
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE
Assessing the impact of CD73 inhibition on overcoming anti-EGFR resistance in glioma cells
1Programa de Pós-Graduação em Ciências Biológicas: Bioquímica, Instituto de Ciências Básicas da Saúde, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS 90035-003, Brazil
2Departamento de Bioquímica, Instituto de Ciências Básicas da Saúde, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS 90035-003, Brazil
3 Département de microbiologie-infectiologie et d’immunologie, Faculté de Médecine, Université Laval, Québec City, G1V 0A6, Canada
4 Axe maladies infectieuses et immunitaires, Centre de recherche du CHU de Québec, Université Laval, Québec City, G1V 4G2, Canada
* Corresponding Author: FABRÍCIO FIGUEIRÓ. Email:
# These two authors contributed equally to this work
Oncology Research 2025, 33(4), 951-964. https://doi.org/10.32604/or.2024.055508
Received 28 June 2024; Accepted 19 November 2024; Issue published 19 March 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Objectives: Glioblastoma (GB) is a grade IV glial tumor characterized by high malignancy and dismal prognosis, primarily due to high recurrence rates and therapeutic resistance. The epidermal growth factor receptor (EGFR), a receptor tyrosine kinase (RTK), regulates signaling pathways, including cell growth, proliferation, survival, migration, and cell death. Many cancers utilize immune checkpoints (ICs) to attenuate immune responses. CD73 is an enzyme that functions as an IC by hydrolyzing AMP to adenosine, suppressing immune cells in the tumor microenvironment. However, the role of CD73 in resistance to EGFR inhibitors is poorly understood. This study aims to elucidate the resistance mechanisms induced by anti-EGFR treatment and to evaluate an anti-CD73 approach to overcome resistance mediated by anti-EGFR monotherapy. Methods: The U251 GB cell line was treated with AG1478, an EGFR inhibitor, and the resistance markers MRP-1, PD-L1, and CD73 were evaluated using flow cytometry. Additionally, we assessed the combination effects of AG1478 and APCP (an EGFR and a CD73 inhibitor, respectively) on cell cycle progression, proliferation, apoptosis, and migration in vitro. Results: We observed high EGFR, PD-L1, and CD73 expression in human GB cells. The treatment with AG1478 increased the expression of resistance markers MRP-1, PD-L1, and CD73, whereas it decreased CTLA-4. The combination of AG1478 and APCP did not alter proliferation or apoptosis but interfered with cell cycling, arresting the cells in the G1 phase, decreasing cell motility and partially reversing MRP-1 overexpression. Conclusion: In summary, our findings indicate that CD73 inhibition has a modest effect in overcoming resistance to EGFR monotherapy in vitro. Thus, further in vivo studies are needed, as the inhibition of both EGFR and CD73 affects cells in the tumor microenvironment and could potentially enhance anti-tumor immunity.Keywords
Glioblastoma (GB) is the most prevalent and aggressive primary tumor of the central nervous system (CNS), accounting for 50.9% of all malignant brain tumors [1]. The World Health Organization (WHO) classifies GB as a grade IV malignancy, underscoring its highly invasive nature and poor prognosis [2]. Standard treatment protocols for GB typically include maximal surgical resection followed by radio- and chemotherapy with temozolomide (TMZ) [3]. Notwithstanding, the median survival for GB patients remains less than 15 months post-diagnosis [3,4]. Over the years, the therapeutic landscape for GB has seen minimal evolution despite the numerous clinical trials [5,6] conducted since establishing the current treatment paradigm in 2005 [4]. The rapid progression of tumors, the blood-brain barrier, the neuroplasticity and regenerative capacity of treatment-resistant GB stem cells, and the intricate cellular and molecular heterogeneity within the tumor microenvironment are significant obstacles to current treatment approaches [7–9].
Genome instability and sustained proliferative signaling are GB hallmarks, driving its aggressive behavior [10]. Among the genetic alterations in GB, the epidermal growth factor receptor (EGFR) is one of the most mutated genes (approximately 50% of cases) [11]. These alterations typically manifest as gene amplification (40%) or overexpression (60%) [12]. The most prevalent mutation, EGFR variant III (EGFRvIII), is a constitutively active form of the receptor, which is associated with enhanced tumor aggressiveness and a worse prognosis for GB patients [13]. Given the prominence of EGFR and EGFRvIII in GB pathophysiology, these molecules have been primary targets for therapeutic intervention. In this context, the tyrosine kinase inhibitor, tyrphostin AG1478, has been reported to effectively impair EGFR autophosphorylation and signaling in experimental GB [14–16]. Unfortunately, despite their theoretical promise, clinical trials targeting EGFR in GB have yielded disappointing results, with only a small subset of patients deriving meaningful therapeutic benefits [17,18]. This limited response is often linked to resistance emergence throughout treatment.
EGFR inhibitors have shown promising results in preclinical studies combined with other therapeutic agents [16,19,20]. Combining EFGR inhibitors with other target therapies might be an interesting strategy to minimize the acquired resistance and improve clinical outcomes through synergistic inhibition of downstream growth pathways [21]. In light of this, a significant association between EGFR and CD73 signaling pathways has been observed in several tumors [22,23]. CD73, also known as ecto-5′-nucleotidase, is a cell membrane protein responsible for hydrolyzing AMP into adenosine (ADO), an immunosuppressive molecule. CD73 overexpression has been identified in several cancer cells and tumor biopsies and is associated with reduced disease-free survival in GB patients [24,25]. CD73 interacts with extracellular matrix components to promote adhesion, cell growth, epithelial-mesenchymal transition (EMT), invasion and to assist in pro-metastatic processes [26–28]. Furthermore, extracellular ADO production mediated by CD73 activity contributes to immunosuppression [29,30], cell proliferation [26], angiogenesis [31], and chemoresistance [32]. Efforts to target CD73 as a potential cancer therapy have primarily aimed at inhibiting its immunomodulatory activity to render immunologically “cold” tumors sensitive to checkpoint inhibitors [33–37].
Therefore, in light of CD73’s role in cancer cell survival and progression, it would be insightful to explore whether inhibiting CD73 in combination with the EGFR inhibitor tyrphostin AG1478 could enhance therapeutic responses in a GB in vitro model. Thus, this study aimed to explore the potential of an anti-CD73 approach to overcome resistance mediated by anti-EGFR monotherapy.
Dulbecco’s modified Eagle’s medium (DMEM, cat#12100046), Fetal Bovine Serum (FBS, cat#26140079), Amphotericin B (cat#15290026), penicillin/streptomycin (cat#15140148), and trypsin-EDTA (cat#15400054) solution were acquired from Gibco (Gibco BRL, Grand Island, NY, USA). Dimethylsulfoxide (DMSO, cat#D2438), Tyrphostin AG1478 (AG1478, cat#T4182), adenosine 5′-(α, β-methylene) diphosphate (APCP, cat# M3763), temozolomide (TMZ, cat#T2577) and Trypan Blue (cat# T8154) were obtained from Sigma-Aldrich (Burlington, MA, USA). AnnexinV-APC and PI, Active Caspase-3, anti-human EGFR, anti-human MRP-1, anti-human PD-L1, anti-CTLA-4, anti-human CD73 were purchased from BD Biosciences (BD Bioscience, Franklin Lakes, NJ, USA).
The U251-MG, U87-MG, U138-MG, T98G, and A172 human GB cell lines were acquired from American Type Culture Collection (ATCC, Rockville, MD, USA). Cells were cultured and maintained in DMEM containing 10% FBS, 0.5 U/mL penicillin/streptomycin and 100 g/mL Amphotericin B, under humidified conditions (37°C, 5% CO2), for up to 30 passages. The cell lines were routinely monitored for Mycoplasma sp. contamination by PCR, following the method described by Fan et al. [38] and using the following primer sequences: F: ACACCATGGGAGCTGGTAAT; R: CGTAGGTTGTACTCCGTAGAAAGG.
The U251-MG cell line was seeded in 24-well plates (1.1 × 104 cells/well). When semi-confluence was reached, cells were treated with AG1478 (EGFR inhibitor, Sigma-Aldrich, Burlington, MA, USA) in concentrations of 5 to 80 µM for 48 h, under standard cell culture conditions (37°C in 5% CO2). Following 48 h of treatment, cells were washed twice with 1 × Phosphate Buffered Saline (1 × PBS, pH 7.4) and detached with 0.5% trypsin-EDTA solution. Then, cells were stained with Trypan Blue (0.1%) and immediately counted in a hemocytometer. In experiments investigating the role of the purinergic pathway, cells were plated and treated for 48 h with 35 μM AG1478 and/or 100 μM APCP (CD73 inhibitor, Sigma-Aldrich, Burlington, MA, USA) under standard cell culture conditions (37°C in 5% CO2). In TMZ experiments, the U251-MG were plated and treated for 48 h with 75, 250, and 500 µM of TMZ alone or in cotreatment with 35 μM AG1478.
The hemolytic capability of AG1478 was performed as previously described [39]. For this, peripheral blood samples of healthy donors (4 mL) were collected after a signed consent. The blood was centrifugated at 2500 g for 10 min (25°C, Eppendorf® Centrifuge 5804-R), and the erythrocytes were separated and washed three times with 1 × PBS (pH 7.0). Red blood cells were plated in 96-well plates (5 × 105 cells/well) and treated with 10–250 μM AG1478 for 48 h, under usual cell culture conditions (37°C, 5% CO2). All controls necessary for the experiment were used (negative control, vehicle control, and positive control-Triton X-100 0.1%, Sigma-Aldrich, Burlington, MA, USA). After the end of treatment, the plate was centrifuged at 2250× g per 10 min (25°C, Eppendorf® Centrifuge 5804-R) and the hemoglobin detected into the culture supernatant was quantified using absorbance at 540 nm (SpectraMax® M5, Molecular Devices). The degree of hemolysis was calculated using the hemolytic ratio. The data acquired from the samples treated with AG1478 (ODtest) were normalized to positive (Triton X-100 0.1%, ODpos) and negative (untreated, ODneg) control samples to provide the hemolytic ratio (HR), as defined by the formula (1):
Assessment of EGFR protein expression in GB cell lines
All flow cytometry experiments were conducted as previously reported by our group [40]. We assessed EGFR expression, in untreated samples, of the U251-MG, U87-MG, U138-MG, T98G, and A172 human GB cell lines. Briefly, cells were cultured in 24-well plates (1.1 × 104 cells/well), under typical cell culture conditions (37°C, 5% CO2), until they reached semi-confluence. Next, cells were washed with 1 × PBS (pH 7.4), trypsinized, and centrifuged twice at 400 × g for 6 min at 25°C. Following centrifugation, the cells were stained with anti-human EGFR (1:30, clone EGFR.1, cat#555997, BD Biosciences, Franklin Lakes, NJ, USA) for 30 min in the dark, on ice, and washed twice. Data acquisition was performed using flow cytometry (FACSCalibur, BD Biosciences, USA), and the results were assessed with FlowJo® v10.9.0 software (BD Biosciences, Franklin Lakes, NJ, USA). The most suitable GB cell line was chosen for subsequent experiments based on EGFR expression levels (U251-MG).
Protein expression by flow cytometry
For the protein expression assessment, U251-MG was seeded in 24-well plates (1.1 × 104 cells/well) and grown until semi-confluence, under standard cell culture conditions (37°C, 5% CO2). After, U251-MG was treated with AG1478 (35 µM) and/or 100 μM APCP for 48 h, also in standard in vitro culture settings (37°C, 5% CO2). At the end of treatments, cells were washed with blocking buffer (1 × PBS + 2% FBS) and stained with anti-human ecto-5′-nucleotidase/CD73 (1:200, cat. h5′NT-1L (I4,I5), J. Sevigny’s research lab, Canada, http://ectonucleotidases-ab.com, accessed on 18 November 2024 [41], FITC-conjugated goat anti-rabbit IgG secondary Ab (1:100, cat. 65-6111, Invitrogen, Austin, TX, USA), anti-human MRP-1 (1:50, clone QCRL-3, cat#557594), goat anti-mouse Alexa Fluor™ 488 (1:100, ThermoFisher, cat#A-11001), anti-Ki-67 (clone SolA15, 1:200; eBioscience, Cat# 11-5698-82), anti-human EGFR (1:30, clone EGFR.1, cat#555997), anti-human PD-L1 (1:100, clone MIH1, cat#558017), anti-CTLA-4 (1:10, clone BNI3, cat#560938) and/or anti-human CD73 (1:30, clone AD2, cat#550257) for 30 min on ice. Data were collected using flow cytometry (BD Accuri™ or FACSCalibur, BD Biosciences, USA) and analyzed with FlowJo® v10.9.0 software (BD Biosciences, Franklin Lakes, NJ, USA). A secondary antibody was used as a non-specific binding control (FITC-conjugated goat anti-rabbit IgG secondary Ab (1:100, cat. 65-6111, Invitrogen, USA).
Annexin V-APC/PI and cell cycle
To evaluate cell death and cell cycle, 1.3 × 104 cells/well of U251-MG cells were cultured in 24-well plates. After 24 h, cells were treated with IC50 values of AG1478 (35 μM) and/or 100 μM APCP for 48 h, under standard cell culture conditions (37°C, 5% CO2). Apoptotic and/or necrotic cells were quantified with Annexin V APC/Propidium Iodide (PI) double staining kit following the manufacturer’s guidelines (BD Biosciences, Franklin Lakes, NJ, USA). The U251-MG cells (105 cells/mL) were suspended in a buffer comprising Annexin V-APC and PI for 15 min (BD Biosciences, San Diego, CA, USA, cat#550474). For cell cycle analysis, U251-MG cells (106 cells/mL) were suspended in 300 µL staining solution containing Tris-HCl (0.5 mM, pH 7.6), trisodium citrate (3.5 mM), Nonidet P 40 (0.1% v/v), RNase (100 µg/mL, cat#R6513, Sigma-Aldrich, Burlington, MA, USA) and PI (50 µg/mL, cat#P4170 Sigma-Aldrich, Burlington, Massachusetts, USA), for 15 min in the dark. Data were acquired by a flow cytometrer (BD Accuri™ or FACSCalibur, BD Biosciences, USA), and analyzed using FlowJo® v10.9.0 software (BD Biosciences, Franklin Lakes, NJ, USA). For apoptotic cell death, cells were classified as follows: viable cells (Annexin V and PI negative), early apoptotic cells (Annexin V positive and PI negative), and late apoptotic or necrotic cells (Annexin V and PI positive or Annexin V negative and PI positive).
Chemoresistance protein functional assay—CFDA
ThermoFisher® CFDA AM (5-Carboxyfluorescein Diacetate, Acetoxymethyl Ester, cat#C1354, ThermoFisher®, Franklin, MA, USA) probe was used to assess the capacity of some transmembrane transporters, which can be used by neoplastic cells as efflux pumps for antineoplastic drugs [42]. In the experiment that investigated the acute effect of treatment under efflux pumps, shown in Fig. 4C, cells were treated with IC50 values (35 µM) of AG1478 only in the CFDA incubation period, at the influx time. In the experiment that investigated the chronic effect of co-treatment, U251-MG cells were cultured in 6-well plates (8 × 104 cells/well) and, after 24 h, were treated with IC25 values of AG1478 and 100 µM APCP for 48 h. Next, the cells were trypsinized and washed with 1 × PBS to initiate the chemoresistance functional assay, shown in Fig. 4D. To measure influx, cells were incubated with the probe (2 µM) in medium without FBS for 30 min. For the efflux, cells were incubated in media without FBS or CFDA for 90 min. During the influx and efflux, aliquots were taken to detect CFDA probe fluorescence and, consequently, the ability of the cells to extrude the probe by flow cytometry, using the cytometer BD Accuri C6™.
Active caspase-3 immunocontent
We analyzed the immunocontent of active caspase-3 per the manufacturer’s instruction (PE Active Caspase-3 kit, 1:30, cat#550914, BD Biosciences, USA). In summary, U251-MG (106 cells/mL) were incubated in BD Cytofix/Cytoperm™ solution (20 min on ice). Following this step, cells were washed with BD Perm/Wash™ buffer (1×) and stained with active caspase-3 antibody (PE Active Caspase-3 kit, cat#550914, BD Biosciences, USA) for 30 min at room temperature (RT) and then analyzed by the flow cytometer FACSCalibur (BD Biosciences, Franklin Lakes, NJ, USA). The results were assessed using FlowJo® v10.9.0 software (BD Biosciences, Franklin Lakes, NJ, USA).
To assess the indication of cell migration, we performed the in vitro scratch assay following standard methods with modifications [43]. In brief, U251-MG were cultured in a 6-well plate (7 × 104 cells/well) for 24 h under standard cell culture conditions (37°C, 5% CO2). A straight-edged and cell-free zone across the monolayer was established by employing a 200 µL sterile plastic micropipette tip to create a cross-wound in the bottom of the well. Next, the medium was replaced, containing AG1478 (35 μM) and/or 100 μM APCP and cells were incubated for 48 h in typical in vitro conditions (37°C, 5% CO2). Wound healing was analyzed by the Nikon Eclipse TE300 phase contrast microscopy (Melville, NY, USA) and images of each well were acquired at 0, 24, and 48 h with a Nikon Digital Camera DXM1200C (Düsseldorf, Germany). The scratch closure rate was analyzed by digitally drawing lines to represent the position of the migrating cells at the wounded edges with ImageJ® software (NIH, Bethesda, MD, USA).
To assess the migratory capacity of U251-MG cells, we performed the transwell migration assay following standard methods, with modifications [44]. The cell migration was evaluated using 24-well plates with Transwell inserts (8-μm pore size, cat#353097, Falcon®). Briefly, U251-MG cells (5 × 104) were resuspended in 200 μL serum-free DMEM and added into the upper chambers. A total volume of 600 µL DMEM supplemented with 10% FBS was added to the lower chamber. The treatments were added in the suspension of U251MG cells at the concentration of 100 μM for APCP, 35 μM for AG1478, and co-treatment with APCP+AG1478 was also added. Following 48 h of treatment, the inserts were removed and cells remaining on the upper surface of the inserts were carefully removed using a cotton swab. Cells that spread to the underside surface were fixed with 20% methanol (cat#67-56-1, LiChrosolv®, Merck, Burlington, Massachusetts, USA) for 15 min (RT) and stained with 1% crystal violet for 20 min. Following the staining, five fields were randomly chosen and photographed using a microscope (EVOS XL Core system, Invitrogen™, Oslo-Norway) for cell counting.
The Shapiro-Wilk test was carried out to verify multivariate normality. Kruskal-Wallis, a non-parametric test, was performed for data displaying non-normal distribution. Conversely, for data with normal distribution, a parametric test (one-way analysis of variance-ANOVA), followed by Tukey post-test was performed. All statistical data were carried out GraphPad Prism v8.0 software® (San Diego CA, USA). Data were expressed as the mean ± SD of at least three independent experiments. Statistical significance was determined as p < 0.05.
AG1478 reduces glioblastoma cell viability without affecting healthy cell viability
First, EGFR expression was analyzed by flow cytometry in different human GB cell lines. U251-MG cells exhibited more EGFR-positive cells and a higher EGFR expression level than the other cell lines (Fig. 1A,B). Based on cell viability assay, 48 h of AG1478 treatment resulted in an IC50 value of 35 μM (Fig. 1C), accompanied by morphological changes indicative of cell death (Fig. 1D). To analyze potential cytotoxicity to healthy cells, a dose curve of AG1478 was carried out in the presence of red blood cells. There was no increase in hemolysis at any drug concentrations compared to the vehicle group (Fig. 1E). Also, regardless of concentration, the combination of AG1478 with TMZ has no synergistic effects on cell viability (Fig. 1F).
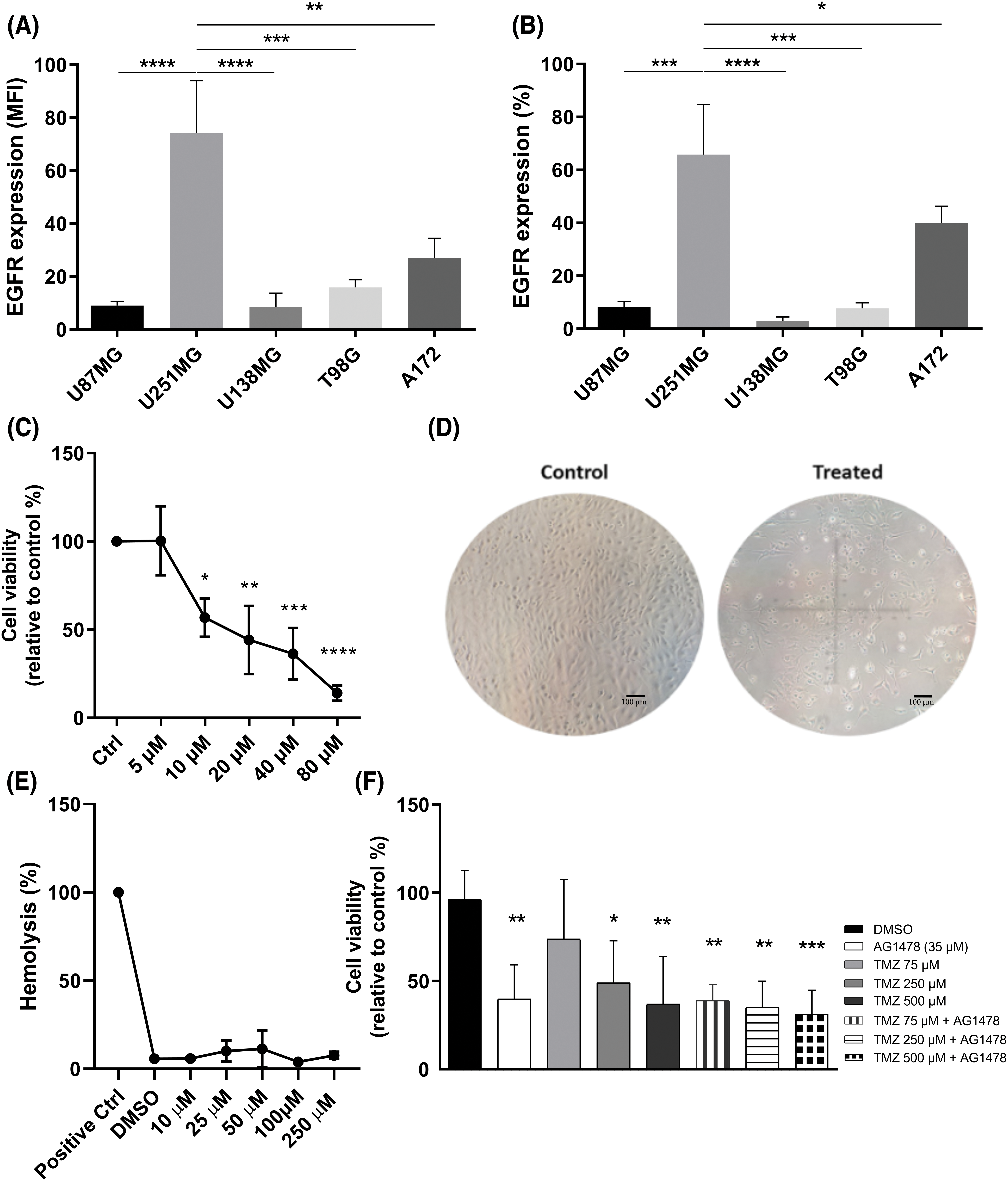
Figure 1: EGFR expression in human glioblastoma lines. (A) median fluorescence intensity (MFI) (B) percentage of cells expressing EGFR. Expression analysis was performed by flow cytometry. (C) Viable U251-MG cells exposed to treatment with Tyrphostin AG1478 for 48 h. (D) Cell morphology of U251-MG cells in the control group (Scale bar = 100 µm) (left) and in the AG1478-treated cells (right). Image obtained by microscopy. (E) Hemolysis of red blood cells treated with Tyrphostin AG1478 for 48 h. (F) U251-MG cells treated with Tyrphostin AG1478 and Temozolomide for 48 h. Cell viability analyses were performed by counting with Trypan Blue. *p < 0.05; **p < 0.01: ***p < 0.001; ****p < 0.0001.
Expression of CD73, PD-L1 and EGFR in U251MG
The expressions of essential proteins for glioma progression, CD73, PD-L1, and EGFR in U251MG cells were also evaluated. There was a high percentage of cells expressing CD73 and EGFR markers, and around 50% of cells expressing PD-L1 (Fig. 2A). Regarding the fluorescence intensity, GB cells overexpress CD73 compared to EGFR and PD-L1 (Fig. 2B). Afterwards, the total number of CD73-positive U251MG cells was divided into low expression of CD73 (up to 20% of CD73+ cells), intermediate expression of CD73 (the middle 50% of CD73+ cells) and high expression of CD73 (20% of CD73+ overexpressing cells) (Fig. 2C). EGFR and PD-L1 expression was not different in these three distinct subsets of CD73+ cells (Fig. 2D,E).
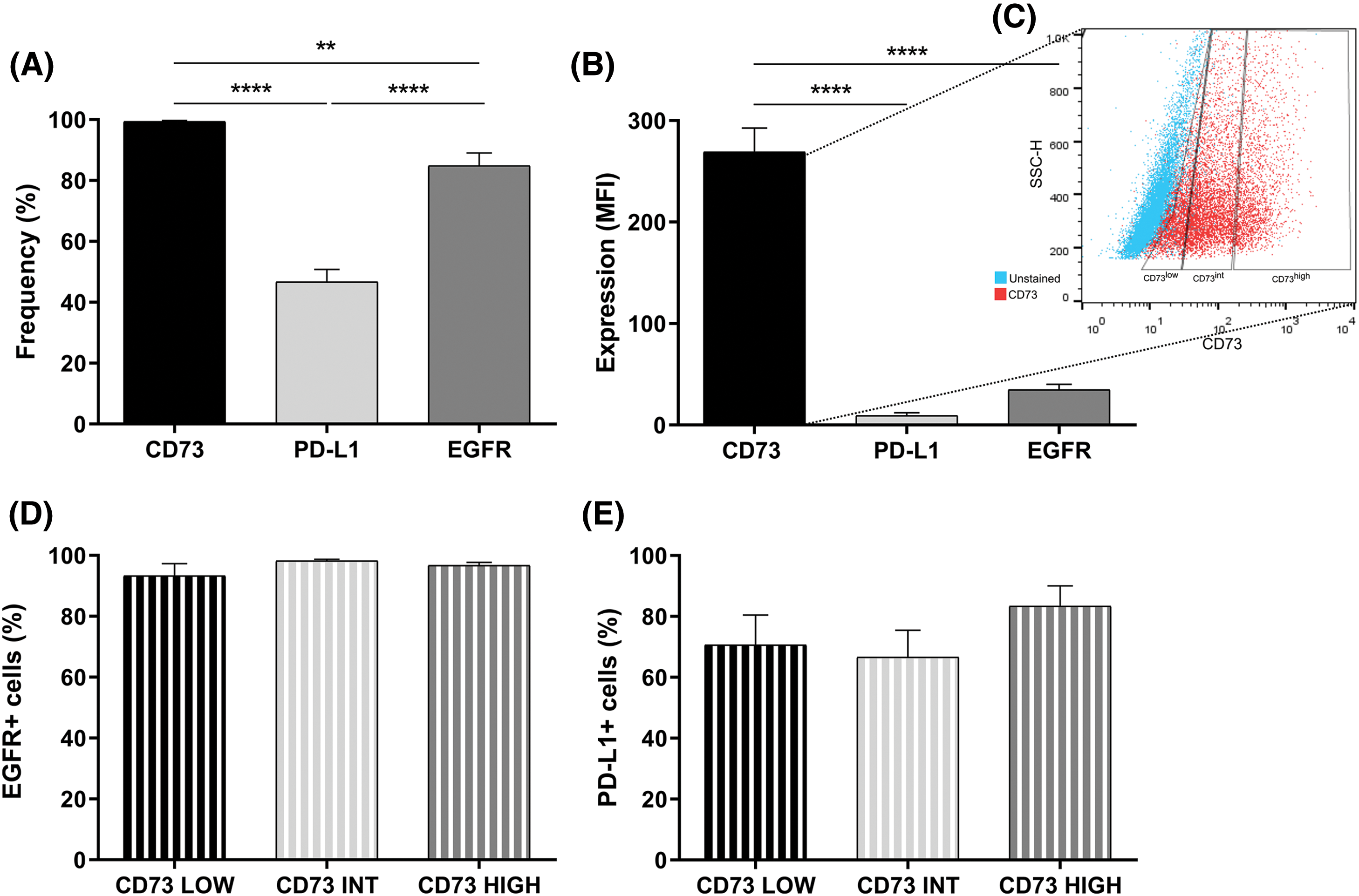
Figure 2: CD73, PD-L1, and EGFR expression in U251-MG cells. (A) Percentage and (B) median fluorescence intensity (MFI) of cells expressing CD73, PD-L1, and EGFR. (C) Subdivision of U251-MG cells in low, intermediate, and high CD73 expression. (D) EGFR expression in U251-MG cells with low (20%), intermediate (50%) or high expression of CD73 (20%) (E) Expression of PD-L1 in U251MG cells with low (20%), intermediate (50%) or high expression of CD73 (20%). Data determined by flow cytometry, n = 3. Data are displayed as means ± SD (**p < 0.01, ****p < 0.0001).
Effect of anti-EGFR treatment on immune checkpoints and resistance
To evaluate the effect of the EGFR inhibitor AG1478 in inducing resistance through immunological and tumoral pathways, the markers CTLA-4, PD-L1, CD73, and MRP-1 were assessed. AG1478 partially decreased CTLA-4 expression (Fig. 3A,B). However, the treatment increased PD-L1, CD73, and MRP-1 protein expression (Fig. 3C–H).
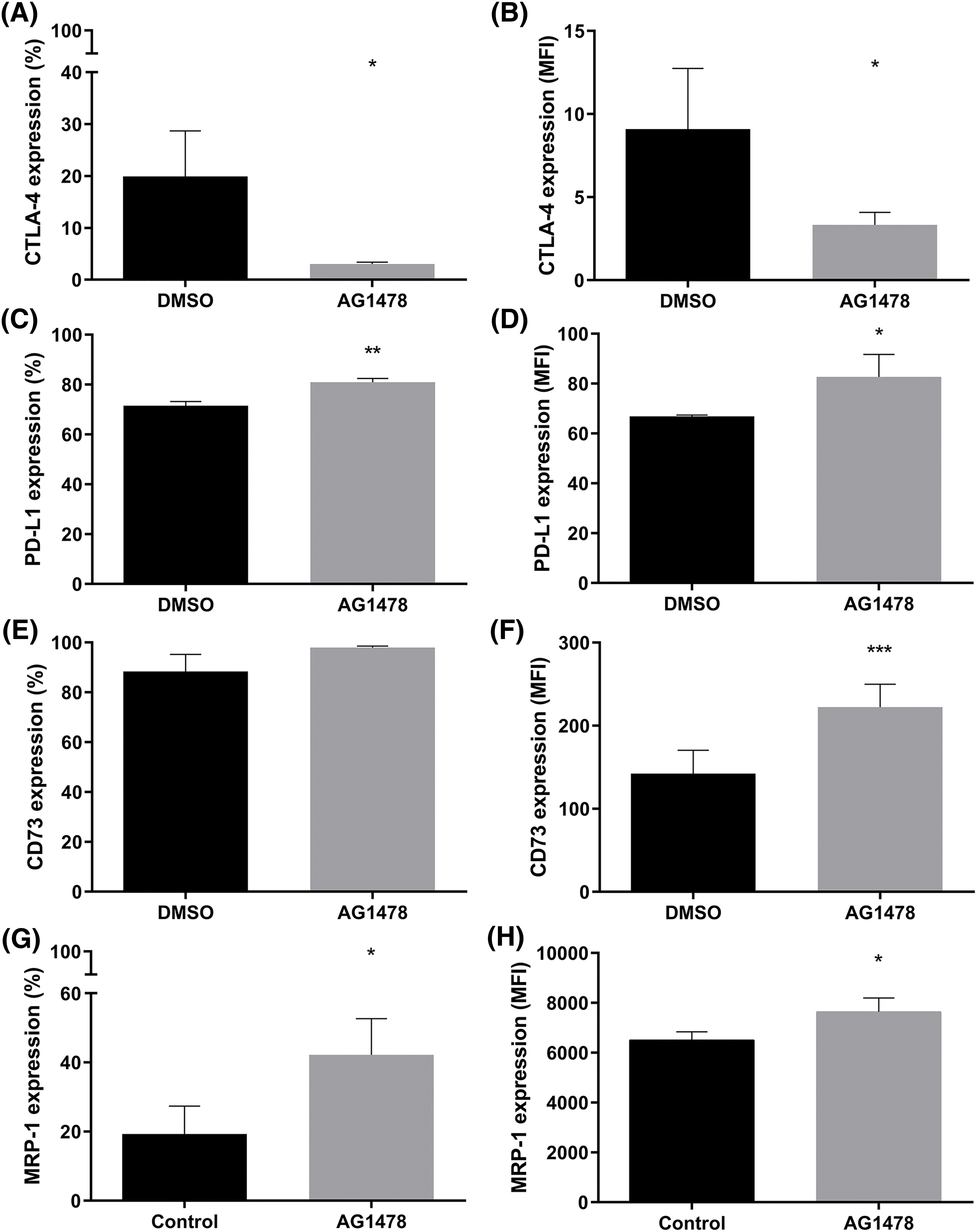
Figure 3: Resistance markers in the EGFR inhibitor AG1478-treated GB cells. (A) Percentage and (B) median fluorescence intensity (MFI) of cells expressing CTLA-4 treated with AG1478. (C) Percentage and (D) MFI of cells expressing PD-L1 treated with AG1478. (E) Percentage and (F) MFI of cells expressing CD73 treated with AG1478. (G) Percentage and (H) MFI of MRP-1 expressing cells treated with AG1478. Data obtained by flow cytometry, n = 3. Data are shown as means ± SD (*p < 0.05, **p < 0.01, ***p < 0.001).
The influence of EGFR inhibition on the purinergic pathway
To further explore the interaction between EGFR and CD73 inhibition, we sought to analyze the effects of these combined treatments on the expression of EGFR and MRP-1. The percentage of cells expressing MRP-1 was not significantly changed when comparing AG1478 and AG1478+the CD73 inhibitor APCP (Fig. 4A). On the other hand, APCP partially reversed the upregulation on MRP1 induced by AG1478 (Fig. 4B). However, both acute exposure and 48-h treatment with AG1478 did not change the functional activity of drug efflux pumps (Fig. 4C), even in the presence of APCP (Fig. 4D). In addition, AG1478 was able to reduce EGFR expression, apart from its well-known antagonistic effect (Fig. 4E). Interestingly, EGFR expression after combined treatment was reverted to control parameters (Fig. 4F).
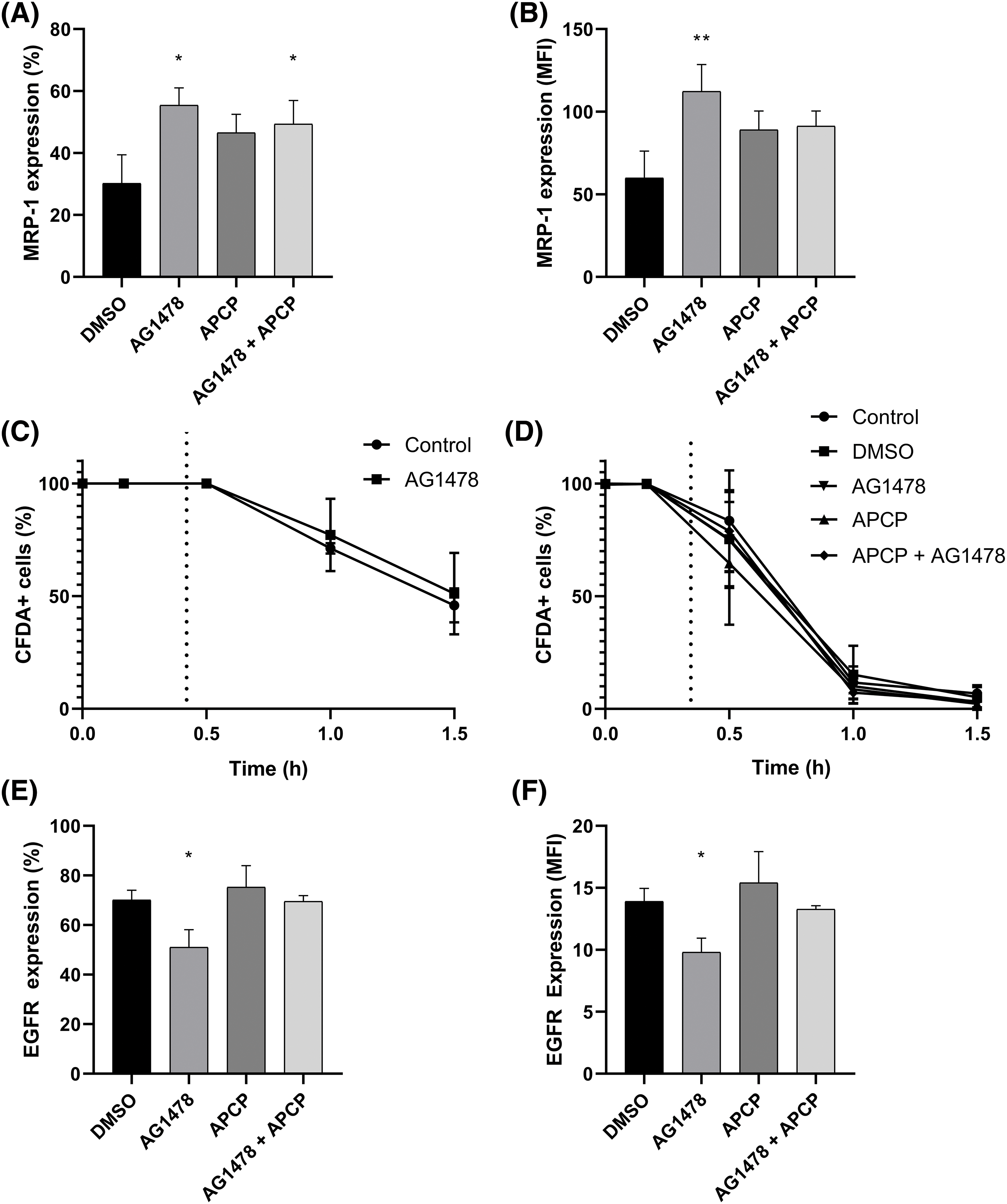
Figure 4: Cell viability and expression of MRP-1 and EGFR U251MG cells treated with Tyrphostin AG1478 associated or not with APCP for 48 h. (A) Percentage and (B) median fluorescence intensity (MFI) of cells expressing MRP-1 compared to control DMSO. (C) Activity of efflux drugs from cells treated with AG1478 acutely and (D) chronically—after 48 h as determined by flow cytometry. (E) percentage and (F) Median fluorescence intensity (MFI) of cells expressing from cells expressing EGFR compared to control DMSO. Data was collected by flow cytometry. Data are shown as mean ± SD (*p < 0.05, **p < 0.01).
The cell death characteristics after co-inhibition of EGFR and CD73
Next, we shifted to the perspective of resistance emergence. As shown in Fig. 5A, APCP did not change the cytotoxic effect of AG1478. Moreover, as shown in Fig. 5B,C, AG1478 and AG1478+APCP did not alter the percentage of cells expressing active caspase-3 or affect apoptosis/necrosis parameters differently. Likewise, there were no significant differences in Ki-67 expression among the groups (Fig. 5D). AG1478 arrested the cells in the G1 phase compared to controls with a consequent reduction of cells in the S phase. This arrest in the G1 phase was even greater in the combined treatment group results (Fig. 5E).
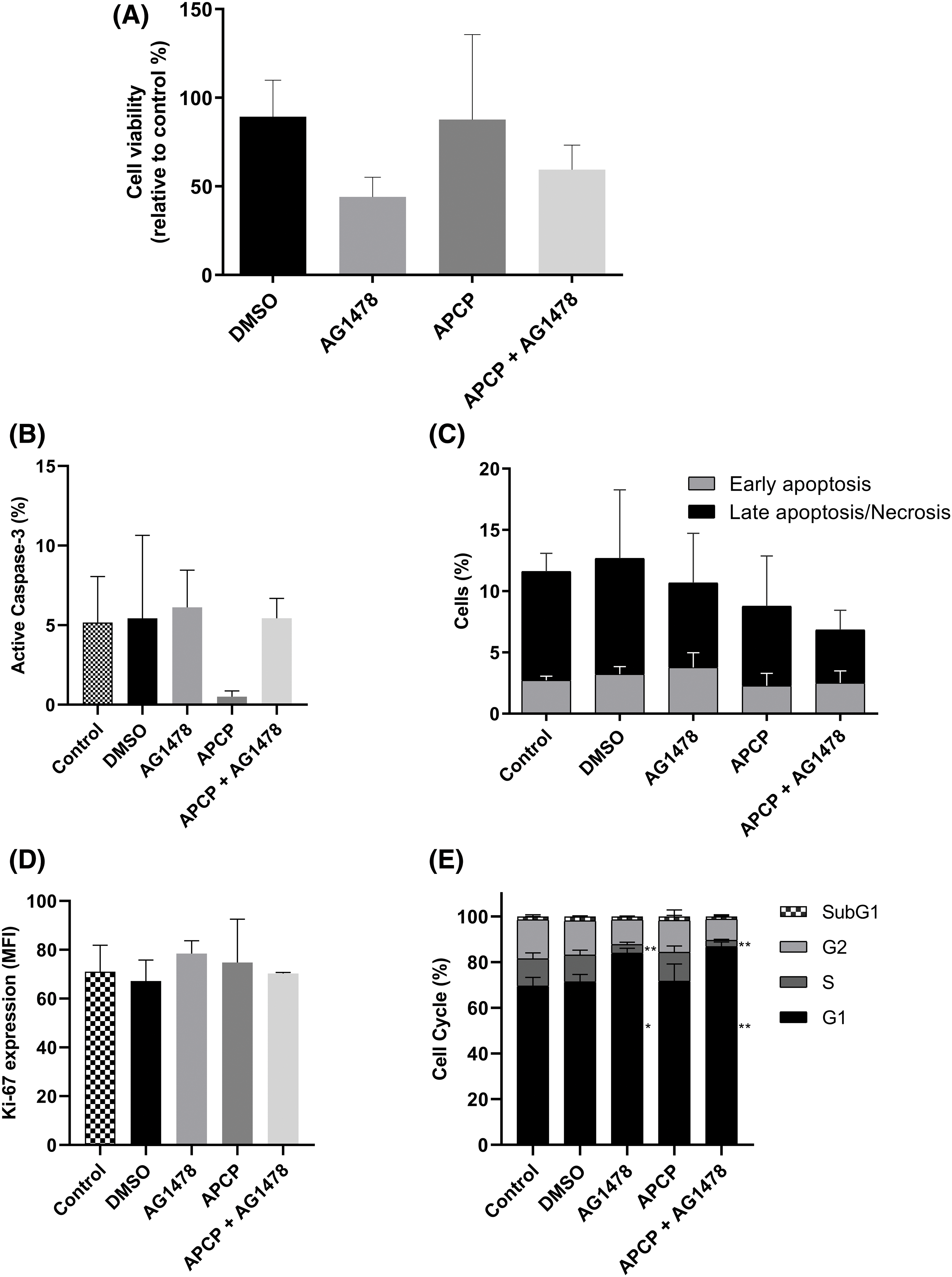
Figure 5: Cell death of GB cells after treatments with APCP and AG1478. (A) Viability of cells treated with AG1478 and APCP combined or not. Data determined by Trypan blue exclusion count. (B) Percentage of cells expressing active caspase-3. (C) Cells undergoing apoptosis or necrosis as determined by flow cytometry. (D) Expression of Ki-67 by median fluorescence intensity (MFI). (E) Percentage of cells in each cell cycle stage. Data are shown as means ± SD (*p < 0.05, **p < 0.01).
AG1478 and AG1478+APCP interfere in GB cells’ motility
To explore the effect of the dual inhibition of CD73 and EGFR on cell malignancy, we performed cell migration assays. Considering the transwell assay, AG1478 and AG1478+APCP reduced cell migration after 48 h of treatment (Fig. 6A,B), whereas only AG1478+APCP presented statistically significant changes in the wound healing assay (Fig. 6C,D).
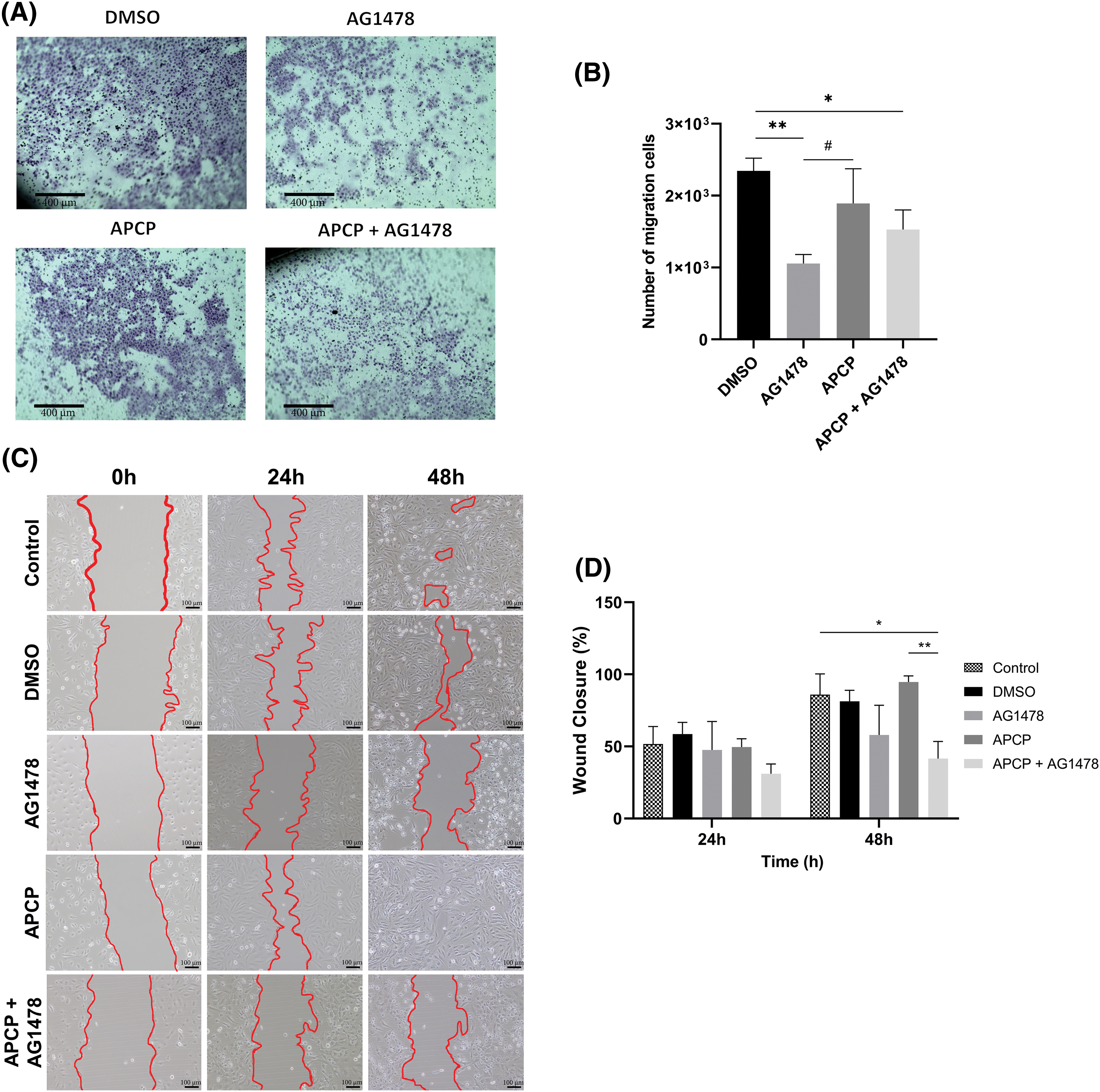
Figure 6: Migration of GB cells after APCP and AG1478 treatments. (A) Representative images of crystal violet staining cells after transwell assay (Scale bar = 400 µm). (B) Quantitative analysis of staining cells following AG1478 and APCP treatments (C) Microscopy image representative of wound closure at 0, 24 and 48 h of exposure to treatments (Scale bar = 100 µm) (D) Wound closure rate at 24 and 48 h of treatments. Data are shown as means ± SD (#p < 0.05, *p < 0.05, **p < 0.01).
GB is the most prevalent primary brain tumor in adults, characterized by significant heterogeneity and a poor prognosis. Although extensive research has focused on uncovering the intracellular mechanisms driving GB aggressiveness and therapeutic resistance, an effective cure remains elusive. The development of new drugs for GB is hindered by several challenges, including the tumor’s high phenotypic heterogeneity, proliferative and invasive capacity, the presence of glioblastoma stem cells (GSCs), the immunological suppression, the complex interactions between GB cells and the dynamic tumor microenvironment, and acquired resistance [45]. Combining targeted therapies that address multiple cancer drivers may offer an effective strategy to overcoming resistance. Thus, this study aimed to elucidate the effects of combined EGFR and CD73 inhibition in comparison to EGFR monotherapy in vitro. Our results revealed elevated EGFR, PD-L1, and CD73 expression levels in the U251-MG cell line. Treatment with AG1478, an EGFR inhibitor, increased resistance markers such as MRP-1, PD-L1, and CD73. On the other hand, the combination of AG1478 with APCP, a CD73 inhibitor, inhibited cell growth through G0/G1 arrest, reduced cell motility, and partially reversed MRP-1 overexpression. To our knowledge, this is the first study to assess the combination of CD73 and EGFR inhibitors in an in vitro GB model.
Firstly, we observed considerable variability of EGFR expression in GB cell lines, which correlates with the heterogeneity of this oncogene’s expression in GB patients [46,47]. The activity of EGFR-mediated kinases is critical to promote greater malignancy, making it a promising target to prevent tumor progression [48]. Tyrphostin (AG1478) is a selective EGFR inhibitor that disrupts EGFR autophosphorylation and signaling in cancer, including GB [14,49,50], inhibiting cell migration and invasion [51,52]. Additionally, it has been reported that AG1478 can cause fragmentation of the Golgi apparatus independently of EGFR [53], which is likely related to the morphological cell alterations observed in this study. Moreover, it is possible to observe that the treatment induced the formation of tumor-like tunneling nanotubes (TNTs). TNTs have been described as an adaptative mechanism in resistance development [54], which can be boosted when using tyrosine kinase inhibitors (TKIs) [55].
Although there is a considerable number of studies on EGFR-targeted therapy, consensus has not been reached due to variability in interpatient responses. Areeb et al. demonstrated that TMZ and radiotherapy reduced EGFR expression and that this treatment selected a population of cells with low activity of this receptor, rendering an ineffective response to EGFR-TKIs [56]. Allied with this statement, we did not find an effective response to the combination of TMZ and AG1478 regarding the viability of GB cells. On the other hand, some studies justify the use of simultaneous treatment with TMZ and TKIs [57,58]. The combination of TMZ and nimotuzumab, an anti-EGFR antibody, increased the antitumor response in GB, especially in those patients expressing the EGFRvIII mutation [59]. This treatment combination improved progression-free survival and overall survival compared to TMZ monotherapy [60].
Tumorigenesis involves a complex array of molecular and cellular dysregulations, with chemotherapy resistance frequently emerging in GB, leading to tumor recurrence. Thus, developing treatments that completely eradicate GB cells remains a significant challenge. Our study reflects this issue, as anti-EGFR monotherapy was found to elevate pro-tumor markers, underscoring the limitations of single-agent therapies. The levels of EGFR, immune checkpoints (ICs), and CD73 (and their co-occurrence) are fundamental to leading the GB microenvironment and to an immunosuppressed state. Considering these aspects, we demonstrated that most GB cells present high CD73, PD-L1, and EGFR levels. In addition, inhibition of EGFR by AG1478 upregulated PD-L1 and CD73 expression. Data suggests that EGFR activation can upregulate PD-L1 via different pathways [61–63]. However, EGFR-TKIs resistance development is reported to elevate PD-L1 levels [64,65], which aligns with our study findings. It has been observed that resistance to EGFR inhibitors promotes immune escape by increasing PD-L1 expression in non-small cell lung cancer (NSCLC) [66]. In breast cancer, it was observed that the PD-1/PD-L1 interaction induced the activation of the PI3K/AKT and MAPK/ERK pathways and enhanced MDR1/P-gp expression [67]. In our study, the increase in MRP-1 expression was accompanied by PD-L1 after treatment with AG1478, suggesting that the change in these markers can be established quickly. However, co-treatment with APCP blocked the induction of MRP-1 expression by AG1478, demonstrating a possible mechanism between CD73 and acquired resistance to TKIs.
It is important to note that CD73 expression is very high in relation to the other markers when comparing the MFI and that CD73+ cells were also EGFR+ and PD-L1+. Several studies have demonstrated a strong link between the EGFR and CD73 signaling pathways in various cancers [22,23,68,69]. In this work, we showed that EGFR inhibition increases CD73 expression. Terp and colleagues report that treatment using MAPK inhibitors upregulated CD73 via the compensatory p38 MAPK pathway, reducing the antitumor response [70]. In line with our findings, Griesing et al. have shown that CD73 expression is modulated by the EGFR-ERK signaling pathway [23]. In NSCLC samples with EGFR mutations, CD73 expression is significantly elevated, accompanied by reduced tumor necrosis factor (TNF) expression compared to wild-type tumors [71]. In breast cancer, CD73 has been shown to regulate EGFR expression [72], while Turcotte et al. demonstrated that CD73 contributes to resistance against human epidermal growth factor receptor 2-based targeted therapy [73]. All studies highlighted the potential of targeting CD73 to enhance the efficacy of EGFR therapies.
Thus, we assessed the in vitro effects of AG1478 with APCP, a CD73 inhibitor. We showed that the combination of these inhibitors could downregulate the expression of MRP-1. However, since the percentage of cells expressing MRP-1 remained unchanged, it suggests this protein is uniformly downregulated across the entire cell population rather than selectively lost in a subset of cells. In line with this, Torres et al. demonstrated that ADO signalling pathway can influence Akt and ERK1/2 activation, decreasing MRP1 expression in glioblastoma stem-like cells [74]. Inhibiting CD73 reduces MRP1 expression, enhancing the sensitivity of cancer cells to chemotherapy and leading to improved treatment outcomes [75].
Interestingly, we did not notice any differences in the cell proliferation or death of the treated cells. This differed from most studies using the small molecule AG1478 [14,51,76]. Kersting et al. demonstrated that AG1478 increased senescence in Ewing Sarcoma cells [77]. Whereas we demonstrated that the combination of CD73 and EGFR inhibitors suppressed cell growth by inducing G0/G1 phase cell cycle arrest, which could be induced by p21 and cyclin D1 regulation, as shown by Kim et al. [19]. Carrasco-García and collaborators have also demonstrated that small-molecule EGFR inhibitors (AG1478, gefitinib, and erlotinib) caused G1 arrest in GB cell lines. The arrest promoted, especially by AG1478, was associated with the upregulation of p27 [14]. In addition, Zhang et al. demonstrated that both CD73 silencing and APCP treatment reduced the phosphorylation of Akt, EGFR, and mTOR.
It is worth noting that CD73 also functions as an adhesion molecule, contributing to the migration of both normal and neoplastic cells [78]. It has been shown that CD73 inhibition significantly reduces cell adhesion, migration, and invasiveness in breast cancer, glioma, hepatocellular, and lung carcinoma in vitro models [26,79]. Our findings did not bring similar effects on the migratory response of GB cells treated with APCP only, but the combination of CD73 and EGFR inhibition presented reduced GB cell motility. In agreement with our data, Ma et al. demonstrated that AG1478 inhibits the migration and invasion via cell cycle regulation by matrix metalloproteinase-9 [52]. Similarly, Xiao et al. showed that EGFR inhibition diminished cell migration and invasion [80]. Conversely, an anti-CD73 antibody could inhibit breast cancer cell motility by modulating autophagy, whereas EGFR inhibitors induce autophagy [81], promoting drug resistance [82]. However, we did not find changes in autophagy by acridine orange staining (data not shown).
Even though our study offers valuable insights into the effects of combining EGFR and CD73 inhibitors, some limitations should be considered. Our study was conducted using a limited number of cancer cell models, which might not fully represent the heterogeneity in clinical practice. Additionally, long-term studies are essential to evaluate the potential for reducing acquired resistance, such as chronically co-treating cells in vitro. The resistance mechanisms between EGFR and CD73 pathways remain poorly understood, as our study focused primarily on the role of CD73 in tumor cells. CD73’s most significant effects may be related to immunomodulation rather than direct tumor cell targeting. Therefore, future investigations should explore the role of this co-treatment in vivo and co-culture models with immune cells to understand its potential therapeutic impact better.
EGFR-targeted therapies can promote resistance in GB. In this sense, we demonstrated that simultaneous inhibition of EGFR and CD73 reverted MRP-1 overexpression induced by anti-EGFR monotherapy, inhibited cell growth by arresting GB cells in the G0/G1 phase and decreased GB cells’ invasiveness. However, the underlying mechanisms still must be elucidated.
Acknowledgement: Not applicable.
Funding Statement: This study was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; n° 406035/2021-0 and PQ n° 311580/2021-1, Figueiró F), and Instituto Nacional de Ciência e Tecnologia-INCT/CNPq/CAPES/FAPERGS Grant no. 465671/2014-4. Jean Sévigny received support from the Natural Sciences and Engineering Research Council of Canada (NSERC; RGPIN-2023-05498).
Author Contributions: The authors confirm their contribution to the paper as follows: Luiz Fernando Lopes Silva: Writing—review & editing, Writing—original draft, Visualization, Methodology, Investigation, Formal analysis, Conceptualization. Juliete Nathali Scholl: Writing—review & editing, Validation, Visualization, Methodology, Investigation, Formal analysis, Data curation. Augusto Ferreira Weber: Writing—review & editing, Methodology, Investigation. Camila Kehl Dias: Writing—review & editing, Methodology, Investigation, Formal analysis. Pauline Rafaela Pizzato: Writing—review & editing, Methodology. Vinícius Pierdoná Lima: Writing—review & editing, Methodology. Jean Sévigny: Writing—review & editing, Resources/Reagents. Ana Maria Oliveira Battastini: Writing—review & editing, Resources. Fabrício Figueiró: Writing—review & editing, Writing—original draft, Validation, Supervision, Resources, Project administration, Funding acquisition, Conceptualization. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: All data generated or analyzed during this study are included in this published article.
Ethics Approval: This study was conducted in accordance with the principles of the Declaration of Helsinki, and all individuals provided written informed consent prior to enrollment. The blood from normal donors was provided by blood bank and utilized following the approval of the informed consent form in compliance with Consolidation Ordinance No. 5 of the Department of Health (PRT MS/GM 158/2016).
Conflicts of Interest: The author declares no conflicts of interest to report regarding the present study.
References
1. Ostrom QT, Price M, Neff C, Cioffi G, Waite KA, Kruchko C, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2016—2020. Neuro Oncol. 2023;25:iv1–99. doi:10.1093/neuonc/noad149. [Google Scholar] [PubMed] [CrossRef]
2. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021;23:1231–51. doi:10.1093/neuonc/noab106. [Google Scholar] [PubMed] [CrossRef]
3. Weller M, Wen PY, Chang SM, Dirven L, Lim M, Monje M, et al. Glioma. Nat Rev Dis Primers. 2024;10:33. doi:10.1038/s41572-024-00516-y. [Google Scholar] [PubMed] [CrossRef]
4. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl J Med. 2005;352:987–96. doi:10.1056/NEJMoa043330. [Google Scholar] [PubMed] [CrossRef]
5. Bagley SJ, Kothari S, Rahman R, Lee EQ, Dunn GP, Galanis E, et al. Glioblastoma clinical trials: current landscape and opportunities for improvement. Clin Cancer Res. 2022;28:594–602. doi:10.1158/1078-0432.CCR-21-2750. [Google Scholar] [PubMed] [CrossRef]
6. Lee EQ, Chukwueke UN, Hervey-Jumper SL, de Groot JF, Leone JP, Armstrong TS, et al. Barriers to accrual and enrollment in brain tumor trials. Neuro Oncol. 2019. doi:10.1093/neuonc/noz104. [Google Scholar] [PubMed] [CrossRef]
7. Horn LA, Fousek K, Palena C. Tumor plasticity and resistance to immunotherapy. Trends Cancer. 2020;6:432–41. doi:10.1016/j.trecan.2020.02.001. [Google Scholar] [PubMed] [CrossRef]
8. Tong S, Wang Y, Wu J, Long J, Zhong P, Wang B. Comprehensive pharmacogenomics characterization of temozolomide response in gliomas. Eur J Pharmacol. 2021;912:174580. doi:10.1016/j.ejphar.2021.174580. [Google Scholar] [PubMed] [CrossRef]
9. Uddin MdS, Mamun AAl, Alghamdi BS, Tewari D, Jeandet P, MdS Sarwar, et al. Epigenetics of glioblastoma multiforme: from molecular mechanisms to therapeutic approaches. Semin Cancer Biol. 2022;83:100–20. doi:10.1016/j.semcancer.2020.12.015. [Google Scholar] [PubMed] [CrossRef]
10. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12:31–46. doi:10.1158/2159-8290.CD-21-1059. [Google Scholar] [PubMed] [CrossRef]
11. Patil V, Mahalingam K. Comprehensive analysis of reverse phase protein array data reveals characteristic unique proteomic signatures for glioblastoma subtypes. Gene. 2020;685:85–95. doi:10.1016/j.gene.2018.10.069. [Google Scholar] [PubMed] [CrossRef]
12. Sareen H, Ma Y, Becker TM, Roberts TL, de Souza P, Powter B. Molecular biomarkers in glioblastoma: a systematic review and meta-analysis. Int J Mol Sci. 2022;23:8835. doi:10.3390/ijms23168835. [Google Scholar] [PubMed] [CrossRef]
13. Hoogstrate Y, Ghisai SA, de Wit M, de Heer I, Draaisma K, van Riet J, et al. The EGFRvIII transcriptome in glioblastoma: a meta-omics analysis. Neuro Oncol. 2022;24:429–41. doi:10.1093/neuonc/noab231. [Google Scholar] [PubMed] [CrossRef]
14. Carrasco-García E, Saceda M, Grasso S, Rocamora-Reverte L, Conde M, Gómez-Martínez Á, et al. Small tyrosine kinase inhibitors interrupt EGFR signaling by interacting with erbB3 and erbB4 in glioblastoma cell lines. Exp Cell Res. 2011;317:1476–89. doi:10.1016/j.yexcr.2011.03.015. [Google Scholar] [PubMed] [CrossRef]
15. Nagane M, Narita Y, Mishima K, Levitzki A, Burgess AW, Cavenee WK, et al. Human glioblastoma xenografts overexpressing a tumor-specific mutant epidermal growth factor receptor sensitized to cisplatin by the AG1478 tyrosine kinase inhibitor. J Neurosurg. 2001;95:472–9. doi:10.3171/jns.2001.95.3.0472. [Google Scholar] [PubMed] [CrossRef]
16. Buendia Duque M, de Pinheiro KV, Thomaz A, da Silva CA, Freire NH, Brunetto AT, et al. Combined inhibition of hdac and egfr reduces viability and proliferation and enhances STAT3 mRNA expression in glioblastoma cells. J Mol Neurosci. 2019;68:49–57. doi:10.1007/s12031-019-01280-5. [Google Scholar] [PubMed] [CrossRef]
17. Reardon DA, Wen PY, Mellinghoff IK. Targeted molecular therapies against epidermal growth factor receptor: past experiences and challenges. Neuro Oncol. 2014;16:viii7–13. doi:10.1093/neuonc/nou232. [Google Scholar] [PubMed] [CrossRef]
18. Chakravarti A, Wang M, Robins HI, Lautenschlaeger T, Curran WJ, Brachman DG, et al. RTOG 0211: a phase 1/2 study of radiation therapy with concurrent gefitinib for newly diagnosed glioblastoma patients. Int J Radiat Oncol. 2013;85:1206–11. doi:10.1016/j.ijrobp.2012.10.008. [Google Scholar] [PubMed] [CrossRef]
19. Kim M, Kim S, Yim J, Keam B, Kim TM, Jeon YK, et al. Targeting CD73 to overcomes resistance to first-generation EGFR tyrosine kinase inhibitors in non-small cell lung cancer. Cancer Res Treat. 2023;55:1134–43. doi:10.4143/crt.2023.311. [Google Scholar] [PubMed] [CrossRef]
20. Xu W, Bi Y, Kong J, Zhang J, Wang B, Li K, et al. Combination of an anti-EGFRvIII antibody CH12 with Rapamycin synergistically inhibits the growth of EGFRvIII+PTEN− glioblastoma in vivo. Oncotarget. 2016;7:24752–65. doi:10.18632/oncotarget.8407. [Google Scholar] [PubMed] [CrossRef]
21. Ezzati S, Salib S, Balasubramaniam M, Aboud O. Epidermal growth factor receptor inhibitors in glioblastoma: current status and future possibilities. Int J Mol Sci. 2024;25:2316. doi:10.3390/ijms25042316. [Google Scholar] [PubMed] [CrossRef]
22. Gao Z, Wang H, Lin F, Wang X, Long M, Zhang H, et al. CD73 promotes proliferation and migration of human cervical cancer cells independent of its enzyme activity. BMC Cancer. 2017;17:135. doi:10.1186/s12885-017-3128-5. [Google Scholar] [PubMed] [CrossRef]
23. Griesing S, Liao B-C, Yang JC-H. CD73 is regulated by the EGFR-ERK signaling pathway in non-small cell lung cancer. Anticancer Res. 2021;41(3):1231–42. doi:10.21873/anticanres.14880. [Google Scholar] [PubMed] [CrossRef]
24. Wang J, Matosevic S. NT5E/CD73 as correlative factor of patient survival and natural killer cell infiltration in glioblastoma. J Clin Med. 2019;8:1526. doi:10.3390/jcm8101526. [Google Scholar] [PubMed] [CrossRef]
25. Gelsleichter NE, Azambuja JH, Rubenich DS, Braganhol E. CD73 in glioblastoma: where are we now and what are the future directions? Immunol Lett. 2023;256–257:20–7. doi:10.1016/j.imlet.2023.03.005. [Google Scholar] [PubMed] [CrossRef]
26. Azambuja JH, Gelsleichter NE, Beckenkamp LR, Iser IC, Fernandes MC, Figueiró F, et al. CD73 downregulation decreases in vitro and in vivo glioblastoma growth. Mol Neurobiol. 2019;56:3260–79. doi:10.1007/s12035-018-1240-4. [Google Scholar] [PubMed] [CrossRef]
27. Tsiampali J, Neumann S, Giesen B, Koch K, Maciaczyk D, Janiak C, et al. Enzymatic activity of CD73 modulates invasion of gliomas via epithelial-mesenchymal transition-like reprogramming. Pharmaceuticals. 2020;13:378. doi:10.3390/ph13110378. [Google Scholar] [PubMed] [CrossRef]
28. Kitabatake K, Kaji T, Tsukimoto M. Involvement of CD73 and A2B receptor in radiation-induced DNA damage response and cell migration in human glioblastoma A172 cells. Biol Pharm Bull. 2021;44:197–210. doi:10.1248/bpb.b20-00654. [Google Scholar] [PubMed] [CrossRef]
29. Zahavi D, Hodge J. Targeting immunosuppressive adenosine signaling: a review of potential immunotherapy combination strategies. Int J Mol Sci. 2023;24:8871. doi:10.3390/ijms24108871. [Google Scholar] [PubMed] [CrossRef]
30. Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer. 2017;17:709–24. doi:10.1038/nrc.2017.86. [Google Scholar] [PubMed] [CrossRef]
31. Ludwig N, Yerneni SS, Azambuja JH, Gillespie DG, Menshikova EV, Jackson EK, et al. Tumor-derived exosomes promote angiogenesis via adenosine A2B receptor signaling. Angiogenesis. 2020;23:599–610. doi:10.1007/s10456-020-09728-8. [Google Scholar] [PubMed] [CrossRef]
32. Yan A, Joachims ML, Thompson LF, Miller AD, Canoll PD, Bynoe MS. CD73 promotes glioblastoma pathogenesis and enhances its chemoresistance via A2B adenosine receptor signaling. J Neurosci. 2019;39:4387–402. doi:10.1523/JNEUROSCI.1118-18.2019. [Google Scholar] [PubMed] [CrossRef]
33. Goswami S, Walle T, Cornish AE, Basu S, Anandhan S, Fernandez I, et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat Med. 2020;26:39–46. doi:10.1038/s41591-019-0694-x. [Google Scholar] [PubMed] [CrossRef]
34. Roh M, Wainwright DA, Wu JD, Wan Y, Zhang B. Targeting CD73 to augment cancer immunotherapy. Curr Opin Pharmacol. 2020;53:66–76. doi:10.1016/j.coph.2020.07.001. [Google Scholar] [PubMed] [CrossRef]
35. Azambuja JH, Schuh RS, Michels LR, Gelsleichter NE, Beckenkamp LR, Lenz GS, et al. CD73 as a target to improve temozolomide chemotherapy effect in glioblastoma preclinical model. Cancer Chemother Pharmacol. 2020;85:1177–82. doi:10.1007/s00280-020-04077-1. [Google Scholar] [PubMed] [CrossRef]
36. Coy S, Wang S, Stopka SA, Lin J-R, Yapp C, Ritch CC, et al. Single cell spatial analysis reveals the topology of immunomodulatory purinergic signaling in glioblastoma. Nat Commun. 2022;13:4814. doi:10.1038/s41467-022-32430-w. [Google Scholar] [PubMed] [CrossRef]
37. Yu MW, Quail DF. Immunotherapy for glioblastoma: current progress and challenges. Front Immunol. 2021;12. doi:10.3389/fimmu.2021.676301. [Google Scholar] [PubMed] [CrossRef]
38. Fan HH, Kleven SH, Jackwood MW. Application of polymerase chain reaction with arbitrary primers to strain identification of Mycoplasma gallisepticum. Avian Dis. 1995;39:729–35. [Google Scholar] [PubMed]
39. Sæbø I, Bjørås M, Franzyk H, Helgesen E, Booth J. Optimization of the hemolysis assay for the assessment of cytotoxicity. Int J Mol Sci. 2023;24:2914. doi:10.3390/ijms24032914. [Google Scholar] [PubMed] [CrossRef]
40. Longaray JB, Dias CK, Scholl JN, Battastini AMO, Figueiró F. Investigation of co-treatment multi-targeting approaches in breast cancer cell lines. Eur J Pharmacol. 2024;966:176328. doi:10.1016/j.ejphar.2024.176328. [Google Scholar] [PubMed] [CrossRef]
41. Pelletier J, Agonsanou H, Manica F, Lavoie G, Salem E, Luyindula M, et al. A Simple and efficient genetic immunization protocol for the production of highly specific polyclonal and monoclonal antibodies against the native form of mammalian proteins. Int J Mol Sci. 2020;21:7074. doi:10.3390/ijms21197074. [Google Scholar] [PubMed] [CrossRef]
42. Matsunaga S, Asano T, Tsutsuda-Asano A, Fukunaga Y. Indomethacin overcomes doxorubicin resistance with inhibiting multi-drug resistance protein 1 (MRP1). Cancer Chemother Pharmacol. 2006;58:348–53. doi:10.1007/s00280-005-0162-9. [Google Scholar] [PubMed] [CrossRef]
43. Grada A, Otero-Vinas M, Prieto-Castrillo F, Obagi Z, Falanga V. Research techniques made simple: analysis of collective cell migration using the wound healing assay. J Investig Dermatol. 2017;137:e11–6. doi:10.1016/j.jid.2016.11.020. [Google Scholar] [PubMed] [CrossRef]
44. Iser IC, Ceschini SM, Onzi GR, Bertoni APS, Lenz G, Wink MR. Conditioned medium from Adipose-Derived Stem Cells (ADSCs) promotes Epithelial-to-Mesenchymal-Like Transition (EMT-Like) in glioma cells In vitro. Mol Neurobiol. 2016;53:7184–99. doi:10.1007/s12035-015-9585-4. [Google Scholar] [PubMed] [CrossRef]
45. Schiffer D, Annovazzi L, Casalone C, Corona C, Mellai M. Glioblastoma: microenvironment and niche concept. Cancers. 2018;11:5. doi:10.3390/cancers11010005. [Google Scholar] [PubMed] [CrossRef]
46. Śledzińska P, Bebyn MG, Furtak J, Kowalewski J, Lewandowska MA. Prognostic and predictive biomarkers in gliomas. Int J Mol Sci. 2021;22:10373. doi:10.3390/ijms221910373. [Google Scholar] [PubMed] [CrossRef]
47. Miratashi Yazdi SA, Bakhshi N, Nazar E, Tabriz HM, Gorji R. Epidermal growth factor receptor (EGFR) expression in high grade glioma and relationship with histopathologic findings, a cross sectional study. Int J Surg Open. 2022;46:100527. doi:10.1016/j.ijso.2022.100527. [Google Scholar] [CrossRef]
48. van Linde ME, Labots M, Brahm CG, Hovinga KE, De Witt Hamer PC, Honeywell RJ, et al. Tumor drug concentration and phosphoproteomic profiles after two weeks of treatment with sunitinib in patients with newly diagnosed glioblastoma. Clin Cancer Res. 2022;28:1595–602. doi:10.1158/1078-0432.CCR-21-1933. [Google Scholar] [PubMed] [CrossRef]
49. Bojko A, Cierniak A, Adamczyk A, Ligeza J. Modulatory effects of curcumin and tyrphostins (AG494 and AG1478) on growth regulation and viability of LN229 human brain cancer cells. Nutr Cancer. 2015;67:1170–82. doi:10.1080/01635581.2015.1073764. [Google Scholar] [PubMed] [CrossRef]
50. Pinheiro KV, Thomaz A, Souza BK, Metcalfe VA, Freire NH, Brunetto AT, et al. Expression and pharmacological inhibition of TrkB and EGFR in glioblastoma. Mol Biol Rep. 2020;47:6817–28. doi:10.1007/s11033-020-05739-2. [Google Scholar] [PubMed] [CrossRef]
51. Zhang Y-G, Du Q, Fang W-G, Jin M-L, Tian X-X. Tyrphostin AG1478 suppresses proliferation and invasion of human breast cancer cells. Int J Oncol. 2008;33:595–602. [Google Scholar] [PubMed]
52. Ma L, Yan H, Zhou Q. AG1478 inhibits the migration and invasion of cisplatin-resistant human lung adenocarcinoma cells via the cell cycle regulation by matrix metalloproteinase-9. Oncol Lett. 2014;8(2):921–7. doi:10.3892/ol.2014.2224. [Google Scholar] [PubMed] [CrossRef]
53. Pan H, Yu J, Zhang L, Carpenter A, Zhu H, Li L, et al. A novel small molecule regulator of guanine nucleotide exchange activity of the adp-ribosylation factor and golgi membrane trafficking. J Biol Chem. 2008;283:31087–96. doi:10.1074/jbc.M806592200. [Google Scholar] [PubMed] [CrossRef]
54. Valdebenito S, Audia A, Bhat KPL, Okafo G, Eugenin EA. Tunneling nanotubes mediate adaptation of glioblastoma cells to temozolomide and ionizing radiation treatment. iScience. 2020;23:101450. doi:10.1016/j.isci.2020.101450. [Google Scholar] [PubMed] [CrossRef]
55. Omsland M, Andresen V, Gullaksen S, Ayuda-Durán P, Popa M, Hovland R, et al. Tyrosine kinase inhibitors and interferon-α increase tunneling nanotube (TNT) formation and cell adhesion in chronic myeloid leukemia (CML) cell lines. FASEB J. 2020;34:3773–91. doi:10.1096/fj.201802061RR. [Google Scholar] [PubMed] [CrossRef]
56. Areeb Z, Stuart SF, West AJ, Gomez J, Nguyen HPT, Paradiso L, et al. Reduced EGFR and increased miR-221 is associated with increased resistance to temozolomide and radiotherapy in glioblastoma. Sci Rep. 2020;10:17768. doi:10.1038/s41598-020-74746-x. [Google Scholar] [PubMed] [CrossRef]
57. da Santos ES, Nogueira KAB, Fernandes LCC, Martins JRP, Reis AVF, de Neto JBV, et al. EGFR targeting for cancer therapy: pharmacology and immunoconjugates with drugs and nanoparticles. Int J Pharm. 2021;592:120082. doi:10.1016/j.ijpharm.2020.120082. [Google Scholar] [PubMed] [CrossRef]
58. Vengoji R, Macha MA, Nimmakayala RK, Rachagani S, Siddiqui JA, Mallya K, et al. Afatinib and temozolomide combination inhibits tumorigenesis by targeting EGFRvIII-cMet signaling in glioblastoma cells. J Exp Clin Cancer Res. 2019;38:266. doi:10.1186/s13046-019-1264-2. [Google Scholar] [PubMed] [CrossRef]
59. Nitta Y, Shimizu S, Shishido-Hara Y, Suzuki K, Shiokawa Y, Nagane M. Nimotuzumab enhances temozolomide-induced growth suppression of glioma cells expressing mutant EGFR in vivo. Cancer Med. 2016;5:486–99. doi:10.1002/cam4.614. [Google Scholar] [PubMed] [CrossRef]
60. She L, Gong X, Su L, Liu C. Radiotherapy plus temozolomide with or without nimotuzumab against the newly diagnosed EGFR-positive glioblastoma: a retrospective cohort study. Oncologist. 2023;28:e45–53. doi:10.1093/oncolo/oyac202. [Google Scholar] [PubMed] [CrossRef]
61. Abdelhamed S, Ogura K, Yokoyama S, Saiki I, Hayakawa Y. AKT-STAT3 pathway as a downstream target of EGFR signaling to regulate PD-L1 expression on NSCLC cells. J Cancer. 2016;7:1579–86. doi:10.7150/jca.14713. [Google Scholar] [PubMed] [CrossRef]
62. Su L, Guo W, Lou L, Nie S, Zhang Q, Liu Y, et al. EGFR-ERK pathway regulates CSN6 to contribute to PD-L1 expression in glioblastoma. Mol Carcinog. 2020;59:520–32. doi:10.1002/mc.23176. [Google Scholar] [PubMed] [CrossRef]
63. Zhang N, Zeng Y, Du W, Zhu J, Shen D, Liu Z, et al. The EGFR pathway is involved in the regulation of PD-L1 expression via the IL-6/JAK/STAT3 signaling pathway in EGFR-mutated non-small cell lung cancer. Int J Oncol. 2016;49:1360–8. doi:10.3892/ijo.2016.3632. [Google Scholar] [PubMed] [CrossRef]
64. Han JJ, Kim D-W, Koh J, Keam B, Kim TM, Jeon YK, et al. Change in PD-L1 expression after acquiring resistance to gefitinib in EGFR-mutant non-small-cell lung cancer. Clin Lung Cancer. 2016;17:263–70.e2. doi:10.1016/j.cllc.2015.11.006. [Google Scholar] [PubMed] [CrossRef]
65. Li X, Lian Z, Wang S, Xing L, Yu J. Interactions between EGFR and PD-1/PD-L1 pathway: implications for treatment of NSCLC. Cancer Lett. 2018;418:1–9. doi:10.1016/j.canlet.2018.01.005. [Google Scholar] [PubMed] [CrossRef]
66. Peng S, Wang R, Zhang X, Ma Y, Zhong L, Li K, et al. EGFR-TKI resistance promotes immune escape in lung cancer via increased PD-L1 expression. Mol Cancer. 2019;18:165. doi:10.1186/s12943-019-1073-4. [Google Scholar] [PubMed] [CrossRef]
67. Liu S, Chen S, Yuan W, Wang H, Chen K, Li D, et al. PD-1/PD-L1 interaction up-regulates MDR1/P-gp expression in breast cancer cells via PI3K/AKT and MAPK/ERK pathways. Oncotarget. 2017;8:99901–12. doi:10.18632/oncotarget.21914. [Google Scholar] [PubMed] [CrossRef]
68. Liu C, Gao Z-W, Wang X, Lin F, Zhang H-Z, Dong K. CD73 promotes cervical cancer growth via EGFR/AKT1 pathway. Transl Cancer Res. 2022;11:1089–98. doi:10.21037/tcr-21-2446. [Google Scholar] [PubMed] [CrossRef]
69. Han Y, Lee T, He Y, Raman R, Irizarry A, Martin ML, et al. The regulation of CD73 in non-small cell lung cancer. Eur J Cancer. 2022;170:91–102. doi:10.1016/j.ejca.2022.04.025. [Google Scholar] [PubMed] [CrossRef]
70. Terp MG, Gammelgaard OL, Vever H, Gjerstorff MF, Ditzel HJ. Sustained compensatory p38 MAPK signaling following treatment with MAPK inhibitors induces the immunosuppressive protein CD73 in cancer: combined targeting could improve outcomes. Mol Oncol. 2021;15:3299–316. doi:10.1002/1878-0261.13046. [Google Scholar] [PubMed] [CrossRef]
71. Park LC, Rhee K, Kim WB, Cho A, Song J, Anker JF, et al. Immunologic and clinical implications of CD73 expression in non-small cell lung cancer (NSCLC). J Clin Oncol. 2018;36:12050. doi:10.1200/JCO.2018.36.15_suppl.12050. [Google Scholar] [CrossRef]
72. Zhi X, Wang Y, Yu J, Yu J, Zhang L, Yin L, et al. Potential prognostic biomarker CD73 regulates epidermal growth factor receptor expression in human breast cancer. IUBMB Life. 2012;64:911–20. doi:10.1002/iub.1086. [Google Scholar] [PubMed] [CrossRef]
73. Turcotte M, Allard D, Mittal D, Bareche Y, Buisseret L, José V, et al. CD73 promotes resistance to HER2/ErbB2 antibody therapy. Cancer Res. 2017;77:5652–63. doi:10.1158/0008-5472.CAN-17-0707. [Google Scholar] [PubMed] [CrossRef]
74. Torres A, Vargas Y, Uribe D, Jaramillo C, Gleisner A, Salazar-Onfray F, et al. Adenosine A3 receptor elicits chemoresistance mediated by multiple resistance-associated protein-1 in human glioblastoma stem-like cells. Oncotarget. 2016;7:67373–86. doi:10.18632/oncotarget.12033. [Google Scholar] [PubMed] [CrossRef]
75. Carrera-Martínez M, de Mora-García ML, García-Rocha R, Weiss-Steider B, Montesinos-Montesinos JJ, Hernández-Montes J, et al. Inhibition of CD73 expression or A2AR blockade reduces MRP1 expression and increases the sensitivity of cervical cancer cells to cisplatin. Cell Biochem Funct. 2023;41:321–30. doi:10.1002/cbf.3784. [Google Scholar] [PubMed] [CrossRef]
76. Wang W, Yan M, Liu C, Wang Y, Wang Y, Wang L, et al. Epidermal growth factor receptor inhibitor AG1478 affects HepG2 cell proliferation, cell cycle, apoptosis and c-Myc protein expression in a dose-dependent manner. Biotechnol Biotechnol Equip. 2019;32:1021–7. doi:10.1080/13102818.2018.1460620. [Google Scholar] [CrossRef]
77. Kersting N, Kunzler Souza B, Araujo Vieira I, Pereira dos Santos R, Brufatto Olguins D, José Gregianin L, et al. Epidermal growth factor receptor regulation of ewing sarcoma cell function. Oncology. 2019;94:383–93. doi:10.1159/000487143. [Google Scholar] [PubMed] [CrossRef]
78. Da M, Chen L, Enk A, Ring S, Mahnke K. The multifaceted actions of CD73 during development and suppressive actions of regulatory T cells. Front Immunol. 2022;13:914799. doi:10.3389/fimmu.2022.914799. [Google Scholar] [PubMed] [CrossRef]
79. Teixeira FC, Bruxel F, Azambuja JH, Berenguer AM, Stefani MA, Sévigny J, et al. Development and characterization of CD73-siRNA-loaded nanoemulsion: effect on C6 glioma cells and primary astrocytes. Pharm Dev Technol. 2020;25:408–15. doi:10.1080/10837450.2019.1705485. [Google Scholar] [PubMed] [CrossRef]
80. Xiao M, Fan J, Li M, Xu F, Zhao X, Xi D, et al. A photosensitizer-inhibitor conjugate for photodynamic therapy with simultaneous inhibition of treatment escape pathways. Biomaterials. 2020;257:120262. doi:10.1016/j.biomaterials.2020.120262. [Google Scholar] [PubMed] [CrossRef]
81. Henson E, Chen Y, Gibson S. EGFR family members’ regulation of autophagy is at a crossroads of cell survival and death in cancer. Cancers. 2017;9:27. doi:10.3390/cancers9040027. [Google Scholar] [PubMed] [CrossRef]
82. Aveic S, Tonini GP. Resistance to receptor tyrosine kinase inhibitors in solid tumors: can we improve the cancer fighting strategy by blocking autophagy? Cancer Cell Int. 2016;16:62. doi:10.1186/s12935-016-0341-2. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools