| Phyton-International Journal of Experimental Botany |
DOI: 10.32604/phyton.2022.019278
ARTICLE
Integrated Analysis of Metabolome and Transcriptome of Bambusa emeiensis Shoots in Response to Bamboo Snout Beetle Cyrtotrachelus buqueti (Coleoptera: Curculionidae)
College of Life Science, Leshan Normal University, Leshan, 614000, China
*Corresponding Author: Chaobing Luo. Email: 13366181512@163.com
#Hao Tang and Yuanqiu Li contributed equally to this work
Received: 13 September 2021; Accepted: 01 November 2021
Abstract: Plants have evolved constitutive and inductive defense mechanisms to protect themselves from herbivorous insects. Metabolites in plants are thought to be involved in response to herbivores. Bambusa emeiensis is an important bamboo species widely distributed in Southwest China. It is easy to attract herbivores such as Cyrtotrachelus buqueti. Through the detection and analysis of metabolites in B. emeiensis metabolomic data, 35 differential metabolites (biomarkers) were finally identified from 206 detection peaks, mainly primary metabolites. Subsequently, we conducted an association analysis between 35 biomarkers that annotated to be involved in 71 metabolic pathways in the metabolome and 324 differentially expressed genes (DEGs) in 207 gene expression pathways distributed in the transcriptome of B. emeiensis after feeding by C. buqueti. We also discussed the relationship between the changes in gene expression levels and metabolite content variation. A total of 47 integrated pathways containing the corresponding DEGs and biomarkers were screened out, with the amino acid synthesis pathway (ko01230) containing the most DEGs and biomarkers. In these integrated pathways, the changes in biomarkers content and the expression levels of the corresponding genes were generally consistent. For example, the increase in tryptophan content was accompanied by an increase in the expression of the corresponding catalase in the tryptophan synthesis pathway. Similar to this was glucose and trehalose in carbohydrate metabolism. Therefore, this study further deepened our understanding of the defense mechanism of B. emeiensis against bamboo pests and provided new insights for the prevention and control of bamboo diseases and pests.
Keywords: Bambusa emeiensis; herbivore; Cyrtotrachelus buqueti; metabolome; transcriptome
In the natural environment, for economically valuable plants, insect feeding is an important constraint that affects plant growth and plant resource exploitation. From the perspective of insects, plants are a source of food for their growth and development, and reproduction, a place to lay eggs, and a shelter against natural enemies [1]. Plants have evolved corresponding mechanisms of resistance to insect feeding to reduce or overcome insect damage [2]. For example, plants form their own physical barriers, including waxy cuticles, spines, and trichomes [3,4]. A representative example is the trichomes of bamboo shoots that effectively avoid feeding by C. buqueti [5]. In addition to this, plants respond to insect feeding stress by producing large amounts of primary and secondary metabolites such as sucrose, saponins, and phenolic compounds [6,7], or by secreting volatile substances to induce natural enemies of insects, thereby directly or indirectly developing resistance to insect feeding [8].
The response of plants to insect feeding involves the regulation of expression of a large number of genes, the regulation of transcription factors, the synthesis of proteins (enzymes) and changes in metabolite content. Omics applications and their associated analyses can help us to better understand the mechanisms of plant-insect interactions. Much of the previous work was carried out in multiple species using single omics techniques, such as the high expression of carbohydrate-related genes in B. emeiensis in response to C. buqueti feeding stress [9] and the significant changes in energy metabolism-related proteins in maize in response to Holotrichia parallela feeding [10]. However, the multi-omics integration analysis can help us understand the mechanisms of plant-insect interactions in greater depth at the molecular level [11–14]. For example, a study showed that ‘Red Russian’ Kale (Brassicae napus var. pabularia) had a common mechanism of induction of changes in metabolites under methyl jasmonate induction and Cabbage Looper (Trichoplusia ni Hübner) feeding stress, implying that it is useful for pest control and nutritional quality of kale [15]
B. emeiensis is widely distributed in Southwest China and is a plant with high economic value that is currently used in papermaking, landscaping and environmental protection [16]. In our previous study, we identified a large number of carbohydrate genes in C. buqueti [17], analyzed the ability of C. buqueti to degrade B. emeiensis lignocellulose, and explored the potential of using B. emeiensis for bioenergy conversion [18]. In the process, we learned that C. buqueti is a B. emeiensis specialist insect that severely damages and inhibits the development of bamboo shoots and affects the bamboo success rate of B. emeiensis. In addition, the expression of related genes and metabolite changes in B. emeiensis after feeding by C. buqueti remain unclear. Therefore, in this paper, we used transcriptome and metabolome integration analysis to investigate the most important gene expression changes and metabolite changes in B. emeiensis in response to C. buqueti infestation. This work will further deepen our understanding of the mechanism of B. emeiensis against pest feeding and provide new clues for the prevention and control of bamboo pests and subsequent exploitation of bamboo resources.
2.1 Plant Samples and Herbivory Treatments
B. emeiensis shoots grew in a bamboo garden at the Bamboo Diseases and Pests Control and Resources Development Key Laboratory of Sichuan Province (103°98′N, 28°96′E), Sichuan Province, China. C. buqueti samples were retrieved from Muchuan in Sichuan in August 2019. They were starved for 24 h prior to use in this experiment. Subsequently, C. buqueti individuals were enclosed around the attached bamboo shoots by cages in the treatments. The samples collection complied with local regulations and did not require specific permission. B. emeiensis shoots that were not subjected to herbivore treatment were used as controls and named as W group, while the B. emeiensis shoots infestation with C. buqueti was labeled with Q group. After bamboo shoots of the Q group were fed for 3 h, the samples in the two groups were immediately frozen in liquid nitrogen and stored at −80°C [19,20].
Previously, we have completed the sequencing of B. emeiensis transcriptome under C. buqueti pressure and the identification of some transcription factors [16,20]. Here, we briefly describe the relevant methods. Total RNA was extracted from the samples and detected for RNA integrity. We did three biological replicates. Subsequently, libraries were constructed and 125 bp/150 bp paired end-reads were generated by Illumina sequencing platform (HiSeqTM 2500) sequencing. Low-quality reads were filtered out from the original data and subsequently assembled to obtain a non-redundant unigenes library. Transcripts were normalized for expression using the Trinity program and FPKM (fragments per million characteristic kilobases) was calculated. Normalized gene expression was performed by centralized and log2 transformation [21]. Gene function was annotated using NR, Swiss-Prot, GO and KEGG databases. The screening and identification of DEGs referred to corresponding standards [22].
Metabolite extraction was performed on 100 mg of B. emeiensis tissue with reference to a previous method [23] and with slight modifications. 100 (±1) mg of plant tissues were placed in 5 mL tubes with steel beads frozen in liquid nitrogen and then placed in a high-throughput tissue grinder (Xinzhi SCIENTZ-48, China) and pulverized at 70 Hz for 1 min. The powder samples were then extracted with 1400 μL of pre-cooled methanol (−20°C) in an ultrasonicator (Shumei KW-100TDV, China) with 750 μL chloroform and 1400 μL ddH2O for 30 min of sonication. The samples were centrifuged at 14,000 rpm for 10 min in a freezing centrifuge (Xiangyi H1650-W, China), and the supernatant was then transferred to a new 1.5 mL Eppendorf tube and concentrated using a vacuum centrifuge concentrator (Eppendorf 53050, Germany). 60 μL of methoxy solution was added and the reaction was carried out for 2 h at 37°C. Finally, 60 μL of BSTFA reagent (containing 1% trimethylchlorosilane) was added and reacted at 37°C for 90 min. The supernatant was removed by centrifugation at 12,000 rpm for 10 min at 4°C. The supernatant of the same sample was subsequently combined, and 60 μL of aqueous ribitol solution (internal standard, 0.2 mg/mL, purchased from Sigma-Aldrich, A5502) was added. In the extraction condition experiments, at least nine replicates were taken for each condition. Quality control (QC) samples were obtained by mixing 20 μL of each sample to be tested and were used to correct for bias in the analytical results of the mixed samples and for errors caused by the analytical instrument itself.
2.4 Gas Chromatography-Mass Spectrometry Analysis
The operation of gas chromatography-mass spectrometry (GC-MS) was referred to the method proposed by Jan et al. [24]. The procedure was as follows: the extracts were analyzed using 7890A-5975C GC-MS (Agilent, Palo Alto, USA) with an HP-5MS capillary column (5% phenyl/95% methylsiloxane 30 m × 250 μm inner diameter, 0.25 μm film thickness; Agilent J&W Scientific, Folsom, USA). Subsequently, 1 μL of sample was injected through the autosampler in a 20:1 split ratio mode. The injector, interface and ion source were maintained at temperatures of 280°C, 150°C and 230°C. A ramp-up procedure was used, holding the initial temperature at 60°C for 2 min, then increasing to 300°C at a rate of 10°C per minute and maintaining it at 300°C for 5 min. The carrier gas was helium at a flow rate of 1 mL/min. The compound entered the electron bombardment ion source (70 eV) and was converted to ions, which then entered the quadrupole mass analyzer. Data were collected in full scan mode with a mass range of 35–750 mass-to-charge ratio (m/z).
Agilent MSD Chemstation (version E.02.00.493) with the default parameters was used to translate the raw data to netCDF to obtain the baseline correction, peak alignment, peak detection, accurate masses, and normalized peak intensity [25]. Then the automatic mass spectrometry deconvolution recognition system (AMDIS) was used for deconvolution and peak acquisition. Subsequently, identification of metabolites was conducted by matching their retention times and mass fragment patterns with several databases, such as the National Institutes of Standards and Technology 8.0 (NIST, USA), the Human Metabolome Database (HMDB; http://www.hmdb.ca/).
Raw data through Pareto scaling, multivariate statistical analysis was conducted by using the ropls package of R language. The principal component analysis (PCA), partial least squares-discriminate analysis (PLS-DA) and orthogonal projections to latent structures discriminant analysis (OPLS-DA) were used to investigate the relationships among the samples. Next, the variable importance in projection (VIP) value was calculated to assess the contribution of each metabolite in B. emeiensis, by comparing the metabolite profiling of samples at the same time. The differential metabolites (biomarkers) were VIP values greater than 1 from each time point and P value < 0.05 [26]. Finally, differential metabolites between Q and W groups were mapped to their respective biochemical pathways by utilizing MetPA online tool (http://metpa.metabolomics.ca/) which was based on KEGG pathway. Through the hypergeometric test, only the metabolic pathway with log (-p) value greater than 1.301 (P < 0.05) was retained.
2.6 Integrated Analysis between DEGs and Biomarkers
The functional annotator KAAS (KEGG Automated Annotation Server) [27] was used to map transcript identifiers to KO numbers and thus assign transcripts to KEGG pathways (single bidirectional best hit approach using KAAS for gene representative sets; default blast score of 60). All biomarkers showing masses that are statistically significantly discriminatory between the treatment and control groups were included in the pathway mapping. MassTRIX reloaded for integrated analysis of transcriptomic and metabolomic data [28]. Log2 fold ratios of mapped transcripts and biomarkers are displayed as color codes on the KEGG pathway.
The relative abundance of each metabolite was log2 transformed before analysis to meet normality. Statistical analyses were performed using the SPSS 22.0 software package (IBM SPSS, Somers, NY, USA).
3.1 Identification and Expression Pattern Analysis of Metabolites
All the samples were analyzed by GC-MS with AMDIS software. A total of 206 peaks were detected and 75 substances were finally identified (Table S1). These metabolites are mainly amino acid, organic acid, polyol, sugar, fatty acid, and phosphoric acid (Fig. S1A). Among these classes, amino acids were the most prevalent (24 members constituting 32% of the metabolites), followed by organic acids (22 members constituting 29% of the metabolites) (Fig. S1A). To evaluate the metabolic pattern of the metabolites in the two groups, the relative value of the metabolites was taken as the metabolic level, and the clustering analysis was carried out. The results of the heat map showed that the metabolic level of the identified metabolites was significantly different between the two groups (Fig. S1B). Subsequently, by Pareto scaling of the metabolite data, PCA results show that the first two components (PC1 and PC2) accounted for 47.8% of the total variation in the PCA score plot of B. emeiensis samples (PC1%, 34%; PC2%, 13.8%). To better screen for differential metabolites between samples in the follow-up process, we used supervised pattern recognition methods (PLS-DA and OPLS-DA), where the PLS-DA score plot explained 42.4% of the variation and the permutation test plot indicated that the model was not overfitted. In addition, the results of the OPLS-DA score plot indicated a 24.6% between-group variation and a 16.8% within-group variation for the W and Q groups (Fig. S2). Also, we calculated the VIP values of metabolites and the results were integrated into Table 1.
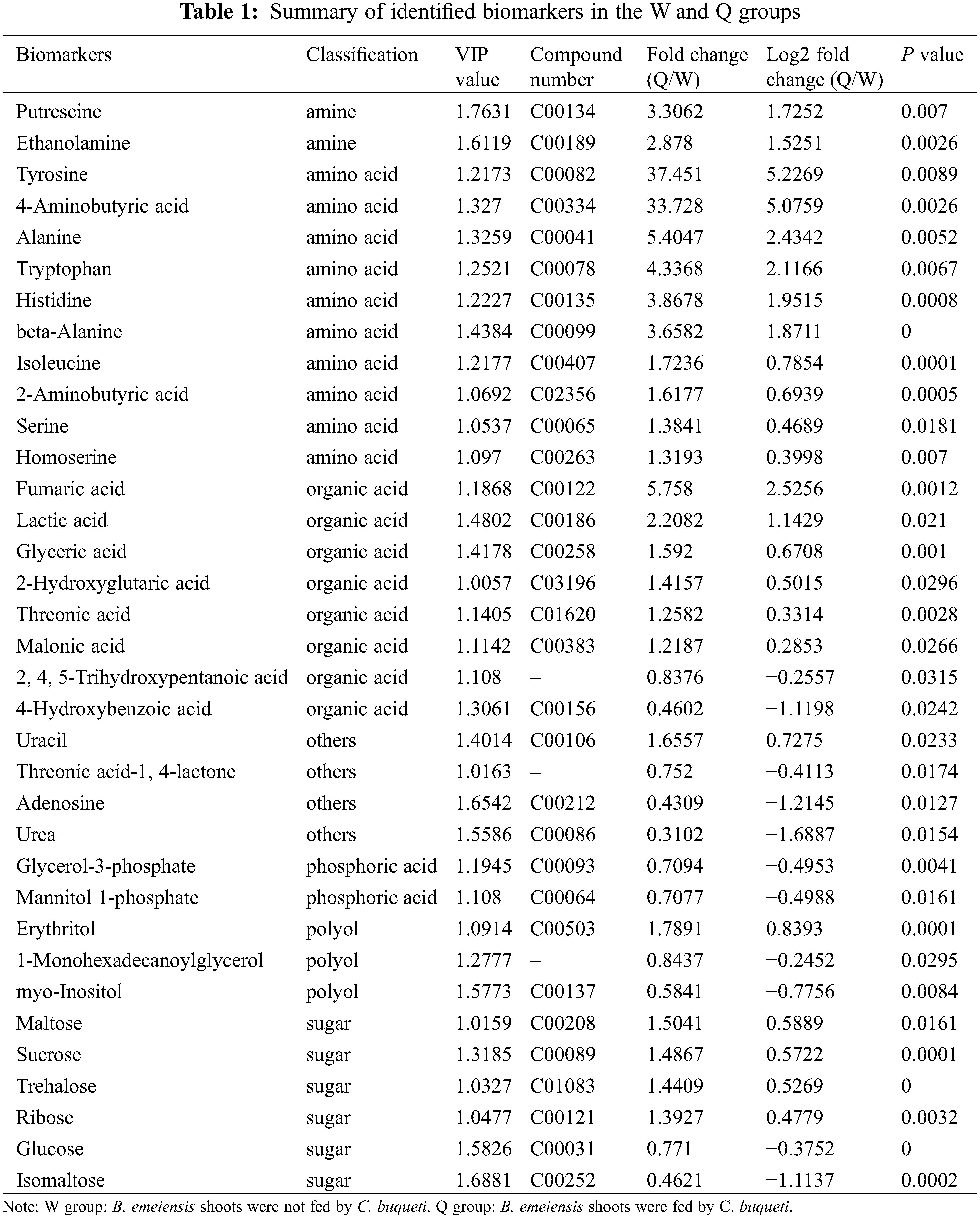
3.2 Screening of Differential Metabolites and Annotation of Metabolic Pathways
35 differential metabolites (biomarkers) between the two groups (Q and W) were screened from the 75 metabolites, and the detailed information was summarized in Table 1. We found that 11 biomarkers predominantly accumulated in the W group and 24 significantly accumulated in the Q group. The biomarkers were mainly classified into several major categories, such as amino acids and sugars. Among them, tyrosine exhibited the maximum differences with fold changes of 37.5 between the W group and Q group. We also investigated the metabolic pathways by using MetPA online tool, and the results showed that the 35 biomarkers were annotated into 71 KEGG pathways, including metabolic pathways, biosynthesis of secondary metabolites, and biosynthesis of amino acids (Table S2).
In this study, we used the transcriptome data of B. emeiensis sequenced in the previous study [16,20], from which 207 KEGG pathways, mainly involved with the biosynthesis of amino acids, starch and sucrose metabolism, carbon metabolism, and tyrosine and tryptophan biosynthesis, were retrieved (Table S3). In total, 324 differentially expressed genes (DEGs) were also retrieved among these pathways and the detailed DEGs information, including fold change values, length, annotations, and P values, were shown in Table S4.
3.4 Integrated Analysis of Metabolomic and Transcriptomic KEGG Pathways
All the possible metabolic pathways provided by KEGG were well annotated with the required catalytic enzymes at almost every metabolic step. We performed an integrated analysis between 71 metabolic pathways (35 biomarkers) and 207 gene expression pathways (324 DEGs). The results showed that only 47 pathways consisting of related DEGs and biomarkers were identified. Biosynthesis of amino acids (ko01230) contained the most biomarkers and DEGs among all pathways. The next were the starch and sucrose metabolism pathways (ko00500), associated with 13 DEGs and 5 biomarkers (Table 2).

3.5 Changes in the Expression Levels of Genes and Metabolites Content Involved in the Amino Acid Biosynthesis Pathway and the Starch and Sucrose Metabolism Pathway in B. emeiensis Shoots Infestion with C. buqueti
The relationship between changes in biomarker content and expression levels of DEGs can be visualized in the same KEGG pathway. Here, we remapped the tryptophan and tyrosine biosynthetic pathways (Table 2, Fig. 1A). We focused on the expression levels of genes associated with the tryptophan biosynthesis pathway in the transcriptome data, and were able to retrieve many DEGs associated with tryptophan synthesis from shikimate to tryptophan, essentially constituting a more complete tryptophan synthesis pathway (Fig. 1A). The expression levels of genes related to tryptophan synthesis in B. emeiensis were upregulated when B. emeiensis was fed by C. buqueti (Fig. 1A, Table S5). Also, combined with the results of our metabolomics data, we found that tryptophan content in B. emeiensis was significantly increased after feeding by C. buqueti (Table 1, Fig. 1B). In addition, tyrosine shares part of the pathway with tryptophan biosynthesis, and the metabolomic results showed that the content of tyrosine increased after feeding by C. buqueti, but the expression of related genes in the biosynthetic pathway of tyrosine increased (comp5435_seq3, comp17012_seq0) and decreased (comp3098_seq0); the study could not explain the phenomenon well (Figs. 1A, 1B). Similar to these were histidine, alanine and serine, which need to be explored in further studies.
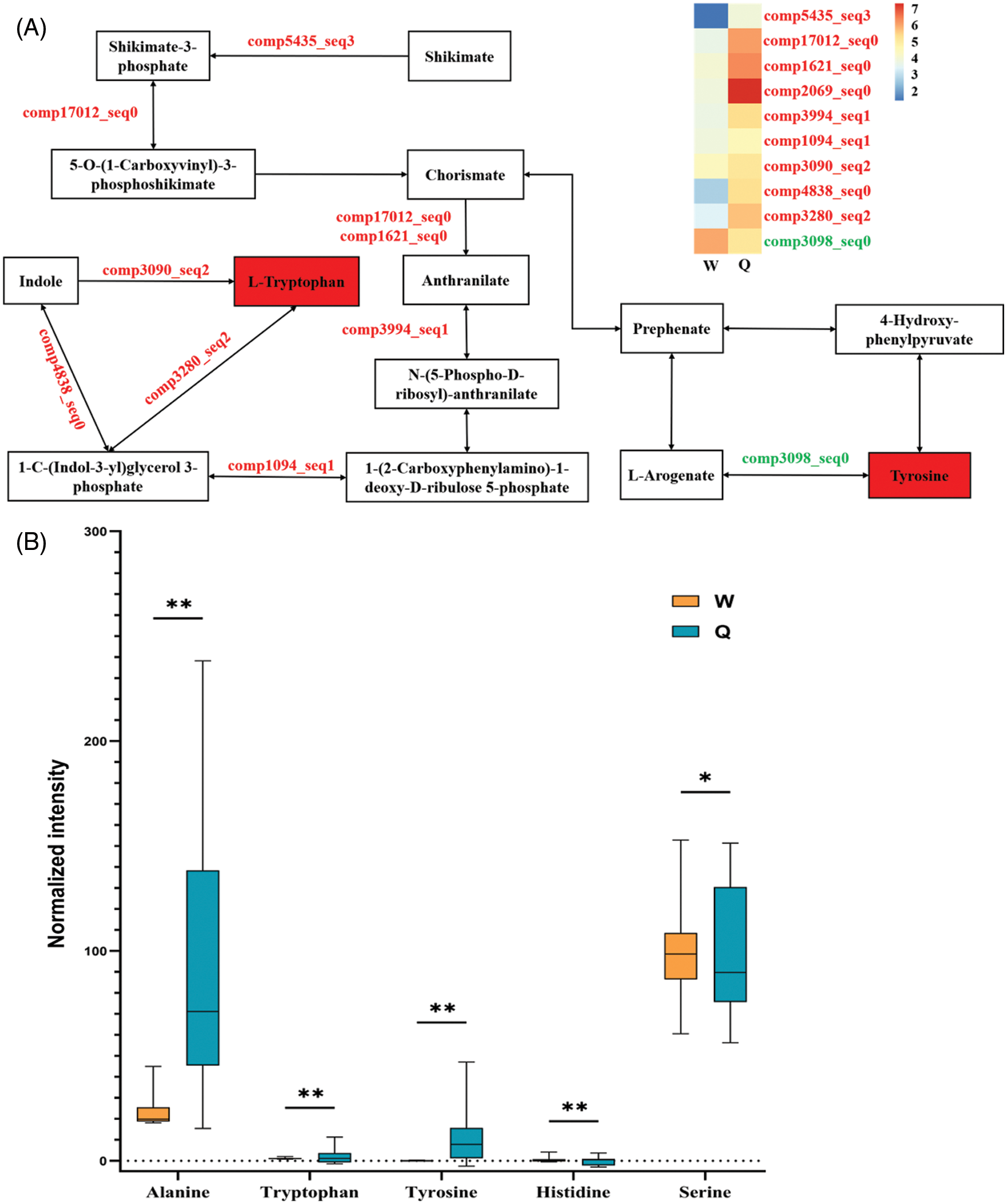
Figure 1: Herbivore-induced changes of genes expression levels and biomarkers content involved in biosynthesis of amino acids in B. emeiensis. (A) A sketch of the tryptophan and tyrosine synthesis pathways in this study and a heat map of the expression levels of the genes involved in this pathway. Metabolites filled with red boxes mean increased metabolite levels. Gene colors indicated in red mean upregulation at the corresponding step, while green is downregulation. The heat map ranges on a logarithmic scale from 0 to 7. The meanings of W and Q are as described in the text. (B) Metabolite normalized intensity in B. emeiensis in the W and Q groups. The X-axis represents the amin acid, while the Y-axis represents the amino acid normalized intensity. Orange and blue boxes represent metabolites in the control (W) and herbivore-treated (Q) groups, respectively. In the panel, only the values from the same amino acid were compared (The asterisks represent different statistical significance, *: P < 0.05, **: P < 0.01). All the values are shown as the mean ± SEM
As mentioned above, through the integration of KEGG annotation pathways of transcriptome and metabolome involved in this study, we sketched the metabolic pathway maps of starch and sucrose to preliminarily reflect the relationship between these metabolites (Table 2, Fig. 2A). This map was associated with 13 DEGS and 5 biomarkers. For metabolite content changes, sucrose, maltose and trehalose increased, while glucose and isomaltose decreased (Fig. 2B). In addition, from the perspective of the changes in gene expression levels, we found that partial genes related to trehalose synthesis (comp7674_seq1, comp9619_seq2 and comp12329_seq1) were up-regulated. However, most of the genes related to the biosynthesis of glucose (comp19053_seq5, comp2133_seq0, comp4551_seq0, etc.) were down-regulated (Fig. 2A, Table S5). The results of gene expression levels could further explain the reasons for the changes in trehalose and glucose contents.
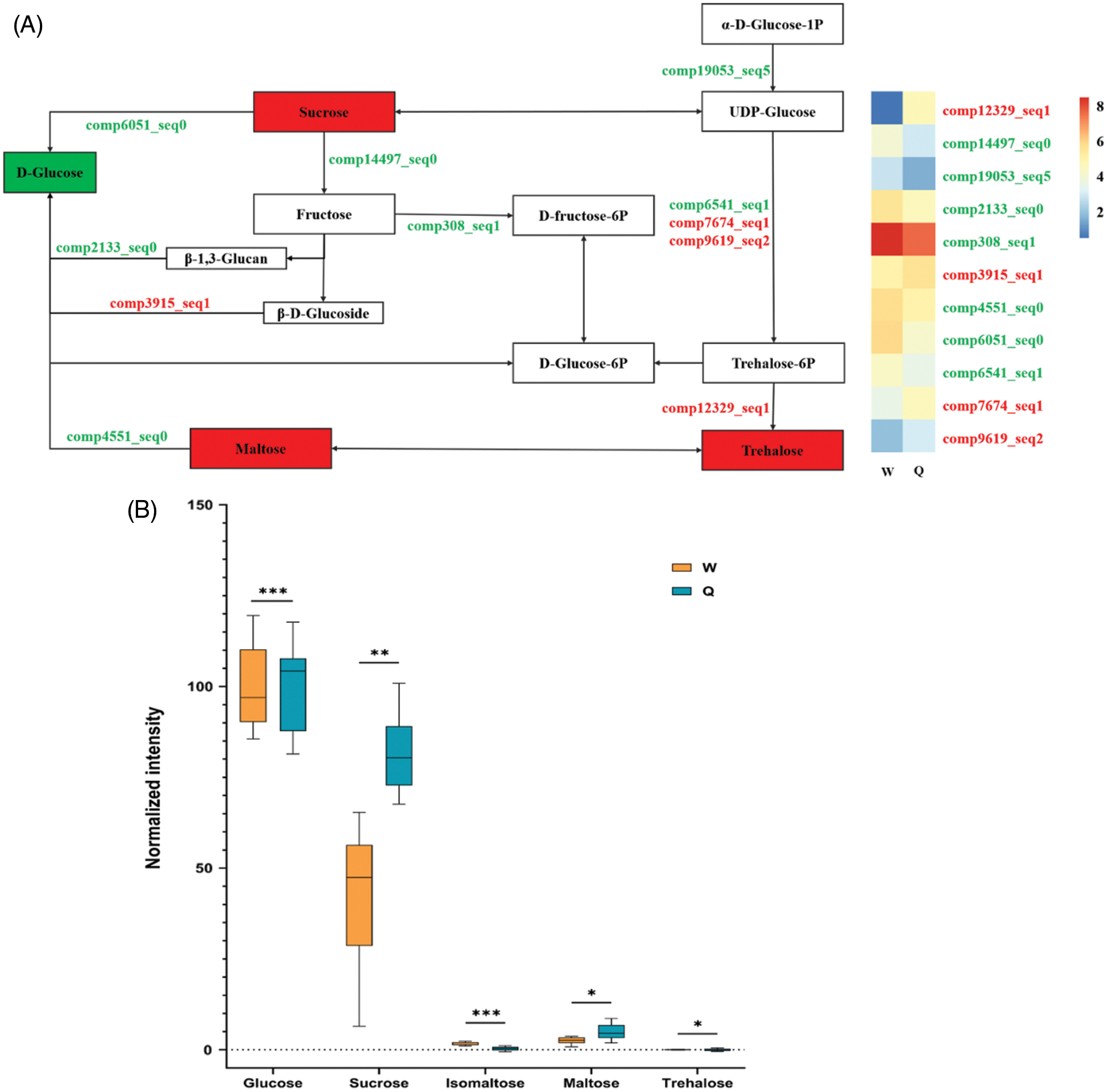
Figure 2: Herbivore-induced changes of gene expression levels and biomarkers content involved in starch and sucrose metabolism in B. emeiensis. (A) A sketch of the starch and sucrose metabolism pathways in this study and a heat map of the expression levels of the genes involved in this pathway. Metabolites filled in red imply an increase in the level of that metabolite and those filled in green indicate downregulation. Other annotations are as described above. (B) Metabolite normalized intensity in B. emeiensis samples. The X-axis represents the sugars, while the Y-axis represents the normalized intensity of the sugars. Orange and blue boxes represent metabolites in the control (W) and herbivore-treated (Q) groups, respectively. In the panel, only the values from the same sugar were compared (The asterisks represent different statistical significance, *: P < 0.05, **: P < 0.01, ***: P < 0.001). All the values are shown as the mean ± SEM
Amino acids are the main form of nitrogen in plants and are not only a growth constraint for insect herbivores but also a precursor to many defense-related plant metabolites, such as phenolics, alkaloids, and some phytohormones [29–32]. Therefore, the regulation of amino acid biosynthesis is an important regulatory mechanism for plant defense against herbivorous pests [33]. In our study, the content of amino acid metabolites in B. emeiensis under insect feeding conditions was generally higher than that in B. emeiensis when it was not fed, and tryptophan was the most representative compound (Fig. 1B). We analyzed the content change of tryptophan and the gene expression patterns in the shikimate pathway and the tryptophan biosynthesis pathway. Among the two associated pathways, chorismate was a key node. In the shikimate pathway, we found that the expression levels of shikimate kinase (comp5435_seq3) and 3-phosphoshikimate 1-carboxyvinyltransferase (comp17012_seq0) were up-regulated, suggesting that chorismate synthesis should be increased, although we did not detect this compound in metabolites. Since then, tryptophan biosynthesis had been initiated from the starting point of chorismate, and the catalytic anthranilate of chorismate should be a key step in the transformation, which is mainly dominated by anthranilate synthase. The genes encoding this enzyme (comp1621_seq0 and comp2069_seq0) were significantly upregulated. In addition, the gene expression levels of other enzymes encoding the synthesis of tryptophan were also up-regulated (Table S5, Fig. 1A). These results further explained the reason why the content of tryptophan in B. emeiensis increased after C. buqueti feeding (Table 1, Fig. 1B), consistent with the other research results [34]. We also found that the concentration of tyrosine increased (Fig. 1B), and Coley et al. [35] reported that when the concentration of tyrosine was 4%, the growth rate of larvae reduced by half, proving that tyrosine was also an integral part of herbivore defense.
As products of photosynthesis, carbohydrates are the main source of stored energy for host plants and insect herbivores. In our previous study, we found that C. buqueti had a large scale of carbohydrate-active enzyme genes and very high degradation efficiency for the lignocellulose of B. emeiensis, and the lignocellulose of B. emeiensis was mainly composed of lignin, cellulose and hemicellulose, the three components of which were mainly sugars and their derivatives [17,18]. This study further explored the role of starch and sucrose metabolism related to carbohydrate in B. emeiensis based on transcriptome and metabolome association analysis. After B. emeiensis was fed by C. buqueti, various sugars content in B. emeiensis were changed and the possible reason was that there existed transformation among these sugars (Table 1, Fig. 2A). We analyzed the expression patterns of relevant DEGs in this process. For example, glucose was an important compound and can be converted from sucrose, maltose and fructose. In terms of metabolic pathway, from sucrose to glucose, this process was catalyzed by sucrose: sucrose 1-fructosyltransferase (comp6051_seq0) and the gene expression of this enzyme was down-regulated. Besides, the expression of 4-α-glucanotransferase (comp4551_seq0), which was involved in the conversion of maltose to glucose, was also down-regulated. According to the other pathway, glucose could also be produced as glucose-6P, which was then converted to other sugars, and the glucose content of B. emeiensis decreased after C. buqueti feeding (Table S5, Figs. 2A, 2B).
Regarding trehalose, it has not been specifically reported in previous studies in relation to insect defense, in contrast to the large number of studies showing the involvement of trehalose accumulation in response to abiotic stresses (e.g., drought and salt stress) and in improving the efficiency of photosynthesis [36,37]. The slight increase in its content after C. buqueti feeding could be due to two reasons. One was the increase in sucrose content, which was converted to trehalose through the UDP-glucose and trehalose-6P pathways. At the same time, the expression of most catalytic enzymes in this pathway was up-regulated, leading to a gradual enrichment of trehalose. However, from another perspective, the content of UDP-glucose as an intermediate might not be high due to the down-regulation of a gene (comp19053_seq5) that regulates UDP-glucose synthesis, which indirectly leads to a decrease in trehalose conversion. In addition, the increase in maltose content could be explained by similar reasons as trehalose, while the increase in sucrose content could not be well explained according to the correlation analysis of this study (Table S5, Figs. 2B, 2C). However, according to the conclusion of Yoon et al. [38], among these soluble sugars, sucrose not only acts as a storage and protective molecule but also plays a central role in plant growth.
The involvement of carbohydrates in the construction of plant defense systems had also been observed in previous studies on plant responses to insect feeding stress. For example, Kang et al. [23] showed that Nilaparvata lugens significantly decreased glucose levels but increased maltose levels after the infestation of rice, in agreement with our results. In addition, they found that most of the sugar levels recovered after 96 h of feeding and noted that the physiological regulation of sugar metabolism in rice is extremely strong. Why is there a huge fluctuation of sugar in plants during insect feeding? Roitsch et al. [39] observed that leaf damage induced an increase in the intensity of carbon sinks by increasing the activity of invertase, which drove the influx of sucrose. Invertases and other enzymes catalyzing carbohydrate degradation were activated and the expression levels of encoded genes, such as α-glucosidase, β-fructofuranosidase and α-galactosidase, tend to be upregulated at the onset of insect damage [33], in slight contrast to our results (Table S5). In addition to directly enhancing insect resistance, rapid redistribution of primary metabolites in plants might also play a role in herbivore tolerance. Specifically, when herbivores remained in plants, plants decomposed primary metabolites in damaged tissues and transferred them to other organs for storage [40]. From a plant redevelopment perspective, this redistribution process prevented herbivores from taking up more resources in the tissues so that the plant could use them in future regrowth processes. Only after the end of the herbivore menace, resources are transferred from storage organs to growing tissues [34], which was also consistent with the recovery of most sugar levels in Nilaparvata lugens-fed rice since 96 h [23]. Another view was that the primary metabolites of plants were precursors of many secondary metabolites and that changes in their levels were related to the synthesis of secondary metabolites with signal transduction and pest resistance [41]. In addition, there was a view that differences in the level of metabolite variation were related to differences in herbivore species, plant species, infested tissues, time of infestation, and techniques used to detect gene expression [13,42].
In this work, we used transcriptome and metabolome integrated analysis to analyze the changes in B. emeiensis metabolite and gene expression levels in response to C. buqueti feeding stress. We first identified 35 biomarkers, which focused on amino acids and sugars, and better explained the changes in tryptophan’s content and gene expression levels during its involvement in resisting the insect, and preliminarily explored the process of tyrosine, glucose, and trehalose content changes. This study will further deepen our understanding of the mechanisms of bamboo resistance to insect feeding and provide some reference guidance for the subsequent prevention and control of bamboo diseases and insects.
Acknowledgement: We thank other members of the laboratory for suggestions and discussion regarding this work and revision of the manuscript.
Author Contribution Statement: H. T. planned and carried out the experiments, data collection, and wrote the manuscript with input from all authors. Y. Q. L. planned and carried out the experiments, performed analyses. C. B. L. conceived and designed the experiments, contributed to implementation and coordination of the research, and wrote the manuscript. All authors discussed the results and contributed to the final manuscript
Funding Statement: This work was supported by Sichuan Science and Technology Program (2019YFG0139).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Mlynarek, J. J., MacDonald, M., Sim, K., Hiltz, K., McDonald, M. R. et al. (2020). Oviposition, feeding preferences and distribution of Delia species (Diptera: Anthomyiidae) in Eastern Canadian onions. Insects, 11, 780. DOI 10.3390/insects11110780. [Google Scholar] [CrossRef]
2. Tlak Gajger, I., Dar, S. A. (2021). Plant allelochemicals as sources of insecticides. Insects, 12, 189. DOI 10.3390/insects12030189. [Google Scholar] [CrossRef]
3. Mitchell, C., Brennan, R. M., Graham, J., Karley, A. J. (2016). Plant defense against herbivorous pests: Exploiting resistance and tolerance traits for sustainable crop protection. Frontiers in Plant Science, 7, 1132. DOI 10.3389/fpls.2016.01132. [Google Scholar] [CrossRef]
4. War, A. R., Paulraj, M. G., Ahmad, T., Buhroo, A. A., Hussain, B. et al. (2012). Mechanisms of plant defense against insect herbivores. Plant Signaling & Behavior, 7, 1306–1320. DOI 10.4161/psb.21663. [Google Scholar] [CrossRef]
5. Li, Y., Lei, L., Luo, R., Li, C., Luo, C. B. (2020). Morphological structures of bamboo (Bambusa emeiensis) shoot shells and trichomes and functions in response to herbivory. Journal of Plant Growth Regulation, 40, 1400–1408. DOI 10.1007/s00344-020-10195-0. [Google Scholar] [CrossRef]
6. Hussain, M., Debnath, B., Qasim, M., Bamisile, B. S., Islam, W. et al. (2019). Role of saponins in plant defense against specialist herbivores. Molecules, 24, 2067. DOI 10.3390/molecules24112067. [Google Scholar] [CrossRef]
7. Singh, S., Kaur, I., Kariyat, R. (2021). The multifunctional roles of polyphenols in plant-herbivore interactions. International Journal of Molecular Sciences, 22, 1442. DOI 10.3390/ijms22031442. [Google Scholar] [CrossRef]
8. Kalske, A., Shiojiri, K., Uesugi, A., Sakata, Y., Morrell, K. et al. (2019). Insect herbivory selects for volatile-mediated plant-plant communication. Current Biology, 29, 3128–3133, e3. DOI 10.1016/j.cub.2019.08.011. [Google Scholar] [CrossRef]
9. Li, Y., Lei, L., Nong, X., Tang, H., Luo, C. B. (2020). Plant carbohydrate-active enzymes in bamboo Neosinocalamus affinis: Identification, classification and function in lignocellulose biosynthesis in herbivore defence. Nordic Journal of Botany, 38. DOI 10.1111/njb.02597. [Google Scholar] [CrossRef]
10. Pan, Y., Zhao, S., Wang, Z., Wang, X., Zhang, X. et al. (2020). Quantitative proteomics suggests changes in the carbohydrate metabolism of maize in response to larvae of the belowground herbivore Holotrichia parallela. PeerJ, 8, e9819. DOI 10.7717/peerj.9819. [Google Scholar] [CrossRef]
11. He, J., Bouwmeester, H. J., Dicke, M., Kappers, I. F. (2020). Transcriptional and metabolite analysis reveal a shift in direct and indirect defences in response to spider-mite infestation in cucumber (Cucumis sativus). Plant Molecular Biology, 103, 489–505. DOI 10.1007/s11103-020-01005-y. [Google Scholar] [CrossRef]
12. Kersten, B., Ghirardo, A., Schnitzler, J. P., Kanawati, B., Schmitt-Kopplin, P. et al. (2013). Integrated transcriptomics and metabolomics decipher differences in the resistance of pedunculate oak to the herbivore Tortrix viridana L. BMC Genomics, 14, 737. DOI 10.1186/1471-2164-14-737. [Google Scholar] [CrossRef]
13. Liu, Q., Wang, X., Tzin, V., Romeis, J., Peng, Y. et al. (2016). Combined transcriptome and metabolome analyses to understand the dynamic responses of rice plants to attack by the rice stem borer Chilo suppressalis (Lepidoptera: Crambidae). BMC Plant Biology, 16, 259. DOI 10.1186/s12870-016-0946-6. [Google Scholar] [CrossRef]
14. Wang, W., Zheng, C., Hao, W., Ma, C., Ma, J. et al. (2018). Transcriptome and metabolome analysis reveal candidate genes and biochemicals involved in tea geometrid defense in Camellia sinensis. PLoS One, 13, e0201670. DOI 10.1371/journal.pone.0201670. [Google Scholar] [CrossRef]
15. Chiu, Y. C., Juvik, J. A., Ku, K. M. (2018). Targeted metabolomic and transcriptomic analyses of “Red Russian” kale (Brassicae napus var. pabularia) following methyl jasmonate treatment and larval infestation by the cabbage looper (Trichoplusia ni Hübner). International Journal of Molecular Sciences, 19, 1058. DOI 10.3390/ijms19041058. [Google Scholar] [CrossRef]
16. Li, Y., Luo, C. B., Chen, Y., Xiao, X., Fu, C. et al. (2019). Transcriptome-based discovery of AP2/ERF tRanscription factors and expression profiles under herbivore stress conditions in bamboo (Bambusa emeiensis). Journal of Plant Biology, 62. 297–306. DOI 10.1007/s12374-019-0059-5. [Google Scholar] [CrossRef]
17. Luo, C., Li, Y., Liao, H., Yang, Y. (2018). De novo transcriptome assembly of the bamboo snout beetle Cyrtotrachelus buqueti reveals ability to degrade lignocellulose of bamboo feedstock. Biotechnology for Biofuels, 11, 292. DOI 10.1186/s13068-018-1291-9. [Google Scholar] [CrossRef]
18. Luo, C., Li, Y., Chen, Y., Fu, C., Nong, X. et al. (2019). Degradation of bamboo lignocellulose by bamboo snout beetle Cyrtotrachelus buqueti in vivo and vitro: Efficiency and mechanism. Biotechnology for Biofuels, 12, 75. DOI 10.1186/s13068-019-1406-y. [Google Scholar] [CrossRef]
19. Barah, P., Bones, A. M. (2015). Multidimensional approaches for studying plant defence against insects: From ecology to omics and synthetic biology. Journal of Experimental Botany, 66, 479–493. DOI 10.1093/jxb/eru489. [Google Scholar] [CrossRef]
20. Li, Y., Luo, C., Chen, Y., Fu, C., Yang, Y. (2020). Transcriptome-wide identification, classification, and characterization of NAC family genes in bamboo Bambusa emeiensis. Acta Physiologiae Plantarum, 42, 75. DOI 10.1007/s11738-020-03051-x. [Google Scholar] [CrossRef]
21. Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D. et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nature Protocols, 8(8), 1494–1512. DOI 10.1038/nprot.2013.084. [Google Scholar] [CrossRef]
22. Li, B., Dewey, C. N. (2011). RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics, 12. 323. DOI 10.1186/1471-2105-12-323. [Google Scholar] [CrossRef]
23. Kang, K., Yue, L., Xia, X., Liu, K., Zhang, W. (2019). Comparative metabolomics analysis of different resistant rice varieties in response to the brown planthopper Nilaparvata lugens hemiptera: delphacidae. Metabolomics, 15, 62. DOI 10.1007/s11306-019-1523-4. [Google Scholar] [CrossRef]
24. Jan, L., Nicolas, S., Joachim, K., Lothar, W., Fernie, A. R. (2006). Gas chromatography mass spectrometry-based metabolite profiling in plants. Nature Protocols, 1(1), 387–396. DOI 10.1038/nprot.2006.59. [Google Scholar] [CrossRef]
25. Smith, C. A., Want, E. J., O’maille, G., Abagyan, R., Siuzdak, G. (2006). XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Analytical Chemistry, 78, 779–787. DOI 10.1021/ac051437y. [Google Scholar] [CrossRef]
26. Kieffer, D. A., Piccolo, B. D., Vaziri, N. D., Liu, S., Lau, W. L. et al. (2016). Resistant starch alters gut microbiome and metabolomic profiles concurrent with amelioration of chronic kidney disease in rats. American Journal of Physiology-Renal Physiology, 310, F857–F887. DOI 10.1152/ajprenal.00513.2015. [Google Scholar] [CrossRef]
27. Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., Kanehisa, M. (2007). KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Research, 35(Web Server issue), W182–W185. DOI 10.1093/nar/gkm321. [Google Scholar] [CrossRef]
28. Wägele, B., Witting, M., Schmitt-Kopplin, P., Suhre, K. (2012). MassTRIX reloaded: Combined analysis and visualization of transcriptome and metabolome data. PLoS One, 7(7), e39860. DOI 10.1371/journal.pone.0039860. [Google Scholar] [CrossRef]
29. Maeda, H., Dudareva, N. (2012). The shikimate pathway and aromatic amino acid biosynthesis in plants. Annual Review of Plant Biology, 63, 73–105. DOI 10.1146/annurev-arplant-042811-105439. [Google Scholar] [CrossRef]
30. Schwachtje, J., Fischer, A., Erban, A., Kopka, J. (2018). Primed primary metabolism in systemic leaves: A functional systems analysis. Scientific Reports, 8, 216. DOI 10.1038/s41598-017-18397-5. [Google Scholar] [CrossRef]
31. Appel, H. M., Fescemyer, H., Ehlting, J., Weston, D., Rehrig, E. et al. (2014). Transcriptional responses of Arabidopsis thaliana to chewing and sucking insect herbivores. Frontier in Plant Science, 5, 565. DOI 10.3389/fpls.2014.00565. [Google Scholar] [CrossRef]
32. Jing, L., Robert, C. M., Michael, R., Marco, C., Laurent, M. S. et al. (2015). Induced jasmonate signaling leads to contrasting effects on root damage and herbivore performance. Plant Physiology, 167, 1100–1116. DOI 10.1104/pp.114.252700. [Google Scholar] [CrossRef]
33. Zhou, S., Lou, Y. R., Tzin, V., Jander, G. (2015). Alteration of plant primary metabolism in response to insect herbivory. Plant Physiology, 169, 1488–1498. DOI 10.1104/pp.15.01405. [Google Scholar] [CrossRef]
34. Steinbrenner, A. D., Gómez, S., Osorio, S., Fernie, A. R., Orians, C. M. (2011). Herbivore-induced changes in tomato (Solanum lycopersicum) primary metabolism: A whole plant perspective. Journal of Chemical Ecology, 37, 1294–1303. DOI 10.1007/s10886-011-0042-1. [Google Scholar] [CrossRef]
35. Coley, P. D., Endara, M., Ghabash, G., Kidner, C. A., Nicholls, J. A. et al. (2019). Macroevolutionary patterns in overexpression of tyrosine: An antiherbivore defence in a speciose tropical tree genus, Inga (Fabaceae). Journal of Ecology, 107, 1620–1632. DOI 10.1111/1365-2745.13208. [Google Scholar] [CrossRef]
36. Redillas, M. R., Park, S. H., Lee, J. W., Kim, Y. S., Jin, S. J. et al. (2011). Accumulation of trehalose increases soluble sugar contents in rice plants conferring tolerance to drought and salt stress. Plant Biotechnology Reports, 6, 89–96. DOI10.1007/s11816-011-0210-3. [Google Scholar] [CrossRef]
37. Garg, A. K., Kim, J. K., Owens, T. G., Ranwala, A. P., Choi, Y. D. et al. (2002). Trehalose accumulation in rice plants confers high tolerance levels to different abiotic stresses. Proceedings of the National Academy of Sciences of the United States of America, 99, 15898–15903. DOI 10.1073/pnas.252637799. [Google Scholar] [CrossRef]
38. Yoon, J., Cho, L. H., Tun, W., Jeon, J. S., An, G. (2021). Sucrose signaling in higher plants. Plant Science, 302, 110703. DOI 10.1016/j.plantsci.2020.110703. [Google Scholar] [CrossRef]
39. Roitsch, T., González, M. C. (2004). Function and regulation of plant invertases: Sweet sensations. Trends in Plant Science, 9, 606–613. DOI 10.1016/j.tplants.2004.10.009. [Google Scholar] [CrossRef]
40. Orians, C. M., Thorn, A., Gómez, S. (2011). Herbivore-induced resource sequestration in plants: Why bother? Oecologia, 167, 1–9. DOI 10.1007/s00442-011-1968-2. [Google Scholar] [CrossRef]
41. Schwachtje, J., Baldwin, I. T. (2008). Why does herbivore attack reconfigure primary metabolism? Plant Physiology, 146, 845–851. DOI 10.1104/pp.107.112490. [Google Scholar] [CrossRef]
42. Yasur, J., Rani, K. U. (2009). Effects of herbivore feeding on biochemical and nutrient profile of castor bean, Ricinus communis L. plants. Allelopathy Journal, 24, 131–142. DOI 10.2134/agronj2008.0180x. [Google Scholar] [CrossRef]
Supplementary material
Figure S1: All metabolites detected and annotated by GC-MS. A. A classification of all metabolites B. A hierarchical clustering heat map of all metabolites. The heatmap scale ranges from −3 to +3 on a log2 scale. The meanings of W and Q are described above
Figure S2: PCA, PLS-DA and OPLS-DA score plot of W and Q group samples
Table S1: All metabolites detected by GC-MS
Table S2: KEGG pathways with biomarkers identified in metabolome
Table S3: KEGG pathway associated with differentially expressed genes (DEGs) in the transcriptome
Table S4: DEGs in transcriptome enriched by metabonomic integration analysis
Table S5: Analysis of DEGs associated with amino acid or glucose metabolic pathways
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |