| Phyton-International Journal of Experimental Botany |
DOI: 10.32604/phyton.2022.017644
ARTICLE
Global Lysine Acetylome Analysis of Flower Bud Development in Catalpa bungei
1State Key Laboratory of Tree Genetics and Breeding, Key Laboratory of Tree Breeding and Cultivation of State Forestry and Grassland Administration, Research Institute of Forestry, Chinese Academy of Forestry, Beijing, 100091, China
2Key Laboratory of Horticulture Science for Southern Mountains Regions of Ministry of Education, College of Horticulture and Landscape Architecture, Academy of Agricultural Sciences, Southwest University, Chongqing, 400715, China
3State Key Laboratory of Tree Genetics and Breeding, Northeast Forestry University, Harbin, 150040, China
*Corresponding Authors: Shougong Zhang. Email: shougong.zhang@caf.ac.cn; Junhui Wang. Email: wangjh@caf.ac.cn
Received: 26 May 2021; Accepted: 09 July 2021
Abstract: Lysine acetylation, a reversible and dynamic post-translational modification, plays pivotal roles in regulating many biological processes in plants. However, the information of lysine acetylation during flower development of woody angiosperm remains unclear. Herein, we identified a total of 667 lysine acetylation sites in 467 proteins in flower buds of Catalpa Bungei. The acetylated proteins were mainly involved in the pathways of carbon metabolism, glycometabolism and oxidative phosphorylation. Using functional enrichment analysis, 61 and 13 acetylated proteins were involved in the glycometabolism and oxidative phosphorylation pathways, respectively. This suggests that lysine acetylated proteins play critical roles in the energy metabolism pathways. Accordingly, the protein–protein interactions were also highly enriched in the carbon metabolic pathway. Integrative analysis of lysine acetylome and transcriptome showed that transcript level changes of seven key lysine-acetylated proteins/genes involved in energy metabolism were significant different during floral transition. Importantly, compared with the normal flowering variety, the expression levels of phosphoribulokinase, phosphoenolpyruvate carboxykinase, ATP synthase and pyruvate dehydrogenase complex genes were significantly up-regulated at the stages of vegetative and transition buds during early flowering of C. bungei. These data represent the first lysine acetylome in the C. bungei flowers, and can serve as an essential basis for exploring the function of lysine acetylated proteins in the flower bud development of plants.
Keywords: Lysine acetylome; Catalpa bungei; flower development; energy metabolism
Abbreviations
| PTMs | Post-translational modifications |
| LC-MS/MS | liquid chromatography-tandem mass spectrometry |
| ppm | parts per million |
| GPI | glucose-6-phosphate isomerase |
| FBP | fructose-1,6-bisphosphatase I |
| pfp | pyrophosphate-fructose-6-phosphate |
| ALDO | fructose-bisphosphate aldolase |
| talA/B | transaldolase |
| PGLS | 6-phosphogluconolactonase |
| PGD | 6-phosphogluconate dehydrogenase |
| RPE | ribulose-phosphate 3-epimerase |
| rpiA | ribose 5-phosphate isomerase A |
| ACO | aconitate hydratase |
| IDH | isocitrate dehydrogenase |
| DLD | dihydrolipoamide dehydrogenase |
| DLST | dihydrolipoamide succinyltransferase |
| LSC | succinyl-CoA synthetase |
| SDH | succinate dehydrogenase |
| DTT | dithiothreitol |
| FA | formic acid |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| FPKM | Fragments Per Kilobase of exon model per Million mapped fragments. |
Post-translational modifications (PTMs) play important roles in modulating diverse protein properties including localization, activity, protein-protein interaction, and protein-nucleic acid interaction [1–3]. To date, more than 630 PTMs have been identified in the Uniprot database. Among them, lysine acetylation, which extensively occurs on the ε-amino groups of lysine residues in both histones and non-histone proteins, is a reversible, dynamic and ubiquitous PTMs [4–6]. Since the first identification in histone proteins [7], lysine acetylation has been extensively investigated in gene expression regulation and metabolic stability [8–10]. In recent years, proteome-wide analysis of lysine acetylation has been present in humans [4], mouse [11], eukaryotes [12,13] and plants [14–19]. These analyses of lysine acetylome revealed that acetylated proteins regulate various cellular processes, such as enzymatic activities, stress responses and metabolic pathways.
The first lysine acetylome on plants was performed in Arabidopsis by Wu et al. [14] and Finkemeier et al. [19]. More recently, lysine acetylome of strawberry leaves indicated important regulatory roles of the acetylated proteins in photosynthesis [18]. Acetylome analysis of grape mesocarp and exocarp revealed that the acetylated proteins are involved in plant defense [15]. Proteomic analysis of lysine-acetylated proteins in rice seeds demonstrated that the proteins are mainly involved in regulating central metabolisms, especially for the citrate cycle [20]. The lysine acetylome study of rice seedlings showed that the acetylated proteins are mainly associated with the regulation of the glyoxylate and dicarboxylate metabolisms, carbon metabolism, and photosynthesis pathways [21]. However, lysine acetylome in flower buds of woody angiosperms remains unclear.
Catalpa bungei, which belongs to the family Bignoniaceae, is not only cultivated broadly for garden landscaping, but also provides ideal materials of flower development for woody angiosperms [22]. Here we used the analyses of anti-acetyllysine-based enrichment and high-sensitive mass spectrometry to investigate the lysine acetylation proteome in the C. bungei flower buds. These identified acetylated proteins are mainly involved in energy metabolism, including the pentose phosphate pathway, glycometabolism, citrate cycle and oxidative phosphorylation pathways. Importantly, transcript level changes of seven key lysine-acetylated proteins/genes showed significant differences at the floral transition stage. With this global proteomic analysis, we revealed important energy metabolism-related functions of lysine acetylated proteins in flower buds.
2.1 Plant Materials and Growth Conditions
Flower buds of 5, 10, 15 and 20 mm length were collected (Figs. 1A and 1B), respectively, from the double-flower C. bungei trees growing in a natural secondary forest in Xiangyang city, Hubei Province, China. These samples were frozen immediately in liquid nitrogen, and stored at −80°C until used.

Figure 1: Morphological observation of C. bungei flowers and global identification of lysine acetylation sites. (A) an inflorescence of C. bungei. (B) full bloom. (C) SDS-PAGE of the proteins. (D) blotting signals and 3-D view of western blotting of the acetylated proteins. The signals of 11 kD and 30–55 kD bands were enhanced for anti-acetyllysine antibody detection at 5 mm flower bud (black arrow). (E) homologous analysis of acetylated proteins in C. bungei, F. ananassa, O. sativa and P. asperata
2.2 Protein Extraction and Western Blotting
The buds were ground in liquid nitrogen. Then, the obtained powder was suspended in extraction buffer containing 8 M urea, 10 mM dithiothreitol (DTT), 50 mM nicotinamide, 3 μM trichostatin A and 0.1% Protease Inhibitor Cocktail Set VI (Calbiochem, Darmstadt, Germany). This was followed by sonication three times on ice using a high intensity ultrasonic processor (scientz-IID, SCIENTZ GmbH, Ningbo, China). After centrifugation at 20,000 g at 4°C for 10 min, the remaining debris was removed. The supernatant containing protein was precipitated with cold 15% trichloroacetic acid at −20°C for 2 h. The precipitated protein was centrifuged again for 10 min at 4°C at 20,000 g, and the pellet was washed three times with cold acetone. The pellet, which was concentrated in a SpeedVac concentrator (Thermo Fisher Scientific, Inc., Waltham, MA, USA), was re-dissolved in buffer (8 M urea, 100 mM (NH4)2CO3, pH 8.0) and sonicated for 5 min. The protein concentration was measured using a 2-D Quant kit (GE Healthcare) following the manufacturer’s instructions.
For western blot, protein was diluted with SDS loading buffer, and 20 μg protein of each sample was separated by 12% SDS-PAGE (Fig. 1C), and transferred to a PVDF (Millipore) membrane. The blot was probed using a pan anti-acetyllysine antibody (PTM Biolabs, Hangzhou, China) in a 1:1000 dilution. Secondary anti-horseradish peroxidase antibody (HuaAn Biotechnology, Hangzhou, China) was used in a 1:10,000 dilution. Plot density of western blot was analyzed using software of Quantity One (Bio-Rad, USA).
Protein sample (∼10 mg) of 5 mm flower bud was reduced using 10 mM DTT at 37°C for 1 h, and then alkylated with 20 mM iodoacetamide in darkness at room temperature for 45 min. To ensure that the urea concentration was less than 2 M, the treated protein was subsequently diluted by adding 100 mM (NH4)2CO3. The re-suspended protein was finally digested with trypsin/P (Promega) (Product code V5111, Promega Corp., Madison, WI, USA) as described previously [23]. Briefly, trypsin was added at a 1:50 mass ratio of trypsin-to-protein for the first digestion at 37°C overnight. Then, a 1:100 trypsin-to-protein mass ratio was used, and 4 h of digestion was conducted at 37°C. After digestion, the peptides were lyophilized using the SpeedVac.
2.4 Acetylated Peptides Enrichment
Lyophilized peptides were firstly re-dissolved in NETN buffer (100 mM NaCl, 1 mM EDTA, 50 mM Tris-HCl, 0.5% NP-40, pH 8.0), and then incubated overnight at 4°C using pre-washed pan-anti-acetyllysine agarose beads (Catalog No. PTM-104, PTM Biolabs, Hangzhou, China). After incubation with gentle shaking, the agarose beads were washed by centrifugation four times with NETN buffer and twice with ddH2O. Then, 0.1% trifluoroacetic acid was used to elute the peptides. The peptides were concentrated again using the SpeedVac concentrator. The collected peptides were finally cleaned by C18 ZipTips (Millipore Corp, billerica, MA, USA).
2.5 High-Resolution LC-MS/MS Analysis
Enriched peptides were re-dissolved in mobile phase A [0.1% formic acid (FA) in 2% acetonitrile (ACN)] and directly loaded onto a reversed-phase pre-column (Acclaim PepMap 100, Thermo Scientific). The peptides were separated via a reversed-phase analytical column (Acclaim PepMap RSLC, Thermo Scientific). A linear gradient of 3% to 22% mobile phase B (0.1% FA in 98% ACN) was applied for 26 min, and followed by 22% to 35% for 8 min and a climb to 80% in 3 min, and then a hold at 80% for the eventual 3 min, all at a fixed flow rate of 300 nl/min in an EASY-nLC 1000 UPLC system (Thermo Fisher Scientific Inc., USA). The obtained peptides were then subjected to a nanospray ion source, followed by MS/MS in Q ExactiveTM hybrid quadrupole-Orbitrap mass spectrometer (ThermoFisher Scientific, Inc, USA.) coupled online to the UPLC. Intact peptides and ion fragments were detected in the Orbitrap at a resolution of 70,000 and 17,500 (m/z 200), respectively. Additionally, peptides were selected for MS/MS using normalized collisional energy setting as 28. A data-dependent procedure that alternated between one MS scan followed by 20 MS/MS scans was used for the top 20 precursor ions above a threshold ion count of 5E3 in the MS survey scan with 30.0 s dynamic exclusion. The electrospray voltage was set at 2.0 kV. Automatic gain control was used to prevent overfilling of the Orbitrap, and 5E4 ions were accumulated to generate the MS/MS spectra. For MS scans, the range of m/z scans was from 350 to 1800.
The resulting MS/MS data were processed by using MaxQuant software with an integrated Andromeda search engine (version 1.5.1.8). Tandem mass spectra were searched against the Catalpa bungei transcriptome database concatenated with a reverse decoy database [24]. Trypsin/P was specified as cleavage enzyme and the search allowed up to four missing cleavages, five charges and five modifications per peptide. The 10 ppm and 0.02 Da of mass error were set for precursor ions and fragment ions, respectively. Carbamidomethylation on cysteine was defined as a fixed modification; the oxidation on methionine, acetylation on lysine, and acetylation at the protein N-terminus were defined as variable modifications. The false discovery rate (FDR) threshold for protein, peptide and modification site were set as 1%. The minimum peptide length was specified at 7 amino acids. Default values were set in other parameters of the MaxQuant: the Min and Max peptide length for unspecific search were 7 and 25, respectively. The top MS/MS peaks per 100 Da was 12, MS/MS deisotoping was set as true. The site localization probability was set at more than 0.75.
Homologs of acetylated proteins were analyzed in Catalpa bungei, Fragaria ananassa [18], Oryza sativa [21] and Picea asperata [23]. Gene Ontology (GO) annotation proteome information was derived from the UniProt-GOA database (https://www.ebi.ac.uk/GOA/). Lysine acetylated protein IDs were converted to UniProt ID and mapped to GO IDs. Then, the remaining lysine acetylation substrates, which were not annotated by UniProt-GOA database, were further annotated GO function by the InterProScan soft based on protein sequence alignment. The identified acetylated proteins were classified by GO terms based on three categories: biological process, cellular component, and molecular function. For each category, a two-tailed Fisher’s exact test was used to test the enrichment of the identified protein against all database proteins. The GO with a corrected P-value < 0.05 was considered significant. Kyoto Encyclopedia of Genes and Genomes (KEGG) [25], domain description, and subcellular localization prediction were performed through Web-based interfaces and services, along with the bioinformatics tools, including KAAS, KEGG mapper, InterProScan, and WoLF PSORT (PSORT/PSORT II). Secondary structures were predicted using NetSurfP. The annotation categories showing P-values < 0.05 were considered to be significantly enriched [26]. The filtered P-value matrix was transformed by the function x = −log10 (P-value). The results were visualized using a “ggplot2” R project (http://cran.r-project.org/web/packages/ggplot2/).
All acetylated protein name identifiers were searched against the STRING database version 9.1 for protein-protein interactions and blast to Solanum lycopersicum species. Interaction networks from STRING database were visualized in Cytoscape software [27,28]. The lysine acetylated proteins of C. bungei flower buds and transcriptome of buds from early flowering (EF) and normal flowering (NF) were analysed. Then, the relative expression levels for differentially expressed proteins were calculated based on the FPKM [29].
3.1 Identification of Lysine Acetylated Proteins in C. bungei Flower
To acquire an initial overview of the extent of lysine acetylation during flower bud development, we detected the lysine acetylated proteins using western blotting analysis. Multiple major protein bands with a molecular weight higher than histones (10–30 kD) were successfully detected (Fig. 1D). Compared with the flower buds of 10, 15 and 20 mm in length, the signals of 11 kD and 30–55 kD bands were enhanced for anti-acetyllysine antibody detection at 5 mm flower buds (Fig. 1D and Tab. S1). Therefore, the flower buds of 5 mm were chosen to profile the acetylated proteins.
To determine the lysine acetylome, we used anti-acetyllysine-based enrichment and high-resolution liquid chromatography-tandem mass spectrometry (LC-MS/MS) to identify acetylated proteins and their modification sites. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://www.proteomexchange.org/) via the PRIDE partner repository with the dataset identifier PXD005121. For the enriched acetylated peptides, the distributions of mass errors were investigated and all of them were less than 5 parts per million (ppm), showing the high accuracy of the MS data which fits the requirement (Fig. S2A). The lengths of the identified peptides were distributed between 7 and 30 amino acids, which were consistent with the property of tryptic peptides (Fig. S2B). In total, 667 lysine acetylation sites in 467 proteins were identified (Tab. S2).
Comparative proteomic analysis of Catalpa bungei, Fragaria ananassa, Oryza sativa and Picea asperata was analysed to investigate the homology and specificity of acetylated proteins (Fig. 1E). We identified 266, 238, and 252 overlapped proteins in C. bungei vs. F. ananassa, C. bungei vs. O. sativa, and C. bungei vs. P. asperata, respectively.
3.2 Distribution Analysis of Lysine Acetylation Sites
To elucidate the distribution of acetylation sites in the identified proteins, we calculated the degree of lysine acetylation. A survey on the 667 acetylation sites in 467 proteins demonstrated that the average number of acetylation was 1.4 sites per protein with the level ranging from 1 to 8 (Fig. 2A). Among these acetylated proteins, 75% contained only one acetylation site; the remaining 25% were modified on multiple lysine residues.

Figure 2: Properties of acetylated sites. (A) acetylated site distribution in the acetylated proteins. (B) probabilities of localization on different protein secondary structures (alpha helix, beta strand and coil)
To explore the relationship between lysine acetylation and protein structure, we performed local secondary structural analysis of all identified proteins. The results demonstrated that 56% of the acetylation sites were detected in regions with ordered secondary structures of proteins (Fig. 2B). Among them, 45% were distributed in alpha-helix and 11% were located in beta-strand; the remaining 44% of the acetylation sites were located at regions with the disordered structures.
3.3 Functional Characterization of Lysine Acetylated Proteins
To acquire a better understanding of the acetylated proteins identified in the flower buds, GO classification analysis was performed. The biological process analysis indicated that the acetylated proteins were involved in metabolic, cellular, and single-organism processes, accounting for 37%, 29% and 23% of all the identified acetylated proteins, respectively (Fig. 3A). For the molecular functions ontology, acetylated proteins were mainly associated with catalytic activity, binding, and structural-molecule activity (Fig. 3B). Consistently, in the cellular components classification, the acetylated proteins mainly focused on cell (39%), macromolecular complex (25%), organelle membrane (24%), and membrane (11%) (Fig. 3C). The GO classification analysis revealed that the proteins mainly involved in various metabolic processes and catalytic activities could be acetylated at lysine sites.
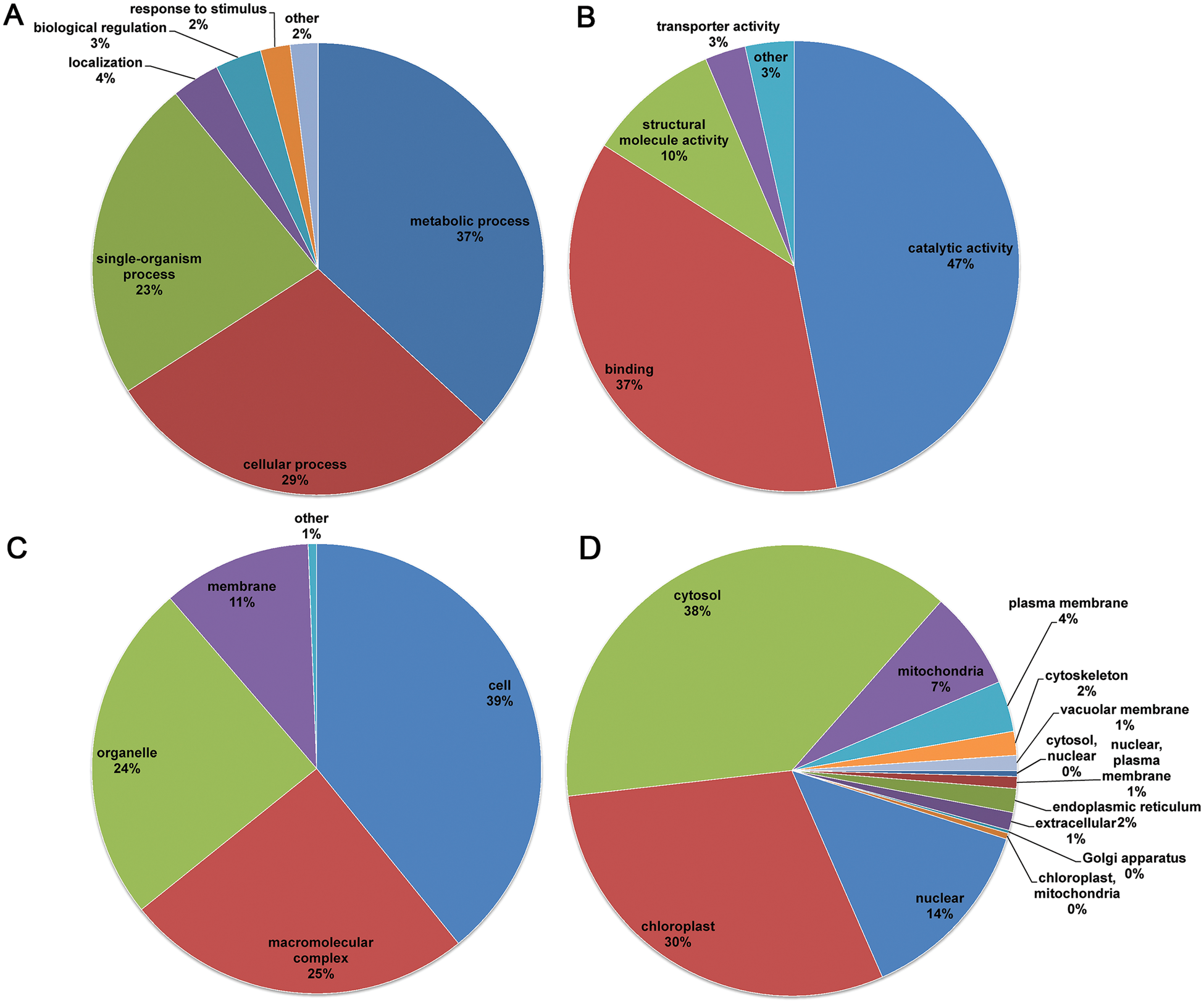
Figure 3: Distribution of the acetylated proteins in the C. bungei flower. (A) Biological process classifications. (B) Molecular function classification. (C) Cellular component classification. (D) Subcellular location predictions
Subcellular localization of the acetylated proteins was performed, and the results showed that a larger proportion of identified acetylated proteins were distributed in the cytosol (38%), indicating that acetylated proteins play important roles in this compartment in flower buds (Fig. 3D). Meanwhile, a number of acetylated proteins were also localized in the chloroplast (30%), nucleus (14%) and mitochondria (7%). Furthermore, some proteins were distributed in other components such as the plasma membrane (4%), cytoskeleton (2%) and endoplasmic reticulum (2%).
To reveal the preferential functional terms of lysine acetylation, the enrichment analysis of all the acetylated proteins was performed. The top significantly enriched GO terms and KEGG pathways were shown in Fig. 4. In the ontology of biological process, many metabolic and biosynthetic processes had a higher tendency to be acetylated (Fig. S3). Consistently, proteins, which were involved in structural molecule activity, structural constituent of ribosome, oxidoreductase activity and cofactor binding, were more likely to be acetylated based on the molecular function analysis. The cellular components of the proteins were mainly classified into cytoplasm, cell, cell part, and intracellular part. Meanwhile, we found that carbon metabolism, carbon fixation in photosynthetic organisms, glycolysis/gluconeogenesis, citrate cycle, pentose phosphate pathway, pyruvate metabolism, fructose and mannose metabolism, and oxidative phosphorylation pathways related terms were significantly enriched on the basis of KEGG pathway enrichment analysis (Fig. 4).

Figure 4: KEGG pathways of acetylated proteins
3.4 Clustering Analysis of Functional Enrichment
To investigate the preferential target substrates of lysine acetylation, we conducted functional enrichment on the basis of clustering analysis of subcellular locations. In KEGG pathway enrichment, the acetylated proteins were mainly focused on the pentose phosphate pathway, photosynthesis, and the fructose and mannose metabolisms in the chloroplast. The proteins were mainly classified into aminoacyl-tRNA biosynthesis in the cytosol. The proteins were significantly enriched in the citrate cycle, oxidative phosphorylation, and carbon metablism in the mitochondria (Fig. S4). Meanwhile, on the basis of biological process category of GO enrichment analysis, the proteins were mainly classified into photosynthesis in the chloroplast. The main proteins were involved in gene expression, macromolecule biosynthetic process, and nucleic acid metabolic process in the cytosol. The proteins were mainly enriched in generation of precursor metabolites and energy, electron transport chain, and oxidation-reduction processes in the mitochondria (Fig. S5). The KEGG pathway enrichment of acetylated proteins corresponded well to the biological process of GO enrichment analysis.
Accordingly, the cellular components of GO enrichment analysis were involved in photosystem I, photosystem, and photosynthetic membrane in the chloroplast. The main cellular components were classified into ribosome in the cytosol. The respiratory chain, oxidoreductase complex, and respiratory chain complex III were also significantly enriched in the cellular component category in the mitochondria (Fig. S6). Furthermore, on the basis of the molecular function category of GO enrichment analysis, iron ion binding, organic acid binding, and pyridoxal phosphate binding were mainly enriched in the chloroplast. The RNA bingding and aminoacyl-tRNA ligase activity were significantly enriched in the cytosol. The transmembrane transporter activity, oxidoreductase activity, antioxidant activity and ion channel activity were mainly enriched in the mitochondria (Fig. S7). Consistently, the major domains of the pyridoxal phosphate-dependent transferase, chlorophyll a/b binding protein, translation protein, ribosomal protein, aminoacyl-tRNA synthetase and ATPase were significantly enriched based on the classification of protein domain (Fig. S8).
3.5 Acetylated Proteins Involved in the Glycometabolism and Oxidative Phosphorylation Pathways
The pathway enrichment analysis of acetylated proteins revealed that lysine acetylation may play important roles in the energy-metabolism pathway. Consistent with this hypothesis, we found that 61 and 13 acetylated proteins were associated with the glycometabolism and oxidative phosphorylation pathways (Tab. S2), respectively. As shown in Fig. 5a wide array of enzymes in glycometabolism were detected as being acetylated.

Figure 5: Representative significantly enriched glycometabolism pathways of the acetylated proteins. The characters in red show acetylated enzymes
In higher plants, the oxidation of carbohydrates involves enzymatic conversion to glucose-6-P, which is further metabolized via the oxidative pentose phosphate or glycolysis pathway [30,31]. In the pentose phosphate pathway, various enzymes were shown to be acetylated. They included the glucose-6-phosphate isomerase (GPI), fructose-1,6-bisphosphatase I (FBP), pyrophosphate-fructose-6-phosphate (pfp), fructose-bisphosphate aldolase (ALDO), transaldolase (talA/B), 6-phosphogluconolactonase (PGLS), 6-phosphogluconate dehydrogenase (PGD), ribulose-phosphate 3-epimerase (RPE), and ribose 5-phosphate isomerase A (rpiA). In addition to the enzymes in the pentose phosphate pathway, many enzymes involved in glycolysis were identified to be acetylated.
The citrate cycle, a series of chemical reactions to generate energy, provides precursors of certain amino acids and the reducing agent NADH that are needed in many important biochemical reactions [32]. In this study, the proteins involved in the citrate cycle, such as the aconitate hydratase (ACO), isocitrate dehydrogenase (IDH), dihydrolipoamide dehydrogenase (DLD), dihydrolipoamide succinyltransferase (DLST), succinyl-CoA synthetase (LSC), and succinate dehydrogenase (SDH), were identified to be acetylated in the flower buds.
The oxidative phosphorylation system is the process in which ATP is synthesized as a result of transfer of electrons from NADH and FADH2 to oxygen [33]. Our data demonstrated that 33 of the acetylated proteins were localized in the mitochondria in the flower buds. Among them, 13 were involved in the oxidative phosphorylation pathway, including the NADH dehydrogenase, succinate dehydrogenase, ubiquinol-cytochrome c reductase complex protein, cytochrome b-c1 complex, and ATPase (Tab. S3 and Fig. S9).
3.6 Interaction Network Analysis of Acetylated Proteins
To better understand the cross-linked pathways of these acetylated proteins, the protein-protein interaction networks were established. In the whole network, a total of 255 acetylated proteins were predicted to be involved in 18 clusters of protein-protein interaction networks, presenting functional diversity of acetylated proteins in the flower development (Tab. S4). We extracted the top three highly interconnected clusters (Fig. 6). The top clusters I, II and III comprised 43, 18 and 13 proteins were associated with ribosome, carbon metabolism and proteasome. Meanwhile, these carbon-metabolism associated proteins involved in the processes of pentose phosphate, glycolysis/gluconeogenesis, fructose and mannose metabolism, and carbon fixation (Table 1).

Figure 6: Protein-protein interaction network of lysine acetylated proteins

3.7 Transcript Level Changes in Important Lysine-Acetylated Proteins of the Energy Metabolism
We further analyzed transcript level changes of important lysine-acetylated proteins in C. bungei buds from early flowering (EF) and normal flowering (NF) varieties at vegetative buds (Vb), transition buds (Tb) and reproductive buds (Rb). We identified 66, 73 and 70 overlapped proteins/genes in EF-Vb vs. NF-Vb, EF-Tb vs. NF-Tb, and EF-Rb vs. NF-Rb (Tab. S5 and Fig. 7). The expression level changes of some lysine-acetylated proteins, such as the phosphoribulokinase, phosphoenolpyruvate carboxykinase, ATP synthase, ribulose bisphosphate carboxylase large chain, pyruvate dehydrogenase complex, H(+)-transporting ATP synthase and fructokinase-1, showed different expression trends in the EF and NF buds (Fig. 7). The expression levels of the phosphoribulokinase, phosphoenolpyruvate carboxykinase, ATP synthase and pyruvate dehydrogenase complex were up-regulated at EF-Vb and EF-Tb of early flowering C. bungei (Fig. 7D and Table 2).

Figure 7: The overlapped proteins and expression levels of some lysine-acetylated proteins in C. bungei buds from early flowering (EF) and normal flowering (NF). (A) The overlapped proteins in EF-Vb vs. NF-Vb. (B) The overlapped proteins in EF-Tb vs. NF-Tb. (C) The overlapped proteins in EF-Rb vs. NF-Rb. (D) The expression levels of some lysine-acetylated proteins. KAc: lysine-acetylated proteins. Vb: vegetative buds. Tb: transition buds. Rb: reproductive buds

4.1 Global Proteomic Analysis of Lysine-Acetylated Proteins in C. bungei Flower Buds
Lysine acetylation is emerging as a dynamic, reversible and highly conserved post-translation modification in both eukaryotes and prokaryotes with multiple biological functions [34]. However, the lysine acetylome has been reported only in leaves [17,18], seeds [20,35] and seedlings [16] of a limited number of plants. In this study, we determined the lysine acetylome of C. bungei flower buds. Compared to lysine acetylome in leaves of F. ananassa, seeds of O. sativa and somatic embryos of P. asperata, the acetylome of the C. bungei flower has a large quantity of unique acetylated proteins, revealing an indispensable role of lysine acetylation in flower development (Fig. 1C). Meanwhile, the acetylated proteins were mainly associated with carbon metabolism, glycometabolism and oxidative phosphorylation pathways. The protein–protein interactions were also highly enriched in the process of carbon metabolism to confirm this hypothesis. Meanwhile, transcript level changes of some lysine-acetylated proteins indicated that these proteins were associated with floral transition. These results reveal that the lysine acetylated proteins play important roles in many regulatory pathways during flower development.
4.2 Lysine Acetylated Proteins Involved in Glycometabolism
In plants, glycometabolism is one of the most important metabolic processes [36,37]. Lysine acetylation of glycometabolism related proteins plays important roles in plant life activities such as plant development and growth [18,20]. In our work, 212 acetylated proteins were localized in the cytosol and the mitochondria, and many of them were involved in glycometabolism including glycolysis/gluconeogenesis, citrate cycle, pentose phosphate pathway, pyruvate metabolism, and fructose and mannose metabolisms. Similarly, acetylated proteins of three glycometabolism related pathways, which is citrate cycle, glycolysis/gluconeogenesis, and pyruvate metabolism, were significantly enriched in strawberry leaves [18]. It was also reported that the enzymes of the citrate cycle were identified to contain lysine acetylation sites in pea seedlings [16]. Meanwhile, Zhang et al. [17] found that the acetylated proteins were involved in the pentose phosphate pathway and glycolysis/gluconeogenesis in common wheat leaves .
4.3 Lysine Acetylated Proteins Involved in Oxidative Phosphorylation
As the primary source of metabolic energy, oxidative phosphorylation is a highly efficient way to form ATP using energy derived from the transfer of electron donors to electron acceptors, and takes place inside the mitochondria in higher plants [38]. Notably, 13 acetylated proteins were significantly enriched in the oxidative phosphorylation pathway in our study. Similarly, several researches have shown that the acetylated proteins of the oxidative phosphorylation pathway are significantly enriched in pea seedlings [16], rice embryos [20], and HL60 cells [39]. These studies may facilitate the revealing of the mechanism of the lysine acetylation regulated oxidative phosphorylation pathway in both plants and human beings.
4.4 Transcript Level Changes of the Acetylated Proteins Involved in Energy Metabolism
The protein-protein interaction networks of the acetylated proteins that were mainly enriched in the pathways of the ribosome and carbon metabolism, might be associated with protein synthesis and energy metabolism. Previously, the ribosome is associated with the efficiency of protein synthesis at the codon level in plants [40]. At the floral anthesis stage of Eriobotrya japonica, the ribosome pathway is significantly enriched [41]. Similarly, carbon metabolism is significantly enriched at the flowering stage in Lonicera japonica [42].
The expression level of some lysine-acetylated proteins, which are involved in energy metabolism such as phosphoribulokinase, phosphoenolpyruvate carboxykinase, ATP synthase, ribulose bisphosphate carboxylase large chain, pyruvate dehydrogenase complex, H(+)-transporting ATP synthase and fructokinase-1, were significantly differentially expressed in the flower buds. Previously, as an important photosynthetic enzyme, phosphoribulokinase is involved in the carbon and energy metabolism by reversible oxidation/reduction [43]. Phosphoenolpyruvate carboxykinase catalyzes the conversion of some amino acids and lipids to sugars by gluconeogenesis, and its activity was significantly higher in the floral buds than that in the leaves in plants [44]. Meanwhile, ATP synthase and ribulose bisphosphate carboxylase large chain and pyruvate dehydrogenase complex are the key and important factors in energy conversion, and thus, they are important for plant growth [45–48]. Furthermore, fructokinase plays key roles in the carbohydrate metabolism by catalyzing fructose phosphorylation, and fructokinase-1 participates in regulating flower development [49,50]. Taken together, the expression level analysis suggests that the lysine acetylation on energy metabolism-related proteins plays important roles in flower bud development.
In the study, we provided the most extensive lysine acetylome in C. bungei flower buds to date. These results reveal that, in flower buds, lysine acetylation might play critical roles in energy metabolism including glycometabolism and oxidative phosphorylation pathway. These data not only expand our knowledge of the physiological role of lysine acetylation, but also provide bioinformatic resources for the functional analysis of lysine acetylation in regulating flower development on C. bungei and in all plants.
Acknowledgement: The experimental materials were provided by the National Catalpa bungei Germplasm Bank.
Availability of Data and Materials: Processed data included within Additional file Tables S1–S4 and Figs. S1–S8.
Author Contributions: D.J. performed the research and drafted the manuscript; D.J., N.W. and N.L. contributed to data analysis; G.Q., G.L. and W.M. advised on experimental design; S.Z. and J.W. provided plant tissue, laboratory facilities, and project supervision.
Ethics Approval and Consent to Participate: Not applicable.
Funding Statement: This work was financially supported by the Open Fund of State Key Laboratory of Tree Genetics and Breeding (Chinese Academy of Forestry) (No. TGB2019005) and Fundamental Research Funds for the Central Universities (XDJK2020B058). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflicts of Interest: The authors declare that they have no conflict of interest.
1. Hart, G. W., Ball, L. E. (2013). Post-translational modifications: A major focus for the future of proteomics. Molecular & Cellular Proteomics, 12(12), 3443. DOI 10.1074/mcp.E113.036491. [Google Scholar] [CrossRef]
2. Witze, E. S., Old, W. M., Resing, K. A., Ahn, N. G. (2007). Mapping protein post-translational modifications with mass spectrometry. Nature Methods, 4(10), 798–806. DOI 10.1038/nmeth1100. [Google Scholar] [CrossRef]
3. Walsh, C. T., Garneau-Tsodikova, S., Gatto, G. J. (2005). Protein posttranslational modifications: The chemistry of proteome diversifications. Angewandte Chemie International Edition, 44(45), 7342–7372. DOI 10.1002/anie.200501023. [Google Scholar] [CrossRef]
4. Choudhary, C., Kumar, C., Gnad, F., Nielsen, M. L., Rehman, M. et al. (2009). Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science, 325(5942), 834–840. DOI 10.1126/science.1175371. [Google Scholar] [CrossRef]
5. Liu, F., Yang, M., Wang, X., Yang, S., Gu, J. et al. (2014). Acetylome analysis reveals diverse functions of lysine acetylation in Mycobacterium tuberculosis. Molecular & Cellular Proteomics, 13(12), 3352–3366. DOI 10.1074/mcp.M114.041962. [Google Scholar] [CrossRef]
6. Kim, S. C., Sprung, R., Chen, Y., Xu, Y., Ball, H. et al. (2006). Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Molecular Cell, 23(4), 607–618. DOI 10.1016/j.molcel.2006.06.026. [Google Scholar] [CrossRef]
7. Phillips, D. M. (1963). The presence of acetyl groups of histones. Biochemical Journal, 87(2), 258–263. DOI 10.1042/bj0870258. [Google Scholar] [CrossRef]
8. Sterner, D. E., Berger, S. L. (2000). Acetylation of histones and transcription-related factors. Microbiology & Molecular Biology Reviews, 64(2), 435–459. DOI 10.1128/MMBR.64.2.435-459.2000. [Google Scholar] [CrossRef]
9. MacDonald, V. E., Howe, L. J. (2009). Histone acetylation: Where to go and how to get there. Epigenetics, 4(3), 139–143. DOI 10.4161/epi.4.3.8484. [Google Scholar] [CrossRef]
10. Oliver, D. J., Nikolau, B. J., Wurtele, E. S. (2009). Acetyl-coA-life at the metabolic nexus. Plant Science, 176(5), 597–601. DOI 10.1016/j.plantsci.2009.02.005. [Google Scholar] [CrossRef]
11. Fritz, K. S., Galligan, J. J., Hirschey, M. D., Verdin, E., Petersen, D. R. (2012). Mitochondrial acetylomic analysis in a mouse model of alcohol-induced liver injury utilizing SIRT3 knockout mice. Free Radical Biology & Medicine, 51, S19–S20. DOI 10.1016/j.freeradbiomed.2011.10.044. [Google Scholar] [CrossRef]
12. Duffy, S. K., Friesen, H., Baryshnikova, A., Lambert, J. P., Chong, Y. T. et al. (2012). Exploring the yeast acetylome using functional genomics. Cell, 149(4), 936–948. DOI 10.1016/j.cell.2012.02.064. [Google Scholar] [CrossRef]
13. Henriksen, P., Wagner, S. A., Weinert, B. T., Sharma, S., Bacinskaja, G. et al. (2012). Proteome-wide analysis of lysine acetylation suggests its broad regulatory scope in saccharomyces cerevisiae. Molecular & Cellular Proteomics, 11 (11), 1510–1522. DOI 10.1074/mcp.M112.017251. [Google Scholar] [CrossRef]
14. Wu, X., Oh, M. H., Schwarz, E. M., Larue, C. T., Sivaguru, M. et al. (2011). Lysine acetylation is a widespread protein modification for diverse proteins in arabidopsis. Plant Physiology, 155(4), 1769–1778. DOI 10.1104/pp.110.165852. [Google Scholar] [CrossRef]
15. Melo-Braga, M. N., Verano-Braga, T., León, I. R., Antonacci, D., Nogueira, F. C. et al. (2012). Modulation of protein phosphorylation, N-glycosylation and lys-acetylation in grape (Vitis vinifera) mesocarp and exocarp owing to Lobesia botrana infection. Molecular & Cellular Proteomics, 11(10), 945–956. DOI 10.1074/mcp.M112.020214. [Google Scholar] [CrossRef]
16. Smith-Hammond, C. L., Hoyos, E., Miernyk, J. A. (2014). The pea seedling mitochondrial Nε-lysine acetylome. Mitochondrion, 19, 154–165. DOI 10.1016/j.mito.2014.04.012. [Google Scholar] [CrossRef]
17. Zhang, Y., Song, L., Liang, W., Mu, P., Wang, S. et al. (2016). Comprehensive profiling of lysine acetylproteome analysis reveals diverse functions of lysine acetylation in common wheat. Scientific Reports, 6, 21069. DOI 10.1038/srep21069. [Google Scholar] [CrossRef]
18. Fang, X., Chen, W., Zhao, Y., Ruan, S., Zhang, H. et al. (2015). Global analysis of lysine acetylation in strawberry leaves. Frontiers in Plant Science, 6, 739. DOI 10.3389/fpls.2015.00739. [Google Scholar] [CrossRef]
19. Finkemeier, I., Laxa, M., Miguet, L., Howden, A. J., Sweetlove, L. J. (2011). Proteins of diverse function and subcellular location are lysine acetylated in arabidopsis. Plant Physiology, 155(4), 1779–1790. DOI 10.1104/pp.110.171595. [Google Scholar] [CrossRef]
20. He, D., Wang, Q., Li, M., Damaris, R. N., Yi, X. et al. (2016). Global proteome analyses of lysine acetylation and succinylation reveal the widespread involvement of both modification in metabolism in the embryo of germinating rice seed. Journal of Proteome Research, 15(3), 879–890. DOI 10.1021/acs.jproteome.5b00805. [Google Scholar] [CrossRef]
21. Xiong, Y., Peng, X., Cheng, Z., Liu, W., Wang, G. L. (2016). A comprehensive catalog of the lysine-acetylation targets in rice (Oryza sativa) based on proteomic analyses. Journal of Proteomics, 138, 20–29. DOI 10.1016/j.jprot.2016.01.019. [Google Scholar] [CrossRef]
22. Jing, D., Xia, Y., Chen, F., Wang, Z., Zhang, S. et al. (2015). Ectopic expression of a Catalpa bungei (Bignoniaceae) PISTILLATA homologue rescues the petal and stamen identities in Arabidopsis pi-1 mutant. Plant Science, 231, 40–51. DOI 10.1016/j.plantsci.2014.11.004. [Google Scholar] [CrossRef]
23. Xia, Y., Jing, D., Kong, L., Zhang, J., Ouyang, F. et al. (2016). Global lysine acetylome analysis of desiccated somatic embryos of picea asperata. Frontiers in Plant Science, 7, 1927. DOI 10.3389/fpls.2016.01927. [Google Scholar] [CrossRef]
24. Wang, Z., Zhu, T., Ma, W., Wang, N., Qu, G. et al. (2018). Genome-wide analysis of long non-coding RNAs in Catalpa bungei and their potential function in floral transition using high-throughput sequencing. BMC Genet, 19(1), 86. DOI 10.1186/s12863-018-0671-2. [Google Scholar] [CrossRef]
25. Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., Tanabe, M. (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Research, 44(D1), D457. DOI 10.1093/nar/gkv1070. [Google Scholar] [CrossRef]
26. Huang, D. W., Sherman, B. T., Lempicki, R. A. (2009). Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Research, 37(1), 1–13. DOI 10.1093/nar/gkn923. [Google Scholar] [CrossRef]
27. Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T. et al. (2003). Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Research, 13(11), 2498–2504. DOI 10.1101/gr.1239303. [Google Scholar] [CrossRef]
28. Szklarczyk, D., Franceschini, A., Kuhn, M., Simonovic, M., Roth, A. et al. (2011). The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Research, 39( suppl_1), D561–D568. DOI 10.1093/nar/gkq973. [Google Scholar] [CrossRef]
29. Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., Wold, B. (2018). Mapping and quantifying mammalian transcriptomes by RNA-Seq.Nature Methods, 5(7621–628. DOI 10.1038/nmeth.1226. [Google Scholar] [CrossRef]
30. Purvis, A. C., Fites, R. C. (1979). Changes in enzyme levels of EMP and pentose phosphate pathways during germination of cottonseed. Botanical Gazette, 140(2), 121–126. DOI 10.1086/337066. [Google Scholar] [CrossRef]
31. Rumpho, M. E., Kennedy, R. A. (1983). Activity of the pentose phosphate and glycolytic pathways during anaerobic germination of Echinochloa crus-galli (barnyard grass) seeds. Journal of Experimental Botany, 34(144), 893–902. DOI 10.1093/jxb/34.7.893. [Google Scholar] [CrossRef]
32. Popova, T. N., de Carvalho, M. Â. P. (1998). Citrate and isocitrate in plant metabolism. Biochimica et Biophysica Acta, 1364(3), 307–325. DOI 10.1016/s0005-2728(98)00008-5. [Google Scholar] [CrossRef]
33. Kushnareva, Y., Newmeyer, D. D. (2010). Bioenergetics and cell death. Annals of the New York Academy of Sciences, 1201(1), 50–57. DOI 10.1111/j.1749-6632.2010.05633.x. [Google Scholar] [CrossRef]
34. Huang, D., Li, Z. H., You, D., Zhou, Y., Ye, B. C. (2015). Lysine acetylproteome analysis suggests its roles in primary and secondary metabolism in Saccharopolyspora erythraea. Applied Microbiology and Biotechnology, 99(3), 1399–1413. DOI 10.1007/s00253-014-6144-2. [Google Scholar] [CrossRef]
35. Smith-Hammond, C. L., Swatek, K. N., Johnston, M. L., Thelen, J. J., Miernyk, J. A. (2014). Initial description of the developing soybean seed protein Lys-n(ε)-acetylome. Journal of Proteomics, 96, 56–66. DOI 10.1016/j.jprot.2013.10.038. [Google Scholar] [CrossRef]
36. Plaxton, W. C. (1996). The organization and regulation of plant glycolysis. Annual Review of Plant Physiology and Plant Molecular Biology, 47(1), 185–214. DOI 10.1146/annurev.arplant.47.1.185. [Google Scholar] [CrossRef]
37. Hu, D., Luo, W., Fan, L. F., Liu, F. L., Gu, J. et al. (2016). Dynamics and regulation of glycolysis-tricarboxylic acid metabolism in the midgut of Spodoptera litura during metamorphosis. Insect Molecular Biology, 25(2), 153–162. DOI 10.1111/imb.12208. [Google Scholar] [CrossRef]
38. Van Dingenen, J., Blomme, J., Gonzalez, N., Inzé, D.(2016). Plants grow with a little help from their organelle friends. Journal of Experimental Botany, 67(22), 6267–6281. DOI 10.1093/jxb/erw399. [Google Scholar] [CrossRef]
39. Zhu, X., Liu, X., Cheng, Z., Zhu, J., Xu, L. et al. (2015). Quantitative analysis of global proteome and lysine acetylome reveal the differential impacts of VPA and SAHA on HL60 cells. Scientific Reports, 6, 19926. DOI 10.1038/srep19926. [Google Scholar] [CrossRef]
40. Fujita, T., Kurihara, Y., Iwasaki, S. (2019). The plant translatome surveyed by ribosome profiling. Plant and Cell Physiology, 60(9), 1917–1926. DOI 10.1093/pcp/pcz059. [Google Scholar] [CrossRef]
41. Jing, D., Chen, W., Hu, R., Zhang, Y., Xia, Y. et al. (2020). An integrative analysis of transcriptome, proteome and hormones reveals key differentially expressed genes and metabolic pathways involved in flower development in loquat. International Journal of Molecular Sciences, 21 (14), 5107. DOI 10.3390/ijms21145107. [Google Scholar] [CrossRef]
42. Yang, B., Zhong, Z., Wang, T., Ou, Y., Tian, J. et al. (2019). Integrative omics of lonicera japonica thunb. flower development unravels molecular changes regulating secondary metabolites. Journal of Proteomics, 208, 103470. DOI 10.1016/j.jprot.2019.103470. [Google Scholar] [CrossRef]
43. Porter, M. A., Stringer, C. D., Hartman, F. C. (1998). Characterization of the regulatory thioredoxin site of phosphoribulokinase. Journal of Biological Chemistry, 263(1), 123–129. DOI 10.1016/S0021-9258(19)57366-5. [Google Scholar] [CrossRef]
44. Malone, S., Chen, Z., Bahrami, A. R., Walker, R. P., Gray, J. E. et al. (2007). Phospho enol pyruvate carboxykinase in arabidopsis: Changes in gene expression, protein and activity during vegetative and reproductive development. Plant and Cell Physiology, 48(3), 441–450. DOI 10.1093/pcp/pcm014. [Google Scholar] [CrossRef]
45. Sun, J., Hu, H., Li, Y., Wang, L., Zhou, Q. et al. (2016). Effects and mechanism of acid rain on plant chloroplast ATP synthase. Environmental Science and Pollution Research, 23(18), 18296–18306. DOI 10.1007/s11356-016-7016-3. [Google Scholar] [CrossRef]
46. Kartashov, I. M., Opanasenko, V. K., Malyan, A. N. (2015). Effects of medium viscosity increasing agents on ATP synthesis in chloroplast thylakoids. Biofizika, 60(3), 481–486. [Google Scholar]
47. Spreitzer, R. J., Thow, G., Zhu, G. (1995). Pseudoreversion substitution at large-subunit residue 54 influences the CO2/O2 specificity of chloroplast ribulose-bisphosphate carboxylase/oxygenase. Plant Physiology, 109(2), 681–685. DOI 10.1104/pp.109.2.681. [Google Scholar] [CrossRef]
48. Turk, H., Genisel, M. (2019). Melatonin-related mitochondrial respiration responses are associated with growth promotion and cold tolerance in plants. Cryobiology, 92, 76–85. DOI 10.1016/j.cryobiol.2019.11.006. [Google Scholar] [CrossRef]
49. Yao, Y., Geng, M., Wu, X., Sun, C., Wang, Y. et al. (2017). Identification, expression, and functional analysis of the fructokinase gene family in cassava. International Journal of Molecular Sciences, 18(11), 2398. DOI 10.3390/ijms18112398. [Google Scholar] [CrossRef]
50. Odanaka, S., Bennett, A. B., Kanayama, Y. (2002). Distinct physiological roles of fructokinase isozymes revealed by gene-specific suppression of Frk1 and Frk2 expression in tomato. Plant Physiology, 129(3), 1119–1126. DOI 10.1104/pp.000703. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |