
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW
Development of PROTACS degrading KRAS and SOS1
Institute of Pharmacology, Medical University of Vienna, Vienna, 1090, Austria
* Corresponding Author: GERHARD HAMILTON. Email:
Oncology Research 2024, 32(8), 1257-1264. https://doi.org/10.32604/or.2024.051653
Received 12 March 2024; Accepted 24 April 2024; Issue published 17 July 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
The Kirsten rat sarcoma virus—son of sevenless 1 (KRAS-SOS1) axis drives tumor growth preferentially in pancreatic, colon, and lung cancer. Now, KRAS G12C mutated tumors can be successfully treated with inhibitors that covalently block the cysteine of the switch II binding pocket of KRAS. However, the range of other KRAS mutations is not amenable to treatment and the G12C-directed agents Sotorasib and Adragrasib show a response rate of only approximately 40%, lasting for a mean period of 8 months. One approach to increase the efficacy of inhibitors is their inclusion into proteolysis-targeting chimeras (PROTACs), which degrade the proteins of interest and exhibit much higher antitumor activity through multiple cycles of activity. Accordingly, PROTACs have been developed based on KRAS- or SOS1-directed inhibitors coupled to either von Hippel-Lindau (VHL) or Cereblon (CRBN) ligands that invoke the proteasomal degradation. Several of these PROTACs show increased activity in vitro and in vivo compared to their cognate inhibitors but their toxicity in normal tissues is not clear. The CRBN PROTACs containing thalidomide derivatives cannot be tested in experimental animals. Resistance to such PROTACS arises through downregulation or inactivation of CRBN or factors of the functional VHL E3 ubiquitin ligase. Although highly active KRAS and SOS1 PROTACs have been formulated their clinical application remains difficult.Graphic Abstract
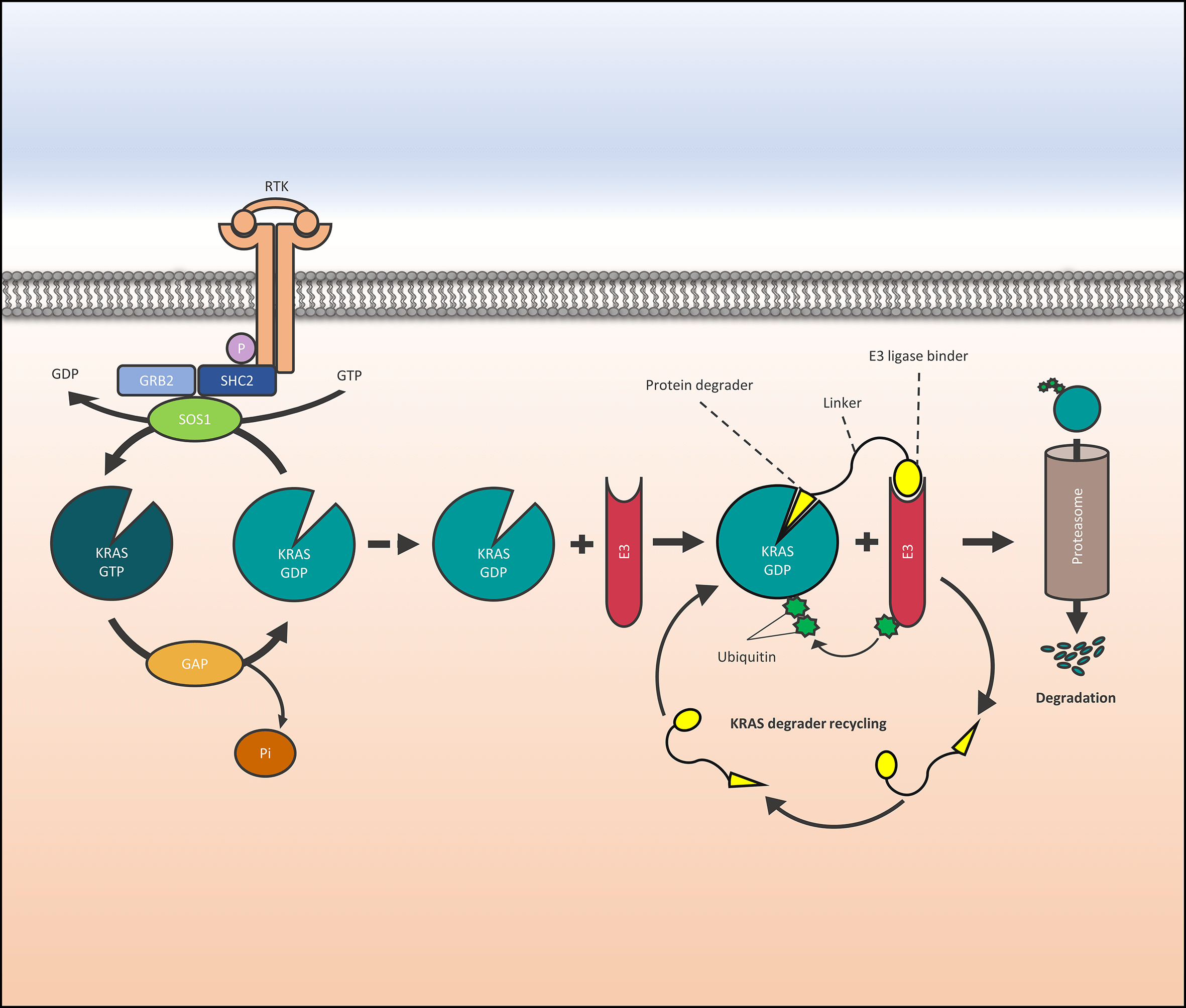
Keywords
Proteolysis-targeting chimeras (PROTACs); Kirsten rat sarcoma virus (KRAS); Son of sevenless 1 (SOS1); Von Hippel-Lindau; Cereblon
Cite This Article
APA Style
HAMILTON, G., EGGERSTORFER, M., STICKLER, S. (2024). Development of PROTACS degrading KRAS and SOS1. Oncology Research, 32(8), 1257–1264. https://doi.org/10.32604/or.2024.051653
Vancouver Style
HAMILTON G, EGGERSTORFER M, STICKLER S. Development of PROTACS degrading KRAS and SOS1. Oncol Res. 2024;32(8):1257–1264. https://doi.org/10.32604/or.2024.051653
IEEE Style
G. HAMILTON, M. EGGERSTORFER, and S. STICKLER, “Development of PROTACS degrading KRAS and SOS1,” Oncol. Res., vol. 32, no. 8, pp. 1257–1264, 2024. https://doi.org/10.32604/or.2024.051653
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools