging-sensitive and waterlogging-tolerant Chrysanthemum morifolium cultivars under waterlogging stress and reoxygenation conditions. International Journal Molecular Sciences, 19(5), 1455. DOI 10.3390/ijms19051455. [Google Scholar] [CrossRef]
9. Asiche, W. O., Mitalo, O. W., Kasahara, Y., Tosa, Y., Mworia, E. G. et al. (2018). Comparative transcriptome analysis reveals distinct ethylene-independent regulation of ripening in response to low temperature in kiwifruit. BMC Plant Biology, 18(1), 47. DOI 10.1186/s12870-018-1264-y. [Google Scholar] [CrossRef]
10. Ma, W., Kim, J. K., Jia, C., Yin, F., Kim, H. J. et al. (2019). Comparative transcriptome and metabolic profiling analysis of buckwheat (Fagopyrum tataricum (L.) Gaertn.) under salinity stress. Metabolites, 9(10), 225. DOI 10.3390/metabo9100225. [Google Scholar] [CrossRef]
11. Cui, D., Hu, C., Zou, Z., Sun, X., Shi, J. et al. (2020). Comparative transcriptome analysis unveils mechanisms underlying the promoting effect of potassium iodide on astaxanthin accumulation in Haematococcus pluvialis under high light stress. Aquaculture, 525, 735279. DOI 10.1016/j.aquaculture.2020.735279. [Google Scholar] [CrossRef]
12. Xiong, H., Guo, H., Xie, Y., Gu, J., Zhao, L. et al. (2020). Comparative transcriptome analysis of two common wheat varieties induced by 7Li-ion beam irradiation reveals mutation hotspot regions and associated pathways. Radiation Physics and Chemistry, 170, 108650. DOI 10.1016/j.radphyschem.2019.108650. [Google Scholar] [CrossRef]
13. Li, H., Qiu, J., Chen, F., Lv, X., Fu, C. et al. (2012). Molecular characterization and expression analysis of dihydroflavonol 4-reductase (DFR) gene in Saussurea medusa. Molecular Biology Reports, 39(3), 2991–2999. DOI 10.1007/s11033-011-1061-2. [Google Scholar] [CrossRef]
14. Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A. et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology, 29(7), 644–652. DOI 10.1038/nbt.1883. [Google Scholar] [CrossRef]
15. Li, L., Stoeckert Jr, C. J., Roos, D. S. (2003). OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Research, 13(9), 2178–2189. DOI 10.1101/gr.1224503. [Google Scholar] [CrossRef]
16. Yang, Z. (2007). PAML 4: Phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution, 24(8), 1586–1591. DOI 10.1093/molbev/msm088. [Google Scholar] [CrossRef]
17. Young, M. D., Wakefield, M. J., Smyth, G. K., Oshlack, A. (2010). Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biology, 11(2), R14. DOI 10.1186/gb-2010-11-2-r14. [Google Scholar] [CrossRef]
18. Kanehisa, M., Araki, M., Goto, S., Hattori, M., Hirakawa, M. et al. (2008). KEGG for linking genomes to life and the environment. Nucleic Acids Research, 36(suppl_1), D480–D484. DOI 10.1093/nar/gkm882. [Google Scholar] [CrossRef]
19. Wadsworth, J., Rettberg, P., Cockell, C. S. (2019). Aggregated cell masses provide protection against space extremes and a microhabitat for hitchhiking co-inhabitants. Astrobiology, 19(8), 995–1007. DOI 10.1089/ast.2018.1924. [Google Scholar] [CrossRef]
20. Wang, W., Vinocur, B., Shoseyov, O., Altman, A. (2004). Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends in Plant Science, 9(5), 244–252. DOI 10.1016/j.tplants.2004.03.006. [Google Scholar] [CrossRef]
21. Imai, J., Yahara, I. (2001). Role of HSP90 in salt stress tolerance via stabilization and regulation of calcineurin. Molecular and Cellular Biology, 20(24), 9262–9270. DOI 10.1128/MCB.20.24.9262-9270.2000. [Google Scholar] [CrossRef]
22. Lee, U., Rioflorido, I., Hong, S. W., Larkindale, J., Waters, E. R. et al. (2007). The Arabidopsis ClpB/Hsp100 family of proteins: Chaperones for stress and chloroplast development. Plant Journal, 49(1), 115–127. DOI 10.1111/j.1365-313X.2006.02940.x. [Google Scholar] [CrossRef]
23. Schenk, P. M., Kazan, K., Wilson, I., Anderson, J. P., Richmond, T. et al. (2000). Coordinated plant defense responses in Arabidopsis revealed by microarray analysis. Plant Biology, 97(21), 11655–11660. DOI 10.1073/pnas.97.21.11655. [Google Scholar] [CrossRef]
24. Che, P., Bussell, J. D., Zhou, W., Estavillo, G. M., Pogson, B. J. et al. (2010). Signaling from the endoplasmic reticulum activates brassinosteroid signaling and promotes acclimation to stress in Arabidopsis. Science Signaling, 3(141). DOI 10.1126/scisignal.2001140. [Google Scholar] [CrossRef]
25. Lee, D. H., Park, S. J., Ahn, C. S., Pai, H. S. (2017). MRF family genes are involved in translation control, especially under energy-deficient conditions, and their expression and functions are modulated by the TOR signaling pathway. Plant Cell, 29(11), 2895–2920. DOI 10.1105/tpc.17.00563. [Google Scholar] [CrossRef]
26. Yadeta, K. A., Elmore, J. M., Creer, A. Y., Feng, B. M., Franco, J. Y. et al. (2017). A cysteine-rich protein kinase associates with a membrane immune complex and the cysteine residues are required for cell death. Plant Physiology, 173(1), 771–787. DOI 10.1104/pp.16.01404. [Google Scholar] [CrossRef]
27. Huang, C., Ding, S., Zhang, H., Du, H., An, L. (2011). CIPK7 is involved in cold response by interacting with CBL1 in Arabidopsis thaliana. Plant Science, 181(1), 57–64. DOI 10.1016/j.plantsci.2011.03.011. [Google Scholar] [CrossRef]
28. Liu, Y., Kao, H. I., Bambara, R. A. (2004). Flap Endonuclease 1: A central component of DNA metabolism. Annual Review of Biochemistry, 73, 589–615. DOI 10.1146/annurev.biochem.73.012803.092453. [Google Scholar] [CrossRef]
29. Tomaz, T., Bagard, M., Pracharoenwattana, I., Lindén, P., Lee, C. P. et al. (2010). Mitochondrial malate dehydrogenase lowers leaf respiration and alters photorespiration and plant growth in Arabidopsis. Plant Physiology, 154(3), 1143–1157. DOI 10.1104/pp.110.161612. [Google Scholar] [CrossRef]
30. Rocha, P. S. C. F., Sheikh, M., Melchiorre, R., Fagard, M., Boutet, S. et al. (2005). The Arabidopsis HOMOLOGY-DEPENDENT GENE SILENCING1 gene codes for an S-adenosyl-L-homocysteine hydrolase required for DNA methylation-dependent gene silencing. Plant Cell, 17, 404–417. DOI 10.1105/tpc.104.028332. [Google Scholar] [CrossRef]
31. Akihiro, T., Koike, S., Tani, R., Tominaga, T., Watanabe, S. et al. (2008). Biochemical mechanism on GABA accumulation during fruit development in tomato. Plant Cell Physiology, 49(9), 1378–1389. DOI 10.1093/pcp/pcn113. [Google Scholar] [CrossRef]
32. Ament, K., Krasikov, V., Allmann, S., Rep, M., Takken, F. L. W. et al. (2010). Methyl salicylate production in tomato affects biotic interactions. Plant Journal, 62, 124–134. DOI 10.1111/j.1365-313x.2010.04132.x. [Google Scholar] [CrossRef]
33. Leng, F., Lin, Q., Wu, D., Wang, S., Wang, D. et al. (2016). Comparative transcriptomic analysis of grape berry in response to root restriction during developmental stages. Molecules, 21(11), 1431. DOI 10.3390/molecules21111431. [Google Scholar] [CrossRef]
34. Takahashi, F., Mizoguchi, T., Yoshida, R., Ichimura, K., Shinozaki, K. (2011). Calmodulin-dependent activation of MAP kinase for ROS homeostasis in Arabidopsis. Molecule Cell, 41(6), 649–660. DOI 10.1016/j.molcel.2011.02.029. [Google Scholar] [CrossRef]
35. Liscum, E., Reed, J. W. (2002). Genetics of Aux/IAA and ARF action in plant growth and development. Plant Molecular Biology, 49, 387–400. DOI 10.1023/a:1015255030047. [Google Scholar] [CrossRef]
36. Yamaguchi, M., Noda, N. N., Nakatogawa, H., Kumeta, H., Ohsumi, Y. et al. (2010). Autophagy-related protein 8 (Atg8) family interacting motif in Atg3 mediates the Atg3-Atg8 interaction and is crucial for the cytoplasm-to-vacuole targeting pathway. Journal of Biological Chemistry, 285(38), 29599–29607. DOI 10.1074/jbc.M110.113670. [Google Scholar] [CrossRef]
37. Xu, W., Dai, W., Yan, H., Li, S., Shen, H. et al. (2015). Arabidopsis NIP3;1 plays an important role in arsenic uptake and root-to-shoot translocation under arsenite stress conditions. Molecular Plant, 8(5), 722–733. DOI 10.1016/j.molp.2015.01.005. [Google Scholar] [CrossRef]
38. Yang, L., You, J., Wang, Y., Li, J., Quan, W. et al. (2017). Systematic analysis of the G-box Factor 14-3-3 gene family and functional characterization of GF14a in Brachypodium distachyon. Plant Physiology and Biochemistry, 117, 1–11. DOI 10.1016/j.plaphy.2017.05.013. [Google Scholar] [CrossRef]
39. Weigel, R. R., Bäuscher, C., Pfitzner, A. J. P., Pfitzner, U. M. (2001). NIMIN-1, NIMIN-2 and NIMIN-3, members of a novel family of proteins from Arabidopsis that interact with NPR1/NIM1, a key regulator of systemic acquired resistance in plants. Plant Molecular Biology, 46, 43–160. DOI 10.1023/a:1010652620115. [Google Scholar] [CrossRef]
40. Song, J., Xing, Y., Munir, S., Yu, C., Song, L. et al. (2016). An ATL78-Like RING-H2 finger protein confers abiotic stress tolerance through interacting with RAV2 and CSN5B in tomato. Frontiers in Plant Science, 7, 1305. DOI 10.3389/fpls.2016.01305. [Google Scholar] [CrossRef]
41. Rivero, F., Kuspa, A., Brokamp, R., Matzner, M., Noegel, A. A. (1998). Interaptin, an actin-binding protein of the α-actinin superfamily in Dictyostelium discoideum, is developmentally and cAMP-regulated and associates with intracellular membrane compartments. Journal of Cell Biology, 142(3), 735–750. DOI 10.1083/jcb.142.3.735. [Google Scholar] [CrossRef]
42. Chang, J., Xu, Z., Li, M., Yang, M., Qin, H. et al. (2019). Spatiotemporal cytoskeleton organizations determine morphogenesis of multicellular trichomes in tomato. PLoS Genetics, 15(10), e1008438. DOI 10.1371/journal.pgen.1008438. [Google Scholar] [CrossRef]
43. Sibéril, Y., Doireau, P., Gantet, P. (2001). Plant bZIP G-box binding factors modular structure and activation mechanisms. European Journal of Biochemistry, 268, 5655–5666. DOI 10.1046/j.0014-2956.2001.02552.x. [Google Scholar]
44. Pertschy, B., Saveanu, C., Zisser, G., Lebreton, A., Tengg, M. et al. (2007). Cytoplasmic recycling of 60S preribosomal factors depends on the AAA protein Drg1. Molecular and Cellular Biology, 27(19), 6581–6659. DOI 10.1128/mcb.00668-07. [Google Scholar] [CrossRef]
45. Ai, B., Gao, Y., Zhang, X., Tao, J., Kang, M. et al. (2015). Comparative transcriptome resources of eleven Primulina species, a group of stone plants from a biodiversity hotspot. Molecular Ecology Resources, 15(3), 619–632. DOI 10.1111/1755-0998.12333. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |