
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW
Research advancements in nanoparticles and cell-based drug delivery systems for the targeted killing of cancer cells
Department of Biology, College of Science, Sultan Qaboos University, Muscat, 123, Oman
* Corresponding Authors: SIRIN A. ADHAM. Email: ,
Oncology Research 2025, 33(1), 27-44. https://doi.org/10.32604/or.2024.056955
Received 04 August 2024; Accepted 07 November 2024; Issue published 20 December 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
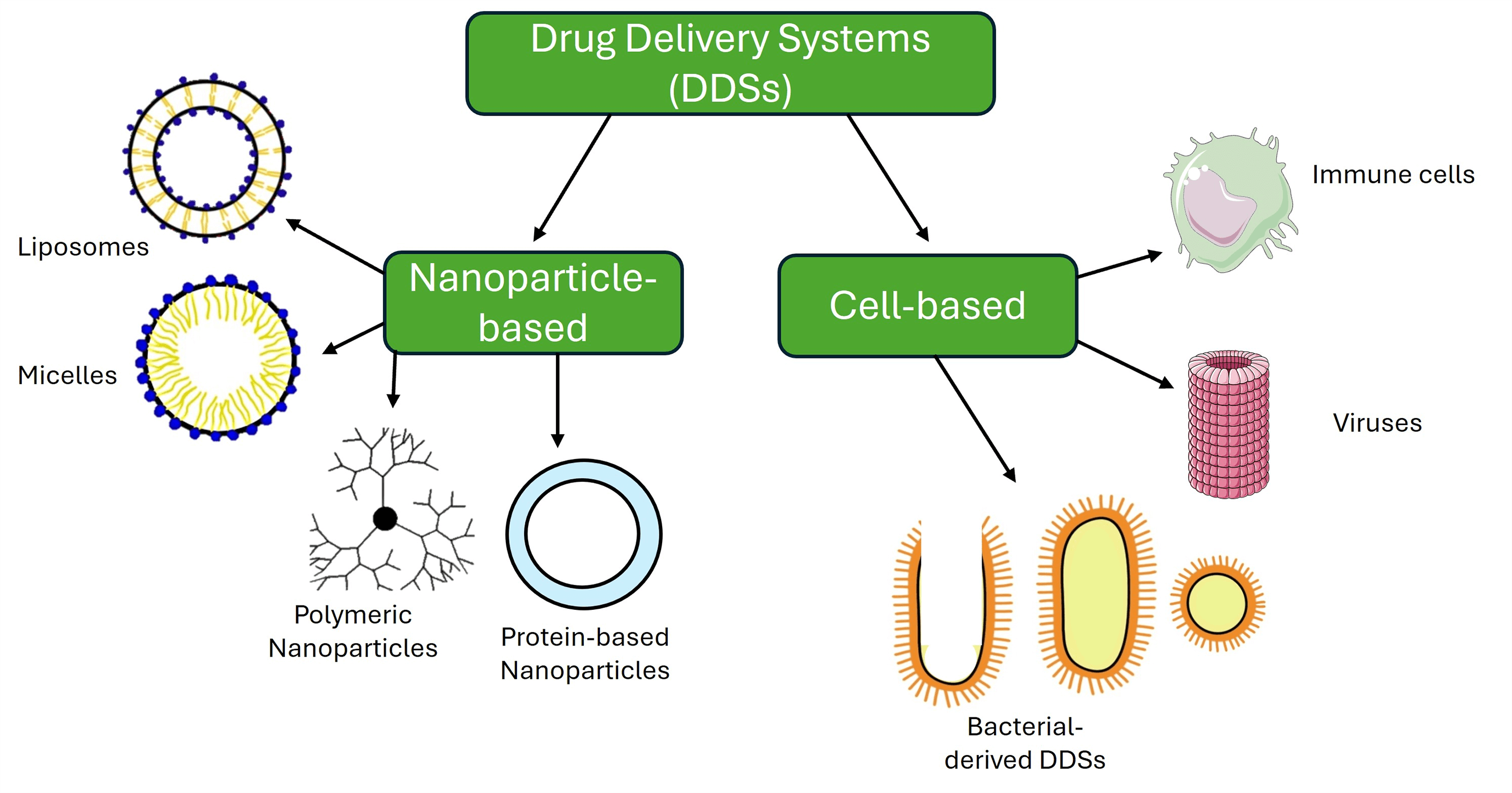
Nanotechnology in cancer therapy has significantly advanced treatment precision, effectiveness, and safety, improving patient outcomes and personalized care. Engineered smart nanoparticles and cell-based therapies are designed to target tumor cells, precisely sensing the tumor microenvironment (TME) and sparing normal cells. These nanoparticles enhance drug accumulation in tumors by solubilizing insoluble compounds or preventing their degradation, and they can also overcome therapy resistance and deliver multiple drugs simultaneously. Despite these benefits, challenges remain in patient-specific responses and regulatory approvals for cell-based or nanoparticle therapies. Cell-based drug delivery systems (DDSs) that primarily utilize the immune-recognition principle between ligands and receptors have shown promise in selectively targeting and destroying cancer cells. This review aims to provide a comprehensive overview of various nanoparticle and cell-based drug delivery system types used in cancer research. It covers approved and experimental nanoparticle therapies, including liposomes, micelles, protein-based and polymeric nanoparticles, as well as cell-based DDSs like macrophages, T-lymphocytes, dendritic cells, viruses, bacterial ghosts, minicells, SimCells, and outer membrane vesicles (OMVs). The review also explains the role of TME and its impact on developing smart DDSs in combination therapies and integrating nanoparticles with cell-based systems for targeting cancer cells. By detailing DDSs at different stages of development, from laboratory research to clinical trials and approved treatments, this review provides the latest insights and a collection of valuable citations of the innovative strategies that can be improved for the precise treatment of cancer.Graphic Abstract
Keywords
Abbreviations
| ADXS11-001 | Axalimogene filolisbac |
| AXAL | Axalimogene filolisbac |
| BGs | Bacterial Ghosts |
| CAR-T | Chimeric Antigen Receptor T-Cell |
| Ce6 | Chlorin e6 |
| CEA | Carcinoembryonic Antigen |
| cRGD | Cyclic arginine-glycine-aspartate |
| DC | Dendritic Cell |
| DDSs | Drug Delivery Systems |
| DNA | Deoxyribonucleic Acid |
| EGFR | Epidermal Growth Factor Receptor |
| EPR | Enhanced Permeability and Retention |
| EVs | Extracellular Vesicles |
| FDA | Food and Drug Administration |
| HER-2 | Human Epidermal Growth Factor Receptor 2 |
| HPV | Human Papillomavirus |
| HAS | Human Serum Albumin |
| IFN-γ | Interferon-γ |
| LPS | Lipopolysaccharide |
| miRNA | Micro RNA |
| mRNA | Messenger RNA |
| NDH-II | Alternative NADH Dehydrogenase |
| NIR | Near Infrared Radiation |
| NLRs | NOD-Like Receptors |
| NSCLC | Non-Small Cell Lung Carcinoma |
| OMVs | Outer Membrane Vesicles |
| PAMAM | Polyamidoamine |
| PD-1 | Programmed Death 1 |
| PD-L1 | Programmed Death Ligand 1 |
| PEG | Polyethylene Glycol |
| PMMoV | Pepper Mild Mottle Virus |
| rGO | Reduced Graphene Oxide |
| RLRs | RIG-I-Like Receptors |
| RNA | Ribonucleic Acid |
| siRNA | Small Interfering RNA |
| TAMs | Tumor-Associated Macrophages |
| TCF-7 | Transcription Factor 7 |
| TLRs | Toll-Like Receptors |
| TME | Tumor Microenvironment |
| TNBC | Triple Negative Breast Cancer |
| VLPs | Viral-Like Particles |
| VNPs | Viral Nanoparticles |
According to Globocan’s 2022 cancer statistics, there were over 20 million new cases of cancer worldwide, with 9.7 million deaths attributed to the disease [1]. Despite the advancement in cancer therapies, there are still many challenges to achieving a complete response in patients [2]. The difficulty in cancer treatment is rooted in the heterogenic nature of the tumors that contain different types of rapidly growing cells with genetic and epigenetic instability and mutations [3,4], the surrounding TME, and their ability to evade the immune system [5,6]. Besides the side effects and toxicity due to cancer treatments, certain chemotherapies can also enhance metastasis [1]. Mechanisms of drug resistance vary from tumor cells’ ability to efflux the drugs [2] to alter their metabolism enhanced by the surrounding hypoxic TME [3–5]. Other mechanisms include cell cycle regulation that targets drugs against specific cell cycle phases [6], bypassing pathways to evade the effects of targeted therapies [7], and DNA repair mechanisms that aid the cancer cells in repairing changes in DNA caused by the drug [8]. Adding to the list of treatment challenges is the development of stubborn cancer stem cells that can alter the drug metabolism and reduce efficacy with added hurdles to all cellular responses [9]. Besides the cellular mechanisms of resistance, patient variability increases the complexity of genetic differences, overall health, and treatment responses, which can affect outcomes, making personalized approaches essential yet challenging to apply [10]. Several treatment approaches were taken to overcome the challenges mentioned above by designing new drugs based on docking to their molecular targets in the cells, giving the term “targeted therapies” to increase specificity and avoid harm to normal cells seen using chemotherapy [7,11,12]. Whether it is chemotherapy or targeted therapy, the same challenge with drug-resistant mechanisms can be seen [13]. Therefore, cancer research in the past two decades focused on developing biotechnological tools based on drug delivery systems (DDS) to overcome the mechanisms of drug resistance essential for administering therapeutic compounds to achieve desired outcomes [14–16]. Conventional drug delivery methods, which are non-targeted, often suffer from significant limitations, including harmful side effects on healthy tissues, low bioavailability, and rapid metabolization of active ingredients upon entering the body [17].
In contrast, smart, targeted DDSs offer a more refined approach by delivering therapeutic agents directly to the site of interest while sparing healthy cells. The precision of drug delivery reduces the required dosage, minimizes side effects, and ensures more uniform drug concentrations in the bloodstream, maintaining therapeutic levels over an extended period [17]. As a result, evolving DDS technology is critical to improving outcomes, particularly for cancer patients. Nanotechnology offered the use of nanoparticles as carriers of single or multiple drugs, and different types of functionalized nanoparticles were proven to increase drug bioavailability, biocompatibility, and pharmacokinetics [18–20]. The success achieved using nanoparticles in improving patients’ survival [21,22] paved the way to develop smart nanocarriers that not only deliver the drug and stabilize its bioavailability but allow the release of their payload in response to specific signals from the TME sensing the TME PH, Hypoxia, levels of enzymes, and adenosine triphosphate, reducing the off-target effects on adjacent cells of the neighboring tissue [23,24]. Smart nanocarriers and many other cell-based delivery systems are developed, and others are in the pipelines to be produced and used to deliver drugs precisely and effectively. An excellent example of a cell-based delivery system is the immune cells equipped and designed to recognize and migrate to sites of infection or inflammation, including the organization of TME through the release of cytokines [25,26]. Since immune cells are naturally part of the immune system, they can be superior in their biocompatibility compared to synthetic carriers, besides their versatility in carrying proteins, small molecules, and gene therapies [27]. Immune cells are used in delivering immunotherapies, or vaccines can be bolstered to fight tumor cells more effectively by enhancing the immune response [28]. Like the smart nanocarriers, engineering immune cells allow for incorporating stimuli-responsive mechanisms, enabling them to release their payload in response to specific signals from the TME [29].
Besides immune cells, nanoscale engineering led to the production of viral-like particles that are investigated for their ability to deliver drugs to cancer cells efficiently [30]. The oncolytic viruses also show promising outcomes when combined with immunotherapy [31]. Not only viruses but also bacteria are engineered with cell-specific antigens for specific recognition and the destruction of tumor cells [32]. Therefore, this review summarizes the latest research output for the available DDSs for the precise targeting of cancer cells and provides the readers easy access to a recent collection of articles that cover DDSs.
The Role of Nanoparticles and Their Mode of Action in Drug Delivery
Nanoparticles in drug delivery began with the discovery of liposomes in the 1960s [33]. Soon after, dendrimers were invented in 1978 [34], followed by PEGylated liposomes [35] and the development of the first Food and Drug Administration (FDA) approved nano-drug called Doxil™, in 1995 [36,37].
Nanoparticles make for excellent drug delivery platforms due to their improved pharmacokinetic properties [38], controlled rate of drug delivery [39], and their ability to deliver drugs in response to certain stimuli [40]. Due to their small size, nanoparticles and other macromolecules can infiltrate the TME via increased leaky vasculature surrounding the tumor [41,42]. Such particles tend to accumulate more in tumor tissue than in normal tissue [41–43], resulting in an effect termed the enhanced permeability and retention (EPR) effect [44], which has been the basis behind the use of nanodrugs as cancer therapeutic agents [44–46]. Various cancer DDSs based their drug-targeting ability on this passive phenomenon, as it has been argued that the EPR effect is sufficient for targeting drugs to the TME. It has also been argued that it is better than active epitope-targeting as it is more generalized and can be applied to a wide range of tumors [47]. However, some studies question the very basis of the EPR effect. In one study, a 10-year analysis revealed that a median of only 0.7% of injected nanoparticles reached tumor cells via the EPR effect [48]. In contrast, Sindhwani et al. found that number to be even less, as their study showed that tumor accumulation of nanoparticles was 0.63% [49]. The small percentage of nanoparticle accumulation in the tumor can be due to the complex microenvironment around the tumor cells, poor blood flow, and their detection by the immune cells and rapid clearance from the bloodstream [50,51]. There are several strategies to improve the physical and chemical properties of nanoparticles for better penetration into the TME and activate them using different ligands that better target them and make them accumulate in tumor cells [52–54].
It was also suggested that active trans-endothelial mechanisms are the primary method of nanoparticles entering tumors [49]. The cells that facilitate nanoparticle transport into tumors are called nanoparticle transport endothelial cells [55]. That contradicts the initial belief that nanoparticles are passively extravasated via inter-endothelial gaps [56]. The EPR effect was further challenged by Nguyen et al., whose study proposed that nanoparticle retention in tumor tissues is not due to damaged lymph nodes, as lymphatic vessels are the primary nanoparticle exiting the tumor [57]. Instead, they proposed that nanoparticle retention was a consequence of the delay in transport from the nanoparticles’ entry to its exit [57]. That is why it is crucial to target therapeutic nanoparticles to the TME or cancer cells actively. Nanoparticles used for drug delivery exist in various forms, each with their properties, advantages, and disadvantages. Examples include liposomes, micelles, protein-based and polymeric nanoparticles [30,58–60]. Complex drug delivery methods also exist, combining nanoparticles with cell-based systems such as immune cells, viruses, bacteria, and their derivatives [61–63].
Types of Nanoparticles as DDSs
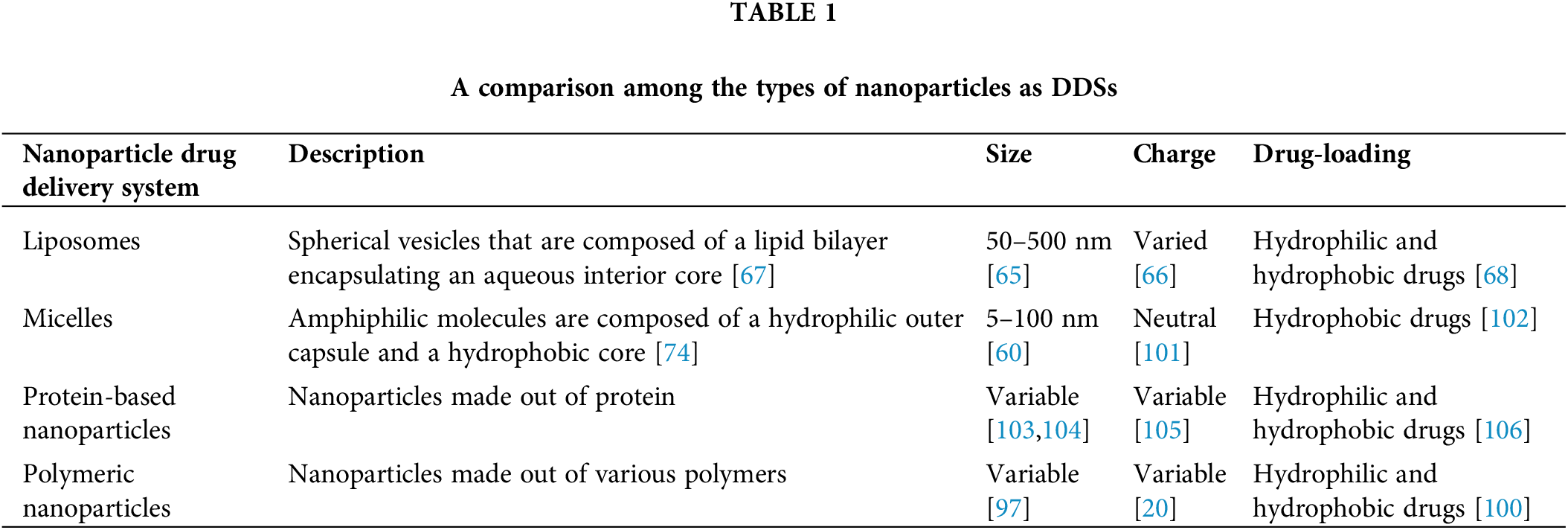
Bangham and Horne described Liposomes in 1964 as one of the first nanoparticles used and most studied as DDSs [64]. Liposomes ranging in size from 50–500 nm [65] can be cationic, anionic, or neutral [66]. These spherical vesicles comprise a lipid bilayer encapsulating an aqueous interior core and are produced when amphiphilic lipids are added to water [67]. They are excellent DDSs because they can stably encapsulate hydrophobic, hydrophilic, and amphiphilic drugs. Furthermore, liposomes are nontoxic, hypoallergenic, non-antigenic, and biodegradable [68].
The first liposomes altered to target specific cells had anti-H-2k antibodies conjugated to them. These liposomes displayed specificity in binding to mouse H-2k cells and did not bind to H-2d cells in vitro [69].
Recent studies have successfully fabricated liposomes conjugated to cyclic arginine-glycine-aspartate (cRGD), which are pH-sensitive and loaded with doxorubicin (Fig. 1A). The cRGD liposomes have been tested both in vitro on the A-549 lung carcinoma cell line and in vivo on nude mice bearing A-549 tumors and have exhibited excellent antitumor activity by inhibiting cell proliferation. The liposomes also displayed a high selectivity towards A-549 cells and low toxicity towards regular HL-7702 cell lines in both in vitro and in vivo studies. These modified liposomes also displayed a rapid cellular uptake, sustained intracellular drug release, greater retention at the tumor site, and notable pH sensitivity as they degraded in the acidic TME, releasing doxorubicin [70].
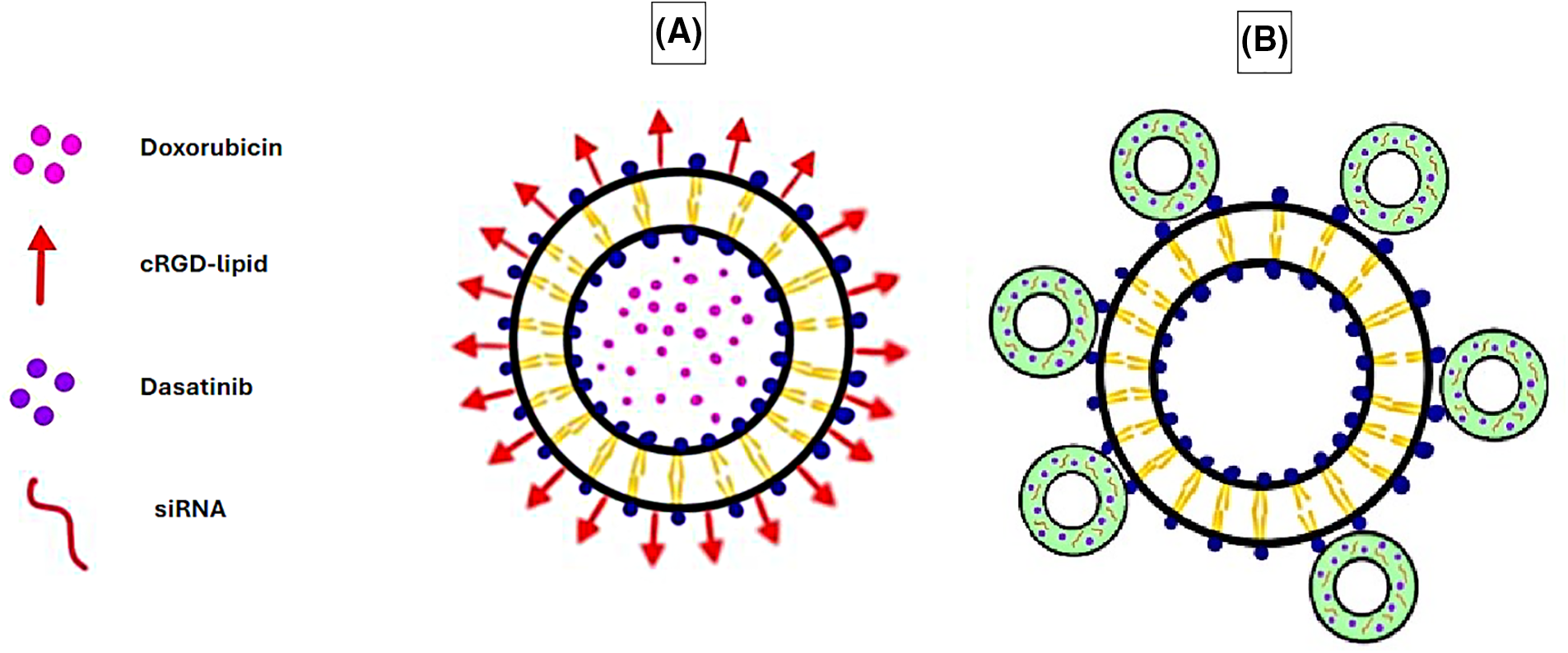
Figure 1: Examples of liposomes used as DDSs in cancer therapy, liposomes vs. microbubble liposomes. (A) Doxorubicin-loaded liposome conjugated with cRGD-lipid. (B) Microbubble-liposome loaded with dasatinib and siRNA. The graphing software HiPaint, by Aige (Wuhan) Technology Co., Ltd. (China), was used to prepare the above figure.
The cRGD liposomes were taken one step further in an orthotopic triple-negative breast cancer (TNBC) mouse model with a 4T1 cell line in vivo, where the liposomes were co-loaded with paclitaxel, a chemotherapeutic drug, and celecoxib, a selective cyclooxygenase-2 inhibitor. The results demonstrated an enhanced tumor targeting efficiency and accumulation, along with dendritic cell (DC)-mediated immunity, which led to immunogenic cell death at the tumor site and the inhibition of tumor rechallenge and metastasis [71].
Additionally, microbubbles (gas bubbles) have been combined with liposomes to form enhanced DDSs. Microbubbles are pressure-sensitive drug carriers that release their contents upon exposure to acoustic radiation, such as ultrasound. Combined with liposomes, they result in efficient drug delivery where the microbubble delivers the drug intracellularly, whereas the liposome enables high drug doses to be loaded [72]. This system has been utilized to co-deliver the multi-kinase inhibitor dasatinib, along with COL11A1-targeting small interfering ribonucleic acid (RNA) (siRNA) to the lung adenocarcinoma A-549 cell line (Fig. 1B). This study’s outcome exhibited how the dasatinib-siRNA microbubble-liposomes significantly inhibited cell proliferation in vitro compared to dasatinib alone or dasatinib microbubble-liposomes lacking siRNA [73].
Micelles are amphiphilic molecules composed of a hydrophilic outer capsule and a hydrophobic core, enabling hydrophobic drugs to be encapsulated within the core [74]. They are formed when lipids or surfactants are dispersed in an aqueous solution, forming aggregates with a size range of 5–100 nm [60,75]. The term micelle was first introduced by James William McBain at a Faraday meeting in 1913 while describing aggregates of soap molecules in aqueous solutions [76,77].
In one study, Fan et al. examined the ability of polyzwitterionic micelles loaded with paclitaxel to inhibit tumor growth after oral administration. The study was conducted in vitro, where the micelles successfully penetrated and accumulated in 4T1 tumor spheroids. Furthermore, three in vivo mouse models were used, consisting of hepatocellular carcinoma HEP G2 cell line, patient-derived hepatocellular carcinoma xenografts, and TNBC 4T1 cell line, where apoptosis-related death was displayed, and tumors were inhibited with low toxicity to the body [78].
In another study, pH-sensitive micelles co-loaded with CD47 antibodies and chlorin e6 (Ce6) were used to target osteosarcomas in the K7M2 cell line in vitro and in vivo (Fig. 2A). The acidic TME triggered the release of the micelles’ cargo, leading to the blockage of the “don’t eat me” signals by cancer cells and inducing damage by the reactive oxygen species generated by Ce6, resulting in apoptosis, necrosis, and immunogenic cell death [79].
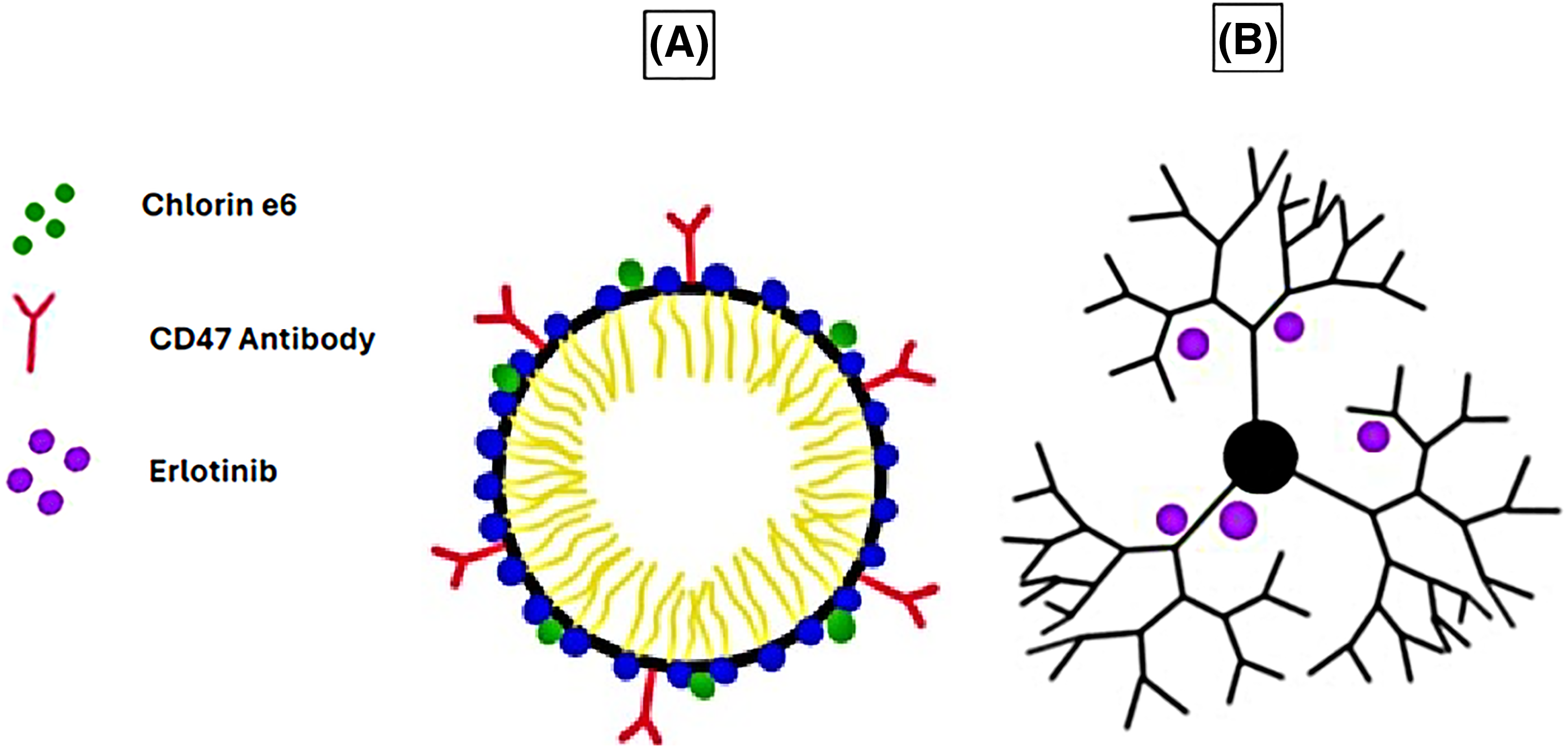
Figure 2: Examples of micelles and dendrimers used as DDSs in cancer therapy. (A) Micelle conjugated with CD47 antibody and loaded with chlorin e6. (B) Erlotinib-loaded G4 PAMAM dendrimer. The graphing software HiPaint by Aige (Wuhan) Technology Co., Ltd. was used to prepare the above figure.
Protein-based nanoparticles involve a protein as a major component of their structure [80]. The synthesis methods of protein-based nanoparticles are discussed by Wang et al. [81]. A successful example used for cancer treatment is Abraxane, which combines the chemotherapeutic drug paclitaxel with human serum albumin (HSA), an approved drug to treat breast cancer by the FDA [82]. Pegaspargase (Oncaspar®), a PEGylated formulation of asparaginase, is an FDA protein-based nanoparticle that is approved for the treatment of acute lymphocytic leukemia in children [83] with low toxicity and enhanced benefit [84]. Gelatin nanoparticles are another example of a protein-based nanoparticle that research evidence is giving promising results in the delivery of the 3-alkyl pyridinium salt, which inhibits the anti-apoptotic and proliferative effects of nicotine on A-549 lung adenocarcinoma cell lines [85]. Silk fibroin, a biopolymer extracted from domestic silkworms approved by the FDA as a safe and nontoxic product, is employed to deliver bioactive compounds [86]. Experiments also provide evidence for the enhanced efficacy of sorafenib when combined with camel-based casein nanoparticles delivered against hepatocarcinoma cells [87].
Polymeric nanoparticles are solid particles that consist of macromolecular polymers [88]. One example of such nanoparticles includes dendrimers, which Fritz Vogtle discovered in 1978 [34]. These are three-dimensional hyperbranched polymers with a size range of 1–15 nm [89] and spherical geometry [90]. They consist of a central core with repeated branches attached and a functional group at the periphery [91], allowing them to encapsulate hydrophilic and hydrophobic drugs. Dendrimers are recognized for their reproducibility at a scalable level due to their mono-dispersity [90].
In one study, polyamidoamine (PAMAM) dendrimers of generation 4.5 (G4.5) conjugated with histidine and cysteine delivered doxorubicin to the cervical carcinoma HeLa cell line in vitro. The results indicated high specificity towards the cancer tissue with reduced off-target side effects, leading to the inhibition of cell proliferation. The dendrimers also inhibited cell proliferation in zebrafish models in vivo [92].
Another study compared the use of two separate erlotinib-loaded PAMAM dendrimer generations, G4 (Fig. 2B) and G5, and their ability to function as DDSs. This study, conducted on non-small cell lung carcinoma (NSCLC) A-549 cell line in vitro, suggested that the erlotinib-loaded G4 dendrimers had a more significant impact on cell viability than the erlotinib-loaded G5 dendrimers. Moreover, the erlotinib-loaded G4 dendrimers had a significant uptake by the cells compared to the G5 dendrimers, which had minimal cellular uptake [93].
Another example of polymeric nanoparticles is chitosan, which is a linear polymer composed of N-acetyl-D-glucosamine and β-(1-4)-linked-D-glucosamine [94] and obtained from the partial deacetylation of the parent polymer, chitin [95]. The synthesis of chitosan nanoparticles from the chitin polysaccharide usually present in exoskeletons of arthropods [96] with various sizes can be done by numerous procedures. Still, the most used methods are ionotropic gelation and polyelectrolyte complex [97]. Functionalizing the chitosan into nanoparticles has shown promising results when used as a DDS against cancer cells [98]. A study has examined the cytotoxicity of black pomegranate-loaded, chitosan-coated magnetic nanoparticles on breast cancer MDA-MB-231 and 4T1 cell lines in vitro. The results suggested that these nanoparticles successfully induced more significant cell line toxicity than the free drug. Furthermore, it was revealed that the nanoparticles had no toxic effect on regular NIH/3T3 cell lines [99]. Other synthetic polymeric nanoparticles showed high specificity to cancer cells, such as the synthetic copolymer that included folic acid with poly (styrene-alt-maleic anhydride) (FA-DABA-SMA) via a biodegradable linker 2,4-aminobutyric acid (DABA) to target folic acid receptors on cancer cells [19,20,100]. A list of the different types of nanoparticles can be found in Table 1 below.
Immune cells are equipped innately with toll-like receptors (TLRs), NOD-like receptors (NLRs), and RIG-I-Like Receptors (RLRs), serving as cell surface proteins that can detect invaders such as bacteria, viruses, or pathogens alerting the immune system to act by releasing the cytokines [107–109], this system is also found to target cancer cells and destroy them however, cancer cells express some of these receptors that can exploit signaling to promote their survival or suppress immune responses [109,110]. Dendritic cells, macrophages, and T lymphocytes, also known as T Cells, infiltrate the TME innately to react with tumor cells [111–113]; this ability of communication between the immune cells and cancer cells allows them to be the best candidates for genetically engineering them with specific chimeric antigen receptors (CARs) that are designed with motifs to recognize the cancer antigens, bind to them and eventually destroy them [114]. Therefore, Encapsulating CAR T cells with liposomes or lentiviruses makes them vehicles to deliver their payloads specifically to the tumor site [115]. In the TME, tumor-associated macrophages (TAMs) exist in one of two polarization forms: the pro-inflammatory M1 form, which targets and engulfs tumor cells [116], or the immunosuppressive M2 form, which promotes cancer progression via the secretion of anti-inflammatory cytokines [117].
Macrophages and their derivatives in drug delivery
Adoptive cell therapy is defined as introducing immune-competent cells as a treatment modality for cancer. Building upon macrophages’ natural affinity to inflammation in the TME, they have been studied as delivery agents for cancer therapeutic drugs. In one report by Qiang et al., a DDS was created by loading doxorubicin-loaded reduced graphene oxide (rGO) into a mouse macrophage-like cell line, RAW264.7. This system utilized both the innate tumor-targeting ability of macrophages, along with ‘rGO’s photothermal properties to synergistically target and inhibit the growth of tumor cells both in vitro in using RM-1, a mouse prostate cancer cell line and in vivo in mice bearing RM-1 tumors, via the rapid heat-induced release of doxorubicin from rGO when exposed to near-infrared (NIR) radiation [118]. A similar study was conducted by Nguyen et al. with macrophages co-loaded with citric acid-coated superparamagnetic nanoparticles and doxorubicin-loaded thermosensitive liposomes. The experiments were performed in vitro on the TNBC 4T1 cell line and in vivo on a mouse model, concluding that the artificially generated magnetic field allowed the macrophages to travel to the tumor’s location [119].
In addition to directly loading macrophages with therapeutic nanoparticles or drugs, some scientists have reported using macrophage byproducts or components to enhance cancer drug delivery. One example includes the use of exosomes obtained from macrophages. The exosomes were used to co-deliver doxorubicin and cholesterol-modified microRNA (miRNA) 159 in vitro to MDA-MB-231 cells and on a TNBC in vivo mouse model, resulting in a synergistic therapeutic effect. Antitumor effects were exhibited, such as reduced cell proliferation and invasion, increased apoptosis, the silencing of transcription factor 7 (TCF7), and the protein levels of both TCF-7 and Myc [120]. In another study, Haney et al. used macrophage-derived extracellular vesicles (EVs) to deliver the chemotherapeutic drugs doxorubicin and paclitaxel to MDA-MB-231 TNBC cells in vitro and in both T11 and MDA-MB-231 mouse models in vivo. The EVs exhibited efficient accumulation and antitumor activity in vitro while inhibiting tumor growth in vivo [121]. Despite their low immunogenicity, high stability, affinity to tumor cells, and ability to fuse with other cells under acidic TME conditions, there are limitations to these EVs. Such limitations include significant alterations to the EV structure needed for efficient drug loading and the restriction of cargo carried by EVs, as they should be hydrophobic [121].
CAR-T and dendritic cell-based vaccines
The success of getting FDA approval for using a CAR-T cell drug named Kymriah to deliver tumor-specific antigens is a milestone in supporting personalized medicine for patients with acute lymphocytic leukemia [122,123]. Infiltrated T-cells can be isolated from tumors and cultured as active tumor targets. Once the T-cells are activated and propagated ex vivo, they will be returned and infused into the same patient from whom they were initially isolated; therefore, they are called autologous antitumor lymphocytes [124]. Recent studies suggested that CAR-T cells overcome the chemotherapy resistance in hard-to-treat cancers and increase overall survival [125]. Like engineered T-cells, DC-based Vaccines are another type of DDS, such as immune cell therapy. DCs are genetically modified to express tumor antigens and are usually more effective at triggering antitumor-immune responses when compared to DCs pulsed with tumor peptides [126]. The DC vaccine named PROVENGE is a successful example of an FDA-approved prostate cancer DC vaccine [127].
Viruses are obligate intracellular parasites [128] that infect and reproduce inside host cells [129]. They consist of short segments of either deoxyribonucleic acids (DNA) or ribonucleic acids (RNA) [130] encapsulated within a protein layer called a capsid [129]. Viruses can further be divided into two groups: enveloped viruses, bound by an outer lipid membrane, and non-enveloped viruses that ‘don’t have a lipid membrane [131].
Viral nanoparticles (VNPs) comprise materials obtained from plant, bacterial, and mammalian viruses to form the drug-enclosed capsid protein [132]. They also retain the innate qualities of viruses, such as their ability to self-assemble into capsids, while carrying artificial cargo, such as therapeutic drugs [129]. On the other hand, virus-like particles (VLPs) are a sub-class of VNPs [133] that are non-infectious as they lack genetic materials [134]. These viral particles can induce immune responses via their antigenic properties and are thus also used in vaccine development [129].
In one study, particles of a plant virus called pH-sensitive pepper mild mottle virus (PMMoV) were conjugated with folate and loaded with paclitaxel to form FA-PMMoVs (Fig. 3A). These viral nanoparticles were then compared with free paclitaxel and targeted toward breast cancer MCF-7 cell line in vitro, with the results suggesting enhanced cellular uptake and apoptotic cell death in the folate receptor-overexpressing MCF-7 [135].
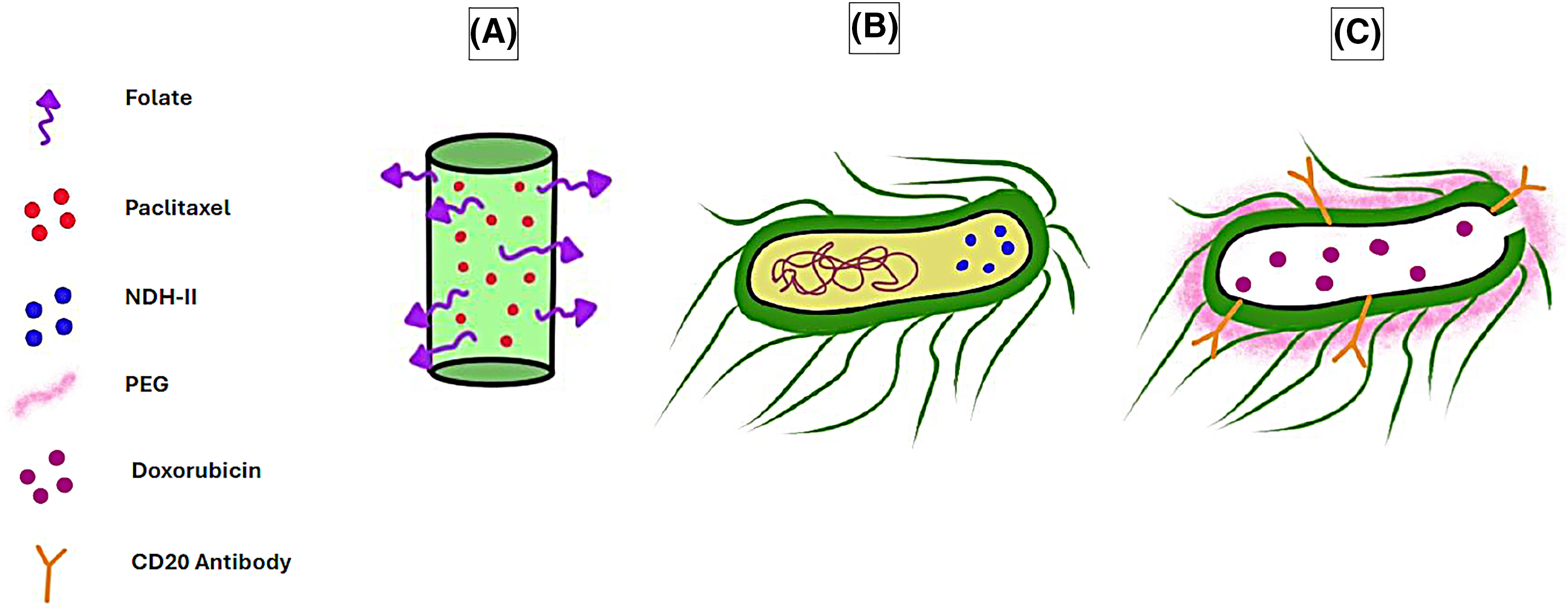
Figure 3: Examples of viruses and bacteria used as DDSs in cancer therapy. (A) Paclitaxel-loaded, folate-conjugated PMMoV. (B) E. coli engineered to overexpress NDH-II. (C) Doxorubicin-loaded PEG-coated bacterial ghost conjugated with CD20 antibody. The graphing software HiPaint, by Aige (Wuhan) Technology Co., Ltd., was used to prepare the above figure.
Moreover, Bao et al. reported using a VNP called reBiosome, packaged with a therapeutic Cas9 messenger RNA (mRNA) gene editing system to delete an oncogenic enhancer of c-Myc in the MCF-7 breast cancer cell line. The outcome indicates successful gene deletion, thus a reduction in c-Myc expression and significant inhibition of the MCF-7 cell line in vitro. Meanwhile, the in vivo results demonstrated tumor growth suppression and apoptotic cell death within the tumor tissue in mice [136].
Bacteria and bacterial-derived methods as DDSs
Bacteria are another example of vehicles that can deliver antitumor drugs.
Utilizing bacteria to fight cancer isn’t new, as it has been practiced and proven efficient when Willian B. Coley injected a patient with Streptococcus to treat their inoperable cancer, leading to tumor shrinkage [137]. He has since treated nearly 1000 patients using bacteria-derived products, termed Coley’s Toxins [138]. Coley’s Toxins stimulate the patient’s immune system, thus helping with tumor destruction via immunotherapy [139]. Although the FDA banned Coleys’ Toxins in 1963 [140], the idea of immunotherapy still sparked in the brains of interested scientists. The immunotherapy drug named axalimogene filolisbac (AXAL, or ADXS11–001) was produced by the attenuation of a strain of Listeria monocytogenes (Lm) that has been engineered to express the Lm-LLO-E7 antigen against human papillomavirus (HPV) infected cancer cells, consequently helping the immune system to target tumor cells infected with HPV [141]. AXAL or ADXS11–001 is currently undergoing clinical trials in various cancer types, including cervical (Phase III) [142–146], head and neck (Phase II) [147,148], and anal cancer (Phase II) [149,150].
Bacteria have been genetically modified by harnessing the power of synthetic biology to target tumor cells and deliver therapeutic agents precisely. They are a great choice due to their innate ability to colonize and proliferate within the hypoxic tumor core [151,152].
In one example, Escherichia coli (E. coli) was engineered to overexpress alternative NADH dehydrogenase (NDH-II) (Fig. 3B), hence enabling the bacteria to function as a bioreactor running a Fenton-like reaction and producing H2O2 and hydroxyl radicals, which induced apoptosis in tumor cells. The bacteria successfully targeted the colon cancer CT 26 cell line both in vitro and in a mouse model in vivo while displaying a reduced accumulation in non-target organs [153].
Additionally, the use of bacteria as DDSs was further employed by Asensio-Calavia et al., where synthetic biology was utilized to program bacteria to target epidermal growth factor receptor (EGFR) on cancer cells and inject catalytic fragments of adenosine diphosphate-ribosyltransferase toxins into colon cancer HCT-116 cell line in vitro and in vivo, leading to cancer cell death and enhanced tumor suppression [154].
BGs are an example where altered bacteria are employed as drug delivery vehicles. BGs, as the name suggests, are empty bacterial ‘shells’ formed from the empty envelopes of Gram-negative bacteria such as E. coli. These BGs are formed by expressing the cloned gene E of bacteriophage PhiX174 origin in E. coli, which leads to bacterial lysis and the subsequent formation of a transmembrane tunnel through the cell, leaving an intact cell envelope consisting of an outer and inner membrane while lacking a cytoplasm [155]. Recombinant BGs can be formed by engineering the bacteria before subjecting them to lysis, such that recombinant proteins are inserted into the inner membrane by N or C terminal anchor sequences [156].
BGs have several advantages when used as drug delivery agents, including their immense safety, as they are essentially membranes with no cytoplasmic or genetic content. Furthermore, they are affordable to produce in large quantities and can be genetically modified to express any antigen of choice. Finally, they are highly immunostimulatory, which can enhance the effectiveness of the engineered antigen [157].
One recent example of BGs used as anti-cancer drug delivery agents has been demonstrated by Li et al., where doxorubicin-loaded E. coli BGs have been modified with polyethylene glycol (PEG) and a CD20 antibody (Fig. 3C) and tested against non-Hodgkin’s lymphoma cells in vitro. The results demonstrated great specificity of the BGs, as they targeted CD20-positive Raji cells and exhibited higher cytotoxic effects than BGs that were not modified with a CD20 antibody. Additionally, the modified BGs exerted minimal cytotoxicity on mouse fibroblast L929 cells, representing non-cancerous cells [158].
Similarly, Klebsiella pneumonia has been used to form BGs co-loaded with doxorubicin and phycocyanin extract, which resulted in a synergistic antitumor effect when used to treat NSCLC cell lines A-549 and NCI-H358 in vitro where these BGs induced apoptosis, inhibited proliferation, modulated oncogenic signaling pathways and increased the cells’ sensitivity to doxorubicin [159].
Minicells, discovered by Adler et al., are miniature, anucleate cells that do not divide and range in size from 100–400 nm [46,160]. Originally obtained from E. coli K12 strains, these cells contain proteins and RNA and can respire due to enzyme activity. They cannot divide due to the lack of chromosomal DNA [46]; however, they often contain plasmids [161]. Minicells are produced in rod-shaped bacteria due to unusual cell division at chromosome-free polar ends [161,162]. Such cell divisions in bacteria are usually caused by alterations in the Min system which is the division leading to unequal size of daughter cells, leading to cell wall formation near the cells’ poles rather than at the center of the cells, therefore resulting in two different daughter cells: one small minicell lacking in chromosomal DNA and one elongated, filamentous cell containing chromosomal DNA [163,164].
In a recent study, minicells derived from the probiotic bacterial strain E. coli Nissle were engineered to produce azurin, a peptide with potent anti-cancer activity [165]. This study conducted various in vitro tests with two colon cancer cell lines: mouse CT-26 and human HT29. The minicells exhibited their ability to preferentially target and bind to the tumor cells compared to the colon epithelial NCM-460 cell line, leading to the release of azurin at the TME and apoptotic cancer cell death. Additionally, the azurin-minicells inhibited cancer cell proliferation, migration, and invasion [164]. Bacteria minicells were tested in Phase I clinical trials, where minicells engineered to target EGFR have been tested on patients with advanced solid tumors and patients with recurrent glioblastoma [166,167].
SimCells are generated novel, simple cells, which are cells that build on the idea of minicells that lack a chromosome. The difference between minicells and SimCells is their size (Fig. 4A–C) and the method by which the chromosomes have been removed. SimCells are normal-sized cells genetically engineered to express I-CeuI, an endonuclease that forms double-stranded breaks in the bacteria’s chromosome. After the formation of the double-stranded breaks, the chromosome will be degraded entirely via the bacterial cell’s endogenous nucleases [168].
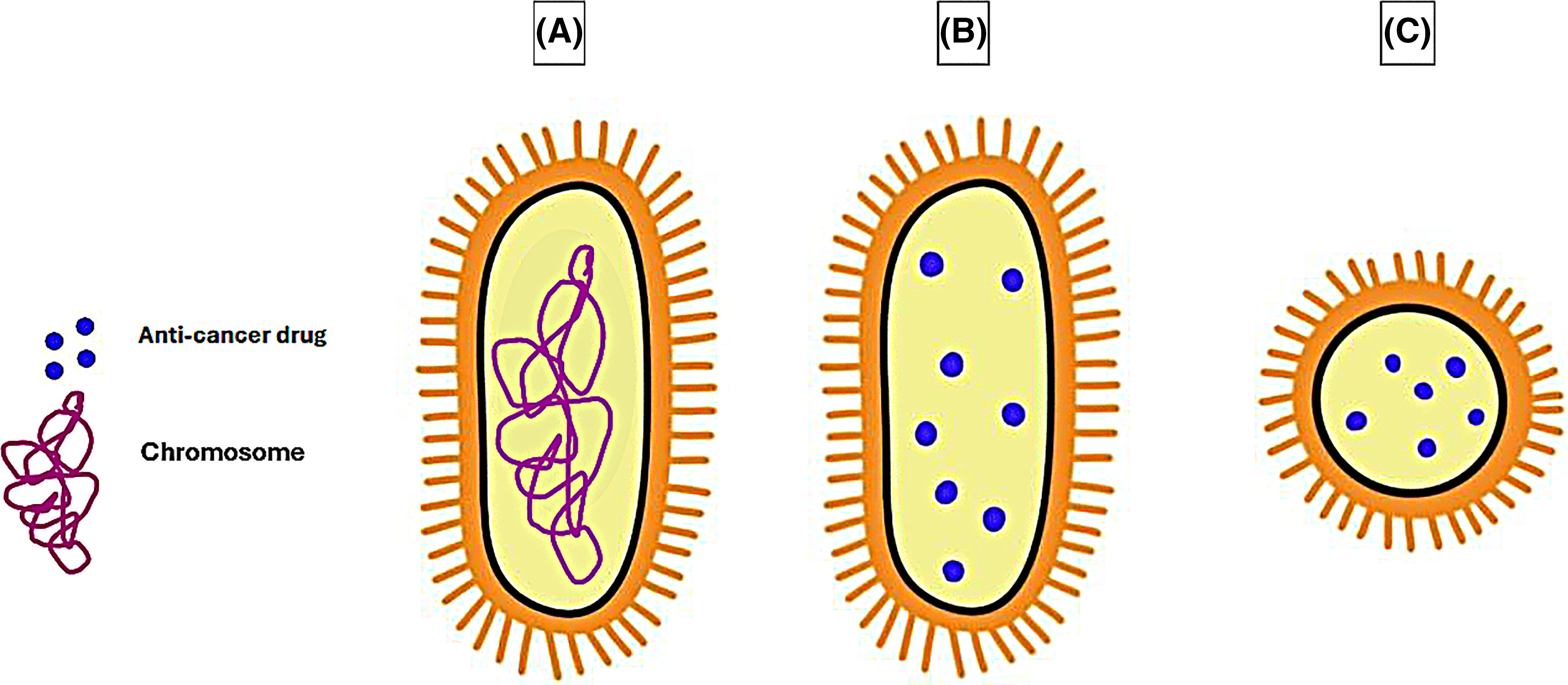
Figure 4: Comparison in the size of a (A) normal bacterial cell, (B) drug-loaded Sim Cell, and (C) drug-loaded minicell. The graphing software HiPaint, by Aige (Wuhan) Technology Co., Ltd., was used to prepare the above figure.
In one study, SimCells displaying anti-carcinoembryonic antigen (CEA) nanobodies demonstrated their ability to target the CEA-expressing colorectal Caco-2 cell line and not the non-CEA-expressing SW-80 cell line. Additionally, these SimCells were engineered with an inducible genetic circuit to produce the anti-cancer drug catechol, which resulted in cancer cell death in vitro and a precise manner [169].
Bacterial outer membrane vesicles (OMVs)
OMVs were first observed by Knox et al. as blebs bound by triple-layer membranes released by E. coli grown under conditions of limited lysine [170]. OMVs are released from gram-negative bacteria during their exponential growth phase [171]; these spherical vesicles have a size range of 20–500 nm [172,173] and are found in the bacterial growth media [171]. OMVs are obtained from the bacteria’s outer membrane, and their contents are similar to those of the bacteria they come from [174], as they contain outer membrane and periplasmic proteins but at a lower concentration when compared to the parent cells [175]. OMVs also contain RNA and chromosomal DNA [176] while lacking cytoplasmic proteins [175].
Compared to other DDSs, OMVs are advantageous due to their innate immunogenicity, as they include both immunogenic endogenous proteins [177] and lipopolysaccharides (LPS) on their surface [178]. Additionally, they preferentially target and accumulate in tumor tissues over healthy ones and exhibit antitumor effects on various cancer types, as suggested by studies on numerous in vivo mouse models with the following cell lines: MC-38 colon cancer, metastatic 4T1, B-16BL6 melanoma, and CT-26 colon adenocarcinoma [179] and in vitro against cell lines including breast cancer MCF-7 and colon cancer Caco-2 [180]; breast cancer SkBr3 and leukemia Nalm6 [181]; and ovarian cancer OV-33 [182]. In contrast, standard cell lines such as human keratinocytes HaCaT and human dermal fibroblast HdFn were less affected by the OMV’s cytotoxicity [182,183].
To date, various studies have utilized OMVs as DDSs. In one example, OMVs were engineered to display human epidermal growth factor receptor-2 (HER-2) affibodies, small proteins that selectively bind to targets with a high affinity [184]. These OMVs, loaded with kinesin spindle protein siRNAs, lead to cell death of various HER-2 expressing cell lines in vitro, including ovarian cancer SKOV-3, breast cancer BT-474, and HCC-1954. Interestingly, only the siRNA-loaded OMVs caused cytotoxicity and not the unloaded OMVs. The tests were conducted on mouse models in vivo, where the affibody siRNA-loaded OMVs displayed more significant tumor inhibition than the non-targeted OMVs [185].
OMVs have been engineered to display various proteins that aid in targeting them towards tumor cells, including programmed death-1 (PD-1) to target tumor cells overexpressing programmed death ligand-1 (PD-L1) in mouse models derived from B-16 melanoma and CT-26 colorectal cell lines. The results were promising as the PD-1 OMVs had more potent tumor inhibition than PD-1 alone or OMVs alone [186].
A similar idea was described in Pan and colleagues’ work, where they developed OMVs displaying LyP-1 to target tumors in vitro and deliver PD-1 plasmids. The three cell lines tested (4T1 breast cancer, B-16 melanoma, and CT-26 colorectal cancer) successfully took up the OMVs and expressed PD-1, which bound to their intrinsic PD-L1, thus leading to self-blockade. The potent anti-tumoral activity was seen both in vitro and in vivo [187].
Another interesting example can be seen in Rezaei Adriani et al.’s study, where OMVs were engineered with scFv proteins to target EGFR on various cell lines in vitro, such as HT29 colorectal cancer cells, HCT-116 colon cancer cells, A-549 lung cancer cells, 4T1 breast cancer cells, and normal kidney HEK-293 cells; and in 4T1 mouse models in vivo. The in vitro results did not display any cytotoxicity. In contrast, the in vivo results showed significant antitumor effects explained by the OMVs’ activation of natural killer (NK) cells that skewed M2 macrophages towards an M1 phenotype in mice, and these NK cells are not present in the in vitro cell culture model [188].
Finally, one interesting article described macrophage-encapsulated OMVs used to deliver Ce6 and doxorubicin to breast cancer 4T1 cell lines in vitro and in a mouse model in vivo. The therapeutic outcome consisted of shifting macrophages from anti-inflammatory M2 to pro-inflammatory M1 and pyroptosis, a form of microbial-initiated inflammatory cell death [189,190]. The list of cell-based drug delivery systems is listed in Table 2 below.

Nanoparticles combined with cell-based DDS results in better therapeutic effect
Combination therapies employ multiple therapeutic agents to treat cancer [198] and reduce the drug resistance commonly caused by monotherapies [199]. Since drug combinations are effective in cancer treatment, using a good DDS is needed to carry multiple drugs to the TME, which is one of the significant objectives of current research. Many of the laboratory studies discussed in this review utilized the co-delivery of drugs (combination therapy) that showed better results in combating cancer cells, such as combining microbubbles with liposomes [71–73].
Similarly, the co-delivery of paclitaxel and resveratrol combination using liposomes and macrophages showed a better ability to target tumor cells when compared with using liposomes alone in an in vivo mouse study that is attributed to the innate homing ability of the macrophages [200]. Drug-loaded nanoparticles maintain macrophages’ viability and homing ability, as the free drugs could be toxic. Moreover, the macrophages should be derived from the same patient to minimize harmful side effects [118]. Interestingly, combining bacterial ghosts with nanoparticles gives promising results, as shown by Ling et al., who examined the use of liposomal paclitaxel-loaded BGs coated with cancer cell membranes. This combination therapy was tested on the 4T1 cell line in vitro and on a metastatic lung cancer mouse model in vivo, leading to positive results that demonstrated synergistic chemo-immunotherapy. Furthermore, the cancer cell membrane enabled a faster fusion of the BGs with tumor cells compared to the BGs on their own and a higher cancer cell targeting by the nanoparticles [201].
Indeed, recent developments in personalized and precision medicine have been achieved due to the development of smart delivery systems. Research and development are improving the DDSs based on the challenges faced at the laboratory or clinical levels. All indicators show that the future of cancer therapy may lie in combining nanotechnology and cellular engineering, bringing hope for better treatment efficacy with minimum toxicity for such heterogeneous diseases. The diversity of DDSs discussed in this review provides a collective view of the different types and options for researchers to choose from to deliver drugs effectively to tumor cells. Each DDS has its mechanism of action, and despite the diverse DDSs we see, the ones first invented are still in use and show effectiveness, such as the FDA-approved Doxil and Onivyde [202]. Research and development continue to improve liposomes to customize and enhance their interactions with negatively charged cancer cells while reducing their toxicity to healthy cells bearing a neutral charge [203]. The drug-loading capacity of liposomes is also relatively large—more significant than that of micelles and similar to that of dendrimers, allowing a larger quantity of drugs to be loaded if needed [204].
Concerning using micelles as DDSs, there are a few disadvantages, including their comparatively low drug-loading capacity [203], their ability to carry only hydrophobic drugs efficiently [102], their poor stability in the blood [205], and the insufficient interactions between micelles and malignant cells due to their neutral charge [203]. Despite these drawbacks, the main benefit of micelles is their low toxicity [206].
Dendrimers are another type of DDS discussed in this review. Although dendrimers have advantages, such as their high drug-loading capacity [203] and reproducibility at a scalable level [90], they also have a few downsides. Their main issue is their toxicity, especially for dendrimers containing amine groups, such as PAMAM dendrimers. Dendrimers with a higher generation tend to be more toxic than their lower generation counterparts, as amine groups increase as a dendrimer’s generation rises [207]. Another thing to consider is the dendrimer’s charge, which can be altered to carry either a positive, negative, or neutral charge [208]. Notably, positively charged dendrimers function as a double-edged sword because although they induce cytotoxicity in negatively charged cancer cells [209], they are also toxic to normal cells [210] and erythrocytes [211].
Similar to other bacterial-derived DDSs, such as BGs, the surface of OMVs is decorated with LPS. The benefits of the LPS displayed on OMVs are a topic of great controversy among scientists. In one aspect, the lipid A component of LPS is responsible for its endotoxicity and can lead to sepsis and toxic shock [178]. On the other hand, the LPS also leads to beneficial immunostimulatory effects in the TME. Therefore, a balance has to be reached between these two effects [212].
On the bright side, scientists have devised ways to reduce the LPS cytotoxicity in bacteria [213], enabling the OMVs derived from them to be used in cancer therapy [188,189]. The need to reduce LPS toxicity was further demonstrated in a study on the human monocyte THP-1 cell line. This study examined the difference in cytotoxicity between OMVs obtained from msbB E. coli mutants vs. wild-type strains, and the results indicated that the wild-type strains induced toxicity in vitro and even mortality in mice in vivo [185].
OMVs are also advantageous due to their innate cytotoxic effects against cancer cells [179,214]. However, a contradicting study suggests that OMVs obtained from pathogenic strains of E. coli have no cytotoxic effects on Caco-2 colon carcinoma cell lines and even cause a significant increase in their growth [215]. One issue with OMVs is their contribution to the immune evasion of cancer cells [216], as OMVs induce interferon-γ (IFN-γ) production by T-cells [179], which leads to the overexpression of PD-L1. That, in turn, binds to PD-1 on T-cells, hence suppressing its immune response [186]. Interestingly, two studies indicate that OMVs do not elicit antitumor effects in vitro but may do so in vivo [186,188]. That might suggest that their in vivo toxicity mainly stems from the surrounding immune system’s reaction to OMVs.
Finally, an emerging enhancement to DDSs involves using neoantigens, newly formed antigens from genetic mutations typically found in cancer cells. These mutations produce abnormal proteins not found in normal cells, allowing the immune system to recognize them as foreign. Since neoantigens are unique to the tumor, they are considered promising targets for cancer immunotherapy, as they help the immune system to distinguish between healthy and cancerous cells [217].
Yet, there are hurdles and limitations to the research in targeted drug delivery systems. Factors such as TME barriers represented by the heterogeneity of solid tumors limit the proper delivery of drugs to all areas of the tumor since some areas can be hypoxic, areas with poor blood flow, areas with different interstitial fluid pressure, and regions with varying densities of cells [218–220]. Even though immune cells can target tumor cells, a solid tumor can be immunosuppressive for the immune cells to penetrate and target tumor cells, a limitation scientists are trying to solve using different strategies [220,221]. One difficulty scientists face in targeting nanoparticle use is the circulation clearance by the macrophages in the liver and spleen, which decreases the availability of nanoparticles in the tumor tissue [222,223]. Although PEGylated nanocarriers have shown better efficacy, they face challenges in producing anti-PEGylated antibodies that target these nano-carriers and clear them [224]. Tumor cells adapted to immune cell therapeutics by decreasing the target antigens’ presentation and initiating anti-apoptotic pathways, making it harder for the immune cells to recognize them [5]. In addition to tumor interaction with DDSs, the variability among patients adds another layer of challenges based on the patient’s metabolic activity, immune system function, and tumor characteristics. Therefore, active research looks for all the strategies available based on the DDSs they used to overcome these limitations and challenges. Future meta-analysis for each DDS and the corresponding strategy for improvement is recommended to comprehend all the findings in one place.
In conclusion, the diverse nature of DDSs indicates a positive sign of hope for better treating the different types of cancer precisely with the most negligible toxicity possible. More efficient cancer treatment can be designed and developed by combining the biocompatibility feature of immune cells and the increase in drug bioavailability of nanoparticles. Strategies to increase the bioavailability and enhance nanoparticle accumulation and action in tumors are under intense research. The active targeting of nanoparticles by ligands, such as peptides, antibodies, or host cell membranes, increases the efficacy of the nanoparticle availability in the tumor tissue. The addition of ligands will add superiority to the nanoparticles by either attaching them to the cells’ membrane or even internalizing them inside the tumor cells. Similar to the ligand binding for active targeting, modification to the physicochemical properties of nanoparticles is critical for the best performance against tumor tissue. Whether it is a nanoparticle, immune cell, virus, or bacteria-derived DDSs, all these vehicles will be designed and chemically or genetically modified to target tumors precisely based on tumor cell surface antigens and their TME. While there are approved drugs using nanoparticles and immune cells for certain types of cancer, the bacterial ghosts, Minicells, SimCells, and bacterial outer membrane vesicles (OMVs) are still under research focus. Since next-generation sequencing technology is accelerating, we believe more DDSs will come along for future trials. Finally, combining nanoparticles with cell-based delivery methods such as immune cells might be a good strategy for the stability and specificity of these delivery vehicles by functioning as a Trojan horse and evading detection by the host’s defense systems.
Acknowledgement: None.
Funding Statement: This research review is prepared using the fund obtained by Sirin A. Adham from Sultan Qaboos University internal grant through the College of Science through the Deanship of Research # IG/SCI/BIOL/24/01.
Author Contributions: Meryem A. Abdessalem: Writing—original draft. Sirin A. Adham: Supervision, editing, and reviewing. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: No data was used for the research described in the article.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74(3):229–63. doi:10.3322/caac.v74.3. [Google Scholar] [CrossRef]
2. Moitra K, Lou H, Dean M. Multidrug efflux pumps and cancer stem cells: insights into multidrug resistance and therapeutic development. Clin Pharmacol Ther. 2011;89(4):491–502. doi:10.1038/clpt.2011.14. [Google Scholar] [PubMed] [CrossRef]
3. Mansoori B, Mohammadi A, Davudian S, Shirjang S, Baradaran B. The different mechanisms of cancer drug resistance: a brief review. Adv Pharm Bull. 2017;7(3):339. doi:10.15171/apb.2017.041. [Google Scholar] [PubMed] [CrossRef]
4. McAleese CE, Choudhury C, Butcher NJ, Minchin RF. Hypoxia-mediated drug resistance in breast cancers. Cancer Lett. 2021;502:189–99. doi:10.1016/j.canlet.2020.11.045. [Google Scholar] [PubMed] [CrossRef]
5. Li Y-R, Halladay T, Yang L. Immune evasion in cell-based immunotherapy: unraveling challenges and novel strategies. J Biomed Sci. 2024;31(1):5. doi:10.1186/s12929-024-00998-8. [Google Scholar] [PubMed] [CrossRef]
6. Beaumont KA, Hill DS, Daignault SM, Lui GY, Sharp DM, Gabrielli B, et al. Cell cycle phase-specific drug resistance as an escape mechanism of melanoma cells. J Invest Dermatol. 2016;136(7):1479–89. doi:10.1016/j.jid.2016.02.805. [Google Scholar] [PubMed] [CrossRef]
7. Bhullar KS, Lagaron NO, McGowan EM, Parmar I, Jha A, Hubbard BP, et al. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer. 2018;17(1):48. doi:10.1186/s12943-018-0804-2. [Google Scholar] [PubMed] [CrossRef]
8. Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8(3):193–204. doi:10.1038/nrc2342. [Google Scholar] [PubMed] [CrossRef]
9. Chae YC, Kim JH. Cancer stem cell metabolism: target for cancer therapy. BMB Rep. 2018;51(7):319–26. doi:10.5483/BMBRep.2018.51.7.112. [Google Scholar] [PubMed] [CrossRef]
10. Verma M. Personalized medicine and cancer. J Pers Med. 2012;2(1):1–14. doi:10.3390/jpm2010001. [Google Scholar] [PubMed] [CrossRef]
11. Shaikh N, Linthoi RK, Swamy KV, Karthikeyan M, Vyas R. Comprehensive molecular docking and dynamic simulations for drug repurposing of clinical drugs against multiple cancer kinase targets. J Biomol Struct Dyn. 2023;41(16):7735–43. doi:10.1080/07391102.2022.2124453. [Google Scholar] [PubMed] [CrossRef]
12. Baudino TA. Targeted cancer therapy: the next generation of cancer treatment. Curr Drug Discov Technol. 2015;12(1):3–20. doi:10.2174/1570163812666150602144310. [Google Scholar] [PubMed] [CrossRef]
13. Sarmento-Ribeiro AB, Scorilas A, Goncalves AC, Efferth T, Trougakos IP. The emergence of drug resistance to targeted cancer therapies: clinical evidence. Drug Resist Updat. 2019;47:100646. doi:10.1016/j.drup.2019.100646. [Google Scholar] [PubMed] [CrossRef]
14. Adepu S, Ramakrishna S. Controlled drug delivery systems: current status and future directions. Molecules. 2021;26(19):5905. doi:10.3390/molecules26195905. [Google Scholar] [PubMed] [CrossRef]
15. Kandula S, Singh PK, Kaur GA, Tiwari A. Trends in smart drug delivery systems for targeting cancer cells. Mater Sci Eng: B. 2023;297:116816. doi:10.1016/j.mseb.2023.116816. [Google Scholar] [CrossRef]
16. Felice B, Prabhakaran MP, Rodriguez AP, Ramakrishna S. Drug delivery vehicles on a nano-engineering perspective. Mater Sci Eng C Mater Biol Appl. 2014;41:178–95. doi:10.1016/j.msec.2014.04.049. [Google Scholar] [PubMed] [CrossRef]
17. Vikal A, Maurya R, Bhowmik S, Patel P, Gupta GD, Kurmi BD. From conventional to cutting-edge: a comprehensive review on drug delivery systems. Drug Deliv Lett. 2024;14(3):226–43. doi:10.2174/0122103031304556240430161553. [Google Scholar] [CrossRef]
18. Kalaydina RV, Bajwa K, Qorri B, Decarlo A, Szewczuk MR. Recent advances in “smart” delivery systems for extended drug release in cancer therapy. Int J Nanomed. 2018;13:4727–45. doi:10.2147/IJN. [Google Scholar] [CrossRef]
19. DeCarlo A, Malardier-Jugroot C, Szewczuk MR. Folic acid-functionalized nanomedicine: folic acid conjugated copolymer and folate receptor interactions disrupt receptor functionality resulting in dual therapeutic anti-cancer potential in breast and prostate cancer. Bioconjugate Chem. 2021;32(3):512–22. doi:10.1021/acs.bioconjchem.0c00625. [Google Scholar] [PubMed] [CrossRef]
20. Li X, Szewczuk MR, Malardier-Jugroot C. Folic acid-conjugated amphiphilic alternating copolymer as a new active tumor targeting drug delivery platform. Drug Des Devel Ther. 2016;10:4101–10. doi:10.2147/DDDT. [Google Scholar] [CrossRef]
21. Girigoswami A, Girigoswami K. Potential applications of nanoparticles in improving the outcome of lung cancer treatment. Genes. 2023;14(7):1370. doi:10.3390/genes14071370. [Google Scholar] [PubMed] [CrossRef]
22. Ghazal H, Waqar A, Yaseen F, Shahid M, Sultana M, Tariq M, et al. Role of nanoparticles in enhancing chemotherapy efficacy for cancer treatment. Next Mater. 2024;2(8):100128. doi:10.1016/j.nxmate.2024.100128. [Google Scholar] [CrossRef]
23. Wang Y, Deng T, Liu X, Fang X, Mo Y, Xie N, et al. Smart nanoplatforms responding to the tumor microenvironment for precise drug delivery in cancer therapy. Int J Nanomed 2024;19:6253–77. doi:10.2147/IJN.S459710. [Google Scholar] [PubMed] [CrossRef]
24. Kaushik N, Borkar SB, Nandanwar SK, Panda PK, Choi EH, Kaushik NK. Nanocarrier cancer therapeutics with functional stimuli-responsive mechanisms. J Nanobiotechnol. 2022;20(1):152. doi:10.1186/s12951-022-01364-2. [Google Scholar] [PubMed] [CrossRef]
25. Mempel TR, Lill JK, Altenburger LM. How chemokines organize the tumour microenvironment. Nat Rev Cancer. 2024;24(1):28–50. doi:10.1038/s41568-023-00635-w. [Google Scholar] [PubMed] [CrossRef]
26. Yang W, Liu S, Mao M, Gong Y, Li X, Lei T, et al. T-cell infiltration and its regulatory mechanisms in cancers: insights at single-cell resolution. J Exp Clin Cancer Res. 2024;43(1):38. doi:10.1186/s13046-024-02960-w. [Google Scholar] [PubMed] [CrossRef]
27. Zhang S-S, Li R-Q, Chen Z, Wang X-Y, Dumont AS, Fan X. Immune cells: potential carriers or agents for drug delivery to the central nervous system. Mil Med Res. 2024;11(1):19. [Google Scholar] [PubMed]
28. Gong N, Alameh MG, El-Mayta R, Xue L, Weissman D, Mitchell MJ. Enhancing in situ cancer vaccines using delivery technologies. Nat Rev Drug Discov. 2024;23(8):607–25. doi:10.1038/s41573-024-00974-9. [Google Scholar] [PubMed] [CrossRef]
29. Castilla-Casadiego DA, Loh DH, Pineda-Hernandez A, Rosales AM. Stimuli-responsive substrates to control the immunomodulatory potential of stromal cells. Biomacromolecules. 2024;25(10):6319–37. doi:10.1021/acs.biomac.4c00835. [Google Scholar] [PubMed] [CrossRef]
30. Abbasi H, Kouchak M, Mirveis Z, Hajipour F, Khodarahmi M, Rahbar N, et al. What we need to know about Liposomes as drug nanocarriers: an updated review. Adv Pharm Bull. 2023;13(1):7–23. [Google Scholar] [PubMed]
31. Shi T, Song X, Wang Y, Liu F, Wei J. Combining oncolytic viruses with cancer immunotherapy: establishing a new generation of cancer treatment. Front Immunol. 2020;11:683. doi:10.3389/fimmu.2020.00683. [Google Scholar] [PubMed] [CrossRef]
32. Cao Z, Liu J. Bacteria and bacterial derivatives as drug carriers for cancer therapy. J Control Release. 2020;326:396–407. doi:10.1016/j.jconrel.2020.07.009. [Google Scholar] [PubMed] [CrossRef]
33. Gregoriadis G, Ryman BE. Liposomes as carriers of enzymes or drugs: a new approach to the treatment of storage diseases. Biochem J. 1971;124(5):58P. doi:10.1042/bj1240058P. [Google Scholar] [PubMed] [CrossRef]
34. Vögtle F, Richardt G, Werner N. Dendrimer chemistry: concepts, syntheses, properties, applications. Weinheim: Wiley-VCH; 2009, 10.1002/9783527626953 [Google Scholar] [CrossRef]
35. Dang Y, Guan J. Nanoparticle-based drug delivery systems for cancer therapy. Smart Mater Med. 2020;1:10–9. doi:10.1016/j.smaim.2020.04.001. [Google Scholar] [PubMed] [CrossRef]
36. Rodriguez F, Caruana P, De la Fuente N, Espanol P, Gamez M, Balart J, et al. Nano-based approved pharmaceuticals for cancer treatment: present and future challenges. Biomolecules. 2022;12(6):784. doi:10.3390/biom12060784. [Google Scholar] [PubMed] [CrossRef]
37. Barenholz Y. Doxil®—the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–34. doi:10.1016/j.jconrel.2012.03.020. [Google Scholar] [PubMed] [CrossRef]
38. Abdifetah O, Na-Bangchang K. Pharmacokinetic studies of nanoparticles as a delivery system for conventional drugs and herb-derived compounds for cancer therapy: a systematic review. Int J Nanomed. 2019;14:5659–77. doi:10.2147/IJN. [Google Scholar] [CrossRef]
39. Nikolova MP, Kumar EM, Chavali MS. Updates on responsive drug delivery based on liposome vehicles for cancer treatment. Pharmaceutics. 2022;14(10):2195. doi:10.3390/pharmaceutics14102195. [Google Scholar] [PubMed] [CrossRef]
40. Sun L, Liu H, Ye Y, Lei Y, Islam R, Tan S, et al. Smart nanoparticles for cancer therapy. Signal Transduct Target Ther. 2023;8(1):418. doi:10.1038/s41392-023-01642-x. [Google Scholar] [PubMed] [CrossRef]
41. Dvorak HF. Leaky tumor vessels: consequences for tumor stroma generation and for solid tumor therapy. Prog Clin Biol Res. 1990;354A:317–30. [Google Scholar] [PubMed]
42. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12):6387–92. [Google Scholar] [PubMed]
43. Gerlowski LE, Jain RK. Microvascular permeability of normal and neoplastic tissues. Microvasc Res. 1986;31(3):288–305. doi:10.1016/0026-2862(86)90018-X. [Google Scholar] [PubMed] [CrossRef]
44. Matsumura Y, Oda T, Maeda H. General mechanism of intratumor accumulation of macromolecules: advantage of macromolecular therapeutics. Gan To Kagaku Ryoho. 1987;14(3):821–9 (In Japanese). [Google Scholar] [PubMed]
45. Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev. 2011;63(3):131–5. doi:10.1016/j.addr.2010.03.011. [Google Scholar] [PubMed] [CrossRef]
46. Adler HI, Fisher WD, Cohen A, Hardigree AA. Miniature Escherichia coli cells deficient in DNA. Proc Natl Acad Sci U S A. 1967;57(2):321–6. doi:10.1073/pnas.57.2.321. [Google Scholar] [PubMed] [CrossRef]
47. Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev. 2013;65(1):71–9. doi:10.1016/j.addr.2012.10.002. [Google Scholar] [PubMed] [CrossRef]
48. Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, et al. Analysis of nanoparticle delivery to tumours. Nat Rev Mater. 2016;1(5):16014. doi:10.1038/natrevmats.2016.14. [Google Scholar] [CrossRef]
49. Sindhwani S, Syed AM, Ngai J, Kingston BR, Maiorino L, Rothschild J, et al. The entry of nanoparticles into solid tumours. Nat Mater. 2020;19(5):566–75. doi:10.1038/s41563-019-0566-2. [Google Scholar] [PubMed] [CrossRef]
50. Engin AB, Nikitovic D, Neagu M, Henrich-Noack P, Docea AO, Shtilman MI, et al. Mechanistic understanding of nanoparticles’ interactions with extracellular matrix: the cell and immune system. Part Fibre Toxicol. 2017;14:22. doi:10.1186/s12989-017-0199-z. [Google Scholar] [PubMed] [CrossRef]
51. Munir MU. Nanomedicine penetration to tumor: challenges, and advanced strategies to tackle this issue. Cancers. 2022;14(12):2904. doi:10.3390/cancers14122904. [Google Scholar] [PubMed] [CrossRef]
52. Yang T, Zhai J, Hu D, Yang R, Wang G, Li Y, et al. “Targeting design” of nanoparticles in tumor therapy. Pharmaceutics. 2022;14(9):1919. doi:10.3390/pharmaceutics14091919. [Google Scholar] [PubMed] [CrossRef]
53. Hu J, Yuan X, Wang F, Gao H, Liu X, Zhang W. The progress and perspective of strategies to improve tumor penetration of nanomedicines. Chin Chem Lett. 2021;32(4):1341–7. doi:10.1016/j.cclet.2020.11.006. [Google Scholar] [CrossRef]
54. Yoo J, Park C, Yi G, Lee D, Koo H. Active targeting strategies using biological ligands for nanoparticle drug delivery systems. Cancers. 2019;11(5):640. doi:10.3390/cancers11050640. [Google Scholar] [PubMed] [CrossRef]
55. Kingston BR, Lin ZP, Ouyang B, MacMillan P, Ngai J, Syed AM, et al. Specific endothelial cells govern nanoparticle entry into solid tumors. ACS Nano. 2021;15(9):14080–94. doi:10.1021/acsnano.1c04510. [Google Scholar] [PubMed] [CrossRef]
56. Subhan MA, Parveen F, Filipczak N, Yalamarty SSK, Torchilin VP. Approaches to improve EPR-based drug delivery for cancer therapy and diagnosis. J Pers Med. 2023;13(3):389. doi:10.3390/jpm13030389. [Google Scholar] [PubMed] [CrossRef]
57. Nguyen LNM, Lin ZP, Sindhwani S, MacMillan P, Mladjenovic SM, Stordy B, et al. The exit of nanoparticles from solid tumours. Nat Mater. 2023;22(10):1261–72. doi:10.1038/s41563-023-01630-0. [Google Scholar] [PubMed] [CrossRef]
58. Jafernik K, Ladniak A, Blicharska E, Czarnek K, Ekiert H, Wiacek AE, et al. Chitosan-based nanoparticles as effective drug delivery systems–a review. Molecules. 2023;28(4):1963. doi:10.3390/molecules28041963. [Google Scholar] [PubMed] [CrossRef]
59. An H, Deng X, Wang F, Xu P, Wang N. Dendrimers as nanocarriers for the delivery of drugs obtained from natural products. Polymers. 2023;15(10):2292. doi:10.3390/polym15102292. [Google Scholar] [PubMed] [CrossRef]
60. Bose A, Roy Burman D, Sikdar B, Patra P. Nanomicelles: types, properties and applications in drug delivery. IET Nanobiotechnol. 2021;15(1):19–27. doi:10.1049/nbt2.12018. [Google Scholar] [PubMed] [CrossRef]
61. Zheng J, Jiang J, Pu Y, Xu T, Sun J, Zhang Q, et al. Tumor-associated macrophages in nanomaterial-based anti-tumor therapy: as target spots or delivery platforms. Front Bioeng Biotechnol. 2023;11:1248421. doi:10.3389/fbioe.2023.1248421. [Google Scholar] [PubMed] [CrossRef]
62. Li H, Zhu Y, Wang X, Feng Y, Qian Y, Ma Q, et al. Joining forces: the combined application of therapeutic viruses and nanomaterials in cancer therapy. Molecules. 2023;28(22):7679. doi:10.3390/molecules28227679. [Google Scholar] [PubMed] [CrossRef]
63. Cheng W, He L, Ren W, Yue T, Xie X, Sun J, et al. Bacteria-nanodrug cancer treatment system: the combination of dual swords and the confrontation of needle tips. Nano TransMed. 2023;2(2–3):100008. [Google Scholar]
64. Nsairat H, Khater D, Sayed U, Odeh F, Al Bawab A, Alshaer W. Liposomes: structure, composition, types, and clinical applications. Heliyon. 2022;8(5):e09394. doi:10.1016/j.heliyon.2022.e09394. [Google Scholar] [PubMed] [CrossRef]
65. Lombardo D, Kiselev MA. Methods of liposomes preparation: formation and control factors of versatile nanocarriers for biomedical and nanomedicine application. Pharmaceutics. 2022;14(3):543. doi:10.3390/pharmaceutics14030543. [Google Scholar] [PubMed] [CrossRef]
66. Tseu GYW, Kamaruzaman KA. A review of different types of liposomes and their advancements as a form of gene therapy treatment for breast cancer. Molecules. 2023;28(3):1498. doi:10.3390/molecules28031498. [Google Scholar] [PubMed] [CrossRef]
67. Pande S. Liposomes for drug delivery: review of vesicular composition, factors affecting drug release and drug loading in liposomes. Artif Cells Nanomed Biotechnol. 2023;51(1):428–40. doi:10.1080/21691401.2023.2247036. [Google Scholar] [PubMed] [CrossRef]
68. Ta T, Porter TM. Thermosensitive liposomes for localized delivery and triggered release of chemotherapy. J Control Release. 2013;169(1–2):112–25. [Google Scholar] [PubMed]
69. Huang A, Huang L, Kennel SJ. Monoclonal antibody covalently coupled with fatty acid. A reagent for in vitro liposome targeting. J Biol Chem. 1980;255(17):8015–8. doi:10.1016/S0021-9258(19)70595-X. [Google Scholar] [CrossRef]
70. Fu S, Zhao Y, Sun J, Yang T, Zhi D, Zhang E, et al. Integrin αvβ3-targeted liposomal drug delivery system for enhanced lung cancer therapy. Colloids Surf B Biointerfaces. 2021;201:111623. doi:10.1016/j.colsurfb.2021.111623. [Google Scholar] [PubMed] [CrossRef]
71. Qian X, Yang H, Ye Z, Gao B, Qian Z, Ding Y, et al. Celecoxib Augments paclitaxel-induced immunogenic cell death in triple-negative breast cancer. ACS Nano. 2024;18(24):15864–77. doi:10.1021/acsnano.4c02947. [Google Scholar] [PubMed] [CrossRef]
72. Malik R, Pancholi K, Melzer A. Microbubble-liposome conjugate: payload evaluation of potential theranostic vehicle. Nanobiomedicine. 2016;3:1849543516670806. doi:10.1177/1849543516670806. [Google Scholar] [PubMed] [CrossRef]
73. Nannan M, Venkatabalasubramanian S. Microbubble-based liposomal delivery of dasatinib and COL11A1siRNA for enhanced combination therapy against lung adenocarcinoma. Appl Nanosci. 2024;14(8):931–41. doi:10.1007/s13204-024-03057-8. [Google Scholar] [CrossRef]
74. Viswanathan VK, Rajaram Manoharan SR, Subramanian S, Moon A. Nanotechnology in spine surgery: a current update and critical review of the literature. World Neurosurg. 2019;123:142–55. doi:10.1016/j.wneu.2018.11.035. [Google Scholar] [PubMed] [CrossRef]
75. Fujii S, Yamada S, Matsumoto S, Kubo G, Yoshida K, Tabata E, et al. Platonic micelles: monodisperse micelles with discrete aggregation numbers corresponding to regular polyhedra. Sci Rep. 2017;7:44494. doi:10.1038/srep44494. [Google Scholar] [PubMed] [CrossRef]
76. Vincent B. McBain and the centenary of the micelle. Adv Colloid Interface Sci. 2014;203:51–4. doi:10.1016/j.cis.2013.11.012. [Google Scholar] [PubMed] [CrossRef]
77. Schryver SB, Ramsden W, Cross CF, Schidrowitz P, Dreaper WP, McBain JW, et al. Discussion. Trans Faraday Soc. 1913;9:93–107. doi:10.1039/tf9130900093. [Google Scholar] [CrossRef]
78. Fan W, Wei Q, Xiang J, Tang Y, Zhou Q, Geng Y, et al. Mucus penetrating and cell-binding polyzwitterionic micelles as potent oral nanomedicine for cancer drug delivery. Adv Mater. 2022;34(16):e2109189. doi:10.1002/adma.202109189. [Google Scholar] [PubMed] [CrossRef]
79. Xiao J, Xiao H, Cai Y, Liao J, Liu J, Yao L, et al. Codelivery of anti-CD47 antibody and chlorin e6 using a dual pH-sensitive nanodrug for photodynamic immunotherapy of osteosarcoma. Oncol Res. 2024;32(4):691–702. doi:10.32604/or.2023.030767. [Google Scholar] [PubMed] [CrossRef]
80. Hong S, Choi DW, Kim HN, Park CG, Lee W, Park HH. Protein-based nanoparticles as drug delivery systems. Pharmaceutics. 2020;12(7):604. doi:10.3390/pharmaceutics12070604. [Google Scholar] [PubMed] [CrossRef]
81. Wang G, Uludag H. Recent developments in nanoparticle-based drug delivery and targeting systems with emphasis on protein-based nanoparticles. Expert Opin Drug Deliv. 2008;5(5):499–515. doi:10.1517/17425247.5.5.499. [Google Scholar] [PubMed] [CrossRef]
82. Cho H, Jeon SI, Ahn CH, Shim MK, Kim K. Emerging albumin-binding anticancer drugs for tumor-targeted drug delivery: current understandings and clinical translation. Pharmaceutics. 2022;14(4):728. doi:10.3390/pharmaceutics14040728. [Google Scholar] [PubMed] [CrossRef]
83. Dinndorf PA, Gootenberg J, Cohen MH, Keegan P, Pazdur R. FDA drug approval summary: pegaspargase (Oncaspar®) for the first-line treatment of children with acute lymphoblastic leukemia (ALL). Oncologist. 2007;12(8):991–8. doi:10.1634/theoncologist.12-8-991. [Google Scholar] [PubMed] [CrossRef]
84. Riley DO, Schlefman JM, Vitzthum Von Eckstaedt VHC, Morris AL, Keng MK, El Chaer F. Pegaspargase in practice: minimizing toxicity, maximizing benefit. Curr Hematol Malig Rep. 2021;16(3):314–24. doi:10.1007/s11899-021-00638-0. [Google Scholar] [PubMed] [CrossRef]
85. Ozhava D, Winkler P, Mao Y. Enhancing antimicrobial activity and reducing cytotoxicity of silver nanoparticles through gelatin nanoparticles. Nanomed. 2024;19(3):199–211. doi:10.2217/nnm-2023-0246. [Google Scholar] [PubMed] [CrossRef]
86. Hcini K. Silk Fibroin Nanoparticles (SFNs) for nanoencapsulation of bioactive molecules. Int J Nanomater Nanotechnol Nanomed. 2024;10(1):12–5. doi:10.17352/ijnnn. [Google Scholar] [CrossRef]
87. Mittal A, Mahala N, Dhanawade NH, Dubey SK, Dubey US. Evaluation of the cytotoxic activity of sorafenib-loaded camel milk casein nanoparticles against hepatocarcinoma cells. Biotechnol J. 2024;19(3):2300449. doi:10.1002/biot.v19.3. [Google Scholar] [CrossRef]
88. Jiang T, Jin K, Liu X, Pang Z. Nanoparticles for tumor targeting. In: Jana S, Maiti S, Jana S, editors. Biopolymer-based composites. Cambridge: Woodhead Publishing; 2017. p. 221–67. doi:10.1016/B978-0-08-101914-6.00008-9. [Google Scholar] [CrossRef]
89. Mittal P, Saharan A, Verma R, Altalbawy FM, Alfaidi MA, Batiha GE-S, et al. Dendrimers: a new race of pharmaceutical nanocarriers. Biomed Res Int. 2021;2021:8844030. [Google Scholar] [PubMed]
90. Ambekar RS, Choudhary M, Kandasubramanian B. Recent advances in dendrimer-based nanoplatform for cancer treatment: a review. Eur Polym J. 2020;126:109546. doi:10.1016/j.eurpolymj.2020.109546. [Google Scholar] [CrossRef]
91. Morikawa A. Comparison of properties among dendritic and hyperbranched poly(ether ether ketone)s and linear poly(ether ketone)s. Molecules. 2016;21(2):219. doi:10.3390/molecules21020219. [Google Scholar] [PubMed] [CrossRef]
92. Wu SY, Chou HY, Tsai HC, Anbazhagan R, Yuh CH, Yang JM, et al. Amino acid-modified PAMAM dendritic nanocarriers as effective chemotherapeutic drug vehicles in cancer treatment: a study using zebrafish as a cancer model. RSC Adv. 2020;10(35):20682–90. doi:10.1039/D0RA01589J. [Google Scholar] [PubMed] [CrossRef]
93. Fatani WK, Aleanizy FS, Alqahtani FY, Alanazi MM, Aldossari AA, Shakeel F, et al. Erlotinib-Loaded dendrimer nanocomposites as a targeted lung cancer chemotherapy. Molecules. 2023;28(9):3974. doi:10.3390/molecules28093974. [Google Scholar] [PubMed] [CrossRef]
94. Tian B, Hua S, Liu J. Multi-functional chitosan-based nanoparticles for drug delivery: recent advanced insight into cancer therapy. Carbohydr Polym. 2023;315:120972. doi:10.1016/j.carbpol.2023.120972. [Google Scholar] [PubMed] [CrossRef]
95. Kumar A, Kumar A. Chitosan-based drug conjugated nanocomposites: advances and innovation in cancer therapy. Regen Eng Transl Med. 2024;10(1):1–8. doi:10.1007/s40883-023-00310-4. [Google Scholar] [CrossRef]
96. Moussian B. Chitin: structure, chemistry and biology. Adv Exp Med Biol. 2019;1142:5–18. doi:10.1007/978-981-13-7318-3. [Google Scholar] [CrossRef]
97. Divya K, Jisha M. Chitosan nanoparticles preparation and applications. Environ Chem Lett. 2018;16:101–12. doi:10.1007/s10311-017-0670-y. [Google Scholar] [CrossRef]
98. Grewal AK, Salar RK. Chitosan nanoparticle delivery systems: an effective approach to enhancing efficacy and safety of anticancer drugs. Nano TransMed. 2024;3:100040. doi:10.1016/j.ntm.2024.100040. [Google Scholar] [CrossRef]
99. Taherian A, Esfandiari N, Rouhani S. Breast cancer drug delivery by novel drug-loaded chitosan-coated magnetic nanoparticles. Cancer Nano. 2021;12(1):15. doi:10.1186/s12645-021-00086-8. [Google Scholar] [CrossRef]
100. Li X, Sambi M, DeCarlo A, Burov SV, Akasov R, Markvicheva E, et al. Functionalized folic acid-conjugated amphiphilic alternating copolymer actively targets 3D multicellular tumour spheroids and delivers the hydrophobic drug to the inner core. Nanomater. 2018;8(8):588. doi:10.3390/nano8080588. [Google Scholar] [PubMed] [CrossRef]
101. Mella M, Tagliabue A, Izzo L. On the distribution of hydrophilic polyelectrolytes and their counterions around zwitterionic micelles: the possible impact on the charge density in solution. Soft Matter. 2021;17(5):1267–83. doi:10.1039/D0SM01541E. [Google Scholar] [PubMed] [CrossRef]
102. Hussein YHA, Youssry M. Polymeric micelles of biodegradable diblock copolymers: enhanced encapsulation of hydrophobic drugs. Materials. 2018;11(5):688. doi:10.3390/ma11050688. [Google Scholar] [PubMed] [CrossRef]
103. Habibi N, Mauser A, Ko Y, Lahann J. Protein nanoparticles: uniting the power of proteins with engineering design approaches. Adv Sci. 2022;9(8):e2104012. doi:10.1002/advs.v9.8. [Google Scholar] [CrossRef]
104. Budi S, Suliasih BA, Rahmawatia I, Erdawati E. Size-controlled chitosan nanoparticles prepared using ionotropic gelation. Sci Asia. 2020;46(4):457–61. doi:10.2306/scienceasia1513-1874.2020.059. [Google Scholar] [CrossRef]
105. Pustulka SM, Ling K, Pish SL, Champion JA. Protein nanoparticle charge and hydrophobicity govern protein corona and macrophage uptake. ACS Appl Mater Interfaces. 2020;12(43):48284–95. doi:10.1021/acsami.0c12341. [Google Scholar] [PubMed] [CrossRef]
106. Shin J, Cole BD, Seyedmohammad M, Lim SI, Jang Y. Protein nanocarriers capable of encapsulating both hydrophobic and hydrophilic drugs. Methods Mol Biol. 2024;2720:143–50. doi:10.1007/978-1-0716-3469-1. [Google Scholar] [CrossRef]
107. Fleer A, Krediet TG. Innate immunity: toll-like receptors and some more: a brief history, basic organization and relevance for the human newborn. Neonatol. 2007;92(3):145–57. doi:10.1159/000102054. [Google Scholar] [PubMed] [CrossRef]
108. Wicherska-Pawłowska K, Wróbel T, Rybka J. Toll-like receptors (TLRsNOD-like receptors (NLRsand RIG-I-like receptors (RLRs) in innate immunity. TLRs, NLRs, and RLRs ligands as immunotherapeutic agents for hematopoietic diseases. Int J Mol Sci. 2021;22(24):13397. doi:10.3390/ijms222413397. [Google Scholar] [PubMed] [CrossRef]
109. Huang B, Zhao J, Unkeless J, Feng Z, Xiong H. TLR signaling by tumor and immune cells: a double-edged sword. Oncogene. 2008;27(2):218–24. doi:10.1038/sj.onc.1210904. [Google Scholar] [PubMed] [CrossRef]
110. Chen R, Alvero AB, Silasi DA, Steffensen KD, Mor G. Cancers take their Toll--the function and regulation of Toll-like receptors in cancer cells. Oncogene. 2008;27(2):225–33. doi:10.1038/sj.onc.1210907. [Google Scholar] [PubMed] [CrossRef]
111. Katopodi T, Petanidis S, Charalampidis C, Chatziprodromidou I, Eskitzis P, Tsavlis D, et al. Tumor-infiltrating dendritic cells: decisive roles in cancer immunosurveillance, immunoediting, and tumor T cell tolerance. Cells. 2022;11(20):3183. doi:10.3390/cells11203183. [Google Scholar] [PubMed] [CrossRef]
112. Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol. 2020;11:583084. doi:10.3389/fimmu.2020.583084. [Google Scholar] [PubMed] [CrossRef]
113. Yu P, Fu Y-X. Tumor-infiltrating T lymphocytes: friends or foes? Lab Invest. 2006;86(3):231–45. doi:10.1038/labinvest.3700389. [Google Scholar] [PubMed] [CrossRef]
114. Hong M, Clubb JD, Chen YY. Engineering CAR-T cells for next-generation cancer therapy. Cancer cell. 2020;38(4):473–88. doi:10.1016/j.ccell.2020.07.005. [Google Scholar] [PubMed] [CrossRef]
115. Li D, Li X, Zhou W-L, Huang Y, Liang X, Jiang L, et al. Genetically engineered T cells for cancer immunotherapy. Sig Transduct Target Ther. 2019;4:35. doi:10.1038/s41392-019-0070-9. [Google Scholar] [PubMed] [CrossRef]
116. Zhang X, Liu Y, Jiang M, Mas-Rosario JA, Fedeli S, Cao-Milan R, et al. Polarization of macrophages to an anti-cancer phenotype through in situ uncaging of a TLR 7/8 agonist using bioorthogonal nanozymes. Chem Sci. 2024;15(7):2486–94. doi:10.1039/D3SC06431J. [Google Scholar] [PubMed] [CrossRef]
117. Lin Y, Xu J, Lan H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol Oncol. 2019;12(1):76. doi:10.1186/s13045-019-0760-3. [Google Scholar] [PubMed] [CrossRef]
118. Qiang L, Cai Z, Jiang W, Liu J, Tai Z, Li G, et al. A novel macrophage-mediated biomimetic delivery system with NIR-triggered release for prostate cancer therapy. J Nanobiotechnol. 2019;17(1):83. doi:10.1186/s12951-019-0513-z. [Google Scholar] [PubMed] [CrossRef]
119. Nguyen VD, Min HK, Kim HY, Han J, Choi YH, Kim CS, et al. Primary macrophage-based microrobots: an effective tumor therapy in vivo by dual-targeting function and near-infrared-triggered drug release. ACS Nano. 2021;15(5):8492–506. doi:10.1021/acsnano.1c00114. [Google Scholar] [PubMed] [CrossRef]
120. Gong C, Tian J, Wang Z, Gao Y, Wu X, Ding X, et al. Functional exosome-mediated co-delivery of doxorubicin and hydrophobically modified microRNA 159 for triple-negative breast cancer therapy. J Nanobiotechnol. 2019;17(1):93. doi:10.1186/s12951-019-0526-7. [Google Scholar] [PubMed] [CrossRef]
121. Haney MJ, Zhao Y, Jin YS, Li SM, Bago JR, Klyachko NL, et al. Macrophage-derived extracellular vesicles as drug delivery systems for triple negative breast cancer (TNBC) therapy. J Neuroimmune Pharm. 2020;15(3):487–500. doi:10.1007/s11481-019-09884-9. [Google Scholar] [PubMed] [CrossRef]
122. Lin X, Lee S, Sharma P, George B, Scott J. Summary of US food and drug administration chimeric antigen receptor (CAR) T-cell biologics license application approvals from a statistical perspective. J Clin Oncol. 2022;40(30):3501–9. doi:10.1200/JCO.21.02558. [Google Scholar] [PubMed] [CrossRef]
123. Mullard A. FDA approves first CAR T therapy. Nat Rev Drug Discov. 2017;16(10):669. doi:10.1038/nrd.2017.196. [Google Scholar] [PubMed] [CrossRef]
124. Dudley ME, Rosenberg SA. Adoptive cell transfer therapy. Semin Oncol. 2007;34(6):524–31. doi:10.1053/j.seminoncol.2007.09.002. [Google Scholar] [PubMed] [CrossRef]
125. Feins S, Kong W, Williams EF, Milone MC, Fraietta JA. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am J Hematol. 2019;94(S1):S3–S9. [Google Scholar] [PubMed]
126. Zhang X, Gordon JR, Xiang J. Advances in dendritic cell-based vaccine of cancer. Cancer Biother Radiopharm. 2002;17(6):601–19. [Google Scholar] [PubMed]
127. Cheever MA, Higano CS. PROVENGE (Sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clin Cancer Res. 2011;17(11):3520–6. doi:10.1158/1078-0432.CCR-10-3126. [Google Scholar] [PubMed] [CrossRef]
128. Aljabali AA, Hassan SS, Pabari RM, Shahcheraghi SH, Mishra V, Charbe NB, et al. The viral capsid as novel nanomaterials for drug delivery. Future Sci OA. 2021;7(9):FSO744. doi:10.2144/fsoa-2021-0031. [Google Scholar] [PubMed] [CrossRef]
129. Shaik S, Kumar R, Chaudhary M, Kaur C, Khurana N, Singh G. Artificial viruses: a nanotechnology based approach. DARU. 2024;32(1):339–52. doi:10.1007/s40199-023-00496-6. [Google Scholar] [PubMed] [CrossRef]
130. Reta DH, Tessema TS, Ashenef AS, Desta AF, Labisso WL, Gizaw ST, et al. Molecular and immunological diagnostic techniques of medical viruses. Int J Microbiol. 2020;2020:8832728. [Google Scholar] [PubMed]
131. Lucas W. Viral capsids and envelopes: structure and function. Available from: https://doi.org/10.1002/9780470015902.a0001091.pub2 [Accessed 2010]. [Google Scholar]
132. Arul SS, Balakrishnan B, Handanahal SS, Venkataraman S. Viral nanoparticles: current advances in design and development. Biochimie. 2024;219:33–50. doi:10.1016/j.biochi.2023.08.006. [Google Scholar] [PubMed] [CrossRef]
133. Yildiz I, Shukla S, Steinmetz NF. Applications of viral nanoparticles in medicine. Curr Opin Biotechnol. 2011;22(6):901–8. doi:10.1016/j.copbio.2011.04.020. [Google Scholar] [PubMed] [CrossRef]
134. Chung YH, Cai H, Steinmetz NF. Viral nanoparticles for drug delivery, imaging, immunotherapy, and theranostic applications. Adv Drug Deliv Rev. 2020;156:214–35. doi:10.1016/j.addr.2020.06.024. [Google Scholar] [PubMed] [CrossRef]
135. Peng J, Yin Y, Liang H, Lu Y, Zheng H, Wu G, et al. Tumor microenvironment responsive pepper mild mottle virus-based nanotubes for targeted delivery and controlled release of paclitaxel. Front Bioeng Biotechnol. 2021;9:763661. doi:10.3389/fbioe.2021.763661. [Google Scholar] [PubMed] [CrossRef]
136. Bao CJ, Duan JL, Xie Y, Feng XP, Cui W, Chen SY, et al. Bioorthogonal engineered virus-like nanoparticles for efficient gene therapy. Nanomicro Lett. 2023;15(1):197. doi:10.1007/s40820-023-01153-y. [Google Scholar] [PubMed] [CrossRef]
137. Coley WB II. Contribution to the Knowledge of Sarcoma. Ann Surg. 1891;14(3):199–220. doi:10.1097/00000658-189112000-00015. [Google Scholar] [PubMed] [CrossRef]
138. McCarthy EF. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop J. 2006;26:154–8. [Google Scholar] [PubMed]
139. Redelman-Sidi G. Microbial agents to treat cancer. In: M.Schmidt T, editor. Encyclopedia of microbiology (fourth edition). Cambridge: Academic Press; 2019. p. 103–9. doi:10.1016/B978-0-12-801238-3.64159-4. [Google Scholar] [CrossRef]
140. Zacharski L, Sukhatme V. Coley’s toxin revisited: immunotherapy or plasminogen activator therapy of cancer? J Thromb Haemost. 2005;3(3):424–7. doi:10.1111/j.1538-7836.2005.01110.x. [Google Scholar] [PubMed] [CrossRef]
141. Galicia-Carmona T, Arango-Bravo E, Serrano-Olvera JA, Flores-de La Torre C, Cruz-Esquivel I, Villalobos-Valencia R, et al. ADXS11-001 LM-LLO as specific immunotherapy in cervical cancer. Hum Vaccin Immunother. 2021;17(8):2617–25. doi:10.1080/21645515.2021.1893036. [Google Scholar] [PubMed] [CrossRef]
142. Maciag PC, Radulovic S, Rothman J. The first clinical use of a live-attenuated Listeria monocytogenes vaccine: a Phase I safety study of Lm-LLO-E7 in patients with advanced carcinoma of the cervix. Vaccine. 2009;27(30):3975–83. doi:10.1016/j.vaccine.2009.04.041. [Google Scholar] [PubMed] [CrossRef]
143. Huh WK, Brady WE, Moore KN, Lankes HA, Monk BJ, Aghajanian C, et al. A phase 2 study of live-attenuated listeria monocytogenes cancer immunotherapy (ADXS11-001) in the treatment of persistent or recurrent cancer of the cervix (GOG-0265). J Clin Oncol. 2014;32(15_suppl):TPS5617. doi:10.1200/jco.2014.32.15_suppl.tps5617. [Google Scholar] [CrossRef]
144. Ghamande S, Mauro D, Price C, Wheatley D, Janik J, Khleif SN. High-dose treatment with ADXS11-001, a listeria monocytogenes (Lm)-listeriolysin O (LLO) immunotherapy, in women with cervical cancer. J Immunother Cancer. 2015;3(Suppl 2):P151. doi:10.1186/2051-1426-3-S2-P151. [Google Scholar] [CrossRef]
145. Petit RG, Mehta A, Jain M, Gupta S, Nagarkar R, Kumar V, et al. ADXS11-001 immunotherapy targeting HPV-E7: final results from a Phase II study in Indian women with recurrent cervical cancer. J Immunother Cancer. 2014;2(Suppl 3):P92. doi:10.1186/2051-1426-2-S3-P92. [Google Scholar] [CrossRef]
146. Herzog T, Backes FJ, Copeland L, Diz MDPE, Hare TW, Huh W, et al. AIM2CERV: a randomized phase III study of adjuvant AXAL immunotherapy following chemoradiation in patients who have high-risk locally advanced cervical cancer (HRLACC). J Immunother Cancer. 2016;4(Suppl 1):82. doi:10.1186/s40425-016-0172-7. [Google Scholar] [CrossRef]
147. Cohen EEW, Moore KN, Slomovitz BM, Chung CH, Anderson ML, Morris SR, et al. Phase I/II study of ADXS11-001 or MEDI4736 immunotherapies alone and in combination, in patients with recurrent/metastatic cervical or human papillomavirus (HPV)-positive head and neck cancer. J Immunother Cancer. 2015;3(Suppl 2). [Google Scholar]
148. Krupar R, Imai N, Miles B, Genden E, Misiukiewicz K, Saenger Y, et al. Abstract LB-095: hPV E7 antigen-expressing Listeria-based immunotherapy (ADXS11-001) prior to robotic surgery for HPV-positive oropharyngeal cancer enhances HPV-specific T cell immunity. Cancer Res. 2016;76(14_Supplement):LB-095. doi:10.1158/1538-7445.AM2016-LB-095. [Google Scholar] [CrossRef]
149. Miles B, Safran HP, Monk BJ. Therapeutic options for treatment of human papillomavirus-associated cancers—novel immunologic vaccines: aDXS11-001. Gynecol Oncol Res Pract. 2017;4:10. doi:10.1186/s40661-017-0047-8. [Google Scholar] [PubMed] [CrossRef]
150. Eng C, Fakih M, Amin M, Morris V, Hochster HS, Boland PM, et al. A phase II study of axalimogene filolisbac for patients with previously treated, unresectable, persistent/recurrent loco-regional or metastatic anal cancer. Oncotarget. 2020;11(15):1334–43. doi:10.18632/oncotarget.v11i15. [Google Scholar] [CrossRef]
151. Zheng DW, Chen Y, Li ZH, Xu L, Li CX, Li B, et al. Optically-controlled bacterial metabolite for cancer therapy. Nat Commun. 2018;9(1):1680. doi:10.1038/s41467-018-03233-9. [Google Scholar] [PubMed] [CrossRef]
152. Duong MT, Qin Y, You SH, Min JJ. Bacteria-cancer interactions: bacteria-based cancer therapy. Exp Mol Med. 2019;51(12):1–15. doi:10.1038/s12276-019-0297-0. [Google Scholar] [PubMed] [CrossRef]
153. Fan JX, Peng MY, Wang H, Zheng HR, Liu ZL, Li CX, et al. Engineered bacterial bioreactor for tumor therapy via fenton-like reaction with localized H2O2 generation. Adv Mater. 2019;31(16):e1808278. doi:10.1002/adma.v31.16. [Google Scholar] [CrossRef]
154. Asensio-Calavia A, Mañas C, Cabrera-Fisac A, Pico-Sánchez E, Seco EM, Kolodziej S, et al. Synthetic bacteria with programmed cell targeting and protein injection suppresstumor growth in vivo. bioRxiv. 2024;9:3184. doi:10.1101/2024.04.22.590337. [Google Scholar] [CrossRef]
155. Witte A, Wanner G, Sulzner M, Lubitz W. Dynamics of PhiX174 protein E-mediated lysis of Escherichia coli. Arch Microbiol. 1992;157(4):381–8. doi:10.1007/BF00248685. [Google Scholar] [PubMed] [CrossRef]
156. Szostak MP, Hensel A, Eko FO, Klein R, Auer T, Mader H, et al. Bacterial ghosts: non-living candidate vaccines. J Biotechnol. 1996;44(1–3):161–70. [Google Scholar] [PubMed]
157. Szostak M, Wanner G, Lubitz W. Recombinant bacterial ghosts as vaccines. Res Microbiol. 1990;141(7–8):1005–7. [Google Scholar] [PubMed]
158. Li X, Wang L, Zhang Y, Zhai Y, Wang L, Li X, et al. A targeted drug delivery system based on E. coli ghost for inhibiting non-Hodgkin’s lymphoma. Colloids Surf A: Physicochem Eng Asp. 2023;678:132504. doi:10.1016/j.colsurfa.2023.132504. [Google Scholar] [CrossRef]
159. Abdelaziz BAE, Mahmoud HE, Sadek OM, Sultan AS. Abstract 7589: the novel complex delivery system of C-phycocyanin-loaded klebsiella pneumoniabacterial ghost induces apoptosis and sensitizes non-small cell lung cancer cell lines to doxorubicin. Cancer Res. 2024;84(6_Supplement):7589. doi:10.1158/1538-7445.AM2024-7589. [Google Scholar] [CrossRef]
160. Kim S-J, Chang W, Oh M-K. Escherichia coli minicells with targeted enzymes as bioreactors for producing toxic compounds. Metab Eng. 2022;73(7):214–24. doi:10.1016/j.ymben.2022.08.006. [Google Scholar] [PubMed] [CrossRef]
161. Frazer AC, Curtiss R 3rd. Production, properties and utility of bacterial minicells. Curr Top Microbiol Immunol. 1975;69:1–84. doi:10.1007/978-3-642-50112-8. [Google Scholar] [CrossRef]
162. Farley MM, Hu B, Margolin W, Minicells Liu J. Back in fashion. J Bacteriol. 2016;198(8):1186–95. doi:10.1128/JB.00901-15. [Google Scholar] [PubMed] [CrossRef]
163. Fralick JA, Fisher WD, Adler HI. Polyuridylic acid-directed phenylalanine incorporation in minicell extracts. J Bacteriol. 1969;99(2):621–2. doi:10.1128/jb.99.2.621-622.1969. [Google Scholar] [PubMed] [CrossRef]
164. Ma Y, Zhu G, Feng L, Jiang S, Xiang Q, Wang J. Efficient cytotoxicity of recombinant azurin in Escherichia coli Nissle 1917-derived minicells against colon cancer cells. Bioengineering. 2023;10(10):1188. doi:10.3390/bioengineering10101188. [Google Scholar] [PubMed] [CrossRef]
165. Huang F, Shu Q, Qin Z, Tian J, Su Z, Huang Y, et al. Anticancer actions of azurin and its derived peptide p28. Protein J. 2020;39(2):182–9. doi:10.1007/s10930-020-09891-3. [Google Scholar] [PubMed] [CrossRef]
166. Solomon BJ, Desai J, Rosenthal M, McArthur GA, Pattison ST, Pattison SL, et al. A first-time-in-human phase I clinical trial of bispecific antibody-targeted, paclitaxel-packaged bacterial minicells. PLoS One. 2015;10(12):e0144559. doi:10.1371/journal.pone.0144559. [Google Scholar] [PubMed] [CrossRef]
167. Whittle JR, Lickliter JD, Gan HK, Scott AM, Simes J, Solomon BJ, et al. First in human nanotechnology doxorubicin delivery system to target epidermal growth factor receptors in recurrent glioblastoma. J Clin Neurosci. 2015;22(12):1889–94. doi:10.1016/j.jocn.2015.06.005. [Google Scholar] [PubMed] [CrossRef]
168. Fan C, Davison PA, Habgood R, Zeng H, Decker CM, Gesell Salazar M, et al. Chromosome-free bacterial cells are safe and programmable platforms for synthetic biology. Proc Natl Acad Sci U S A. 2020;117(12):6752–61. doi:10.1073/pnas.1918859117. [Google Scholar] [PubMed] [CrossRef]
169. Lim B, Yin Y, Ye H, Cui Z, Papachristodoulou A, Huang WE. Reprogramming synthetic cells for targeted cancer therapy. ACS Synth Biol. 2022;11(3):1349–60. doi:10.1021/acssynbio.1c00631. [Google Scholar] [PubMed] [CrossRef]
170. Knox KW, Vesk M, Work E. Relation between excreted lipopolysaccharide complexes and surface structures of a lysine-limited culture of Escherichia coli. J Bacteriol. 1966;92(4):1206–17. doi:10.1128/jb.92.4.1206-1217.1966. [Google Scholar] [PubMed] [CrossRef]
171. Zhou L, Srisatjaluk R, Justus DE, Doyle RJ. On the origin of membrane vesicles in gram-negative bacteria. FEMS Microbiol Lett. 1998;163(2):223–8. doi:10.1111/fml.1998.163.issue-2. [Google Scholar] [CrossRef]
172. Mayrand D, Grenier D. Biological activities of outer membrane vesicles. Can J Microbiol. 1989;35(6):607–13. doi:10.1139/m89-097. [Google Scholar] [PubMed] [CrossRef]
173. Deatherage BL, Cookson BT. Membrane vesicle release in bacteria, eukaryotes, and archaea: a conserved yet underappreciated aspect of microbial life. Infect Immun. 2012;80(6):1948–57. doi:10.1128/IAI.06014-11. [Google Scholar] [PubMed] [CrossRef]
174. Hoekstra D, van der Laan JW, de Leij L, Witholt B. Release of outer membrane fragments from normally growing Escherichia coli. BBA–Biomembranes. 1976;455(3):889–99. doi:10.1016/0005-2736(76)90058-4. [Google Scholar] [PubMed] [CrossRef]
175. Katsui N, Tsuchido T, Hiramatsu R, Fujikawa S, Takano M, Shibasaki I. Heat-induced blebbing and vesiculation of the outer membrane of Escherichia coli. J Bacteriol. 1982;151(3):1523–31. doi:10.1128/jb.151.3.1523-1531.1982. [Google Scholar] [PubMed] [CrossRef]
176. Malabirade A, Habier J, Heintz-Buschart A, May P, Godet J, Halder R, et al. The RNA complement of outer membrane vesicles from salmonella enterica serovar typhimurium under distinct culture conditions. Front Microbiol. 2018;9:2015. doi:10.3389/fmicb.2018.02015. [Google Scholar] [PubMed] [CrossRef]
177. Croia L, Boscato Sopetto G, Zanella I, Caproni E, Gagliardi A, Tamburini S, et al. Immunogenicity of Escherichia coli outer membrane vesicles: elucidation of humoral responses against OMV-associated antigens. Membranes. 2023;13(11):882. doi:10.3390/membranes13110882. [Google Scholar] [PubMed] [CrossRef]
178. Gonzalez-Fernandez C, Basauri A, Fallanza M, Bringas E, Oostenbrink C, Ortiz I. Fighting against bacterial lipopolysaccharide-caused infections through molecular dynamics simulations: a review. J Chem Inf Model. 2021;61(10):4839–51. doi:10.1021/acs.jcim.1c00613. [Google Scholar] [PubMed] [CrossRef]
179. Kim OY, Park HT, Dinh NTH, Choi SJ, Lee J, Kim JH, et al. Bacterial outer membrane vesicles suppress tumor by interferon-gamma-mediated antitumor response. Nat Commun. 2017;8(1):626. doi:10.1038/s41467-017-00729-8. [Google Scholar] [PubMed] [CrossRef]
180. Aly RGO, El-Enbaawy MIH, Abd El-Rahman SS, Ata NS, Elhariri M. The in vitro potential antitumor effect of S. typhimurium outer membrane vesicles on MCF-7 and Caco-2 cell lines compared to chemotherapies. Int J Vet Sci. 2019;8(4):259–66. [Google Scholar]
181. Firth J, Sun J, George V, Huang JD, Bajaj-Elliott M, Gustafsson K. Bacterial outer-membrane vesicles promote Vγ9Vδ2 T cell oncolytic activity. Front Immunol. 2023;14:1198996. doi:10.3389/fimmu.2023.1198996. [Google Scholar] [PubMed] [CrossRef]
182. Esam B, Al Sahlanee R. Cytotoxic Impact of outer membrane nanovesicles (OMVs) on ovarian cancer. Bionatura. 2023;8(4):12023. doi:10.21931/RB/CSS/2023.08.04.51. [Google Scholar] [CrossRef]
183. M.Almashgab A, Yahya EB, Banu A. The cytotoxicity effects of outer membrane vesicles isolated from hospital and laboratory strains of pseudomonas aeruginosa on human keratinocyte cell line. Malays J Sci. 2020;39(3):45–53. doi:10.22452/mjs. [Google Scholar] [CrossRef]
184. Tolmachev V, Orlova A. Affibody molecules as targeting vectors for PET imaging. Cancers. 2020;12(3):651. doi:10.3390/cancers12030651. [Google Scholar] [CrossRef]
185. Gujrati V, Kim S, Kim SH, Min JJ, Choy HE, Kim SC, et al. Bioengineered bacterial outer membrane vesicles as cell-specific drug-delivery vehicles for cancer therapy. ACS Nano. 2014;8(2):1525–37. doi:10.1021/nn405724x. [Google Scholar] [PubMed] [CrossRef]
186. Li Y, Zhao R, Cheng K, Zhang K, Wang Y, Zhang Y, et al. Bacterial outer membrane vesicles presenting programmed death 1 for improved cancer immunotherapy via immune activation and checkpoint inhibition. ACS Nano. 2020;14(12):16698–711. doi:10.1021/acsnano.0c03776. [Google Scholar] [PubMed] [CrossRef]
187. Pan J, Li X, Shao B, Xu F, Huang X, Guo X, et al. Self-blockade of PD-L1 with bacteria-derived outer-membrane vesicle for enhanced cancer immunotherapy. Adv Mater. 2022;34(7):e2106307. doi:10.1002/adma.202106307. [Google Scholar] [PubMed] [CrossRef]
188. Rezaei Adriani R, Mousavi Gargari SL, Bakherad H, Amani J. Anti-EGFR bioengineered bacterial outer membrane vesicles as targeted immunotherapy candidate in triple-negative breast tumor murine model. Sci Rep. 2023;13(1):16403. doi:10.1038/s41598-023-43762-y. [Google Scholar] [PubMed] [CrossRef]
189. Li Y, Wu J, Qiu X, Dong S, He J, Liu J, et al. Bacterial outer membrane vesicles-based therapeutic platform eradicates triple-negative breast tumor by combinational photodynamic/chemo-/immunotherapy. Bioact Mater. 2023;20(41):548–60. doi:10.1016/j.bioactmat.2022.05.037. [Google Scholar] [PubMed] [CrossRef]
190. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):99–109. doi:10.1038/nrmicro2070. [Google Scholar] [PubMed] [CrossRef]
191. Combes F, Meyer E, Sanders NN. Immune cells as tumor drug delivery vehicles. J Control Release. 2020;327:70–87. doi:10.1016/j.jconrel.2020.07.043. [Google Scholar] [PubMed] [CrossRef]
192. Choi A, Javius-Jones K, Hong S, Park H. Cell-based drug delivery systems with innate homing capability as a novel nanocarrier platform. Int J Nanomed. 2023;18:509–25. doi:10.2147/IJN.S394389. [Google Scholar] [PubMed] [CrossRef]
193. Nooraei S, Bahrulolum H, Hoseini ZS, Katalani C, Hajizade A, Easton AJ, et al. Virus-like particles: preparation, immunogenicity and their roles as nanovaccines and drug nanocarriers. J Nanobiotechnol. 2021;19(1):59. doi:10.1186/s12951-021-00806-7. [Google Scholar] [PubMed] [CrossRef]
194. Chen H, Ji H, Kong X, Lei P, Yang Q, Wu W, et al. Bacterial ghosts-based vaccine and drug delivery systems. Pharmaceutics. 2021;13(11):1892. doi:10.3390/pharmaceutics13111892. [Google Scholar] [PubMed] [CrossRef]
195. Kim SK, Cho SW. The evasion mechanisms of cancer immunity and drug intervention in the tumor microenvironment. Front Pharmacol. 2022;13:868695. doi:10.3389/fphar.2022.868695. [Google Scholar] [PubMed] [CrossRef]
196. Rampley CPN, Davison PA, Qian P, Preston GM, Hunter CN, Thompson IP, et al. Development of SimCells as a novel chassis for functional biosensors. Sci Rep. 2017;7(1):7261. doi:10.1038/s41598-017-07391-6. [Google Scholar] [PubMed] [CrossRef]
197. Amano A, Takeuchi H, Furuta N. Outer membrane vesicles function as offensive weapons in host-parasite interactions. Microbes Infect. 2010;12(11):791–8. doi:10.1016/j.micinf.2010.05.008. [Google Scholar] [PubMed] [CrossRef]
198. Ozdemir D, Bussgen M. Effectiveness and cost-effectiveness of combination therapy versus monotherapy in malignant melanoma. J Pharm Policy Pract. 2023;16(1):106. doi:10.1186/s40545-023-00611-7. [Google Scholar] [PubMed] [CrossRef]
199. Bayat Mokhtari R, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B, et al. Combination therapy in combating cancer. Oncotarget. 2017;8(23):38022–43. doi:10.18632/oncotarget.16723. [Google Scholar] [PubMed] [CrossRef]
200. Qiu Y, Ren K, Zhao W, Yu Q, Guo R, He J, et al. A “dual-guide” bioinspired drug delivery strategy of a macrophage-based carrier against postoperative triple-negative breast cancer recurrence. J Control Release. 2021;329:191–204. doi:10.1016/j.jconrel.2020.11.039. [Google Scholar] [PubMed] [CrossRef]
201. Ling D, Jia X, Wang K, Yan Q, Yuan B, Du L, et al. Cancer cell membrane-coated bacterial ghosts for highly efficient paclitaxel delivery against metastatic lung cancer. Acta Pharm Sin B. 2024;14(1):365–77. doi:10.1016/j.apsb.2023.08.012. [Google Scholar] [PubMed] [CrossRef]
202. Jia Y, Jiang Y, He Y, Zhang W, Zou J, Magar KT, et al. Approved nanomedicine against diseases. Pharmaceutics. 2023;15(3):774. doi:10.3390/pharmaceutics15030774. [Google Scholar] [PubMed] [CrossRef]
203. Alexander-Bryant AA, Vanden Berg-Foels WS, Wen X. Bioengineering strategies for designing targeted cancer therapies. Adv Cancer Res. 2013;118(Suppl. 3):1–59. doi:10.1016/B978-0-12-407173-5.00002-9. [Google Scholar] [PubMed] [CrossRef]
204. Maboudi AH, Lotfipour MH, Rasouli M, Azhdari MH, MacLoughlin R, Bekeschus S, et al. Micelle-based nanoparticles with stimuli-responsive properties for drug delivery. Nanotechnol Rev. 2024;13(1):20230218. doi:10.1515/ntrev-2023-0218. [Google Scholar] [CrossRef]
205. Hanafy NAN, El-Kemary M, Leporatti S. Micelles structure development as a strategy to improve smart cancer therapy. Cancers. 2018;10(7):238. doi:10.3390/cancers10070238. [Google Scholar] [PubMed] [CrossRef]
206. Saravanakumar K, Anbazhagan S, Pujani Usliyanage J, Vishven Naveen K, Wijesinghe U, Xiaowen H, et al. A comprehensive review on immuno-nanomedicine for breast cancer therapy: technical challenges and troubleshooting measures. Int Immunopharmacol. 2022;103:108433. doi:10.1016/j.intimp.2021.108433. [Google Scholar] [PubMed] [CrossRef]
207. Li X, Naeem A, Xiao S, Hu L, Zhang J, Zheng Q. Safety challenges and application strategies for the use of dendrimers in medicine. Pharmaceutics. 2022;14(6):1292. doi:10.3390/pharmaceutics14061292. [Google Scholar] [PubMed] [CrossRef]
208. Zhang J, Li M, Wang M, Xu H, Wang Z, Li Y, et al. Effects of the surface charge of polyamidoamine dendrimers on cellular exocytosis and the exocytosis mechanism in multidrug-resistant breast cancer cells. J Nanobiotechnol. 2021;19(1):135. doi:10.1186/s12951-021-00881-w. [Google Scholar] [PubMed] [CrossRef]
209. Cruz A, Barbosa J, Antunes P, Bonifacio VDB, Pinto SN. A glimpse into dendrimers integration in cancer imaging and theranostics. Int J Mol Sci. 2023;24(6):5430. doi:10.3390/ijms24065430. [Google Scholar] [PubMed] [CrossRef]
210. Mukherjee SP, Lyng FM, Garcia A, Davoren M, Byrne HJ. Mechanistic studies of in vitro cytotoxicity of poly(amidoamine) dendrimers in mammalian cells. Toxicol Appl Pharmacol. 2010;248(3):259–68. doi:10.1016/j.taap.2010.08.016. [Google Scholar] [PubMed] [CrossRef]
211. Ziemba B, Matuszko G, Bryszewska M, Klajnert B. Influence of dendrimers on red blood cells. Cell Mol Biol Lett. 2012;17(1):21–35. [Google Scholar] [PubMed]
212. Saha S, Pupo E, Zariri A, van der Ley P. Lipid A heterogeneity and its role in the host interactions with pathogenic and commensal bacteria. Microlife. 2022;3:uqac011. doi:10.1093/femsml/uqac011. [Google Scholar] [PubMed] [CrossRef]
213. Liu Q, Li Y, Zhao X, Yang X, Liu Q, Kong Q. Construction of Escherichia coli mutant with decreased endotoxic activity by modifying lipid A structure. Mar Drugs. 2015;13(6):3388–406. doi:10.3390/md13063388. [Google Scholar] [PubMed] [CrossRef]
214. Jin L, Zhang Z, Tan X, Wang Z, Tang B, Wang Z, et al. Antitumor effect of Escherichia coli-derived outer membrane vesicles on neuroblastoma in vitro and in vivo. Acta Bioch Bioph Sin. 2022;54(9):1301–13. [Google Scholar]
215. Tyrer PC, Frizelle FA, Keenan JI. Escherichia coli-derived outer membrane vesicles are genotoxic to human enterocyte-like cells. Infect Agent Cancer. 2014;9(1):2. doi:10.1186/1750-9378-9-2. [Google Scholar] [PubMed] [CrossRef]
216. Al-Zeheimi N, Adham SA. Modeling Neoadjuvant chemotherapy resistance in vitro increased NRP-1 and HER2 expression and converted MCF7 breast cancer subtype. Br J Pharmacol. 2020;177(9):2024–41. doi:10.1111/bph.v177.9. [Google Scholar] [CrossRef]
217. Kamigaki T, Takimoto R, Okada S, Ibe H, Oguma E, Goto S. Personalized dendritic-cell-based vaccines targeting cancer neoantigens. Anticancer Res. 2024;44(9):3713–24. doi:10.21873/anticanres.17196. [Google Scholar] [CrossRef]
218. Shen X, Pan D, Gong Q, Gu Z, Luo K. Enhancing drug penetration in solid tumors via nanomedicine: evaluation models, strategies and perspectives. Bioact Mater. 2024;32:445–72. [Google Scholar] [PubMed]
219. Rey JA, Spanick KG, Cabral G, Rivera-Santiago IN, Nagaraja TN, Brown SL, et al. Heterogeneous mechanical stress and interstitial fluid flow predictions derived from DCE-MRI for Rat U251N orthotopic gliomas. Ann Biomed Eng. 2024;52(11):3053–66. doi:10.1007/s10439-024-03569-y. [Google Scholar] [PubMed] [CrossRef]
220. Narsinh KH, Perez E, Haddad AF, Young JS, Savastano L, Villanueva-Meyer JE, et al. Strategies to improve drug delivery across the blood-brain barrier for glioblastoma. Curr Neurol Neurosci Rep. 2024;24(5):123–39. doi:10.1007/s11910-024-01338-x. [Google Scholar] [PubMed] [CrossRef]
221. Guo Y, Hu P, Shi J. Nanomedicine remodels tumor microenvironment for solid tumor immunotherapy. J Am Chem Soc. 2024;146(15):10217–33. doi:10.1021/jacs.3c14005. [Google Scholar] [PubMed] [CrossRef]
222. Haute DV, Berlin JM. Challenges in realizing selectivity for nanoparticle biodistribution and clearance: lessons from gold nanoparticles. Ther Deliv. 2017;8(9):763–74. doi:10.4155/tde-2017-0057. [Google Scholar] [PubMed] [CrossRef]
223. Andón FT, Digifico E, Maeda A, Erreni M, Mantovani A, Alonso MJ, et al. Targeting tumor associated macrophages: the new challenge for nanomedicine. Semin Immunol. 2017;34:103–13. doi:10.1016/j.smim.2017.09.004. [Google Scholar] [PubMed] [CrossRef]
224. Fu J, Wu E, Li G, Wang B, Zhan C. Anti-PEG antibodies: current situation and countermeasures. Nano Today. 2024;55:102163. doi:10.1016/j.nantod.2024.102163. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools