DOI:10.32604/phyton.2020.012424
| Phyton-International Journal of Experimental Botany DOI:10.32604/phyton.2020.012424 | |
| Article |
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
Different Decaying Wood Effects on Bacterial Diversity: Insights from Molecular Methods
1College of Life Sciences, Northeast Forestry University, Harbin, 150040, China
2State Key Laboratory of Tree Genetics and Breeding, Northeast Forestry University, Harbin, 150040, China
*Corresponding Author: Shaopeng Yan. Email: ysp_4@126.com
#Mu Peng and Yanli Jing are co-authors with contributing equally to this paper
Received: 30 June 2020; Accepted: 13 August 2020
Abstract: Decaying wood is a novel key factor required for biodiversity and function of a forest, as it provides a good account of substrate and habitats for various organisms. Herein, the bacterial diversity in decaying wood of Betula platyphylla was discussed through high throughput sequencing. Our results showed that most of the obtained sequences belonged to the phyla Firmicutes, Proteobacteria, Bacteroidetes, Actinobacteria, Acidobacteria and Verrucomicrobia. Bacterial community compositions in samples with higher moisture content were obviously different than that with lower content, which could be reflected by richness estimators, diversity indices, and cluster and heatmap analysis. All three networks were non-random and possessed topological features of complex systems such as small-world and modularity features. However, these networks exhibited distinct topological features, indicating the potential ability of extensive cooperative and competitive interactions in the decayed wood microenvironments. Redundant analysis showed that most bacterial phyla were mainly distributed in higher-moisture trunks. The obtained data will increase the knowledge of the complex bacterial diversity associated with dead wood, and lay a foundation for the bioconversion technology of plant cell walls using bacteria.
Keywords: Decaying wood; Betula platyphylla; bacterial diversity; moisture content; network analysis
Decaying wood (DW) or dead wood is mostly caused by fire, wind, snow breakage, drought, competition, insects and pathogens in the natural forest [1,2]. In fact, DW is a key factor required for biodiversity and functioning of forests, as it provides nutrients and habitats for various organisms, such as insects, birds and microorganisms [3,4]. Additionally, DW influences and partakes in numerous ecosystem functions, including carbon sequestration and nutrient cycling in the world. To date, many studies in decaying wood have been researched under laboratory and field conditions [5]. These reports exhibited that DW was a determinant factor for biodiversity in forest ecosystems and took part in the biogeochemical cycle.
Microorganisms play an important role in the forest ecosystems with their large set of enzymes to decompose the different wood components such as cellulose and lignin [6]. They act as plant pathogens, mycorrhizal symbionts, and even they are considered to be the principal decomposers of complex organic matter, such as rotten wood or litter [7]. For the saprophytic bacteria, DW is an important structural component for their survival. Among them, Proteobacteria and Actinobacteria species are the major groups on the importance of maintaining forest ecosystem functioning as well as greatly promoting the biological cycle of nature [8]. Moreover, the highly active ligninolytic, cellulolytic, xylanolytic and pectinolytic enzyme systems produced by bacteria have clearly shown highly decomposability for wood in forests. For example, Sphagnum is able to hydrolyze pectin and holocellulose [9]. Hyperthermophilic bacterium Thermotoga maritima produces potent xylanase [10]. The endoglucanase of the bacterium Cellulomonas is reported to randomly attack the cellulose chain [11]. A wild type strain of Sphingobium sp. SYK-6 has been found to be involved in lignin degradation [12]. Bacteria like those belonging to Actinobacteria also have the ability to degrade cellulose and lignin [13].
Nowadays, numerous investigations have focused on the diversity of fungal communities and insects in DW. Mäkipää et al. [14] found that the species richness in dead wood increased along the decay gradient. Kazartsev et al. [15] detected the composition of fungal communities in different decomposition stages and suggested that the diversity of fungal communities is associated with decomposition. Yuan et al. [16] demonstrated that tree species are the main determinant of fungal composition in the decomposition stages, and that the fungal diversity reaches the highest level in the middle and late decomposition stages. The interactions between various physical and chemical factors and fungal communities have similar regulatory mechanisms and there is no specific influence of tree species [16].
However, the role of bacteria in DW has only been reported in a few case studies such as bacterial diversity in DW of Keteleeria evelyniana [17] and bacterial composition present in Pine-wood decomposition [5]. Apart from these studies, little attention is known about the bacterial community in Betula platyphylla. Therefore, we analyzed the bacterial diversity and communities during the stages of natural decay in an economically important tree, B. platyphylla, using 16S rRNA gene on the sequencing Illumina-Miseq platform. The data will provide a reference for the ecological functions of wood-degrading related bacterial communities and reveal the preliminarily underlying mechanisms of bacterial community changes in decaying wood.
Tree B. platyphylla mainly distributes in eastern Asia. In China, it is a dominant species in Northeast of China. It is traditionally used in forestation, ornamental and officinal purposes. The experimental site of Liangshui National Nature Reserve (E128°53’, N47°10’) is located in Heilongjiang Province, Northeast of China. The elevation is 280–707 m, the annual rainfall is 676 mm and the mean annual temperature is −0.3°C [18]. The native mixed forest is composed of pine (Pinus koraiensis), Mongolian oak (Quercus mongolica) and sporadic birch (B. platyphylla). A total of eighteen different B. platyphylla trunks (50 cm length, 10 cm diameter) were collected from fallen or standing dead trees based on different water content. The eighteen trunks were collected and grouped into six groups. Each group had three samples. Each sample was regarded as one independent experiment. The water content of decaying wood was furthermore used as important index of wood decay stages [19], and it was measured according to Cardias [20]. The bark was removed and slices of wood were collected using clean bags to the laboratory for further experiments. The cellulose, hemicelluloses, lignin and ash contents of samples were detected according to Ververis et al. [21].
2.2 DNA Extraction, PCR and Sequencing
The samples (0.1 g) for DNA extraction were ground and extracted using the protocol of E.Z.N.A. soil DNA Kit (Omega Bio-tek, Norcross, GA, US). DNA concentration was detected by NanoDrop2000 UV-VIS Spectrophotometer (Thermo Fisher Scientific Inc., Wilmington, DE, USA). PCR amplification was carried out using the primer set 806R: GGACTACHVGGGTWTCTAAT; 338F: ACTCCTACGGGAGGCAGCAG. The PCR conditions and Illumina Miseq 16S rRNA gene sequencing protocol were performed according to our previous report [22].
Raw data was optimized according to Peng et al. [22], and only filtered sequences by subsampling of sequences to the sample with the fewest reads (49749 reads) were analyzed in the next step. The 16S rRNA gene sequences were classified into operational taxonomic units (OTUs) within a 0.03 difference. OTUs classified as chimeras, chloroplast or mitochondria were removed using the MOTHUR program [23]. A representative set of sequences was then generated and assigned taxonomy using the SILVA database (Release123, http://www.arb-silva.de). Based on the species diversity of OTUs analysis, we analyzed the Alpha-diversity, rarefaction curve, Beta-diversity, clustering analysis. Alpha-diversity indices, including Chao, Ace, Shannon and Simpson, reflect the richness and diversity of microbial communities in each sample. Beta-diversity indices are based on the analysis of community structure and compare similarity or difference among samples containing Venn diagram, Redundant analysis (RDA), and a hierarchically clustered heatmap. In addition, a heatmap was analyzed through hierarchal clustering performed in the R software environment. The Richness estimators and diversity indices and wood characteristics data were used for multivariate regression tree (MRT) analysis by using the ‘mvpart’ (Multivariate partitioning) package in R. By species difference analysis, we compared the species distribution in different samples and verified its significance using Fisher’s exact test.
Based on OTU generated by QIIME, we constructed the ecological network of microbial communities using molecular ecological network analysis pipeline platform (http://ieg4.rccc.ou.edu/MENA) [24]. Some potential keystone taxa were identified according to the topological role of each node based on within-module (Zi) and among-module connectivity (Pi). When a node has Zi > 2.5 or/and Pi > 0.62, it is considered as a key taxon [25].
Data is the mean value with standard deviation (±SD) from all three independent experiments. Statistical analysis was performed using the ‘vegan’ package in the R software environment. Differences were considered statistically significant at p < 0.05.
All the bacterial raw sequences have been deposited to GenBank Short Read Archive (PRJNA593777).
Classification of the degree of wood decay was based on the increase of wood moisture content indicating progressive decay. The six decaying kinds of wood showed a gradient of moisture content levels with values ranging from 29.1 ± 1.5 to 85.1 ± 1.3% in the DW4 and DW3 samples, respectively (Tab. 1). DW4 and DW7, DW1 and DW6, DW3 and DW8 sample pieces had comparable concentrations of moisture content. The growth of wood-degrading bacteria requires a certain amount of inorganic substances. Therefore, the ash content could reflect the growth conditions of microorganisms. Among those samples, ash content ranged from 0.21% to 0.7% (Tab. 1). The highest value existed in DW4 and DW7, whereas the lowest ratio was found in DW3 and DW8. The cellulose content varied from 27.7% to 37.5%, all of which were lower than that of normal samples (about 45% of the dry weight of wood) [26]. In samples of DW1, DW6, DW4 and DW 7, no significant difference has been detected. The content of pentosan was from 10.26% to 18.37% in all samples, which was lower than that in normal plants (20%–30%), suggesting that all samples were subjected to microbial degradation in varying degrees. A significant difference of lignin content was detected in DW1, DW6 and DW3, DW8, indicating that the degree of decomposition was different. These results indicated that the degree of wood decay could alter wood components. Therefore, we set all samples into three stages, DW4 + DW7 (early), DW1 + DW6 (middle) and DW3 + DW8 (late).
Table 1: The moisture, wood ash, cellulose, pentosan and lignin contents of decayed wood samples

3.2 Species Richness and Diversity
A great amplitude of sequences was obtained for the eighteen samples, ranging from 49749 to 116238. To standardize the raw data, the subsampling of sequences to the sample with the fewest reads (DW3, 49749 reads) was analyzed in the next step. The total numbers of OTUs, Ace, Chao, Shannon and Simpson indexes in the DW3 and DW8 were the highest among all the samples (Tab. 2). The richness indices (ACE and Chao) showed increasing trends in different decaying levels. Additionally, the OTU numbers and bacterial diversity (Shannon index) in the decaying wood varied among the different levels, showing similar trends to richness indices. Rarefaction curves based on OTUs of 97% similarity were shown in Fig. S1; it was clearly observed that the samples had different species richness and the highest value existed in the DW3 and DW8.
Table 2: Richness estimators and diversity indices of each sample (97% similarity)

Additionally, we calculated the number of specific and shared bacterial OTUs among all samples. A network-like Venn diagram indicated that the six different decaying degree level samples shared 730 OTUs with the majority of OTUs identified to Proteobacteria (46.58%) and Actinobacteria (15.48%), suggesting that these two phyla were the core microorganisms in decaying wood (Fig. 1). We observed the highest number of samples specific bacterial OTUs (1370) in DW3 + DW8, suggesting that these two samples harbor the highest bacterial diversity. Shared OTUs (455) between the DW3 + DW8 and DW1 + DW6 accounted for the 15.03% of total OTUs, followed by 145 and 141 OTUs shared across DW1 + DW6 and DW4 + DW7, DW4 + DW7 and DW3 + DW8, respectively.
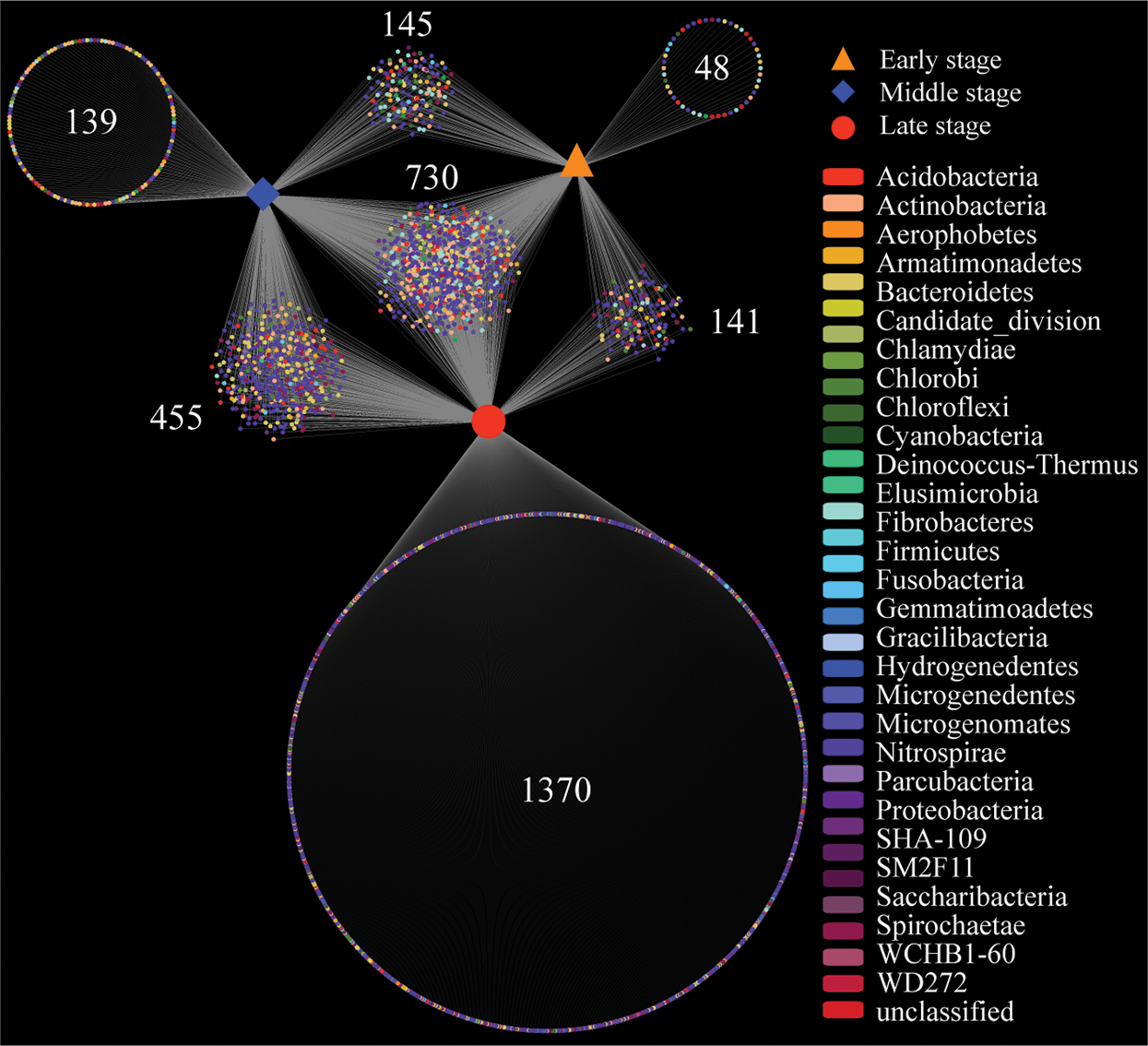
Figure 1: Venn analysis showing the number of specific and shared bacterial OTUs among all samples. Early stage means DW4 and DW7, middle stage means DW1 and DW6, and late stage means DW3 and DW8. Each dot represents bacteria phylum. Number means shared or unique OTU. DW: Decaying wood
3.3 Comparison of Microbial Community Composition
Bacterial community structures in the investigated samples were shown in Figs. 2 and 3. The majority of the obtained sequences, accounting for over 80% of the total sequences, belonged to the phyla Firmicutes, Proteobacteria, Bacteroidetes, Actinobacteria, Acidobacteria and Verrucomicrobia. Although those phyla were present in all samples, Chloroflexi, Chlamydiae, Parcubacteria, Planctomycetes and Saccharibacteria were most highly detected in DW8 and DW3. Firmicutes exhibited the highest abundance in the DW7 decaying wood (66%–70% of the total sequences), whereas it was less than 10% of the total sequences in DW8 and DW3 (Fig. 3A). Firmicutes belonged to the genera Lactococcus and Bacillus and constituted 5%–20% of the total sequences in the DW1, DW4, DW6, DW7, but less than 3% in DW3 and DW8 (Fig. 3B). The majority of Protecbacteria belonged to genus Dyella in DW4 (6%–19% of the total sequences), Pseudomonas and Stenotrophomonas in DW7 (10% and 5%, respectively), Burkholderia in DW6 (>6%) and DW1 (1%–11%), Xanthomonadales, Hyphomicrobium, Nitrosomonadaceae and Methylobacteriaceae in DW3 (<10% each), Acidibacter and Caulobacteraceae in DW8 (<6% each) (Fig. 3B). Bacteroidetes were the most dominant bacterial genus in DW3 (1%) but were rarely detected or absent in the others. Of the total acidobacterial sequences, Terriglobus were mainly encountered in DW4 and DW6, which made up over 2% of the total sequences. Actinobacteria-related sequences of the genus Acidothermus were detected in DW3 and DW1. The actinobacterial sequence was affiliated with the genus Microbacteriaceae, which was just found in DW8 (1%–4% of the total sequences). The bacterial group Verrucomicrobia belonging to genus Methylacidiphilum was detected in DW8 and DW3, with 3% and 0.7% of the total sequences, respectively (Fig. 3B). The remaining bacterial groups Chloroflexi, Parcubacteria, Planctomycetes and Saccharibacteria were detected only in DW8 and DW3 (Fig. 3A). The remaining, less dominant phylum Cyanobacteria was distributed in all samples with a low percentage (<1%).

Figure 2: Bacterial community compositions in decaying wood presented as relative abundance of taxa at the phylum level. Wood samples were grouped according to MRT analysis based on OTUs and wood characteristics. DW: Decaying wood
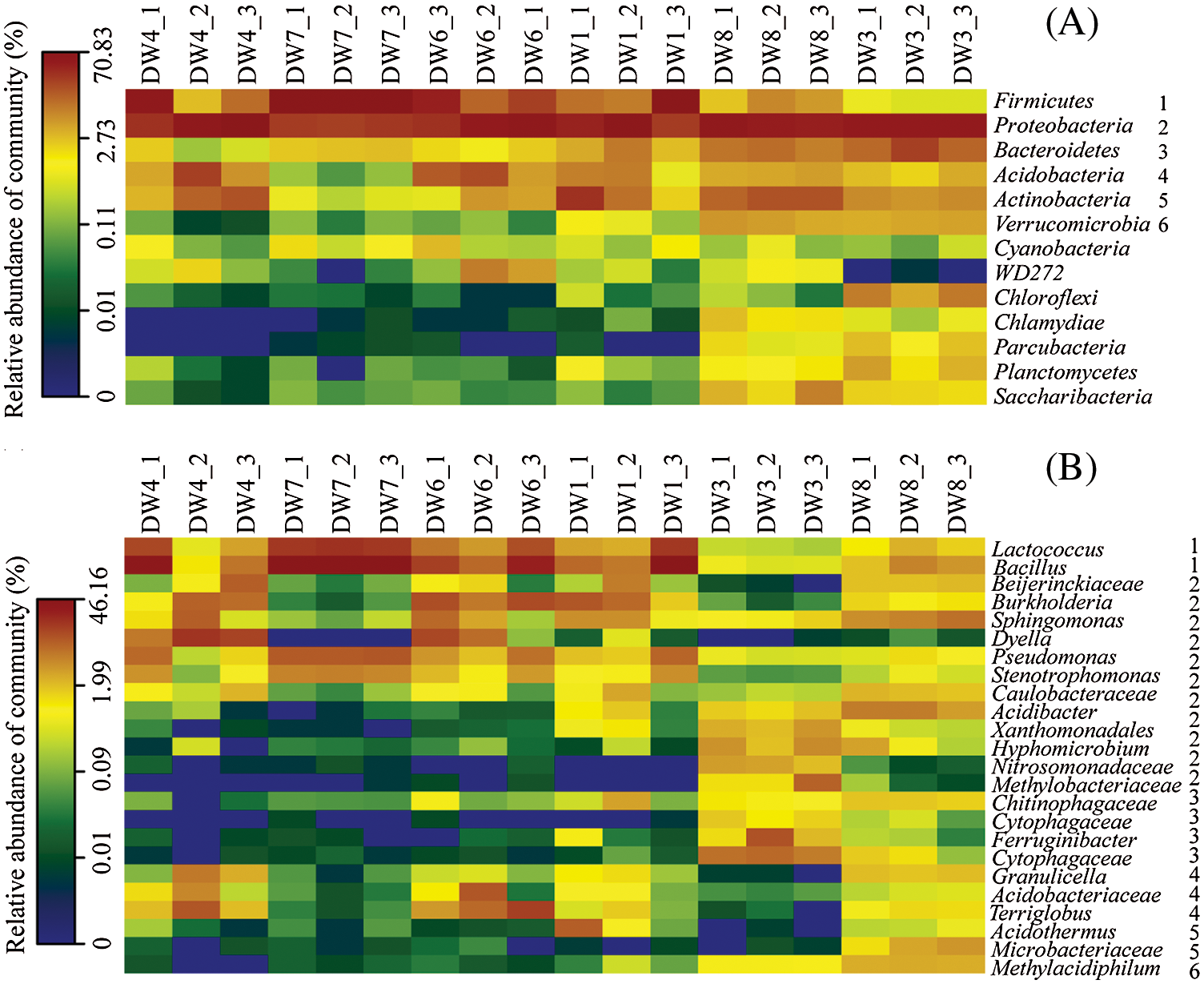
Figure 3: Heatmaps representing a comparison of the relative abundance of bacterial phylum (A) and the major bacterial genera (B) in the most dominant bacterial phylum among the different samples. The right number in B corresponds to phylum in A. Each box represents phylum or genera, and its color represents the relative abundance. DW: Decaying wood
Moreover, three complementary non-parametric statistical methods (Adonis, ANOSIM and MRPP) further revealed that the bacterial communities in different decomposition levels were significantly different (p < 0.05), except for comparison between early and middle (Tab. 3). These results indicated a positive correlation between decomposition levels and bacteria diversity. To support the non-parametric multivariate statistical test results and further ascertain which OTUs are responsible for the observed community differentiation among different decomposition level samples, we used species indicator analyses to discover significant associations between OTUs and decomposition levels. Here, we listed the first three species with highest indicator value in each decomposition levels. We found 3 indicator OTUs in the early stage (Rhodospirillales, Peptoniphilus, Flavobacterium, p < 0.05), 3 in the middle stage (Acetobacteraceae, Burkholderia, Burkholderia, p < 0.05), and 3 in the late stage (Reyranella, Bradyrhizobium, Starkeya, p < 0.01) (Tab. S1). Species indicator analysis revealed all indicator species with a low relative abundance (<1%), suggesting the potentially important role of rare groups in the community.
Table 3: Bacterial community dissimilarity comparison using three different statistical approaches

The OTUs obtained from sequencing were used to construct the bacterial community networks in three stages. The topological traits of each network showed a total of 313, 424, and 1414 nodes connected by dense connections with 2199, 1365 and 82304 edges in early, middle, and late stages, respectively, indicating that the composition of nodes and edges differed strikingly within each network (Tab. S2 and Fig. 4). The average path distance and clustering coefficient of all networks were significantly greater than that their corresponding random network, indicating that the observed networks have small-world properties. The values of network modularity in early and middle stages showed more modular structure due to their larger modularity values (Tab. S2). The modularity values of the three networks were all higher than the corresponding random networks, indicative of the modular feature. The node betweenness and degree were significantly lower and larger, respectively, for the late stage network than the other two networks, suggesting lower centrality and connectedness in the early and middle networks. Each network had a much higher number of strongly positive correlations (85.90%, 61.68%, and 74.13% in early, middle, and late stage network), respectively, than their negative correlations.

Figure 4: Polygenetic molecular ecological networks based on microbial communities in decayed wood and the topological roles of nodes. Green edge represents a negative interaction between two nodes, red edge represents a positive interaction. Each dot indicates an OTU. The color of dot means module. A, B and C represent the network in early (DW4 + DW7), middle (DW1 + DW6) and late (DW3 + DW8) stages, respectively. E, F and G represent the keystone taxa in early (DW4 + DW7), middle (DW1 + DW6) and late (DW3 + DW8) stages, respectively
In the topological role of nodes, most of them were in the peripheral area (Pi < 0.62, Zi < 2.5), and most have no connection with the external modules (Pi = 0) (Fig. 4). In addition, a total of 5, 5, and 4 module hubs (Pi < 0.62, Zi > 2.5) were found in early, middle, and late stages, respectively. A total of 3, 0, and 22 connectors (Pi > 0.62, Zi > 2.5) were found in early, middle, and late stages, respectively. However, no network hubs (Pi > 0.62, Zi > 2.5) were detected in these three networks.
We also used redundant analysis, which examines relationships in ecologic communities, to test for significant differences among the microbial communities and decaying wood traits. Results revealed that the samples markedly partitioned into two distinct groups along the RDA1 axis (Fig. 5). DW3 and DW8 samples were clustered together, revealing that the identified specific bacterial signatures in this group were different than the rest of the samples and formed a completely separate cluster, which was consistent with the dissimilarity comparison analysis (Tab. 3). Except for Acidobacteria and Firmicutes, all bacterial phyla were negatively correlated with most of the wood components, which indicated that the relative abundance of bacteria was higher with higher decay level and lower wood component contents. Meanwhile, Acidobacteria and Firmicutes were positively correlated with most of the wood components, implying that initial stage of wood degradation might induce an increase in the abundance of these two phyla. Furthermore, most of bacterial phyla were mainly distributed in higher-moisture areas.

Figure 5: Redundant analysis based on wood parameters and the relative abundance of bacterial phylum depicting the relationship among all samples. Solid circles represent the decaying wood sample. Arrows indicated the direction and magnitude of variables. DW: Decaying wood
Bacteria were considered to be closely associated with wood decay [19,27]. It has been clearly shown that bacterial communities of decaying wood in forests presented an unexpectedly high diversity [5,17]. In our study, the bacterial community composition of the DW3 and DW8 samples was obviously different from others, which could be reflected by richness estimators and diversity indices, cluster analysis, heatmap and RDA analysis (Tab. 2, Figs. 1–5). The main reason for this difference could relate to the physicochemical properties in each sample. Wood cannot be colonized by microorganisms unless its moisture content is above 25%–30% (as a percentage of oven-dry weight) [20]. At and above this point, the wood cell wall is fully swollen but below this point the wall is less swollen and this poses a greater constraint on accessibility than that at higher moisture contents [28]. Here, the moisture content in our decaying wood pieces ranged with large amplitude from 30% to 85%, suggesting that all samples harbored different bacterial richness and diversity (Tabs. 1 and 2).
In this study, the decay degree of DW3 and DW8 samples was the highest with the content of ash, cellulose, hemicellulose and lignin in these two samples was the lowest, suggesting that the long-term impact of microbial invasion had bad impacts on wood components (Tab. 1). Other researchers suggested that bacterial community assembly were a result of the changes in wood properties during decomposition [5]. The great heterogeneity of microenvironment determined different bacterial diversity of decaying deadwood, these difference includes moisture content and wood density in the dead standing tree and fallen decaying wood [5,29]. In general, wood decay begins with the bacterial invasion then increases the permeability and the moisture uptake of the wood, and then the subsequent fungal degradation can easily go on. In fallen or standing dead tree trunks, the cell lumens at higher moisture contents are filled with water and poor gaseous exchange could create a low oxygen tension, which resulted in the development of a diverse community of microorganisms [30]. Additionally, the moisture content might limit wood decay as in the low moisture content region where decomposer organisms required water for metabolism, whereas decomposition was apparently limited due to poor aeration in the high moisture content region [31]. Therefore, all the evidences could be attributed to the fact that moisture content could indirectly affect the species richness of decaying wood. Except for moisture content, the bulk density of lignin also effect on bacterial degradation, would further reduce the movement of water within the wood cell wall [32]. In addition, wood ash content affected the organic layer, microbial biomass, and microbial activity or community [33].
Here, our study reported the microbial communities of decaying wood was the distinct difference among samples even collected in the same place (Fig. 3). For example, despite the detection of Proteobacteria in all sample pieces, Alphaproteobacteria mainly found in DW3 and DW8 with a higher percentage of total sequences than the other samples, whereas Gammaproteobacteria exhibited the opposite. This was mainly because of the relative moderate humidity in the DW4 and DW7 during the decaying wood, which consequently promoted the growth of bacteria from Gammaproteobacteria. McEldowney et al. [34] have been previously reported the effect of relative humidity was less marked and varied with species and temperature, however, the relative humidity and temperature was 75% and 4°C for the growth of Gammaproteobacteria. In previous reports, Proteobacteria was shown to be members with degrading cellulose, lignocellulose and lignin capabilities [26]. Among the detected proteobacterial genera that potentially contain cellulose-degrading species in our samples were Acidibacter and Pseudomonas. Interestingly, sequences related to Bacillus were prevalent genera in all samples, suggesting that their vast distribution and adaptation to the environmental condition. This genus was frequently isolated from rainforest soils with the ability of lignin degradation [35], which enhanced the release of fermentable sugars from plant cell walls. In addition, cellulose is the most abundant biopolymer in plant biomass; however, it must first be hydrolyzed to a simple soluble substrate that can be used by bacteria. Some researchers have described genus Methylacidiphilum within the phylum Verrucomicrobia with a role in cellulose degradation [36]. In our experiment, Methylacidiphilum mainly detected in DW3 and DW8, which were decaying significantly, demonstrating that different stages of degradation caused by microorganisms could change the components of wood, which further stressed a strong selective pressure on the diversity and distribution of bacteria population in wood. Actinomycetes were considered the efficient cellulose degraders [37,38]. Among the detected lignocellulose-degrading actinobacterial genera in our samples were Acidothermus and Microbacteriaceae, which were consistent with the reports of Svetlitchnyi et al. [39] and Sighania et al. [40]. Most of the sequences detected in our decaying wood samples were related to wood-degrading strains that could contribute to the degradation of three major parts of plant biomass and play a key role in the bioconversion technology of plant cell walls.
Researches established a complex microbial system using five different bacterial genera, including Pseudomonas and Stenotrophomonas [41]. Their results showed that this system could produce different types of cellobiase with the ability to effectively degrade natural cellulose. Therefore, these strains have great potential in cellular-related pollution and waste utilization. Although high cellulase activity was detected in Brevundimonas, including carboxymethyl cellulose and filter paper enzyme, few studies used this strain to handle cellulose [42]. Compared to the bacterial community in the stomach of a different ruminant, researchers isolated a variety of species with the ability of cellulose degradation, such as Pseudomonas aeruginosa, Bacillus, Micrococcus and Streptococcus [43]. Chryseobacterium was isolated from mangrove soil and identified its cellulose ability [44]. All the above bacteria strains could be detected in DW1, DW4, DW6 and DW7 samples with the relatively high abundance, which showed significantly different compared with the abundance in DW3 and DW8 (Figs. 2 and 3). This finding speculated these genera mainly existed in the early or middle stages of wood degradation. In the late stages of degradation, the variety and abundance of wood-rotting bacteria reduced due to decreased wood components and fewer nutrients. Interestingly, we detected some wood-degrading strains in other samples, such as Burkholderia in DW6, which has the catalytic capacity of cellulose with effectively catalyze cellulose into reducing sugar [45]. Cytophagaceae, a family of gliding bacteria with dissolving plant fibers and hydrolyzing cellulose, were mainly found in DW3 samples (Fig. 3). Furthermore, various cellulolytic genus Acidothermus, Caldibacillus, Cellvibrio, Cellulomonas and Erwinia mainly produced endoglucanase, which also detected in our all samples with relatively low dominance.
Network analysis could infer the possible microbial associations in specific samples and detect the complexity of microbial associations [46]. In this study, we found that almost all nodes had a negative or positive relationship, indicating the potential ability of extensive cooperative and competitive interactions in the decaying wood microenvironments (Fig. 4). Note that the negative feedback between nodes in the network tends to stabilize the bacterial community assembly process, while the positive feedback enhances the changes of the ecosystem and destroys the current network structure [47]. All three networks were non-random and possessed topological features of complex systems such as scale-free, small-world and modularity features, which could enable system stability and resilience (Tab. S2). However, those networks exhibited distinct topological features. Similarly, this has been observed in other microbial networks [25,48,49]. In addition, Actinobacteria, Proteobacteria and Bacteroidetes were the keystone taxa, suggesting that these bacteria played an important role in maintaining the structure and function of ecological communities and had a large impact on the community composition [50]. Similar results were found in wood decay of pine, in which the majority of key nodes were Acidobacteria, Actinobacteria and Proteobacteria.
Each ecological network is composed of a certain number of nodes, and the topological role of each node reflects the potential importance of OTU in the bacterial community. Although module hubs or connectors are essential in a network, they do not always exist together [51]. Here, a different number of module hubs and connectors were detected in the networks of the three decomposition stages (Fig. 4). Studies have confirmed that more module hubs can maintain and stabilize the orderliness of microbial community structure [52]. In the late stage network, the highest number of module hubs and connectors were detected, indicating that the bacteria network in the late stage was more orderly. More connectors could organize a series of modules into a complete community, thus improving the efficiency of energy metabolism, nutrient cycling and material transformation in the environment [48]. In addition, the bacterial community based ecological network of decaying wood exhibited a highly modular structure. One possible explanation for these structural characteristics was the absence of key taxa, i.e., no network hub was detected in any network structure. In other taxa, network hubs play a disproportionately important role in maintaining network structure, and therefore, the disappearance of these key taxa might divide the network into more modules [25]. Under certain conditions, modules in the network can be regarded as functional units due to different niche division [53]. The modularization values of the bacterial network in early and middle stages were higher than the late stage network, which might be due to limited available niches in late decomposition stage, resulting in more clustered network structures.
In this study, we estimated the bacterial diversity in decaying wood of B. platyphylla. These findings revealed that moisture content in samples could indirectly affect the bacterial richness of decaying wood. All three networks exhibited distinct topological features, indicating the potential ability of extensive cooperative and competitive interactions in the decayed wood microenvironments. The microbiota of decaying wood remains a lively and underexplored area of ecological research that needs further focus on bacterial-fungi interactions and on assessing the role of these communities in lignocellulose recycling.
Funding Statement: This project was supported by the State Key Laboratory of Tree Genetics and Breeding (Northeast Forestry University), the Fundamental Research Funds for the Central Universities (No. 2572017AA23).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Lupala, A. S., Oh, S. Y., Park, M. S., Kim, T., Yoo, J. S. et al. (2019). Co-occurrence patterns of wood-decaying fungi and ants in dead pines of South Korea. Journal of Asia-Pacific Entomology, 22(4), 1154–1160. DOI 10.1016/j.aspen.2019.10.009.
2. Hood, I. A., McDougal, R. L., Somchit, C., Kimberley, M. O., Lewis, A. S. et al. (2019). Fungi decaying the wood of fallen beech (Nothofagus) trees in the South Island of New Zealand. Canadian Journal of Forest Research, 49(1), 1–17. DOI 10.1139/cjfr-2018-0179. [Google Scholar] [CrossRef]
3. Hawksworth, D. L. (2015). Books on biodiversity and conservation. Biodiversity and Conservation, 24(6), 1553–1566. DOI 10.1007/s10531-015-0860-5. [Google Scholar] [CrossRef]
4. Seibold, S., Bässler, C., Brandl, R., Gossner, M. M., Thorn, S. et al. (2015). Experimental studies of dead-wood biodiversity—A review identifying global gaps in knowledge. Biological Conservation, 191, 139–149. DOI 10.1016/j.biocon.2015.06.006. [Google Scholar] [CrossRef]
5. Kielak, A. M., Scheublin, T. R., Mendes, L. W., Van Veen, J. A., Kuramae, E. (2016). Bacterial community succession in pine-wood decomposition. Frontiers in Microbiology, 7, 231. [Google Scholar]
6. Janusz, G., Pawlik, A., Sulej, J., Świderska-Burek, U., Jarosz-Wilkołazka, A. et al. (2017). Lignin degradation: Microorganisms, enzymes involved, genomes analysis and evolution. FEMS Microbiology Reviews, 41(6), 941–962. DOI 10.1093/femsre/fux049. [Google Scholar] [CrossRef]
7. Molina, R., Horton, T. R. (2015). Mycorrhiza specificity: Its role in the development and function of common mycelial networks. Berlin: Springer. [Google Scholar]
8. Hervé, V., Le Roux, X., Uroz, S., Gelhaye, E., Frey-Klett, P. (2014). Diversity and structure of bacterial communities associated with Phanerochaete chrysosporium during wood decay. Environmental Microbiology, 16(7), 2238–2252. DOI 10.1111/1462-2920.12347. [Google Scholar] [CrossRef]
9. Bengtsson, F., Rydin, H., Hájek, T. (2018). Biochemical determinants of litter quality in 15 species of Sphagnum. Plant and Soil, 425(1–2), 161–176. DOI 10.1007/s11104-018-3579-8. [Google Scholar] [CrossRef]
10. Yang, J., Ma, T., Fang, S. G., Han, Z. (2020). Improving the catalytic activity of thermostable xylanase from Thermotoga maritima via mutagenesis of non-catalytic residues at glycone subsites. Enzyme and Microbial Technology, 139, 109579. DOI 10.1016/j.enzmictec.2020.109579. [Google Scholar] [CrossRef]
11. Kane, S. D., French, C. E. (2018). Characterisation of novel biomass degradation enzymes from the genome of Cellulomonas fimi. Enzyme and Microbial Technology, 113, 9–17. DOI 10.1016/j.enzmictec.2018.02.004. [Google Scholar] [CrossRef]
12. Meux, E., Prosper, P., Masai, E., Mulliert, G., Dumarçay, S. et al. (2012). Sphingobium sp. SYK-6 LigG involved in lignin degradation is structurally and biochemically related to the glutathione transferase omega class. FEBS Letters, 586(22), 3944–3950. DOI 10.1016/j.febslet.2012.09.036. [Google Scholar] [CrossRef]
13. Salwan, R., Sharma, V. (2018). The role of actinobacteria in the production of industrial enzymes. Netherlands: Elsevier. [Google Scholar]
14. Mäkipää, R., Rajala, T., Schigel, D., Rinne, K. T., Pennanen, T. et al. (2017). Interactions between soil- and dead wood-inhabiting fungal communities during the decay of Norway spruce logs. ISME Journal, 11(9), 1964–1974. DOI 10.1038/ismej.2017.57. [Google Scholar] [CrossRef]
15. Kazartsev, I., Shorohova, E., Kapitsa, E., Kushnevskaya, H. (2018). Decaying Picea abies log bark hosts diverse fungal communities. Fungal Ecology, 33, 1–12. DOI 10.1016/j.funeco.2017.12.005. [Google Scholar] [CrossRef]
16. Yuan, J., Zheng, X., Cheng, F., Zhu, X., Hou, L. et al. (2017). Fungal community structure of fallen pine and oak wood at different stages of decomposition in the Qinling Mountains, China. Scientific Reports, 7(1), 1–11. DOI 10.1038/s41598-016-0028-x. [Google Scholar] [CrossRef]
17. Zhang, H. B., Yang, M. X., Tu, R. (2008). Unexpectedly high bacterial diversity in decaying wood of a conifer as revealed by a molecular method. International Biodeterioration & Biodegradation, 62(4), 471–474. DOI 10.1016/j.ibiod.2008.06.001. [Google Scholar] [CrossRef]
18. Liu, M., Mao, Z., Li, Y., Li, X., Liu, R. et al. (2014). Climate effects on radial growth of Korean pines with different bark forms in Liangshui Natural Reserve, Northeast China. Chinese Journal of Applied Ecology, 25(9), 2511–2520. [Google Scholar]
19. Tláskal, V., Zrůstová, P., Vrška, T., Baldrian, P. (2017). Bacteria associated with decomposing dead wood in a natural temperate forest. FEMS Microbiology Ecology, 93(12). [Google Scholar]
20. Cardias Williams, F., Hale, M. D. (2003). The resistance of wood chemically modified with isocyanates: the role of moisture content in decay suppression. International Biodeterioration & Biodegradation, 52(4), 215–221. DOI 10.1016/S0964-8305(03)00070-2. [Google Scholar] [CrossRef]
21. Ververis, C., Georghiou, K., Danielidis, D., Hatzinikolaou, D. G., Santas, P. et al. (2007). Cellulose, hemicelluloses, lignin and ash content of some organic materials and their suitability for use as paper pulp supplements. Bioresource Technology, 98(2), 296–301. DOI 10.1016/j.biortech.2006.01.007. [Google Scholar] [CrossRef]
22. Peng, M., Zi, X., Wang, Q. (2015). Bacterial community diversity of oil-contaminated soils assessed by high throughput sequencing of 16S rRNA genes. International Journal of Environmental Research and Public Health, 12(10), 12002–12015. DOI 10.3390/ijerph121012002. [Google Scholar] [CrossRef]
23. Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M. et al. (2009). Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology, 75(23), 7537–7541. DOI 10.1128/AEM.01541-09. [Google Scholar] [CrossRef]
24. Deng, Y., Jiang, Y. H., Yang, Y., He, Z., Luo, F. et al. (2012). Molecular ecological network analyses. BMC Bioinformatics, 13(1), 113. DOI 10.1186/1471-2105-13-113. [Google Scholar] [CrossRef]
25. Shi, S., Nuccio, E. E., Shi, Z. J., He, Z., Zhou, J. et al. (2016). The interconnected rhizosphere: High network complexity dominates rhizosphere assemblages. Ecology Letters, 19(8), 926–936. DOI 10.1111/ele.12630. [Google Scholar] [CrossRef]
26. Pérez, J., Muñoz-Dorado, J., de la Rubia, T., Martínez, J. (2002). Biodegradation and biological treatments of cellulose, hemicellulose and lignin: An overview. International Microbiology, 5(2), 53–63. DOI 10.1007/s10123-002-0062-3. [Google Scholar] [CrossRef]
27. Johnston, S. R., Boddy, L., Weightman, A. J. (2016). Bacteria in decomposing wood and their interactions with wood-decay fungi. FEMS Microbiology Ecology, 92(11). DOI 10.1093/femsec/fiw179. [Google Scholar] [CrossRef]
28. Brischke, C., Soetbeer, A., Meyer-Veltrup, L. (2017). The minimum moisture threshold for wood decay by basidiomycetes revisited. A review and modified pile experiments with Norway spruce and European beech decayed by Coniophora puteana and Trametes versicolor. Holzforschung, 71(11), 893–903. DOI 10.1515/hf-2017-0051. [Google Scholar] [CrossRef]
29. Lin, P., Li, G. F. (2013). Diversity of rotten wood insects lived in Bombax ceibe in Mengla County. Forest Inventory and Planning, 28, 45–49. [Google Scholar]
30. Zhang, J., Presley, G. N., Hammel, K. E., Ryu, J. S., Menke, J. R. et al. (2016). Localizing gene regulation reveals a staggered wood decay mechanism for the brown rot fungus Postia placenta. Proceedings of the National Academy of Sciences of the United States of America, 113(39), 10968–10973. DOI 10.1073/pnas.1608454113. [Google Scholar] [CrossRef]
31. Zelinka, S. L., Kirker, G. T., Bishell, A. B., Glass, S. V. (2020). Effects of wood moisture content and the level of acetylation on brown rot decay. Forests, 11(3), 299. DOI 10.3390/f11030299. [Google Scholar] [CrossRef]
32. Williams, F. C., Hale, M. (2003). The resistance of wood chemically modified with isocyanates: The role of moisture content in decay suppression. International Biodeterioration & Biodegradation, 52(4), 215–221. DOI 10.1016/S0964-8305(03)00070-2. [Google Scholar] [CrossRef]
33. Fritze, H., Smolander, A., Levula, T., Kitunen, V., Mälkönen, E. (1994). Wood-ash fertilization and fire treatments in a Scots pine forest stand: Effects on the organic layer, microbial biomass, and microbial activity. Biology and Fertility of Soils, 17(1), 57–63. DOI 10.1007/BF00418673. [Google Scholar] [CrossRef]
34. McEldowney, S., Fletcher, M. (1988). The effect of temperature and relative humidity on the survival of bacteria attached to dry solid surfaces. Letters in Applied Microbiology, 7(4), 83–86. DOI 10.1111/j.1472-765X.1988.tb01258.x. [Google Scholar] [CrossRef]
35. Huang, X., Santhanam, N., Badri, D. V., Vivanco, J. M., Manter, D. K. et al. (2013). Isolation and characterization of lignin-degrading bacteria from rainforest soils. Biotechnology and Bioengineering, 110(6), 1616–1626. DOI 10.1002/bit.24833. [Google Scholar] [CrossRef]
36. Ranjan, K. (2011). Verrucomicrobia: a model phylum to study the effects of deforestation on microbial diversity in Amazon forest (Master of Science). The University of Texas, USA. [Google Scholar]
37. Qin, W. (2016). Recent developments in using advanced sequencing technologies for the genomic studies of lignin and cellulose degrading microorganisms. International Journal of Biological Sciences, 12(2), 156. DOI 10.7150/ijbs.11051. [Google Scholar] [CrossRef]
38. Saini, A., Aggarwal, N. K., Sharma, A., Yadav, A. (2015). Actinomycetes: A source of lignocellulolytic enzymes. Enzyme Research, 2015(20), 279381. DOI 10.1155/2015/279381. [Google Scholar] [CrossRef]
39. Svetlitchnyi, V. A., Kensch, O., Falkenhan, D. A., Korseska, S. G., Lippert, N. et al. (2012). Single-step ethanol production from lignocellulose using novel extremely thermophilic bacteria. Biotechnology for Biofuels, 6(1), 1–15. [Google Scholar]
40. Singhania, R. R., Sukumaran, R. K., Patel, A. K., Larroche, C., Pandey, A. (2010). Advancement and comparative profiles in the production technologies using solid-state and submerged fermentation for microbial cellulases. Enzyme and Microbial Technology, 46(7), 541–549. DOI 10.1016/j.enzmictec.2010.03.010. [Google Scholar] [CrossRef]
41. Ping, L., Wang, Y., Liu, K., Lei, T. (2009). Construction of a microbial system for efficient degradation of cellulose. Earth Science, 2, 344–347. [Google Scholar]
42. Zhang, J., Miao, T., Hou, H., Zhou, Q., Sun, H. et al. (2016). Isolation and identification of a cellulose-degrading strain T-6 and characterization of its enzyme production. Journal of Jilin Agricultural University, 38(1), 32–37. [Google Scholar]
43. Oyeleke, S. B., Okusanmi, T. A. (2008). Isolation and characterization of cellulose hydrolysing microorganism from the rumen of ruminants. African Journal of Biotechnology, 7(10), 1503–1504. [Google Scholar]
44. Behera, B., Parida, S., Dutta, S., Thatoi, H. (2014). Isolation and identification of cellulose degrading bacteria from mangrove soil of Mahanadi river delta and their cellulase production ability. American Journal of Microbiological Research, 2(1), 41–46. DOI 10.12691/ajmr-2-1-6. [Google Scholar] [CrossRef]
45. Matsumoto, K., Kobayashi, H., Ikeda, K., Komanoya, T., Fukuoka, A. et al. (2011). Chemo-microbial conversion of cellulose into polyhydroxybutyrate through ruthenium-catalyzed hydrolysis of cellulose into glucose. Bioresource Technology, 102(3), 3564–3567. DOI 10.1016/j.biortech.2010.09.098. [Google Scholar] [CrossRef]
46. Pérez-Valera, E., Goberna, M., Faust, K., Raes, J., García, C. et al. (2017). Fire modifies the phylogenetic structure of soil bacterial co-occurrence networks. Environmental Microbiology, 19(1), 317–327. DOI 10.1111/1462-2920.13609. [Google Scholar] [CrossRef]
47. Simard, S. W., Beiler, K. J., Bingham, M. A., Deslippe, J. R., Philip, L. J. et al. (2012). Mycorrhizal networks: Mechanisms, ecology and modelling. Fungal Biology Reviews, 26(1), 39–60. DOI 10.1016/j.fbr.2012.01.001. [Google Scholar] [CrossRef]
48. Wan, X., Gao, Q., Zhao, J., Feng, J., van Nostrand, J. D.et al. (2020). Biogeographic patterns of microbial association networks in paddy soil within Eastern China. Soil Biology and Biochemistry, 142, 107696. DOI 10.1016/j.soilbio.2019.107696. [Google Scholar] [CrossRef]
49. Banerjee, S., Kirkby, C. A., Schmutter, D., Bissett, A., Kirkegaard, J. A. et al. (2016). Network analysis reveals functional redundancy and keystone taxa amongst bacterial and fungal communities during organic matter decomposition in an arable soil. Soil Biology and Biochemistry, 97, 188–198. DOI 10.1016/j.soilbio.2016.03.017. [Google Scholar] [CrossRef]
50. Chao, Y., Liu, W., Chen, Y., Chen, W., Zhao, L. et al. (2016). Structure, variation, and co-occurrence of soil microbial communities in abandoned sites of a rare earth elements mine. Environmental Science & Technology, 50(21), 11481–11490. DOI 10.1021/acs.est.6b02284. [Google Scholar] [CrossRef]
51. Guimera, R., Mossa, S., Turtschi, A., Amaral, L. N. (2005). The worldwide air transportation network: Anomalous centrality, community structure, and cities’ global roles. Proceedings of the National Academy of Sciences of the United States of America, 102(22), 7794–7799. DOI 10.1073/pnas.0407994102. [Google Scholar] [CrossRef]
52. Xun, W., Huang, T., Li, W., Ren, Y., Xiong, W. et al. (2017). Alteration of soil bacterial interaction networks driven by different long-term fertilization management practices in the red soil of South China. Applied Soil Ecology, 120, 128–134. DOI 10.1016/j.apsoil.2017.08.013. [Google Scholar] [CrossRef]
53. Wu, L., Yang, Y., Chen, S., Zhao, M., Zhu, Z. et al. (2016). Long-term successional dynamics of microbial association networks in anaerobic digestion processes. Water Research, 104, 1–10. DOI 10.1016/j.watres.2016.07.072. [Google Scholar] [CrossRef]
Supplementary Files
Figure S1: Rarefaction curves based on the 16S rRNA gene sequencing of the decayed wood samples. DW: Decaying wood
Table S1: Indicator species analysis in decaying wood samples

Table S2: Key topological properties of the bacterial communities in the decaying wood samples
