International Journal of
Experimental Botany
| Phyton- International Journal of Experimental Botany |
DOI: 10.32604/phyton.2021.015229
ARTICLE
Exploration and Validation of the Potential Downstream Genes Underlying ipa1-2D Locus for Rice Panicle Branching
1Joint International Research Laboratory of Agriculture and Agri-Product Safety of the Ministry of Education, Jiangsu Key Laboratory of Crop Genomics and Molecular Breeding, Yangzhou University, Yangzhou, 225009, China
2Jiangsu Co-Innovation Center for Modern Production Technology of Grain Crops, Jiangsu Key Laboratory of Crop Genetics and Physiology, Yangzhou University, Yangzhou, 225009, China
*Corresponding Authors: Lin Zhang. Email: zhangl@yzu.edu.cn; Qiaoquan Liu. Email: qqliu@yzu.edu.cn
Received: 02 December 2020; Accepted: 07 January 2021
Abstract: In recent years, some super hybrid rice varieties were bred with strong culms and large panicles, which are mainly contributed by the ipa1-2D locus. A gain-of-function allele of OsSPL14 is the ipa1-2D and it can greatly increase the panicle primary branch number. However, the key downstream genes mediating this trait variation are not fully explored. In this study, we developed high-quality near-isogenic lines (NILs) with a difference of only 30 kb chromosomal segment covering the ipa1-2D locus. Using the NILs, we explored the impact of ipa1-2D on five sequential stages of early inflorescence development, and found that the locus can greatly enhance the initiation of primary branch meristems. A transcriptomic analysis was performed to unveil the downstream molecular network of ipa1-2D, and 87 genes were found differentially expressed, many of which are involved in metabolism and catalysis processes. In addition, transgenic lines of overexpression and RNA interference were generated to shape different levels of OsSPL14. They were also used to validate the expression variation explored by transcriptome. Based on the gene annotation, twelve potential downstream targets of ipa1-2D were selected, and their expression variation was confirmed by qRT-PCR analysis both in NILs and transgenic lines. This research expands the molecular network underlying ipa1-2D and provides novel gene information which might be involved in the control of panicle branching. We discussed the potential function of identified genes and highlighted their values for future function exploration and breeding application.
Keywords: Rice; ipa1-2D; panicle branching; inflorescence meristems; gene expression
Rice is the major cereal crop that feeds about one half of the world population, and it is important to develop rice varieties with high yield potential to fulfill the demands of increasing populations. Rice yield is contributed mainly by three components, namely, panicle number, grain number per panicle and grain weight [1]. In recent years, some super hybrid rice varieties have been bred in China, and a super rice called Yongyou 12 presented a yield potential of more than 14.5 ton per hectare [2]. For these super rice varieties, grain number contributes largely to their yield potential. The higher grain number of these varieties is determined by the large panicle morphology with more panicle branches, either primary or secondary branches. In the two-line hybrid rice Liang-you-pei 9, its grain number was far more than that of its two parents, and so did the panicle branch number. This indicated that these traits are involved in heterosis [3]. It is therefore of great importance to dissect the developmental process and molecular mechanism of panicle branch formation in rice.
The morphogenesis of rice panicle branches is largely determined at the early stages of inflorescence meristems (IMs), and shoot apical meristem (SAM), primary branch meristem (PBM), secondary branch meristem (SBM), spikelet meristem (SM) and floret meristem (FM) are initiated in a sequential manner [4]. Up to now, several key genes involved in the initiation or maintenance of IMs in rice have been cloned. LAX1 encodes a basic helix-loop-helix transcription factor, and lax1 mutant exhibited a great reduction of panicle primary and secondary branches, indicating that the gene is involved in the formation of all types of axillary meristems [5]. LAX2 encodes a nuclear protein with a plant-specific conserved domain, and its mutation leads to reduction of only secondary branches, different from that of lax1. The lax1 lax2 double mutant synergistically enhanced the reduced-branching phenotype, indicating the presence of multiple pathways for panicle branching [6]. APO1 encodes an F-box protein, an ortholog of Arabidopsis UFO gene. In apo1 mutant, the branch meristems become spikelet meristems earlier, leading to the reduction of both primary and secondary panicle branches. Overexpression of APO1 caused an increase in branch and spikelet number, but the branches were undulated and spikelets were underdeveloped, making it impossible for yield improvement [7]. SCM2/PBN6/qPL6 is a quantitative trait locus (QTL) with a slightly higher expression of APO1, and it can increase both panicle branches and stem diameters, providing a new target both for yield improvement and lodging resistance [8–10].
QTLs are better targets for gene/allele mining with the consideration of breeding application, and several important QTLs for panicle size were dissected from different varieties. Gn1a is the first QTL cloned for grain productivity, and it encodes the cytokinin oxidase/dehydrogenase (OsCKX2) that degrades cytokinin. Reduced expression of OsCKX2 caused cytokinin accumulation and increase of panicle secondary branches, resulting in enhanced grain yield [11]. Later, another widely-used QTL dep1 was cloned, and it is a gain-of-function mutation causing truncation of a phosphatidylethanolamine-binding protein-like domain protein. The dep1 allele can enhance the meristematic activity, and increase the panicle branching and grain number but reduce the panicle length. Gn1a was down-regulated in NIL-dep1 compared with the wild type, suggesting a possible interaction between two QTLs [12]. IPA1 encodes the SBP domain transcriptional factor OsSPL14, which can be targeted and degraded by miR156 and miR529, and two gain-of-function alleles have been identified, namely, ipa1-1D and ipa1-2D. ipa1-1D harbored one SNP on the coding region that abolished the recognization of miR156, and ipa1-2D achieved a tandem repeats upstream OsSPL14 promoter, and both mutations led to the upregulation of OsSPL14. The two alleles can greatly increase both panicle primary branch number and stem diameter, and are ideal breeding targets both for yield improvement and lodging resistance [13,14]. The IPA1 gene has been designated as a novel green revolution gene, attracting much attention in later studies [15]. NPT was identified as an independent QTL for enhancing panicle primary branch number and stem diameter, and it encodes a deubiquitinating enzyme with homology to human OTUB1. The NPT protein promotes the K48Ub-dependent proteasomal degradation of OsSPL14, and therefore its mutation generated a phenotype similar to that of ipa1-1D/2D [16]. Similarly, IPI1 encodes a RING-finger E3 ligase that interacts with OsSPL14 in the nucleus, and it promotes the degradation of OsSPL14 in panicles while stabilizes OsSPL14 in shoot apexes by different polyubiquitin chains [17]. The ipi1 mutation leads to reduction of only secondary but not primary branches. This is different from the effect of the npt mutation, suggesting that two genes might affect the OsSPL14 protein to different extents. These studies further highlight the important role of OsSPL14 in shaping panicle morphology. However, the downstream genes of OsSPL14 are not fully explored. The ChIP-seq analysis revealed that Dep1 is one target of the OsSPL14 protein. Nevertheless, it cannot explain the constant panicle branching phenotype in the pyramiding line of dep1 and ipa1-1D, suggesting that other genes mediate the panicle morphogenesis by OsSPL14 [18].
With the development of high-throughput sequencing technology, the RNA-seq has become a routine strategy for exploring molecular networks. It also provides an alternative way to find genes underlying important agronomic traits. In this way, many novel candidate genes were identified to participate in the process of panicle development. A global transcriptome profiling of the early stages of inflorescence development identified 357 out of 22000 genes that were expressed differentially in the successive developmental stages, and provided the most possible genes for panicle branching control in rice. Indeed, one transcriptional factor was identified as a close downstream component of the pathway in which LAX1 acts [19]. By transcriptome sequencing of developing inflorescences before and after the transition from branch to spikelet meristem, a core set of genes associated with panicle branch variation were dissected between the wild and domesticated rice accessions. Also, AP2/EREBP-like genes were identified as key factors for panicle branching [20]. Transcriptomic analysis also facilitates the identification of a downstream molecular network underlying a specific gene/locus. In the inflorescence meristem of transgenic plants overexpressing APO1, 833 and 745 genes were up- and down-regulated, respectively. The candidate gene list covered many known QTLs for panicle branching control, including Gn1a and IPA1 [21].
Previously, we successfully cloned ipa1-2D as a gain-of-function allele of IPA1, which shapes modest upregulation of OsSPL14 by an epigenetic mechanism. The modest expression yielded a better balance between panicle number and panicle grain number than that of ipa1-1D, and contributed significantly to the strong culms and large panicles of the Yongyou 12 super hybrid rice [14]. We performed transcriptomic analysis using IMs from high-quality near-isogenic lines of ipa1-2D to clarify the underlying genes determining the ideal impact of the modest OsSPL14 expression. We tested the responses of several downstream genes in both RNAi and overexpression lines of OsSPL14 to confirm the dosage effect of OsSPL14. The potential target genes underlying ipa1-2D were identified.
2.1 Development of High-Quality Near-Isogenic Lines
A pair of near-isogenic lines with either ipa1-2D or IPA1 alleles were developed following the procedure illustrated in Fig. 1a. In detail, the parent YYP1 with the ipa1-2D allele was crossed with Nipponbare (NIP) to generate F1. Thereafter, F1 plants were backcrossed three times with NIP to generate BC3F1. Then successive self-crossings were performed in the single-seed descent way using heterozygous ipa1-2D plants, and NIL-ipa1-2D and NIL-IPA1 were finally obtained at the BC3F8 generation. All the generations of backcrossing and self-crossing were coupled with molecular marker assisted selection (MAS) of ipa1-2D. In the BC3F4 generation, a whole genome resequencing was performed for one line selected for further self-crossing, and the genotype calling and recombination breakpoint determination were obtained by the sliding window approach as published before [22].
2.2 Measurement of Agronomic Traits
Twenty-four individuals of two NILs were planted in the paddy field, and twelve individuals were randomly selected for trait measurements. Panicle number was counted as the number of fertilized panicles at the mature stage. Main culms were used for the measurement of the other traits. Plant height was measured from the ground to the panicle tip with a ruler. Panicle length was measured from the peduncle to the panicle tip. The major and minor diameters of internodes III and IV were measured using a Vernier caliper, and mean values of the two diameters were used for analysis. Panicle branch number and spikelet number per panicle were counted manually. The student’s t-test was applied to determine the significance of the trait differences between NILs.
2.3 Sampling of IMs and Scanning Electronic Microscopy (SEM) Analysis
The NiL-ipa1-2D and NIL-IPA1 were planted in the paddy field, and sampling of inflorescence meristems started at the booting stage. To cover the sequential stages of early inflorescence development, the sampling was carried out every three days, and sampled inflorescence meristems were fixed in FAA solutions (50% ethanol; 3.65% formaldehyde; 5% glacial acetic acid) immediately. For SEM analysis, the samples were dehydrated in a graded ethanol series, and then critical-point-dried. The samples were fixed to a small cropper bed upward, and the bract tissues around the inflorescence meristems were stripped by a dissecting needle. After sputter-coated with Pt, the samples were observed under a scanning electron microscope (JSM-6360LV, JEOL). In all, IMs in five stages were sampled and observed.
IMs in five stages from the two NILs were also sampled and each mixed together for RNA-seq. The mixed samples were grinded into powder in liquid nitrogen, and the total RNA was extracted by the TRIzol reagent (Invitrogen) following the manufacture’s instructions. To construct the cDNA library, the mRNA was enriched by the beads with oligo (dT), and then fragmented in the fragmentation buffer. The first strand cDNA was synthesized by the random hexamers, and then the second strand cDNA was synthesized after adding buffer, dNTPs and DNA polymerase I. The cDNA was purified by the AMPure XP beads, and an adaptor was added for sequencing. The library was finally obtained by PCR amplification, and subjected to high throughput sequencing (illumina HiSeq2000 platform). After excluding the low-quality reads and adapter sequence, the clean reads were aligned to the genome by TopHat2 tools. The expression level was calculated as RPKM, namely, reads per kilo bases per million reads. The threshold for differentially expressed genes was set as | log2 (fold change) | > 1. Hierarchical clustering was performed based on the normalized log10 values of RPKM for differentially expressed genes, and the heat map was generated accordingly. Six levels of expression differences were generated by H-cluster method based on the log2 values of fold change. The gene ontology (GO) enrichment analysis was performed by the application of GOseq [23].
2.5 Generation of Transgenic Lines of OsSPL14 Overexpression and RNAi
For overexpression of OsSPL14, an 11.9 kb genomic segment covering both the coding and promoter region of OsSPL14 was cloned into pCambia1301 vector, and the construct was transformed into NIP by the agrobacterium tumefaciens-mediated method. Both positive and negative plants were obtained at the T0 generations, and one positive line with single copy and one negative line were selected from the T2 generation as previously reported [14]. The RNAi construct for OsSPL14 followed exactly the same way as shown in a previous publication [13], and it was transformed into NIP in the same way as showed above. All the positive and negative lines were planted in the paddy field, and the IMs were sampled at the booting stage for RNA extraction and qRT-PCR analysis. To facilitate citation, OsSPL14-OVE(+/−) and OsSPL14-RNAi (+/−) were used to designate the different transgenic lines.
2.6 Reverse Transcription and Quantitative PCR Analysis
The reverse transcription was carried out by the iScriptTM one-step RT-PCR kit (Bio-Rad), and about 0.5 μg RNA was added for the reaction. The synthesized cDNA was diluted by four fold and stored at −20°C. Twelve differentially expressed genes were selected for qRT-PCR validation, and the primers specially amplifying the cDNA of each gene were designed by the web-based tool QuantPrime (Tab. S1). A 20 μl system was amplified for fluorescence detection on the Mastercycler ep realplex (Eppedorf), including 0.4 μl of each primer (10 μM), 2 μl of cDNA template, 7.2 μl of distilled water and 10 μl of 5 × PrimeScript RT Master Mix (Takara). The two-step PCR program was used as follows: An initial denaturation at 95°C for 15 s, 40 cycles of denaturation at 95°C for 5 s and annealing and extension at 60°C for 20 s. The actin gene was amplified as an internal control. CT values of target genes and actin from three technical repeats were obtained and the relative expression values of target genes were calculated as 2−(CT value of target gene – CT value of actin). To facilitate comparison, the values of the wild type (NIL-IPA1, OsSPL14-OVE (−) and OsSPL14-RNAi (−)) were normalized to one.
3.1 Development of High-Quality NILs for Trait Evaluation
The best way to evaluate the effect of a QTL is developing high-quality NILs, and we therefore carried out a routine crossing and backcrossing to introgress the ipa1-2D locus from YYP1 to NIP (Fig. 1a). During the process, markers linked with ipa1-2D were used to select heterozygous lines. In the BC3F2 population, we performed phenotypic selection in the paddy field, and several individuals with heterozygous ipa1-2D were selected for further self-crossing and planted for trait comparison. In the BC3F4 generation, we identified one heterozygous line with overall better performance than NIP, and this line was selected further for NIL development. By whole genome resequencing, we clarified the genetic background of the BC3F4 line, and found that most of the chromosomal segments belonged to the NIP type, and the introgression with ipa1-2D was very small (~30 kb), confirming the success of our backcrossing (Fig. 1b). To exclude the effect of unrelated heterozygous segments, successive self-crossing was performed to fix the genetic background but retain the heterozygous ipa1-2D by MAS. In the BC3F8 generation, we obtained one pair of NILs with homozygous ipa1-2D and IPA1 allele, respectively, and named them NIL-ipa1-2D and NIL-IPA1. It could be observed that the overall plant height of two lines was similar, but NIL-ipa1-2D was stronger than NIL-IPA1 (Fig. 1c). Moreover, the primary branches of mature panicles in NIL-ipa1-2D were far more than those in NIL-IPA1 (Fig. 1d), and this difference was also found in the young panicle (Fig. 1e), suggesting a different status of early inflorescence development in the two lines. The traits variation was confirmed by the statistical analysis (Tab. 1). In addition to the panicle primary branch number, the stem diameter was also increased, but panicle number was slightly reduced in NIL-ipa1-2D. No significant difference was found for plant height, panicle length and panicle secondary branch number between the two lines, and the spikelet number per panicle increased greatly in NIL-ipa1-2D, which should be contributed mainly by the increase of panicle primary branches.
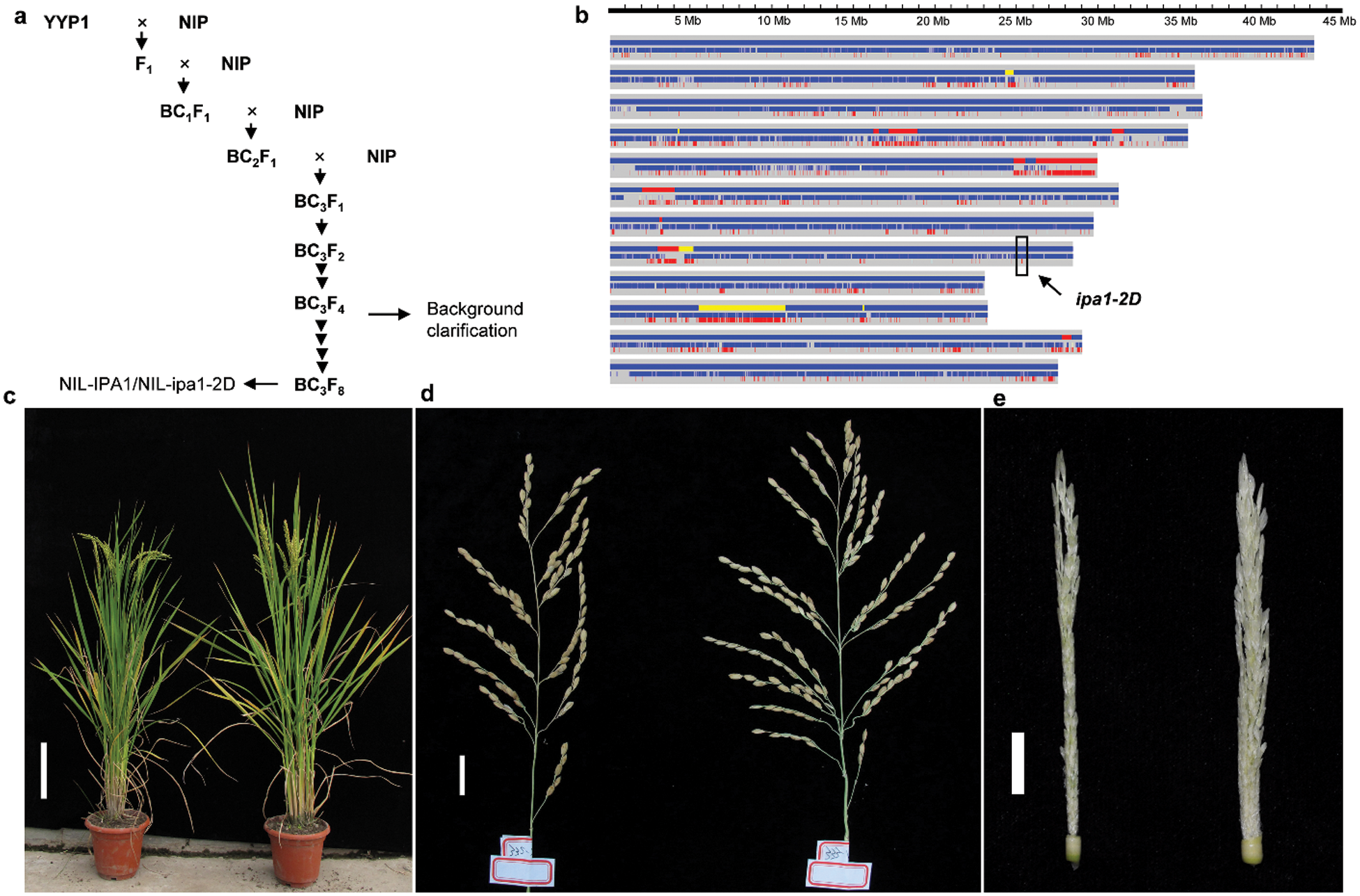
Figure 1: Development of high-quality NILs and their trait performance. a. Sketch map of NIL development by sequential backcrossing and self-crossing. b. High throughput genotyping of one BC3F4 line. Red and blue bars indicate the genotype of YYP1 and NIP, respectively, and the yellow bar indicates the heterozygous region. The location of ipa1-2D was denoted by the arrow. c–e. The morphology of whole plants, mature panicles and young panicles of NILs. NIL-IPA1 and NIL-ipa1-2D are shown on the left and right, respectively. Bar = 20 cm (c), 2 cm (d) and 1 cm (e)
Table 1: Statistical analysis of different traits between NILs
3.2 Comparison of IMs in Five Sequential Stages between NILs
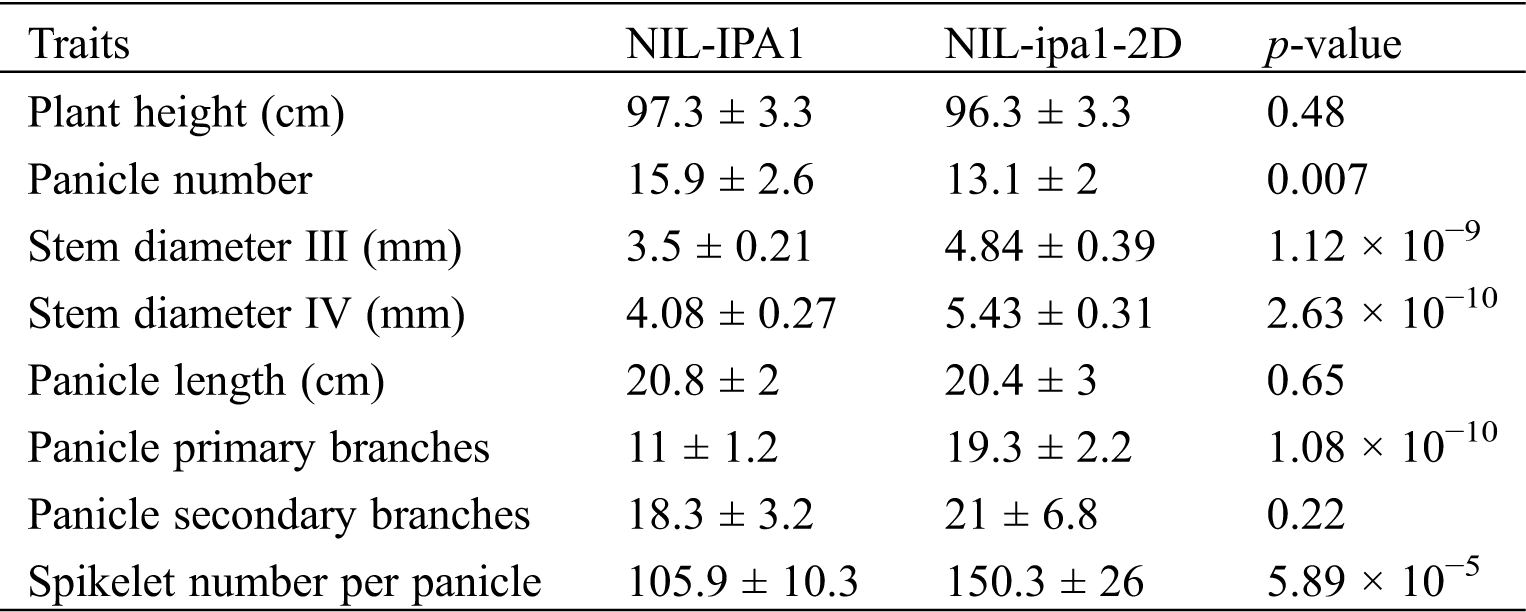
We planted the NILs to sample the IMs in sequential developmental stages, and successfully got the scanning electron microscope images of the tissues in five stages (Fig. 2). At the very early stage, the meristems were domelike and still in the status of SAM (Stage 1). Then, several PBMs initiated in spiral arrangement from the SAM, which was the early status of the PBM stage and could be designated as the PBM1 stage (Stage 2). Later, more PBMs initiated and some hairs formed around them, which was designated as the PBM2 stage (Stage 3). After that, the PBMs started to elongate and either SBMs or SMs initiated from the elongated PBMs (Stage 4). Finally, the FMs generated from the SMs, which further developed into primordia of different floral organs (Stage 5). We found that both NILs took about two weeks to finish the five stages, indicating a similar speed of IM development. Nevertheless, the domelike SAM of NIL-ipa1-2D was larger than that of NIL-IPA1, and it might support the initiation of more PBMs as found both in the PBM1 and PBM2 stages. The significant difference of PBM number should contribute to the obviously different panicle morphology found in the mature panicles between NILs. Moreover, the wider diameter of IMs could explain the phenotype of strong culms found in NIL-ipa1-2D.
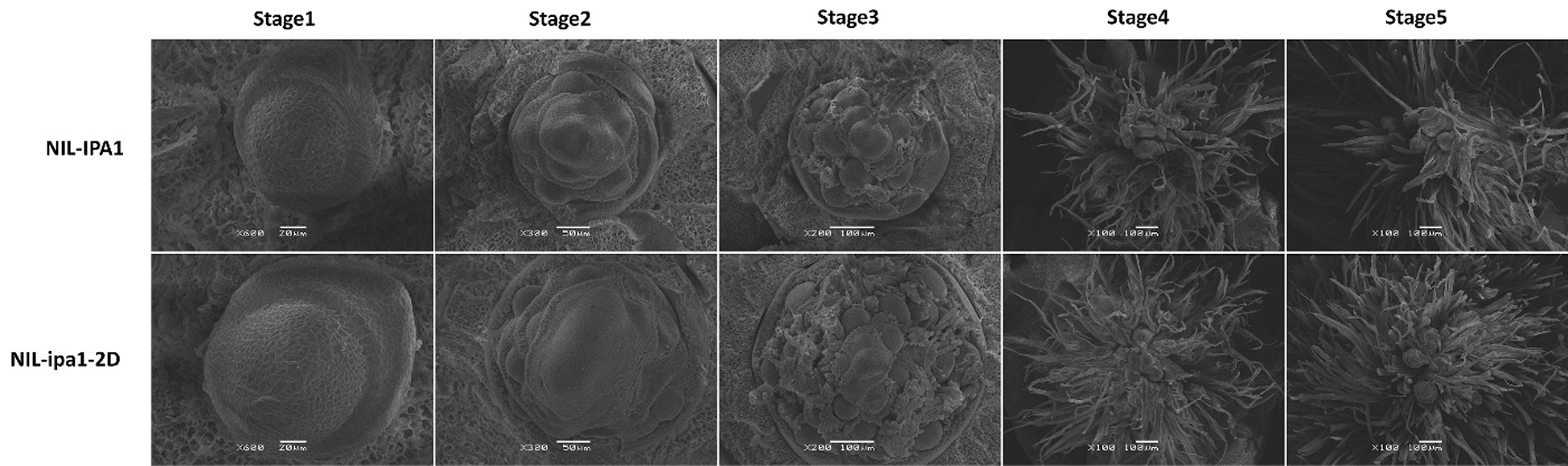
Figure 2: The scanning electron microscope images of five developmental stages of IMs in NILs
3.3 Identification of the Key Molecular Network in IMs Underlying ipa1-2D
As the morphology difference was mainly determined by the IMs development, the sampled IMs covering the five stages were pooled and subjected to RNA-seq analysis. Based on the RPKM values, all the annotated genes were categorized into non-expressed (RPKM < 1), low-expressed (1 < RPKM < 50) and high-expressed (RPKM > 50) genes, respectively. Accordingly, the 17636 and 17612 low-expressed genes and the 2463 and 2499 high-expressed genes were identified from the NIL-IPA1 and NIL-ipa1-2D, respectively, while a large proportion of annotated genes were not expressed (Fig. 3a). We concluded that some of the high-expressed genes might have important roles for the IM development, and found that 2293 of such genes were commonly identified from the two NILs (Fig. 3b and Tab. S2), including OsSPL14. The RPKM value of OsSPL14 in NIL-ipa1-2D was about three fold to that in NIL-IPA1 (Fig. 3c), indicating that the RNA samples can effectively reflect the variation of the two alleles. The expression difference of OsSPL14 was checked by qRT-PCR and similar results were obtained (Fig. 3d). In addition to OsSPL14, 86 genes were identified to have a more than two fold difference between the NILs, and 12 genes were up-regulated and 74 genes were down-regulated in the NIL-ipa1-2D, respectively (Fig. 3e and Tab. S3). These genes covered both high and low-expressed genes, and some genes belonged to a low-expressed gene in one line but a high-expressed gene in another line, and were designated as partial type. According to the log2 value of fold change, these differentially expressed genes could be further categorized into six levels, and most of the genes were in the range of Level 1 (down-regulation of 2–3 fold). As the levels of fold change increased, the gene number was decreased, and both high- and low-expressed genes and partial type could be found in the Levels 1–3 and Level 5 (Fig. 3f). One gene in Level 4 and two genes in Level 6 had more than tenfold differences, and belonged to low-expressed genes. The enrichment of differentially expressed genes in specific GO terms was identified, and we found that most of the enriched terms were involved in the functions of metabolism and catalysis (Fig. 3g), suggesting possible roles of these pathways in panicle development.
Figure 3: Transcriptomic analysis of the dominantly and differentially expressed genes in IMs of NILs. a. The gene number of three expression levels between NILs. b. The venn diagram showing the number and percentage of high-expressed genes between NILs. c. The RPKM values of OsSPL14 between NILs. d. The expression level of OsSPL14 revealed by qRT-PCR. e. The heat map showing the clustering and contrast of differentially expressed genes. The blue and red colors indicate the levels of low- and high-expressed genes, respectively. f. Gene number with different levels of fold change (log2 values) is shown, and three types of expression status are illustrated by different colors. g. The top 29 enriched GO terms of differentially expressed genes. *denotes the significantly enriched terms
3.4 Creating the Panicle Diversity by RNAi and Overexpression of OsSPL14
The differentially expressed genes from transcriptomic analysis provided potential targets mediating the allele contribution of ipa1-2D, and it was worth testing their responses to OsSPL14 dosage in different genetic backgrounds. Therefore, the overexpression and RNAi lines of OsSPL14 were generated to perform such analysis. In the T2 generation, both positive and negative lines of two transgenic constructs were obtained and became ideal materials for expression analysis. We then compared the expression level of OsSPL14 in the IMs of transgenic lines, and a significant expression difference was identified between positive and negative lines of both constructs. In the overexpression lines, the OsSPL14 expression was about 2.5 fold that of the control line, approaching the expression level found in the NIL-ipa1-2D (Fig. 4a). The overall plant morphology of the overexpression line was stronger, and its panicle primary branch number also greatly increased from about 11 to 16, similar to the effect of ipa1-2D (Figs. 4b, 4c). For the RNAi lines, the OsSPL14 expression decreased by about 20 fold, and bushy tillers with smaller panicles were generated (Figs. 4d, 4e). Both the panicle primary branches and panicle length were greatly reduced in the RNAi line (Fig. 4f), indicating that OsSPL14 was indispensable to sustain panicle branching. The transgenic lines provided an additional opportunity to clarify the impact of OsSPL14 on downstream gene expression.
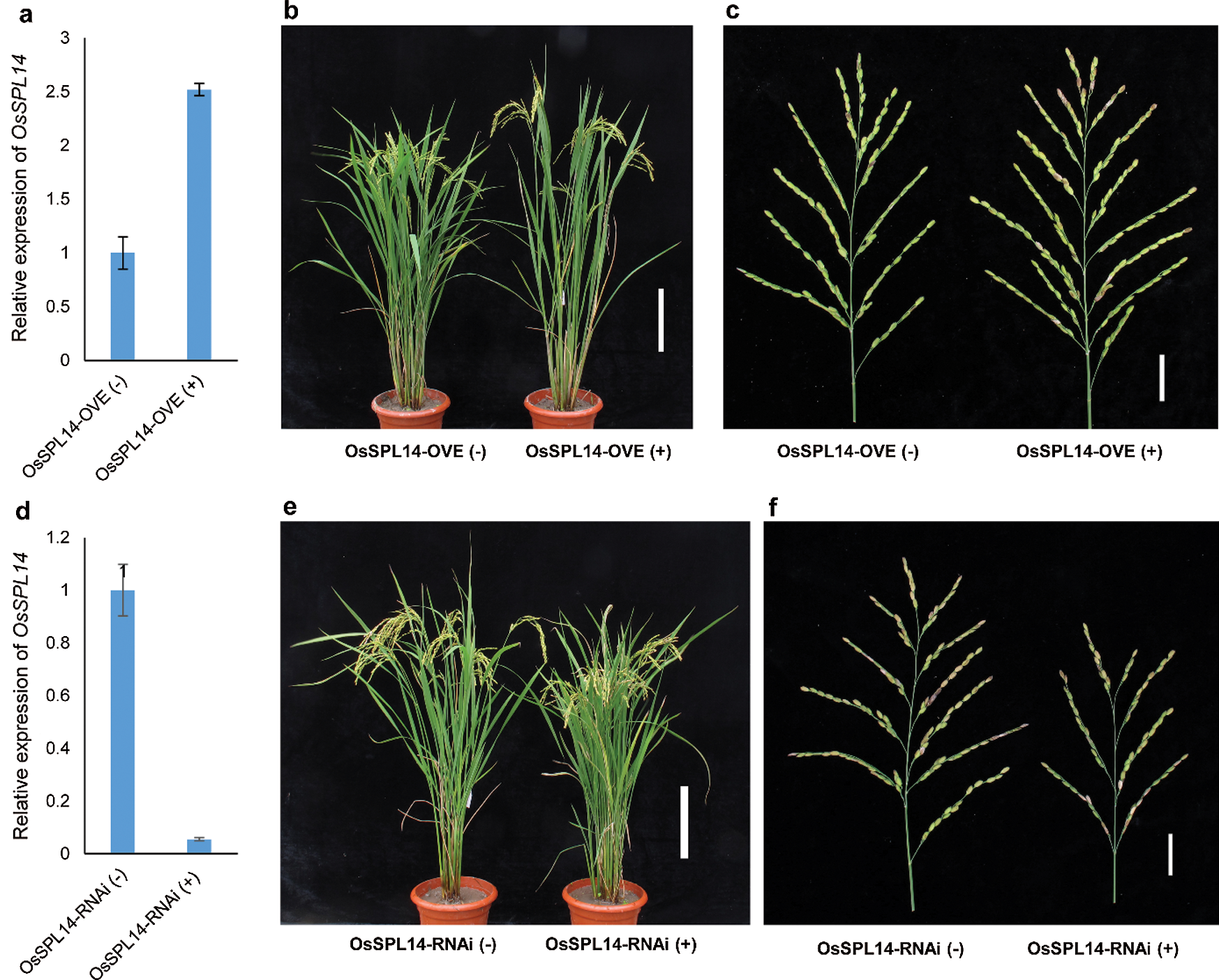
Figure 4: Creation of lines with overexpression and RNAi of OsSPL14, and their trait performance. a,d. The relative expression of OsSPL14 in IMs of overexpression and RNAi lines. The mean of three technical repeats is shown, and bars represent SD. b,e. The overall plant morphology of overexpression and RNAi lines. Bar = 20 cm. c,f. The panicle morphology of overexpression and RNAi lines. Bar = 3 cm
3.5 Clarifying the Responses of 12 Genes to Different OsSPL14 Dosages
Based on the rice genome annotation, twelve genes were selected from the 86 differentially expressed genes for qRT-PCR validation, with two up-regulated and ten down-regulated genes (Tab. 2). The protein families of some selected genes have been suggested to participate in the process of panicle development, such as the AP2-domain and MADS-box proteins. The expression profiles of the selected genes in the NILs and transgenic lines were clarified by the qRT-PCR, and most genes showed consistent results with those revealed by RNA-seq, confirming the effectiveness of the transcriptomic analysis (Fig. 5). Although the mRNA levels of LOC_Os01g64790 and LOC_Os09g36300 in NILs were not significantly different as expected, their down-regulations were validated in overexpression lines (Figs. 5d, 5i). In these later lines, most genes showed a similar expression profile to that found between NILs except for LOC_Os06g40360. In theory, contrast expression profiles in RNAi lines should be identified for each gene, and this was confirmed for 11 genes except for LOC_Os01g21590. Among the 12 genes, six genes showed a consistent expression profile with an obvious fold change among the three sets of comparisons, including five down-regulated genes (LOC_Os01g03680, LOC_Os02g48770, LOC_Os03g46860, LOC_Os04g44820 and LOC_Os09g26780) and one up-regulated gene (LOC_Os07g01820). LOC_Os07g01820 encodes the MADS-box protein OsMADS15, and we conclude that the gene might function as a potential positive regulator for panicle branching, and the other five genes might contain the key negative regulators for panicle branching.
Table 2: Gene information of twelve selected genes for qRT-PCR validation
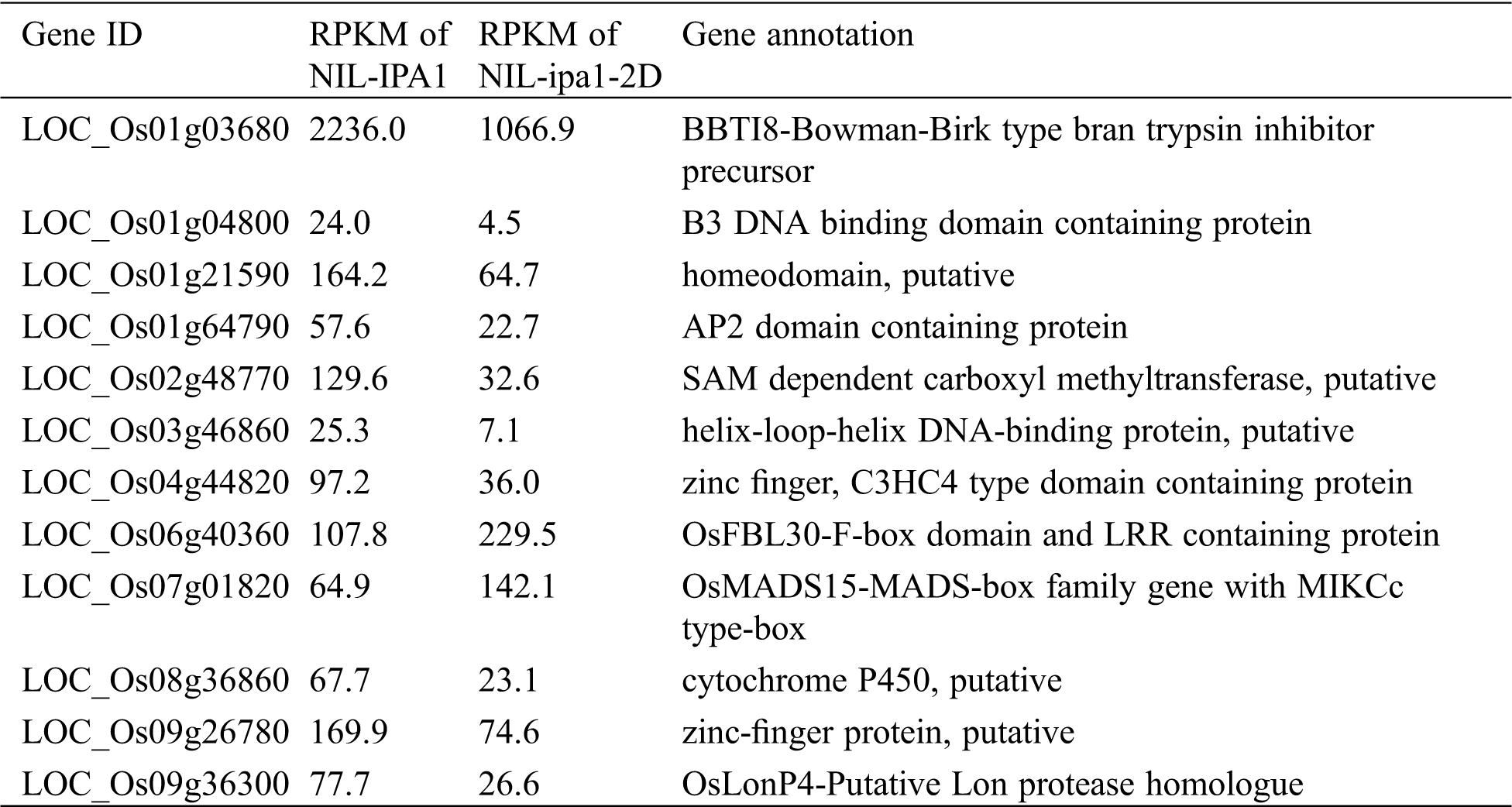
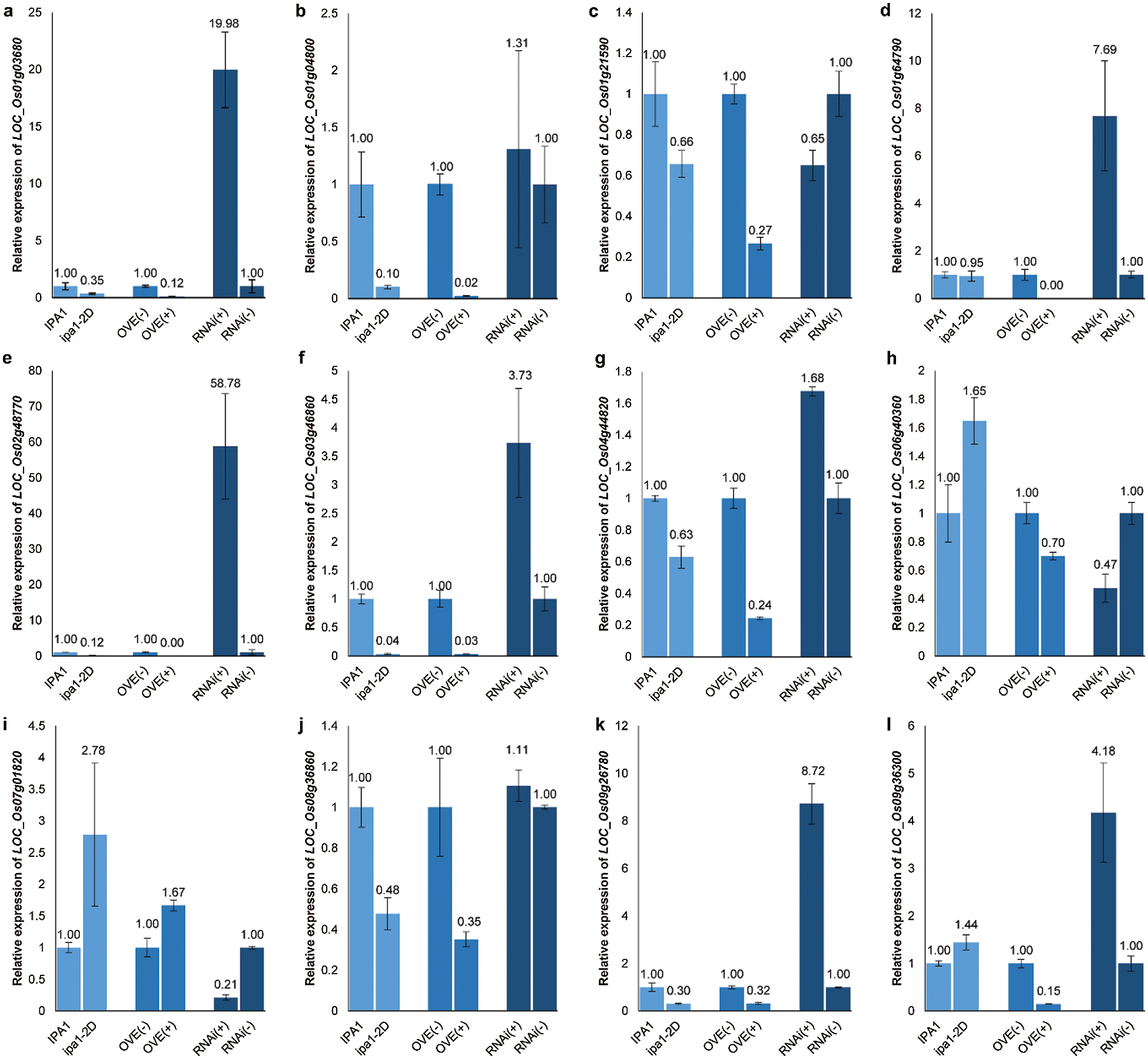
Figure 5: Validation of 12 selected, differentially expressed genes between NILs and positive and negative lines of OsSPL14 overexpression and RNAi. The relative expression values from three technical repeats were labelled, and bars represent SD
Panicle branching is the key determinant for grain number per panicle and therefore contributes to grain yield. ipa1-2D was the major locus that explained nearly one half of the phenotypic variation of panicle primary branch number in the super hybrid rice Yongyou 12. ipa1-2D allele shaped a modest expression of OsSPL14 and therefore created a balance between panicle number and grain number per panicle [14]. OsSPL14 functions as a transcriptional factor, which should drive the expression of downstream genes to coordinate panicle development. In this study, we developed high-quality near-isogenic lines and confirmed that ipa1-2D had a large impact on the initiation of the PBMs. By transcriptomic strategy, we got the candidate gene list for further investigation, and validated the result in different genetic backgrounds.
Near-isogenic lines are an ideal material for QTLstudies, and we introgressed a very small segment with ipa1-2D during the backcrossing, which can exclude the disruption of linked genes. Several other QTLs for panicle size have been introgressed into different backgrounds, including SCM2, SCM3 and Gn1a, but the smallest introgressed QTL region was 163 kb, covering many unrelated genes [11,21]. Therefore, the obtained NILs of ipa1-2D were of high quality both for genetic and molecular study. Heterozygous inbred family (HIF) was an alternative strategy for NIL development, and HIF-NILs for the natural FZP allele have been developed in this way [24,25]. We also took a similar strategy to exclude the disruption of genetic background starting from BC3F2 generation. In this way, a pair of sister lines were selected from a single heterozygous plant in high generation, which should share exactly the same genetic background. Therefore, combination of backcrossing and self-crossing might facilitate the development of high-quality NILs.
The panicle morphology is determined by the differentiation of IMs, which can be further categorized into several stages with initiation of different types of meristems. Using the NILs, we successfully captured all the stages of IMs and found that the SAM with ipa1-2D was larger than that of the wild type. A similar condition was found in dep1 and npt lines [12,16], indicating that enlarged SAM is necessary for generating more panicle branches. We also clarified the impact of ipa1-2D on PBMs initiation, which could facilitate the understanding of the diversity of primary branch numbers found in mature panicles. The previous study showed that Ghd7 would also increase the panicle branching and grain number, and its effect on panicle size was related to the duration of panicle differentiation [1,26]. The increased duration of panicle differentiation might provide more time for PBM and SBM initiation. Differently, we found that the duration of panicle differentiation was similar between NIL-ipa1-2D and NIL-IPA1, indicating that a different mechanism should be involved in shaping panicle branching between ipa1-2D and Ghd7. Therefore, it is worth clarifying the molecular mechanism that determines the difference in future studies.
The previous transcriptomic analysis usually identified a large portion of differentially expressed genes between the wild type and the mutant, ranging from several hundreds to thousands [16,18,21]. Interestingly, we only found 87 genes that were differentially expressed between the NILs, making the selection of candidate genes easier. One possible reason could be that the high-quality NILs excluded the disruption between the genetic background and linked genes. Another possibility could be that the modest up-regulation of OsSPL14 in NIL-ipa-2D had a slight impact on the overall gene network in IMs. Among the limited candidate genes, we found that most of them were involved in the function of metabolism and catalysis. Consistently, such type of differentially expressed genes were also enriched in the young panicle of knocking down lines of RFL, a regulator of the meristem fate particularly during the formation of the branched inflorescence and spikelet [27]. Therefore, such type of genes might have unknown roles in shaping the panicle morphology.
The genes identified by transcriptome might vary in different genetic backgrounds, and we therefore confirmed the expression profiles of 12 selected genes in transgenic lines of OsSPL14. Interestingly, most of the selected genes showed a consistent pattern in matching the level of OsSPL14, increasing their possibility as OsSPL14 targets. The potential functions of several protein families on organ development have been suggested in previous reports, including the B3 DNA-binding domain-containing protein, homeodomain protein, cytochrome P450 protein, AP2 domain containing protein, helix-loop-helix (HLH) DNA-binding protein, zinc finger protein, F-box protein and MADS-box protein. RAV6 encodes a B3 DNA-binding domain-containing protein, and Epi-rav6 mutant plants caused ectopic expression of RAV6 and showed a larger lamina inclination and a smaller grain size [28]. GLR1 encodes a homeodomain protein that regulates trichomes development in rice, and its mutant showed a significant decrease in trichome number of leaves and glumes [29]. CL4 encodes a putative cytochrome P450 protein that involved in brassinosteroid biosynthesis, and cl4 mutant generated panicles with all primary branches clustered on the base of the main rachis [30]. FZP is a well-known gene that determines the transition from spikelet meristem to floret meristem, and it contains an EREBP/AP2 domain [31]. The natural allele with a decreased expression of FZP can increase the panicle secondary branch number [25]. OsBUL1 belongs to a rice atypical HLH protein, and it controls cell elongation and positively affects leaf angles [32]. DST is a zinc finger transcription factor that directly regulates Gn1a expression in the reproductive meristem, and therefore determines panicle branching and grain number [33]. F-box proteins constitute a large family in eukaryotes, and 687 potential F-box proteins were identified in rice [34]. Two F-box proteins APO1 and LARGE PANICLE have been reported to determine panicle branching [7,35]. Differently, mutation of APO1 led to the reduction of panicle branches while the LARGE PANICLE mutation increased the panicle branches. This suggests that F-box proteins could function as either positive or negative regulators of panicle branching. MADS-box proteins are another large protein family in plants, and 75 MADS-box genes have been identified in rice [36]. Mutation of OsMADS34 led to an increase of the panicle primary branch number but to a reduction of the secondary branch number, suggesting that this gene might be involved in the balance of two types of meristems (PBMs and SBMs). OsMADS15 acts downstream of Hd3a and RFT1, which might mediate the flowering signals of two florigens [37]. Moreover, OsMADS15 is indispensable for sexual reproduction together with OsMADS1, and its ectopic overexpression led to a great decrease of the panicle branches [38,39]. Nevertheless, we found that the gene was up-regulated by ipa1-2D, suggesting that its native overexpression might have different effects. We highlighted the potential roles of identified genes for rice branching. With the advantage and convenience of CRISPER/CAS9 gene editing technology, it is worth testing functions of these genes by targeting the either coding or uORF region [40,41]. Also, it is possible to generate better panicle morphology with mutated or accumulated proteins of these genes, providing new targets for breeding selection.
In this study, we aimed to explore the downstream genes underlying the control of panicle branching by ipa1-2D. A pair of high-quality near-isogenic lines were developed with a difference of only 30 kb chromosomal segment covering ipa1-2D. The developmental status of the inflorescence meristems in five stages was clarified, and it showed that more PBMs initiated from a larger SAM in the line with ipa1-2D, providing a deep understanding about branch formation in rice panicles. By the transcriptomic analysis, 2293 genes were identified as high-expressed genes in IMs, and 87 genes including OsSPL14 were found differentially expressed between NILs. The responses of twelve potential downstream genes to OsSPL14 were confirmed by the transgenic lines of OsSPL14 overexpression and RNAi. We proposed that these genes could be ideal targets for further function validation.
Funding Statement: This work was supported by grants from the National Natural Science Foundation of China (31600990, 31871217 and 32072037), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (20KJA210002), Project of Special Funding for Crop Science Discipline Development (yzuxk202006), the open funds of the State Key Laboratory of Crop Genetics and Germplasm Enhancement (ZW202010) and the Key Research and Development Program of Jiangsu Province (BE2018357).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Xing, Y. Z., Zhang, Q. F. (2010). Genetic and molecular bases of rice yield. Annual Review of Plant Biology, 61(1), 421–442. DOI 10.1146/annurev-arplant-042809-112209. [Google Scholar] [CrossRef]
2. Qian, Q., Guo, L. B., Smith, S. M., Li, J. Y. (2016). Breeding high-yield superior quality hybrid super rice by rational design. National Science Review, 3(3), 283–294. DOI 10.1093/nsr/nww006. [Google Scholar] [CrossRef]
3. Li, D. Y., Huang, Z. Y., Song, S. H., Xin, Y. Y., Mao, D. H. et al. (2016). Integrated analysis of phenome, genome, and transcriptome of hybrid rice uncovered multiple heterosis-related loci for yield increase. Proceedings of the National Academy of Sciences of the United States of America, 113(41), E6026–E6035. DOI 10.1073/pnas.1610115113. [Google Scholar] [CrossRef]
4. Caselli, F., Zanarello, F., Kater, M. M., Battaglia, R., Gregis, V. (2020). Crop reproductive meristems in the genomic era: A brief overview. Biochemical Society Transactions, 48(3), 853–865. DOI 10.1042/BST20190441. [Google Scholar] [CrossRef]
5. Komatsu, K., Maekawa, M., Ujiie, S., Satake, Y., Furutani, I. et al. (2011). LAX and SPA: Major regulators of shoot branching in rice. Proceedings of the National Academy of Sciences of the United States of America, 100(20), 11765–11770. DOI 10.1073/pnas.1932414100. [Google Scholar] [CrossRef]
6. Tabuchi, H., Zhang, Y., Hattori, S., Omae, M., Shimizu-Sato, S. et al. (2011). LAX PANICLE2 of rice encodes a novel nuclear protein and regulates the formation of axillary meristems. Plant Cell, 23(9), 3276–3287. DOI 10.1105/tpc.111.088765. [Google Scholar] [CrossRef]
7. Ikeda, K., Ito, M., Nagasawa, O. N., Kyozuka, J., Nagato, Y. (2007). Rice ABERRANT PANICLE ORGANIZATION 1, encoding an F-box protein, regulates meristem fate. Plant Journal, 51(6), 1030–1040. DOI 10.1111/j.1365-313X.2007.03200.x. [Google Scholar] [CrossRef]
8. Ookawa, T., Hobo, T., Yano, M., Murata, K., Ando, T. et al. (2010). New approach for rice improvement using a pleiotropic QTL gene for lodging resistance and yield. Nature Communications, 1(1), 580. DOI 10.1038/ncomms1132. [Google Scholar] [CrossRef]
9. Terao, T., Nagata, K., Morino, K., Hirose, T. (2010). A gene controlling the number of primary rachis branches also controls the vascular bundle formation and hence is responsible to increase the harvest index and grain yield in rice. Theoretical and Applied Genetics, 120(5), 875–893. DOI 10.1007/s00122-009-1218-8. [Google Scholar] [CrossRef]
10. Zhang, L., Wang, J. J., Wang, J. M., Wang, L. Y., Ma, B. et al. (2015). Quantitative trait locus analysis and fine mapping of the qPL6 locus for panicle length in rice. Theoretical and Applied Genetics, 128(6), 1151–1161. DOI 10.1007/s00122-015-2496-y. [Google Scholar] [CrossRef]
11. Ashikari, M., Sakakibara, H., Lin, S. Y., Yamamoto, T., Takashi, T. et al. (2005). Cytokinin oxidase regulates rice grain production. Science, 309(5735), 741–745. DOI 10.1126/science.1113373. [Google Scholar] [CrossRef]
12. Huang, X. Z., Qian, Q., Liu, Z. B., Sun, H. Y., He, S. Y. et al. (2009). Natural variation at the DEP1 locus enhances grain yield in rice. Nature Genetics, 41(4), 494–497. DOI 10.1038/ng.352. [Google Scholar] [CrossRef]
13. Jiao, Y. Q., Wang, Y. H., Xue, D. W., Wang, J., Yan, M. X. et al. (2010). Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nature Genetics, 42(6), 541–544. DOI 10.1038/ng.591. [Google Scholar] [CrossRef]
14. Zhang, L., Yu, H., Ma, B., Liu, G. F., Wang, J. J. et al. (2017). A natural tandem array alleviates epigenetic repression of IPA1 and leads to superior yielding rice. Nature Communications, 8(1), 20260. DOI 10.1038/ncomms14789. [Google Scholar] [CrossRef]
15. Wang, B. B., Wang, H. Y. (2017). IPA1: A new “green revolution” gene?. Molecular Plant, 10(6), 779–781. [Google Scholar]
16. Wang, S. S., Wu, K., Qian, Q., Liu, Q., Li, Q. et al. (2017). Non-canonical regulation of SPL transcription factors by a human OTUB1-like deubiquitinase defines a new plant type rice associated with higher grain yield. Cell Research, 27(9), 1142–1156. DOI 10.1038/cr.2017.98. [Google Scholar] [CrossRef]
17. Wang, J., Yu, H., Xiong, G. S., Lu, Z. F., Jiao, Y. Q. et al. (2017). Tissue-specific ubiquitination by IPA1 INTERACTING PROTEIN1 modulates IPA1 protein levels to regulate plant architecture in rice. Plant Cell, 29(4), 697–707. DOI 10.1105/tpc.16.00879. [Google Scholar] [CrossRef]
18. Lu, Z. F., Yu, H., Xiong, G. S., Wang, J., Jiao, Y. Q. et al. (2013). Genome-wide binding analysis of the transcription activator IDEAL PLANT ARCHITECTURE1 reveals a complex network regulating rice plant architecture. Plant Cell, 25(10), 3743–3759. DOI 10.1105/tpc.113.113639. [Google Scholar] [CrossRef]
19. Furutani, I., Sukegawa, S., Kyozuka, J. (2006). Genome-wide analysis of spatial and temporal gene expression in rice panicle development. Plant Journal, 46(3), 503–511. DOI 10.1111/j.1365-313X.2006.02703.x. [Google Scholar] [CrossRef]
20. Harrop, T. W. R., Mantegazza, O., Luong, A. M., Bethune, K., Lorieux, M. et al. (2019). A set of AP2-like genes is associated with inflorescence branching and architecture in domesticated rice. Journal of Experimental Botany, 70(20), 5617–5629. DOI 10.1093/jxb/erz340. [Google Scholar] [CrossRef]
21. Yano, K., Ookawa, T., Aya, K., Ochiai, Y., Hirasawa, T. et al. (2015). Isolation of a novel lodging resistance QTL gene involved in strigolactone signaling and its pyramiding with a QTL gene involved in another mechanism. Molecular Plant, 8(2), 303–314. DOI 10.1016/j.molp.2014.10.009. [Google Scholar] [CrossRef]
22. Huang, X. H., Feng, Q., Qian, Q., Zhao, Q., Wang, L. et al. (2009). High-throughput genotyping by whole-genome resequencing. Genome Research, 19(6), 1068–1076. DOI 10.1101/gr.089516.108. [Google Scholar] [CrossRef]
23. Young, M. D., Wakefield, M. J., Smyth, G. K., Oshlack, A. (2010). Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biology, 11(2), R14. DOI 10.1186/gb-2010-11-2-r14. [Google Scholar] [CrossRef]
24. Bai, X. F., Wu, B., Xing, Y. Z. (2012). Yield-related QTLs and their applications in rice genetic improvement. Journal of Integrative Plant Biology, 54(5), 300–311. DOI 10.1111/j.1744-7909.2012.01117.x. [Google Scholar] [CrossRef]
25. Bai, X. F., Huang, Y., Hu, Y., Liu, H. Y., Zhang, B. et al. (2017). Duplication of an upstream silencer of FZP increases grain yield in rice. Nature Plants, 3(11), 885–893. DOI 10.1038/s41477-017-0042-4. [Google Scholar] [CrossRef]
26. Xue, W. Y., Xing, Y. Z., Weng, X. Y., Zhao, Y., Tang, W. J. et al. (2008). Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nature Genetics, 40(6), 761–767. DOI 10.1038/ng.143. [Google Scholar] [CrossRef]
27. Rao, N. N., Prasad, K., Kumar, P. R., Vijayraghavan, U. (2008). Distinct regulatory role for RFL, the rice LFY homolog, in determining flowering time and plant architecture. Proceedings of the National Academy of Sciences of the United States of America, 105(9), 3646–3651. DOI 10.1073/pnas.0709059105. [Google Scholar] [CrossRef]
28. Zhang, X. Q., Sun, J., Cao, X. F., Song, X. W. (2015). Epigenetic mutation of RAV6 affects leaf angle and seed size in rice. Plant Physiology, 169(3), 2118–2128. [Google Scholar]
29. Li, J. J., Yuan, Y. D., Lu, Z. F., Yang, L. S., Gao, R. C. et al. (2012). Glabrous rice 1, encoding a homeodomain protein, regulates trichome development in rice. Rice, 5(1), 32. DOI 10.1186/1939-8433-5-32. [Google Scholar] [CrossRef]
30. Guo, M., Yang, Y. H., Liu, M., Meng, Q. C., Zeng, X. H. et al. (2014). Clustered spikelets 4, encoding a putative cytochrome P450 protein CYP724B1, is essential for rice panicle development. Chinese Science Bulletin, 59(31), 4050–4059. DOI 10.1007/s11434-014-0568-z. [Google Scholar] [CrossRef]
31. Komatsu, M., Chujo, A., Nagato, Y., Shimamoto, K., Kyozuka, J. (2003). FRIZZY PANICLE is required to prevent the formation of axillary meristems and to establish floral meristem identity in rice spikelets. Development, 130(16), 3841–3850. DOI 10.1242/dev.00564. [Google Scholar] [CrossRef]
32. Jang, S., An, G., Li, H. Y. (2017). Rice leaf angle and grain size are affected by the OsBUL1 transcriptional activator complex. Plant Physiology, 173(1), 688–702. DOI 10.1104/pp.16.01653. [Google Scholar] [CrossRef]
33. Li, S. Y., Zhao, B. R., Yuan, D. Y., Duan, M. J., Qian, Q. et al. (2013). Rice zinc finger protein DST enhances grain production through controlling Gn1a/OsCKX2 expression. Proceedings of the National Academy of Sciences of the United States of America, 110(8), 3167–3172. DOI 10.1073/pnas.1300359110. [Google Scholar] [CrossRef]
34. Jain, M., Nijhawan, A., Arora, R., Agarwal, P., Ray, S. et al. (2007). F-box proteins in rice. Genome-wide analysis, classification, temporal and spatial gene expression during panicle and seed development, and regulation by light and abiotic stress. Plant Physiology, 143(4), 1467–1483. DOI 10.1104/pp.106.091900. [Google Scholar] [CrossRef]
35. Li, M., Tang, D., Wang, K. J., Wu, X. R., Lu, L. L. et al. (2011). Mutations in the F-box gene LARGER PANICLE improve the panicle architecture and enhance the grain yield in rice. Plant Biotechnology Journal, 9(9), 1002–1013. DOI 10.1111/j.1467-7652.2011.00610.x. [Google Scholar] [CrossRef]
36. Arora, R., Agarwal, P., Ray, S., Singh, A. K., Singh, V. P. et al. (2007). MADS-box gene family in rice: genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genomics, 8(1), 242. DOI 10.1186/1471-2164-8-242. [Google Scholar] [CrossRef]
37. Komiya, R., Ikegami, A., Tamaki, S., Yokoi, S., Shimamoto, K. (2008). Hd3a and RFT1 are essential for flowering in rice. Development, 135(4), 767–774. DOI 10.1242/dev.008631. [Google Scholar] [CrossRef]
38. Wang, K. J., Tang, D., Hong, L. L., Xu, W. Y., Huang, J. et al. (2010). DEP and AFO regulate reproductive habit in rice. PLoS Genetics, 6(1), e1000818. DOI 10.1371/journal.pgen.1000818. [Google Scholar] [CrossRef]
39. Lu, S. J., Wei, H., Wang, Y., Wang, H. M., Yang, R. F. et al. (2012). Overexpression of a transcription factor OsMADS15 modifies plant architecture and flowering time in rice (Oryza sativa L.). Plant Molecular Biology Reporter, 30(6), 1461–1469. DOI 10.1007/s11105-012-0468-9. [Google Scholar] [CrossRef]
40. Lin, Q. P., Zong, Y., Xue, C. X., Wang, S. X., Jin, S. et al. (2020). Prime genome editing in rice and wheat. Nature Biotechnology, 38(5), 582–585. DOI 10.1038/s41587-020-0455-x. [Google Scholar] [CrossRef]
41. Zhang, H. W., Si, X. M., Ji, X., Fan, R., Liu, J. X. et al. (2018). Genome editing of upstream open reading frames enables translational control in plants. Nature Biotechnology, 36(9), 894–898. DOI 10.1038/nbt.4202. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |