
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW
Aldo-keto reductases: Role in cancer development and theranostics
1 World Neem Organization, Mumbai, 400101, India
2 Department of Biochemistry, ICMR-National Institute of Nutrition, Hyderabad, 500007, India
3 Department of Pathology and Cell Biology, Columbia University Irving Medical Center, New York, 10032, USA
* Corresponding Authors: GEEREDDY BHANUPRAKASH REDDY. Email: ,
Oncology Research 2024, 32(8), 1287-1308. https://doi.org/10.32604/or.2024.049918
Received 22 January 2024; Accepted 08 May 2024; Issue published 17 July 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Aldo-keto reductases (AKRs) are a superfamily of enzymes that play crucial roles in various cellular processes, including the metabolism of xenobiotics, steroids, and carbohydrates. A growing body of evidence has unveiled the involvement of AKRs in the development and progression of various cancers. AKRs are aberrantly expressed in a wide range of malignant tumors. Dysregulated expression of AKRs enables the acquisition of hallmark traits of cancer by activating oncogenic signaling pathways and contributing to chemoresistance. AKRs have emerged as promising oncotherapeutic targets given their pivotal role in cancer development and progression. Inhibition of aldose reductase (AR), either alone or in combination with chemotherapeutic drugs, has evolved as a pragmatic therapeutic option for cancer. Several classes of synthetic aldo-keto reductase (AKR) inhibitors have been developed as potential anticancer agents, some of which have shown promise in clinical trials. Many AKR inhibitors from natural sources also exhibit anticancer effects. Small molecule inhibitors targeting specific AKR isoforms have shown promise in preclinical studies. These inhibitors disrupt the activation of oncogenic signaling by modulating transcription factors and kinases and sensitizing cancer cells to chemotherapy. In this review, we discuss the physiological functions of human AKRs, the aberrant expression of AKRs in malignancies, the involvement of AKRs in the acquisition of cancer hallmarks, and the role of AKRs in oncogenic signaling, and drug resistance. Finally, the potential of aldose reductase inhibitors (ARIs) as anticancer drugs is summarized.Keywords
Supplementary Material
Supplementary Material FileAbbreviation
| AKR | Aldo-keto reductase |
| AKT | Protein kinase B |
| ARE | Antioxidant response element |
| AR | Aldose reductase |
| ARI | Aldose reductase inhibitor |
| COX | Cyclooxygenase |
| CRC | Colorectal cancer |
| CYP | Cytochrome P450 |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial-mesenchymal transition |
| FGF | Fibroblast growth factor |
| GSH | Reduced glutathione |
| GS-HNE | 3-Glutathionyl-4-hydroxynonenal |
| GSSG | Oxidized glutathione |
| HCC | Hepatocellular carcinoma |
| HIF-1α | Hypoxia-inducible factor-1α |
| HNE-4 | Hydroxytrans-2-nonenal |
| HO-1 | Heme oxygenase-1 |
| IGF-1 | Insulin-like growth factor-1 |
| Keap1 | Kelch-like ECH-associated protein 1 |
| mTOR | Mammalian target of rapamycin |
| NAD | Nicotinamide-adenine dinucleotide |
| NADP | Nicotinamide-adenine dinucleotide phosphate |
| NF-κB | Nuclear factor-κB |
| NRF2 | Nuclear factor erythroid 2 p45-related factor 2 |
| NSAIDS | Non-steroidal anti-inflammatory drugs |
| ORE | Osmotic response element |
| PCNA | Proliferating cell nuclear antigen |
| PI3K | Phosphoinositide 3-kinase |
| PKC | Protein kinase C |
| ROS | Reactive oxygen species |
| TNF | Tumor necrosis factor |
| VEGF | Vascular endothelial growth factor |
Cancer, a multifactorial heterogeneous group of diseases, is the second leading cause of mortality worldwide [1]. The process of neoplastic transformation involves the progressive accumulation of genetic and epigenetic changes in vital genes that regulate cell survival, proliferation, differentiation, apoptosis, and genome integrity. Dysregulated cellular signaling plays a key role in enabling the acquisition of hallmark traits of cancer [2,3]. Despite the development of state-of-the-art technologies in diagnosis and novel therapeutic modalities, morbidity, and mortality due to cancer continue to be a major challenge to oncologists due to the high rate of recurrence and chemoresistance [4,5]. Several mechanisms contribute to therapy resistance such as xenobiotic-metabolism, drug efflux, and apoptosis evasion, among several others [6].
Both cytochrome P450 (CYP) and non-CYP pathways are involved in phase I xenobiotic metabolism. Although a vast majority of xenobiotics are primarily metabolized by the CYP enzymes, flavin monooxygenase, esterases, and aldo-keto reductases (AKRs) are also involved in the detoxication of drugs and xenobiotics. The CYP monooxygenases are membrane-bound hemeproteins that act on a wide variety of xenobiotics, whereas the AKRs are cytosolic oxidoreductases that primarily catalyze redox transformations of carbonyl-containing xenobiotics [7]. CYPs as well as AKRs that play a critical role in regulating the efficacy of drugs are also implicated in chemoresistance [8,9]. Inhibitors of CYPs and AKRs have therefore emerged as attractive candidate agents for cancer therapy and to counter chemoresistance [10].
The AKR protein superfamily of nicotinamide-adenine dinucleotide phosphate (NADP)-dependent enzymes play a central role in the reduction of a variety of aldehydes and carbonyls formed during drug metabolism and detoxification of xenobiotics. The AKR superfamily consists of over 190 members grouped into 16 distinct families, AKR1 to AKR16 based on amino acid sequence identity, with each family comprised of many subfamilies. In humans, 15 different types of AKRs have been identified that belong to three main families, AKR1, AKR6, and AKR7. Of these, the AKR1 family is the most well-characterized and is comprised of three members, AKR1B1, AKR1B10, and AKR1B15, which share over 70% sequence identity and substrate specificity [11].
The nomenclature of AKR proteins begins with the root symbol of AKR, followed by an Arabic number that indicates the family, a letter indicating the subfamily, and another Arabic number for the specific protein. For instance, in AKR1B1, the first Arabic number (AKR1) represents the family, the letter following the first number (AKR1B) indicates the subfamily, while the second Arabic number (AKR1B1) represents the individual member protein [12].
The AKRs are ubiquitously distributed in both prokaryotes and eukaryotes. AKR1B1 also known as aldose reductase (AR, EC 1.1.1.21) is expressed in many tissues, with high expression in the adrenal gland, seminal vesicles, placenta, eye lens, retina, kidney, and cells of the gastrointestinal tract. AKR1B1 expression was found to be low in the liver, prostate, testes, and thymus, and absent in most normal human tissues [13,14]. High expression of AKR1B10, also known as aldose reductase-like-1 (ARL-1), is seen in the adrenal glands, small intestine, and colon [11,15]. The expression of AKR1B15, a novel enzyme that shares 92% amino acid sequence homology with AKR1B10, is generally low in most tissues and limited to the adipose tissue, testis, and placenta [16].
AKR1B1 (AR), the most extensively studied AKR, catalyzes the nicotinamide adenine dinucleotide phosphate reduced (NADPH)-dependent reduction of aldehydes and ketones to their corresponding alcohols. The reduction of glucose to sorbitol followed by conversion to fructose is depicted as the polyol pathway. The reduction of glucose to fructose by AR was first identified in the seminal vesicle by Hers in 1956 [17]. Subsequently, van Heyningen [18] demonstrated the accumulation of galactitol and sorbitol in the rat lens during diabetic cataractogenesis. Accumulation of polyols in the lens was believed to enhance intracellular osmotic pressure leading to over-hydration, and imbalance in Na+/K+ homeostasis [19]. Based on these observations, AR was implicated as the main facilitator of hyperglycemic injury. A growing body of evidence recognized AR as a key mediator of secondary diabetic complications such as retinopathy, cataracts, microangiopathy, neuropathy, and nephropathy [20,21].
AKRs are overexpressed in several inflammation-associated pathological conditions such as alcoholic liver cirrhosis, atherosclerosis, asthma, gout, osteoporosis, uveitis, sepsis, and cancer [22–24]. While AKR1B1 is implicated in diabetic complications, AKR1C1-1C3 are involved in steroid hormone-dependent tumors, AKR1D1 in bile acid deficiency, and AKR1B10 in defects in retinoic acid signaling [11,15,25,26]. Overexpression of AKRs has been extensively reported in several malignant tumors [15,27–31].
Several studies support the tenet that inhibition of AR ameliorates inflammatory diseases besides diabetic complications [22,32,33]. Consequently, several synthetic AR inhibitors (ARIs) such as sorbinil, zopolrestat, fidarestat, and epalrestat have been tested for their efficacy in addressing these conditions [33,34]. While numerous synthetic ARIs have demonstrated excellent inhibition and shown promising results in animal studies, their efficacy in clinical trials was not very encouraging with some ARIs exhibiting adverse side effects. Hence, of late, natural products are being extensively investigated for their ability to inhibit AR to overcome toxicity and adverse side effects of synthetic agents [34,35].
In this review, we discuss the physiological functions of human AKRs, the aberrant expression of AKRs in malignancies, the involvement of AKRs in the acquisition of cancer hallmarks, and the role of AKRs in oncogenic signaling, and drug resistance. Finally, the potential of ARIs as anticancer drugs is summarized.
The AKR1B1 gene (18 kb) localized on chromosome 7q33 contains 10 exons and 9 introns. The promoter region consists of a TATA (TATTTA) box at −37 and a CCAAT box at −104. Two Alu repeats are present in intron 1, 4, and 9, respectively. Additionally, an androgen-like response element (ALRE), three osmotic response elements (ORE), an activator protein 1 (Ap-1) binding site, and a thyroid receptor element (TRE) are located upstream of the gene. The AKR1B1 gene is translated to a monomeric protein containing 316 amino acid residues [26,36]. Fig. 1A provides a schematic illustration of the structure of the AKR1B1 gene.
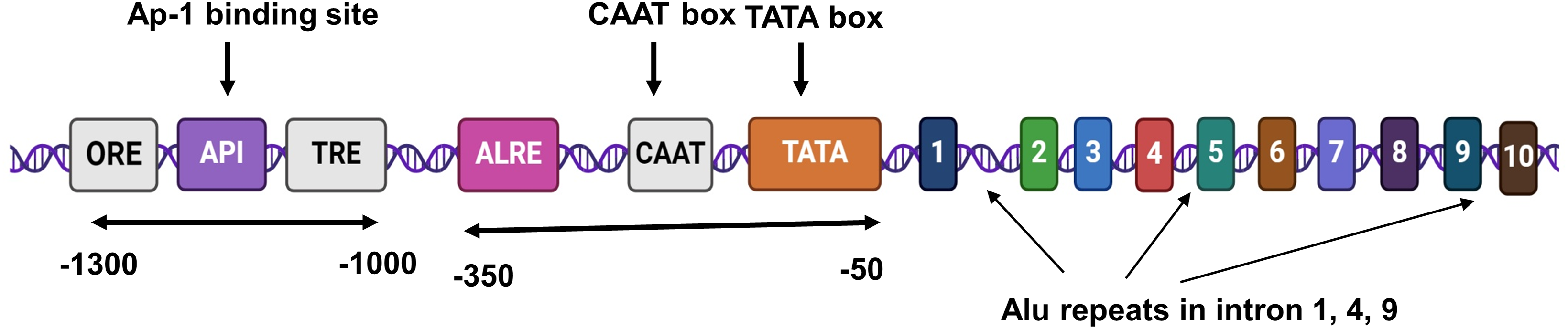
Figure 1A: A schematic representation of AKR1B1 gene structure.
The AKR1B10 gene (13.8 kb) contains 10 exons and 9 introns and shares close structural similarities with the AKR1B1 gene [37]. However, unlike the AKR1B1 gene, the AKR1B10 gene promoter contains a TATA-like (TAATAA) box from −145 to −140 bp and ORE is absent. The CAAT box is present in the region spanning from −193 to −190 bp. Multiple protein binding sites including those for p53, c-Ets-1, C/EBP, and AP-1 have been identified in the AKR1B10 promoter. Luciferase reporter analyses revealed that the sequences between −530 and −520 bp contain functional ARE sequences in tandem with the AP-1 site [38].
The AKRs are cytosolic monomeric proteins of molecular weight 30–40 kDa. Almost all AKRs share a common protein structure, comprised of an (α/β)8-barrel with alternating eight α-helices and eight β-strands and a pyridine nucleotide binding site [39]. The α/β barrel motif is essential for oligomerization as well as for binding of cofactors and metals, thereby generating an active site geometry (Fig. 1B). The back of the barrel consists of three large loops that are responsible for substrate specificity [25]. The binding site of NADPH is highly conserved and involves the hydrophobic amino acids Tyr-48, His-110, Cys-298, Trp-20, and Phe-122 in the active site of the enzyme [40–42]. Both AKR1B1 and AKR1B10 possess a negatively charged acidic hydroxyl group (Tyr48) in the catalytic site that may be important in structure-based drug design [43].
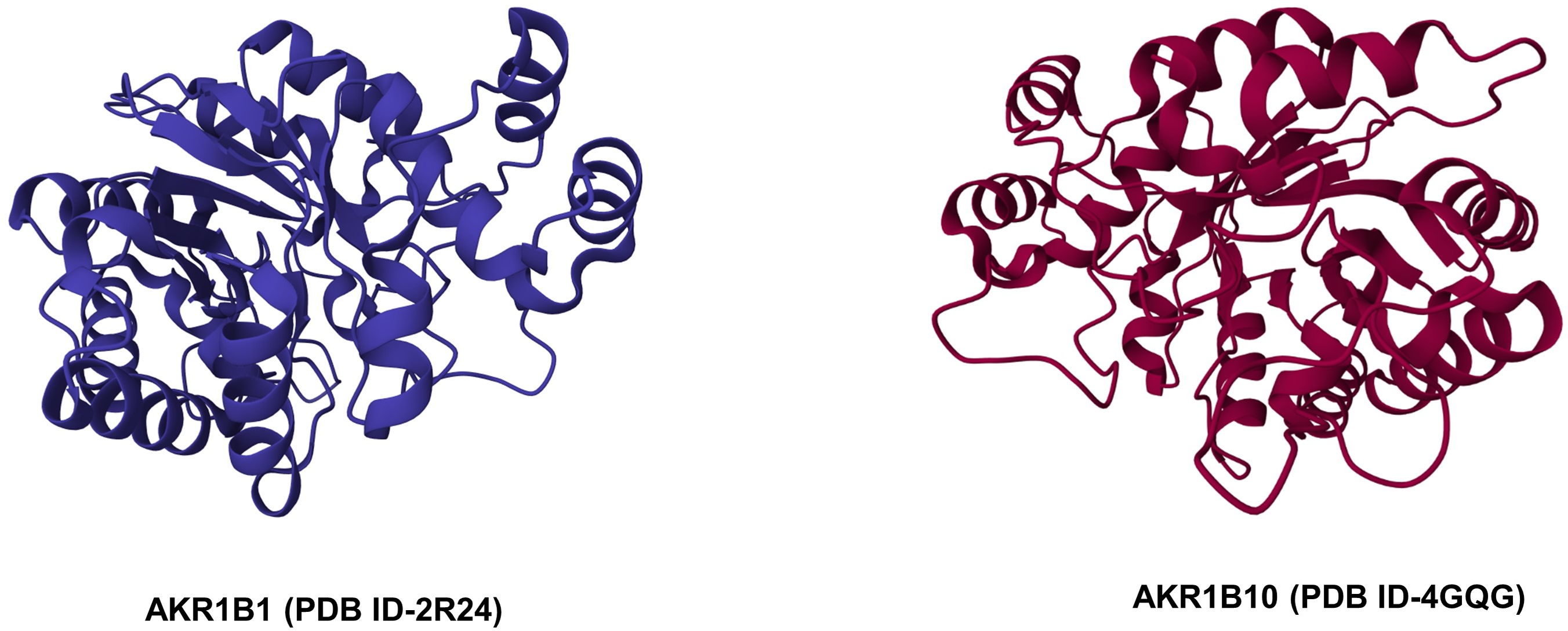
Figure 1B: Crystal structures of AKR1B1 and AKR1B10 (Source: RCSB PDB).
Physiological Functions of Human AKRs
AKRs are pluripotent enzymes that act on diverse substrates and catalyze redox transformations during intermediary metabolism and detoxification [11,44]. AKRs primarily function as reductases by utilizing the reducing equivalents from NADPH. AKRs are involved in the metabolism of sugar aldehydes, lipid aldehydes, keto-prostaglandins, retinals, quinones, keto-steroids, carcinogens, and by-products of lipid peroxidation. AKRs also catalyze the reduction of steroid double bonds (AKR1D, 5β-reductases), and control voltage-gated K+ channels. Since AKRs are involved in the detoxification of drugs and chemical carcinogens, they are generally classified as phase I xenobiotic-metabolizing enzymes [11,15,44].
AKR1B1 catalyzes the reduction of glucose to sorbitol in the presence of NADPH via the polyol pathway. This is followed by the oxidation of sorbitol to fructose using NAD as a cofactor. Under euglycemic conditions, most of the glucose undergoes phosphorylation to glucose-6-phosphate in a reaction catalyzed by hexokinase and less than 3% of glucose is channelized into the polyol pathway. However, under hyperglycemic conditions, the channeling into the polyol pathway is significantly increased to almost 30% glucose with excessive generation of sorbitol implicated in diabetic complications and non-alcoholic fatty liver disease [25,32]. However, there is growing evidence to suggest that glucose is probably not the main substrate of AR, and the enzyme is more efficient in reducing a wide range of aldehydes and their glutathione conjugates formed from lipid peroxidation [21,45]. Reactive oxygen species (ROS) generated during normal metabolism and in pathological conditions induce lipid peroxidation with consequent formation of reactive carbonyl species (RCS) such as 4-hydroxytrans-2-nonenal (HNE) that can deplete reductants including glutathione resulting in damage to cellular macromolecules such as lipids, proteins, and nucleic acids eventually culminating in tissue injury. Although AKRs together with other oxidoreductases can detoxify the RCS to prevent tissue injury, they can trigger ROS signaling [46]. Furthermore, AR-mediated increased flux of sorbitol via the polyol pathway during hyperglycemia can alter NADPH/NADP as well as reduce glutathione/oxidized glutathione (GSH/GSSG) ratios leading to oxidative stress [21,25].
AKR1B10 functions as a multifunctional NADPH-dependent reductase and catalyzes the reduction of a wide range of carbonyl compounds, aldehydes, and xenobiotics including drugs and carcinogenic chemicals such as polycyclic aromatic hydrocarbons (PAH) [47–51]. AKR1B10 catalyzes the transformation of PAH trans-dihydrodiols to highly reactive O-quinones, with the generation of ROS [52]. Despite its sequence similarity with AKR1B10, AKR1B15 is unique concerning to substrate specificity. AKR1B15 was found to be more catalytically active against a range of ketone and carbonyl substrates compared to AKR1B10. Interestingly, AKR1B15 exhibited high catalytic efficiency with 9-cis-retinaldehyde unlike AKR1B10, which prefers all-trans-retinaldehyde as a substrate [53].
Benzo[α]pyrene, a PAH, undergoes metabolic activation by the phase I enzymes cytochrome P450 (CYP)1A1/B1 to the highly carcinogenic benzo[α]pyrene-7,8-dihydrodiol, which is oxidized by AKR1A1 to form the corresponding ketol followed by rearrangement and oxidation to O-quinone with concomitant generation of ROS [47,54]. Jiang et al. [55] provided experimental evidence to demonstrate the competing roles of CYPs and AKRs in the metabolic activation of (+/−)-7,8-dihydroxy-7,8-dihydrobenzo[a]pyrene (BP-7,8-diol) in human bronchoalveolar H358 cell extracts. While CYP1A1/1B1 induction resulted in the formation of (+/−)-trans-7,8-dihydroxy-9alpha,10alpha-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene (anti-BPDE), AKR1A1 overexpressing cells produced benzo [a] pyrene-7,8-dione (BP-7,8-dione). Co-expression of CYP1A1/1B1 and AKR1A1 generated both the products. Furthermore, when NADPH concentration was increased, anti-BPDE was formed by the action of CYP1A1/1B1, whereas under conditions of higher NAD concentration, The system with a relatively higher concentration of NADPH favored the formation of anti-BPDE via P450 1A1/P450 1B1, AKR1A1 mediated formation of BP-7,8-dione was favored. However, equal amounts of both products were formed under conditions of normal cellular redox state. These findings indicate that metabolic activation of BP-7,8-diol depends on the cellular redox status.
Human AKRs regulate the expression of several genes via cis-elements by activating the various response elements in the promoter regions including the osmotic response element (ORE), phorbol ester response (AP-1) element, and the antioxidant response element (ARE) [56–58]. The AKRs respond to different types of stress such as electrophilic stress, oxidative stress, osmotic stress, and steroid hormone stress, and are therefore regarded as stress response genes [59]. AKR1C enzymes are recognized to regulate the expression of several genes via the steroid hormone receptors. AKR1C1 (20α-hydroxysteroid dehydrogenase) reduces progesterone to inactive 20α-hydroxyprogesterone and other progesterones in the endometrium thereby limiting the binding of progestin to the progesterone receptor [60]. AKR1C2 (type 3 3α-HSD/bile acid binding protein) reduces the potent androgen, 5α-dihydrotestosterone (5α-DHT) to the weak androgen 3α-androstanediol and prevents the binding of androgen to the androgen receptor. AKR1C3 catalyzes the activation of androgen Δ4-androstene-3,17-dione, a weak androgen to testosterone, which is then converted to dihydrotestosterone, with resultant enhanced androgen receptor activity in the prostate. AKR1C3 also catalyzes the activation of estrone to 17β-estradiol, and the inactivation of progesterone to 20α-hydroxyprogesterone in the breast with consequent transactivation of the estrogen receptor [61,62].
Aberrant expression of AKRs has been reported in a broad range of malignancies (Table S1). These include cancers of the adrenal cortex [63], breast [64], cervix [65], colorectum [66–68], liver [69], lung [27,70,71], oral cavity [28], stomach [29], pancreas [72] and glioma [73] to name a few. These studies lend credence to the proposition that AKRs are promising biomarkers for tumor diagnosis and prognosis [26,43,69].
The AKR1B and AKR1C family of proteins have been more strongly associated with cancer development and worse outcomes relative to the other isoforms [11]. Analysis of the Oncomine cancer gene expression database revealed wide variations in the expression of AKR1B1 and AKR1B10 in different cancer types [74]. Overexpression of AKR1B10 was seen in cancers of the lungs and liver associated with a less aggressive clinical course, whereas AKR1B1 was more widely overexpressed in several cancers and associated with shortened patient survival. Overexpression of AKRB10 has been documented in several cancer cell lines including pancreatic adenocarcinoma, medulloblastoma D341, and colon cancer HT29. High expression of AKR1B1 was observed in bladder and renal carcinomas with decreased expression in invasive breast cancer, colorectal cancer (CRC), adrenal cortex, and prostate cancer. On the other hand, high protein expression of AKR1B1 was observed in basal-like breast cancer [30], ovarian, pancreatic, and cervical cancer [75] which may be attributed to post-transcriptional regulation or differences in the clinical status of the patients evaluated. Similarly, overexpression of AKR1B10 was documented in hepatocellular carcinoma [76], breast [31,64,77], pancreatic [78], and lung cancer [79] with significant downregulation in CRC [80].
There are very few reports on the involvement of AKR1B15 in cancer [16,81]. Yuan et al. [82] developed a novel metabolism-related gene signature with AKR1B15 as one of the six genes with the potential to predict overall survival, prognosis, and treatment efficiency for HCC. More research on the role of AKR1B15 in different types of cancer is warranted.
Overexpression of AKR1B10 was first observed in hepatocellular carcinoma (HCC) and subsequently reported in a wide range of malignant cell lines and tumor tissues. Increased AKR1B10 expression has been documented in serum and tissue samples of HCC patients [83–85]. High expression of AKR1B10 during the early stages of HCC with subsequent decrease as metastasis supervenes has been documented [86]. Interestingly, overexpression of AKR1B10 in HCC tissues was associated with a favorable prognosis, longer recurrence-free survival (RFS), and disease-specific survival (DSS) [84]. A meta-analysis of 11 studies that included 2747 HCC patients and 2053 controls revealed the high sensitivity and specificity of AKR1B10 as a diagnostic marker for HCC. Notably, unlike AFP, AKR1B10 could predict overall survival and distinguish HCC from benign liver diseases [87]. A large-scale multicenter study involving 273 naïve HCC patients indicated high serum AKR1B10 as an unfavorable prognostic marker that was more sensitive than AFP both independently as well as in combination with TNM stage [69]. While AKR1B10 expression was significantly higher in HCC tumor tissues relative to normal tissues, AKR1B1 expression was not remarkably different between tumor and normal tissues.
Increased expression of AKRs has been reported in tobacco-induced cancers. AKRs are known to activate PAH trans-dihydrodiol proximate carcinogens from tobacco smoke to toxic and highly reactive O-quinones leading to mutational inactivation of p53 [47–50,88]. Increased expression of both AKR1B1 and AKR1B10 was observed in smokers compared to non-smokers. Upregulation of these AKRs has been linked to lung cancer development and progression [89]. Fukumoto et al. [90] demonstrated overexpression of AKR1B10 in smoking-induced non-small cell lung cancer (NSCLC), Kang et al. [91] reported significantly higher expression of AKR1B10 in tumor samples from NSCLC patients compared to patients with benign lung disease and normal controls. They also found a positive correlation between high AKR1B10 levels and smoking history in NSCLC patients. AKR1B10 expression was also higher in the airways of smokers relative to non-smokers as well as in specimens from NSCLC of non-smokers [92]. Simultaneous expression of AKR1B1 was noticed in individual mixed-lineage tumor cells comprised of different histological subtypes of NSCLC underscoring the value of this protein as a candidate target for therapeutic intervention of these tumors [27]. Using a multi-organ microfluidic bionic chip platform, Liu et al. [93] convincingly showed a significant upregulation of AKR1B10 in bone metastasis of NSCLC verifying its efficacy as a valuable serum biomarker of bone metastasis.
Dysregulation of AKRs expression has been documented in hormone-dependent tumors including prostate cancer and breast cancer. AKR1 and AKR7 have been implicated in the development of prostate cancer [94]. AKR1C3 which plays a key role in peripheral androgen biosynthesis was found to be increased by androgen deprivation in castrate-resistant prostate cancer [95–97]. Furthermore, AKR1C3 which functions as a prostaglandin F (PGF) synthase as well as its isoforms PGF2α and 11β-PGF2α are reported to induce prostate cancer development by preventing the activation of peroxisome proliferator-activated receptor gamma (PPARγ) [98].
Overexpression of AKR1C3 is believed to be involved in hormone-dependent breast cancer by catalyzing the conversion of 4-androstene-3,17-dione to testosterone, which functions as a substrate for the aromatase-mediated synthesis of 17β-estradiol [99,100]. In estrogen-dependent malignancies, AKR1C1 and AKR1C3 catalyze the conversion of progesterone to 20α-hydroxyprogesterone thereby increasing the 17β-estradiol: progesterone ratio [60,101]. Li et al. [102] observed a strong correlation between AKR1B10 and HER2 positivity in ductal carcinoma in situ (DCIS), indicating the value of AKR1B10 as a potential diagnostic and prognostic marker for early breast cancers. AKR1B10 is also overexpressed in infiltrating and recurrent breast tumors. The expression of AKR1B10 correlated positively with breast tumor size and nodal metastasis and inversely with disease-free survival (DFS). A positive correlation was seen between high levels of AKR1B10 in serum and tumor tissues in breast cancer patients. Tissue microarray analysis revealed overexpression of AKR1B10 in 84% of breast tumors. Further, AKR1B10 expression was significantly increased in postmenopausal breast tumors, while AKR1B1 was not altered [31]. Serum AKR1B10 is regarded as a novel prognostic marker for breast cancer. AKR1B10 is also overexpressed in cervical, uterine, and endometrial cancers. Furthermore, AKR1B10 expression is associated with keratinization and tumor recurrence in cervical cancer [103].
In gastric cancer, AKR1B1 expression was significantly high and associated with TNM staging indicating its prognostic significance [104]. AKR1B1 expression correlated positively with age, invasion, metastasis, staging, and prognosis of gastric cancer [105]. Elevated expression of AKR1B1 was associated with metastasis and lower overall survival in gastric cancer patients [106]. AKR1B1 was identified as a core gene for the regulation of the immune microenvironment, a biomarker for prognosis, and a potential target for gastric cancer treatment based on weighted gene co-expression network analysis, and data from The Cancer Genome Atlas-stomach adenocarcinoma and GSE62254 [107].
In contrast to AKR1B1, AKR1B10 expression was significantly decreased in gastric cancer and associated with tumor size, stage, and metastasis, suggesting that AKR1B10 may be a reliable prognostic indicator [108]. Using bioinformatic methods, Zhou et al. [29] observed differential gene expression and gene methylation of AKRs in gastric cancer patients based on data from various databases. While the expression levels of AKR1B10, AKR1C1, AKR1C2, and AKR7A3 were significantly low in gastric cancer tissues, AKR6A5 expression was higher relative to normal tissues. AKRB1 expression as well as methylation at cg13801416 was of prognostic value. Using bioinformatics analysis, real-time quantitative polymerase chain reaction (qPCR), and co-immunoprecipitation, assays, Yao et al. [108] convincingly demonstrated that AKR1B10 functions as a tumor suppressor in gastric cancer by inhibiting integrin subunit alpha 5.
Taskoparan et al. [80] demonstrated that the expression levels of AKR1B1 and AKR1B10 are divergent in colorectal cancer (CRC). AKR1B1 expression was unaltered in primary CRC and increased with disease progression. AKR1B1 has also been implicated in the progression of colitis to colon cancer [109]. In cell lines as well as in mouse models of CRC, the expression of AKR1B1 is believed to differ based on the stage and invasiveness of the tumors. For instance, AKR1B1 expression was higher in invasive tumor cells with Trp53 deletion in CRC mice models compared to non-invasive models [69]. Likewise, AKR1B1 expression was higher in the metastatic SW620 CRC cell line compared to the non-metastatic SW480 and HT29 cells [110]. Low AKR1B1 associated with high AKR1B10 expression was indicative of a good prognosis for CRC. Results from four different CRC datasets revealed that decreased AKR1B10 expression is a good predictor of poor overall survival and DFS [111]. Kropotova et al. [67] reported a significant decrease in AKR1B1 mRNA levels in 88 percent of colorectal tumor samples analyzed. qRT-PCR analysis of metastatic lymph nodes from CRC patients revealed a significantly higher expression of AKR1C3 especially in Dukes’ stage C compared to controls indicating that expression of AKR1C3 is a reliable marker for lymph node metastasis [68].
Overexpression of AKR1B10 is recognized as an independent biomarker of poor prognosis in oral squamous cell carcinoma (OSCC) [112]. Ko et al. [113] reported high salivary AKR1B10 levels in OSCC patients that correlated with areca quid chewing habit, tumor size, and advanced clinical stage of the disease. Overexpression of AKR1B10 was associated with tumor size, invasion, recurrence, and poor overall survival as well as DFS [28]. In addition to AKR1B10, AKR1C1 and AKR1C3 were also overexpressed in oral cancer cell lines [114]. Overexpression of AKR1B10 and AKR1C2 has been reported in Barrett’s esophagus with further increase with progression to esophageal cancer [115].
Analysis of the Oncomine database revealed significantly elevated expression of AKR1B1 in leukemias, lymphomas, and melanomas. Overexpression of AKR1B1 in leukemias was associated with TCF3-PBX1 gene fusion and rearrangements of the MLL gene located on chromosome 11q23. A significant overexpression of AKR1B10 was also seen in leukemias [74].
Differential expression of AKR1B1 was seen in benign and malignant tumors of the adrenal cortex [63]. AKR1B1 expression was significantly low in adrenocortical carcinomas compared to adenomas. In human glioma cells, high expression of AKR1B1 was found to be associated with the upregulation of NORAD (non-coding RNA activated by DNA damage) [116]. The expression of AKR1B10 was 20-fold higher in the cyclophosphamide-resistant D341 MED (4-HCR) medulloblastoma cell line compared to the parental cell line [117]. AKR1B10 was found to be upregulated in pancreatic adenocarcinomas and pancreatic intraepithelial neoplasia [78]. A positive correlation was seen between the high expression of AKR1C3 and the risk of bladder cancer progression and mortality [118].
Tumor development is a multistep process that involves the acquisition of eight biological capabilities, referred to as cancer hallmarks, namely sustained proliferation, circumventing growth suppressors, cell death evasion, replicative immortality, stimulating angiogenesis, invasion, metastasis, reprogramming cellular metabolism, and overcoming immune destruction. Currently, phenotypic plasticity, epigenetic programming, polymorphic microbiomes, senescent cells, genome instability and mutation, and tumor-promoting inflammation have been incorporated as emerging hallmarks and ‘enabling characteristics’ that facilitate the acquisition of cancer hallmarks. [3]. AR ablation by siRNA or inhibition by pharmacological agents has unraveled the oncogenic functions and involvement of the enzyme in the acquisition of several hallmark capabilities of cancer (Fig. 2 and Table S2).
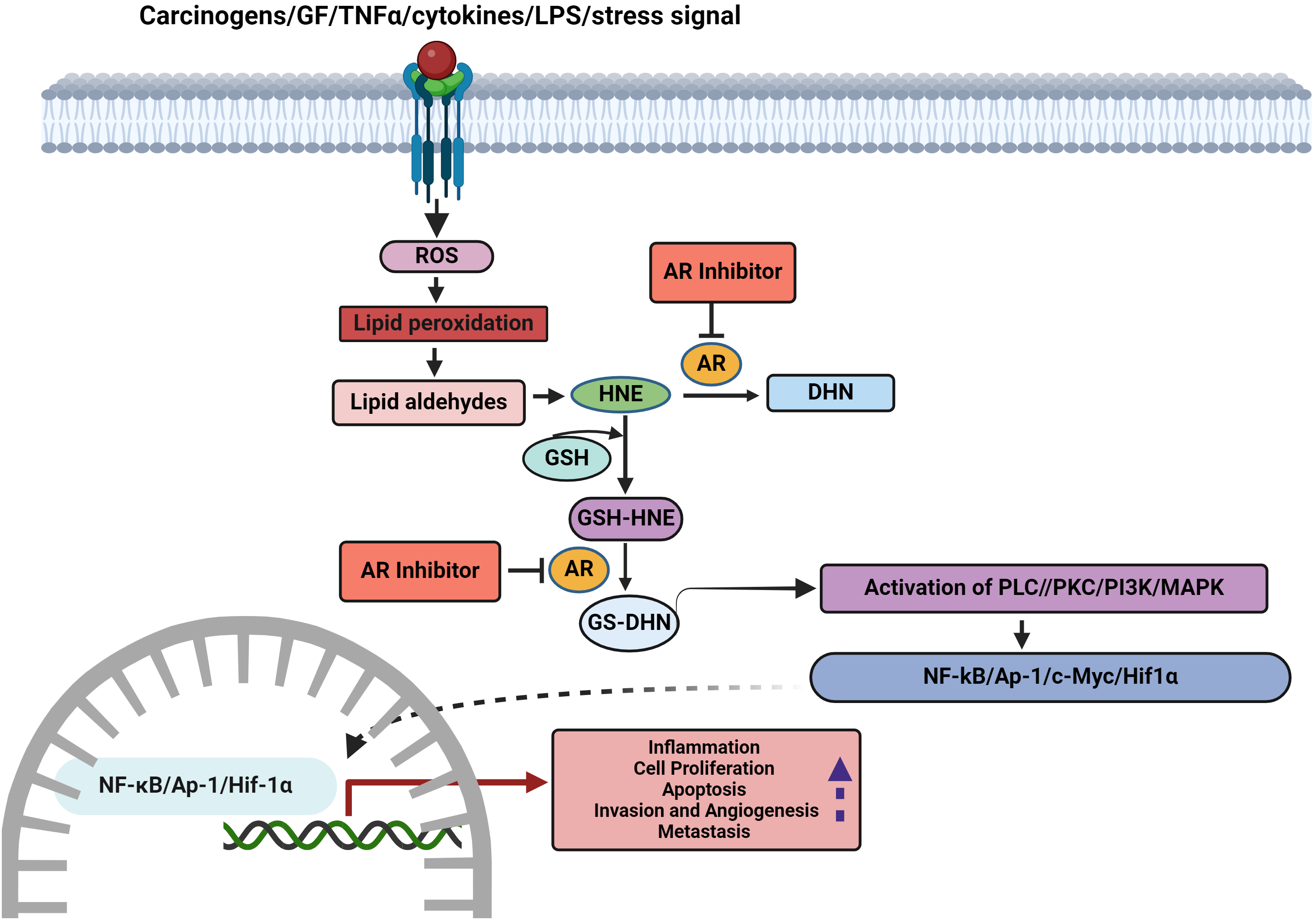
Figure 2: Role of AKRs in the acquisition of hallmarks and scope for therapeutic targeting (LPS-lipopolysaccharide, ROS-reactive oxygen species, HNE-4-hydroxytrans-2-nonenal, GSH-reduced glutathione, PLC-phosphatidylcholine by phospholipase C, PKC-protein kinase C, PI3K-phosphoinositide 3-kinase, MAPK-mitogen-activated protein kinases, NF-κB-nuclear factor-κB, AP1-activator protein-1, HIF1α-hypoxia-inducible factor-1α).
Tumor development is characterized by dysregulated cell cycle control leading to sustained proliferation [119]. Silencing of AKR1B10 inhibited cell growth rate in BT-20 human breast cancer cells and suppressed tumor development in female nude mice [77]. AR inhibition prevented growth factor-induced proliferation of colon cancer cells by arresting cells in the G1 phase, blocking phosphorylation of retinoblastoma protein and cyclin-dependent kinase (cdk)-2, and downregulating the expression of proliferating cell nuclear antigen (PCNA), cyclin D1, cyclin E, cdk4, and c-myc [120,121]. AR inhibition prevented the proliferation of human umbilical vein endothelial cells (HUVECs), as reflected by decreased expression of the proliferation marker, Ki-67 [122]. Both AKR1B1 and AKR1B10 display oncogenic ability. AKR1B1 was shown to induce tumor development by stimulating cell cycle progression in CRC. shRNA knockdown of AKR1B10 in Hep3B HCC cells arrested the cell cycle indicating the role of the enzyme in promoting cell proliferation [123]. Jin et al. [76] provided evidence to show that AKR1B10 facilitates the proliferation of liver cancer cells via phospholipid sphingosine-1-phosphate.
Effects on apoptosis and autophagy
In HCC cells, shRNA knockdown of AKR1B10 induced apoptosis and suppressed proliferation as evidenced by decreased colony formation, and enhanced chemosensitivity to doxorubicin chemotherapy [124]. The AR inhibitor fidarestat potentiated tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) is known to kill tumor cells selectively by inducing the death receptors (DR)-4 and DR-5. This was associated with the downregulation of Bcl-2, Bcl-xL, survivin, XIAP, and FLIP, increased expression of Bax, mitochondrial outer membrane permeabilization (MOMP), resulting in the release of cytochrome c, activation of caspase-3, and cleavage of poly (ADP-ribose) polymerase (PARP). Knockdown experiments revealed that these effects are mediated through activation of the AKT/FOXO3a pathway [125]. In pancreatic adenocarcinoma cell lines, siRNA-knockdown of AKR1B10 induced apoptotic cell death associated with decreased expression of the membrane-bound prenylated protein, KRAS [78].
Autophagy, a cytoprotective mechanism that eliminates misfolded proteins and damaged organelles is intricately linked to apoptosis. In colon cancer, autophagic cell death is recognized to facilitate malignant transformation by enabling cellular nutrient supply, preventing apoptosis, and stimulating drug resistance [126]. Li et al. [127] provided evidence to show that the membrane receptor neuropilin-1 promotes autophagy in colon cancer cells by downregulating AKR1B10 expression indicating that AKR1B10 negatively regulates autophagy. AKR1B10 was demonstrated to repress autophagy in glucose-deprived colon cancer cells by catalyzing the reduction of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) thereby preventing its nuclear import as well as via inhibition of AMPK phosphorylation.
Effects on angiogenesis, invasion, and metastasis
Intratumoral hypoxia, which promotes angiogenesis and invasion is a driving force for tumor progression [128]. Pharmacological or siRNA-mediated inhibition of AR was demonstrated to inhibit the proliferation of colon cancer cells associated with downregulation of hypoxia-inducible factor-1α (HIF-1α) and its downstream target, vascular endothelial growth factor (VEGF), a potent proangiogenic protein. Additionally, AR inhibition also decreased the expression of markers of inflammation (COX-2 and PGE2), and invasion (matrix metalloproteinase (MMP-2), vimentin, urokinase-type plasminogen activator receptor (uPAR), and lysyl oxidase 2) [129]. In HUVECs, AR inhibition or siRNA ablation blocked invasion and migration induced by VEGF and fibroblast growth factor (FGF). This was associated with decreased expression of interleukin-6 (IL-6), MMP-2, MMP-9, vascular cell adhesion molecule-1 (VCAM-1), and intercellular adhesion molecule-1 (ICAM-1). In the Matrigel plug model of angiogenesis in rats, AR inhibition suppressed angiogenesis as evidenced by significantly reduced vascular infiltration, invasion, migration, and formation of capillary-like vessels, coupled with downregulation of the blood vessel markers CD31 and vWF [122].
Several studies have demonstrated the involvement of AKRs in epithelial-mesenchymal transition (EMT), cell migration, and metastasis. A positive correlation was seen in the expression of AKR1B1 and the mesenchymal transcription factor Twist2. AKR1B1 was found to be a direct transcriptional target of Twist2 in basal-like breast cancer [30]. Tammali et al. [130] provided evidence to show that AR inhibition prevents metastasis of colon cancer by blocking cell adhesion, migration, and invasion. Jin et al. [94] provided compelling evidence to show that AKR1B10 promotes the extravasation of lung cancer cells through the blood-brain barrier leading to brain metastasis. Evaluation of the Genomic Data Commons-The Cancer Genome Atlas (GDC-TCGA) and the International Cancer Genome Consortium (ICGC) databases revealed a significant elevation in AKR1B10 expression in HCC. The increase in AKR1B10 was associated with cell proliferation, migration, invasion, and EMT that contribute to metastasis as evidenced by increased expression of CCND1, E-cadherin, N-cadherin, vimentin, and Twist1 via activation of the PI3K/AKT signaling pathway [131]. Cheng et al. [132] reported elevated expression of AKR1B1 in gastric cancer cells associated with enhanced cell proliferation, migration, and invasion. The transcription factor EBF1 was found to negatively regulate AKR1B1 expression. Knockdown of the transcription factor zinc finger protein521 (ZNF521) induced EBF1 which in turn downregulated AKR1B1 leading to suppression of gastric cancer growth and invasiveness. AKR1B1 overexpression rescued the effects of ZNF521. These findings reveal the involvement of the ZNF521/EBF1/AKR1B1 axis in gastric cancer progression.
AKRs and Oncogenic ROS Signalling
There is substantial evidence to indicate that AKR-mediated ROS metabolism activates oncogenic signaling pathways that enable the acquisition of cancer hallmarks [133,134]. Cytokines, growth factors, and lipopolysaccharides trigger ROS-induced lipid peroxidation resulting in the formation of lipid-derived aldehydes such as 4-hydroxynonenal (4-HNE). 4-HNE conjugates with reduced glutathione (GSH) to form 3-glutathionyl-4-hydroxynonenal (GS-HNE). Both HNE and GS-HNE are good substrates for AR. AKRs reduce GS-HNE to the highly reactive GS-4-dihydroxynonane (GS-DHN) that is recognized to activate oncogenic signaling to induce tumorigenesis. AKR1B1 together with GS-DHN is believed to stimulate the phospholipase C/protein kinase C (PLC-PKC) pathway and consequently activate NF-κB. AKR1B1 is also involved in the synthesis of prostaglandins by catalyzing the conversion of PGH2 to prostaglandin F2alpha (PGF2α). Overexpression of AKR1B1 can lead to excessive generation of PGF2α and COX2 resulting in inflammation and tumor development [135,136]. Inhibition of AR prevented growth factor and cytokine-induced cancer cell proliferation, prostaglandin synthesis, COX-2 activity, and activation of NF-κB and PKC. AKR inhibition suppressed ROS generation in cancer cells emphasizing the role of AKR in stimulating ROS-induced oncogenic signaling [25] (Fig. 3). AKR1B1 has been suggested to regulate inflammatory responses and tumorigenesis via activation of ROS, NF-κB, and PGE2 synthesis in CRC [26].
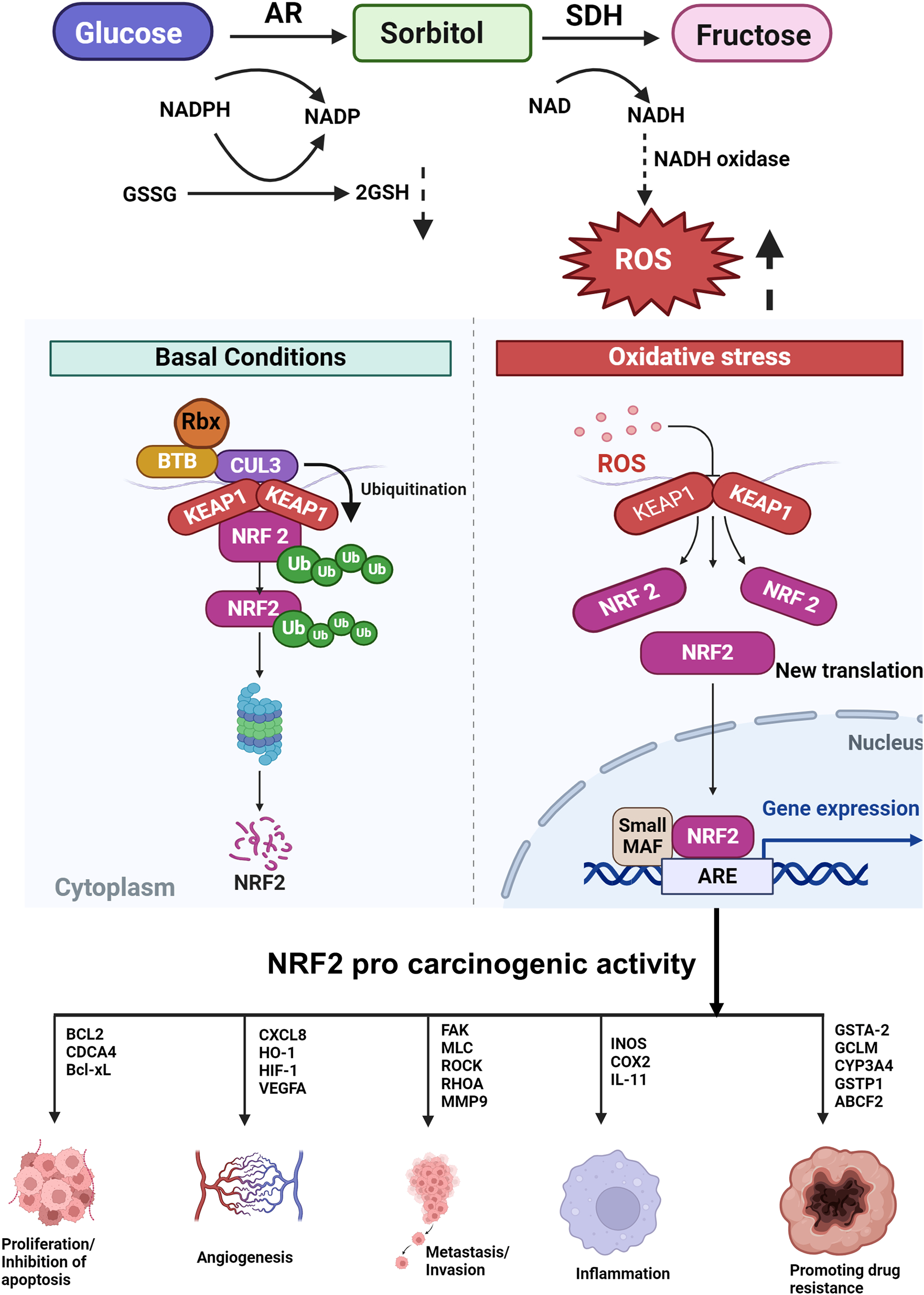
Figure 3: Under normal conditions, there is a basal level of NRF2 expression, while its expression is transiently increased upon exposure to toxic chemicals and ROS as a part of detoxification. However, sustained NRF2 activation confers resistance on cancer cells to anticancer drugs and radiation (SDH-sorbitol dehydrogenase, NADP-nicotinamide-adenine dinucleotide phosphate, NAD-nicotinamide-adenine dinucleotide, ROS-reactive oxygen species, Keap1-Kelch-like ECH-associated protein 1, NRF2-nuclear factor erythroid 2 p45-related factor 2, ARE-antioxidant response element).
Dysregulation of the phosphoinositide 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/AKT/mTOR) signaling pathway, a consistent feature of malignant tumors, is intricately linked to ROS generation [137]. Overexpression of AR was found to promote liver inflammation and HCC development via activation of AKT/mTOR signaling by interaction with the kinase domain of AKT1 [138]. In gastric cancer, AKR1B1 overexpression was associated with activation of the AKT-mTOR pathway [83]. AR has been reported to mediate angiogenesis through ROS/PI3K/AKT/AP-1/NF-κB signaling axis [23,122].
AR expression is tightly regulated by the ROS-sensitive transcription factor nuclear factor erythroid 2 p45-related factor 2 (NRF2). The AR promoter region functions as a binding site for the consensus sequence of NRF2. NRF2 regulates the expression of AKR genes. Under normal conditions, NRF2 is sequestered by Kelch-like ECH-associated protein 1 (Keap1), a component of E3 ubiquitin ligase, which causes ubiquitination and proteasomal degradation of NRF2. Under conditions of electrophilic or oxidative stress, NRF2 is detached from Keap1, translocates to the nucleus where it binds to ARE, and transactivates genes that detoxify the stress signals such as AKR1C [45]. NRF2 is reported to regulate AKR1B10 gene regulation. The AKR genes are among the most overexpressed in response to NRF2/Keap-1 signaling. Despite their protective roles against oxidative stress, overexpression of AKR genes constitutes the “smoking gene” battery as they can result in the activation of PAHs to carcinogenic metabolites (Fig. 3). Additionally, the upregulation of AKRs via the NRF2 pathway can induce drug resistance [139]. McCleod et al. [140] investigated the link between the expression of AKR and NRF2 activity in 23 cell lines. They found high basal levels of AKRs in cell lines that harbored mutations in the NRF2 pathway indicating that AKRs are biomarkers of NRF2 activity. The sedative propofol reduced the proliferation of BGC823 and GES-1 gastric cancer cells by significantly decreasing the expression of both AKR1B1 and AKR1B10 via the NRF2-mediated polyol pathway.
The crosstalk between ROS and the noncoding RNA (ncRNA) is recognized to activate oncogenic signaling and tumor development and progression [141]. Dysregulated microRNA expression has been linked to AKR overexpression. Inhibition of AR was found to prevent the growth of HT29, SW480, and Caco-2 colon cancer cells and nude mice xenografts by downregulating miR-21 expression with concomitant upregulation of programmed cell death 4 (PDCD4), a target of miR-21 by modulation of the ROS/AMPK/mTOR/AP1/4E-BP1 pathway [142]. In addition to PDCD4, AR inhibition also upregulated phosphatase and tensin homolog (PTEN), another miR-21 target, as well as forkhead box O3A (FOXO3a) in HT29, SW480, and Caco-2 colon cancer cells. Silencing of FOXO3a resulted in increasing the expression of AP-1 and miR-21, suggesting that FOXO3a mediates miR-21 repression by inactivating AP-1. Furthermore, AR inhibition prevented growth factor-induced phosphorylation of PI3K/Akt, c-Jun, c-Fos, PTEN, and FOXO3a, as well as DNA-binding activity of AP-1. In nude mice xenografted with human colon adenocarcinoma, AR inhibitor treatment decreased miR-21 expression with a simultaneous increase in the expression of PTEN and FOXO3a [143]. Using integrative bioinformatics mining analysis of NCBI GEO and TCGA databases, Wang et al. [123] identified miR-383-5p as a negative regulator of AKR1B10 in HCC. Dual-luciferase reporter assay provided evidence that miR-383-5p targets the 3’-UTR of AKR1B10 in the post-transcriptional stage leading to its downregulation.
High expression of AKR1B1 and NORAD was seen in glioma cells [116]. Silencing of NORAD prevented proliferation, migration, and invasion, and stimulated apoptosis. On the other hand, co-transfection with AKR1B1 pcDNA3.1 reversed the changes induced by NORAD silencing. In HCC cells, Man et al. [144] demonstrated that downregulation of the epidermal growth factor receptor substrate 15 (EPS15) by the lncRNA EPS15-antisense1 (EPS15-AS1) reduced AKR1B1 expression with consequent stimulation of ferroptosis, inhibition of invasion, and disruption of the redox balance.
Accumulating evidence has implicated AKRs in resistance to cancer chemotherapeutic agents and anti-hormonal drugs [15,145,146]. AKRs are linked to resistance to several major classes of chemotherapeutic agents including anthracyclines, cisplatin, cyclophosphamide, methotrexate, mitomycin, and vinca alkaloids. Epalrestat is a potent ARI-induced chemosensitivity to doxorubicin and sorafenib through inhibition of AKR1B1 and/or AKR1B10 and blockade of the EMT [145]. Upregulation of AKR1B10 in chemotherapy-resistant medulloblastoma D341 and colon cancer HT29 cancer cell lines underscore the utility of this enzyme as a biomarker of drug resistance. AKRs play a central role in the biosynthesis of androgens and estrogens, as well as in hormone-dependent malignancies. They are also upregulated by hormone ablative therapies that provide a basis for drug resistance. AKR1C3 is implicated in resistance to drugs used for castration-resistant prostate cancer therapy such as androgen receptor signaling inhibitors (ARSI) [146,147].
The involvement of AKR in drug resistance may be a consequence of AKR-catalysed metabolism of chemotherapeutic drugs/steroid hormones or because of the need to eliminate drug-induced cellular stress resulting from the generation of electrophiles or ROS. Additionally, drug-induced cellular stress induces activation of the NRF2/Keap1 pathway leading to feed-forward stimulation of AKRs. Inhibitors of the NRF2/Keap1 pathway and pan-AKR inhibitors may be useful in overcoming drug resistance [45].
Several lines of evidence suggest that AKR inhibitors offer promise in surmounting drug resistance [45]. Various AKR inhibitors have been used for overcoming drug resistance in vitro. Although AKR inhibitors potentiate the effect of chemotherapeutic drugs, they are more effective when primed as pretreatment rather than when given concurrently with the chemotherapeutic drug. However, it is important to use pan-AKR inhibitors because inhibition of a single AKR may result in substitution by another AKR. In this context, the non-steroidal anti-inflammatory drugs (NSAIDs) and their analogs have assumed importance in determining the use of AKR inhibitors in surmounting drug resistance. For example, NSAIDs are useful both as isoform-selective inhibitors as well as pan-AKR1C inhibitors. Salicylate is an AKR1C1 selective inhibitor, whereas flufenamic acid, a pan-AKR1C inhibitor can inhibit all four AKR1C isoforms. [148–150]. Recently, Ramana et al. [121] provided evidence to show that AKR1C3 inhibitors restored chemosensitivity in gemcitabine and cisplatin-resistant bladder cancer cells by suppressing androgen signaling.
Accumulating evidence indicates that inhibition of AR is a pragmatic therapeutic option for cancer both alone and in combination with chemotherapeutic drugs. Since AR has a potential role in mediating inflammation, AKR inhibitors are increasingly used to treat inflammatory diseases such as cancer [35,151]. Repurposing of epalrestat (EPA), a potent AKR1B1/AKR1B10 inhibitor appears promising especially for chemoresistant solid tumors [35,145,152]. Novel methods such as computer modeling and molecular dynamics are being used to design and develop potent ARIs. A comparative analysis of high-resolution crystallographic structures of AKR1B1 and AKR1B10 with reversible inhibitors was undertaken. The active site in both enzymes contained an anion-binding pocket. However, two main differences were observed between AKR1B10 and AR concerning to the involvement of external loops in inhibitor binding. The first difference is the alternative conformation of Trp112 (Trp111 in AR). Secondly, loop A in AKR1B10 was relatively large compared to that in AR with a more loosely packed subpocket [153]. In contrast to AKR1B10, AKR1B15 displayed a more narrower inhibitor selectivity. Among several AKR inhibitors tested against AKR1B15, only JF0064 significantly inhibited AKR1B15 while potent inhibitors of AKR1B1 and AKR1B10 such as tolrestat, epalrestat, sulindac, and sorbinil failed to inhibit AKR1B15. Four Phe residues are believed to contribute to the functional uniqueness of AKR1B15 [53].
Based on their structure, the AKR inhibitors are classified into three groups, (i) carboxylic acid derivatives such as epalrestat, tolrestat, ponalrestat, zopolrestat, and zenarestat; (ii) spirohydantoins and related cyclic imides such as fidarestat, minalrestat, ranirestat and sorbinil, (iii) phenolic derivatives such as benzopyran-4-one and chalcone [35,154] as well as natural products such as flavonoids, terpenoids, tannins, etc. (Fig. 4 and Table S3). Several ARIs have successfully passed through phase I and II clinical trials but failed in phase III trials due to toxic side effects.
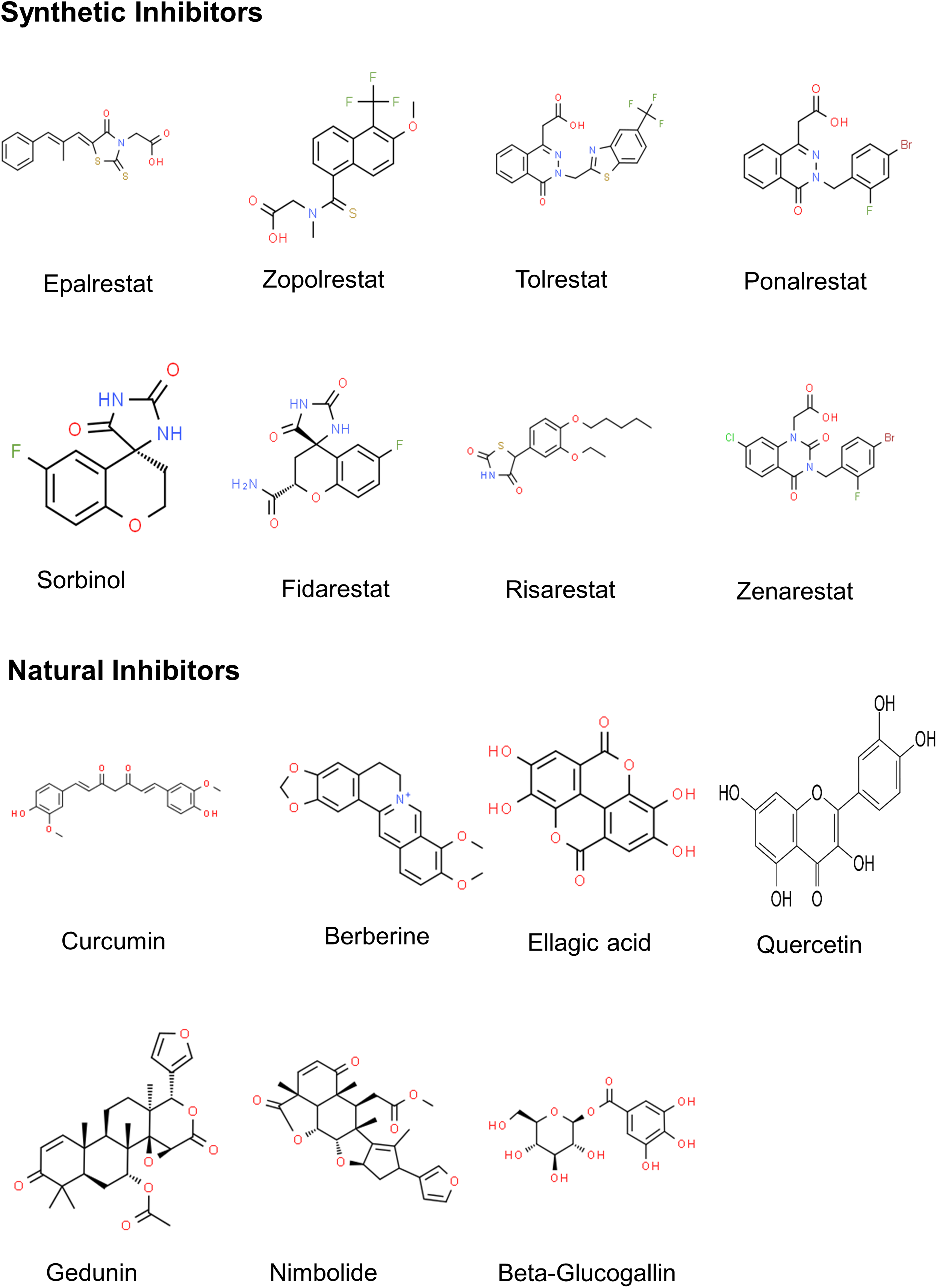
Figure 4: 2D structures of major AR inhibitors.
Epalrestat (ONO-2235) is the only efficacious and safe ARI approved for the clinical management of diabetic neuropathy. Although epalrestat is not approved by the US Food and Drug Administration for reasons unknown, it is marketed and used in Japan, India, and China. Despite not being extensively used in frontline therapy of diabetic neuropathy, epalrestat continues to be a mainstay as a disease-modifying agent [155].
Epalrestat was shown to inhibit the homologous enzymes AKR1B1 and AKR1B10 [145,156]. The compound has an essential 2-thioxo-4-thiazolidinone motif and regulates intracellular carbonyl species. Epalrestat is orally active and capable of penetrating the blood-brain barrier. There is ample experimental evidence for the anticancer effects of epalrestat in a wide range of malignancies alone and combination with chemotherapeutic agents. Epalrestat enhances the chemosensitivity of cancer cells to doxorubicin by inhibiting the activity of AKR1B1 and/or AKR1B10 as well as by preventing epithelial-mesenchymal transition [145]. Epalrestat has been reported to suppress the proliferation and migration of HeLa cervical cancer cells by inhibiting AKR1B1 [65]. Epalrestat restored the chemosensitivity of lung cancer cells resistant to epidermal growth factor receptor (EGFR), and tyrosine kinase inhibitor (TKI), in addition to delaying resistance in a murine lung cancer xenograft model [157]. A two-pronged minimalist nano platform that combined photodynamic therapy (PDT)-mediated tumor killing and epalrestat-mediated EMT blockade was demonstrated to effectively prevent metastasis of basal-like breast cancer [158].
Knockout of AKR1B1 by the CRISPR/Cas9 technology prevented proliferation, migration, and invasion in the HeLa cell line. Interestingly, epalrestat also produced the same effect as AKR1B1 knockout suggesting its therapeutic potential in cervical cancer [65]. However, the short half-life and hydrophobicity of epalrestat are bottlenecks in using it as a standard-of-care cancer treatment. Targeted co-delivery of epalrestat with doxorubicin was able to achieve synergistic chemotherapeutic efficacy and improve half-life and bioavailability. To this end, doxorubicin was co-loaded into redox-sensitive prodrug micelles. The nanoencapsulated formulation when introduced into mammary carcinoma cells induced cell cycle arrest, apoptosis, and significantly downregulated CD44 receptor expression, a major contributing factor of metastasis [159].
Fidarestat is another ARI that has shown a high degree of efficacy as a potential anticancer agent. In SW-480, HT29, and HCT116 CRC cells, fidarestat enhanced the expression of Nrf2. Additionally, in SW480 cells, fidarestat increased Nrf2-DNA binding activity as well as the levels of heme oxygenase-1 (HO-1), NADP(H) quinone oxidoreductase 1 (NQO1), superoxide dismutase, and catalase. In EGF-stimulated CRC cells, fidarestat potentiated EGF-induced Nrf2 and upregulated the expression of peroxisome proliferator-activated receptor gamma coactivator (PGC)-1alpha, transcription factor A, mitochondrial (TFAM), and COX-IV and attenuated DNA damage as seen by decreased levels of 8-hydroxy-2'-deoxyguanosine. Likewise, fidarestat treatment significantly increased Nrf2 and HO-1 levels in nude mice bearing xenografted tumor tissues. Further, fidarestat modulated phosphorylations of AMPK and mTOR as well as p53 expression in EGF-administered cells. Taken together, these results suggest that fidarestat modulates the Nrf2/HO-1/AMPK pathway to regulate mitochondrial biogenesis [160].
Fidarestat administered in drinking water significantly reduced azoxymethane (AOM)-induced premalignant lesions in the colon of C57BL/KsJ-db/db obese mice accompanied by downregulation of PKC-β2, AKT, COX-2, and iNOS in the colonic mucosa, and decreased serum levels of IL-1α, IP-10, MIG, TNF-α and VEGF. Further, in high glucose-treated HT29 colon cancer cells, fidarestat downregulated COX-2, iNOS, XIAP, survivin, β-catenin, and NF-κB [161]. Fidarestat was demonstrated to potentiate TRAIL-induced apoptosis by regulating the AKT/PI3K-dependent activation of FOXO3a [125]. Fidarestat induced autophagy in HT-29 and SW-480 CRC cells as reflected by high levels of LC3 II relative to LC3A/BI [162]. Fidarestat prevented angiogenesis by inhibiting endothelial cell differentiation, invasion, migration, expression of pro-angiogenic factors, and inactivation of AKT [122]. In another study, fidarestat was demonstrated to inhibit metastasis of colon cancer to the liver of nude mice by blockade of invasion, migration, adhesion, and angiogenesis [130].
The AKR inhibitors sorbinil or zopolrestat blocked growth factor-induced binding activity of the transcription factor E2F1, phosphorylation of pRb, expression of cyclins and CDKs, generation of ROS, and activation of the PI3K/AKT pathway in colon cancer cells [121]. The formation of AOM-induced preneoplastic aberrant crypt foci (ACF) was significantly reduced in BALB/c mice treated with sorbinil as well as in AR-null mice. This was accompanied by the downregulation of nitric oxide synthase, cyclooxygenase-2, cyclin D1, and beta-catenin with reduced phosphorylation of PKCβ2 and NF-κB binding protein implicating the involvement of AR in mediating ACF formation [120]. Sorbinil treatment prevented FGF and platelet-derived growth factor (PDGF)-induced growth of Caco-2 colon cancer cells and attenuated COX-2, PGE2, NF-κB activation, and PKC as well as phosphorylation of PKC-beta2. Further, AR inhibition blocked HNE-or GS-HNE-induced upregulation of COX-2 and PGE [12,163].
The AKR inhibitor, ponalrestat (statil) is reported to inhibit cancer cachexia by suppressing IL-1 expression [164] Ponalrestat was found to suppress the proliferation of breast and lung cancer cells by downregulating the expression of AKR1B10 [165]. Kavitha et al. [166] investigated the anticancer effects of spirohydantoin compounds in leukemic cells. They found that these compounds induce mitochondrial apoptosis associated with DNA fragmentation, upregulation of p53, and BAD, downregulation of BCL2, Ku70, and Ku80, activation of caspase-3 and -9, and cleavage of PARP.
Other synthetic small molecular inhibitors
A series of novel quinazolinones (1–21) were synthesized and tested for ALR2 inhibitory activity. Compound 15 exhibited high ALR2 inhibition that was 7.7 fold compared to epalrestat [167]. Of the fifteen tetrazole-hydrazone hybrids designed and synthesized, compound 4 (2-[(1-(4-Hydroxyphenyl)-1H-tetrazol-5-yl)thio]-N'-(4-fluorobenzylidene) acetohydrazide) exhibited strongest AR inhibitory activity comparable to epalrestat without cytotoxicity to normal NIH/3T3 cells [168].
Han et al. [169] designed and synthesized derivatives of quinazolin-4(1H)-one for selective inhibition of AKR1B1. Structure-activity studies revealed that the N1-acetic acid and N3-benzyl groups with electron-withdrawing substituents on the quinazolin-4(1H)-one scaffold enhanced the selectivity and efficiency of AKR1B1 inhibitors. A novel series of 3,4-dihydroquinolin-2(1H)-one derivatives were designed as potent and selective dual inhibitors of AKR1B1 and ROS. Of these, the phenolic 3,5-dihydroxyl compound 8b showed the highest antioxidant activity equivalent to that of Trolox [170].
Novel small molecule derivatives of 2,4-thiazolidinedione (2,4-TZD) synthesized by incorporation of the bioactive scaffolds, benzothiazole heterocycle, and nitrophenacyl moiety were found to exhibit potent AR inhibitory activity [171]. Selective inhibition of AKR1B10 was attempted by synthesizing a series of polyhydroxy steroids. Compound 6 displayed selective inhibition for both AKR1B1 and AKR1B10. Structure-activity studies revealed that the C-19 hydroxyl group can significantly enhance selective inhibition of AKR1B10 [172].
HMPC (7-hydroxy-2-(4-methoxyphenylimino)-2H-chromene-3-carboxylic acid benzylamide) is a potent but nonselective competitive inhibitor of AKR1B10. Derivatives of HPMC were synthesized to enhance selectivity and potency. Among the HMPC derivatives synthesized by removal of the 4-methoxyphenylimino moiety and replacement of the benzylamide with phenylpropylamide, compounds 4c and 4e exhibited greater AKR1B10 inhibitory activity and selectivity compared to HMPC. These compounds significantly suppressed the proliferation, migration, and metastasis of A549 lung cancer cells as well as their cisplatin-resistant counterparts [70].
A series of derivatives containing halophenoxyacetic acid moiety and many bromine (Br) atoms on the aryl moiety were synthesized from the potent AR inhibitor, IDD388. The incorporation of Br atoms reduced AKR1B1 inhibitory activity but enhanced AKR1B10 inhibitory potency. MK204 which forms a strong halogen bond with the enzyme exhibited the highest potency and AKR1B10 selectivity [173].
AKRs are selective targets of cyclopentenone prostaglandin A1 (PGA1), an eicosanoid with anti-inflammatory and antiproliferative properties. PGA1 was shown to covalently bind to Cys299 near the active site of AKR, while His111 and Tyr49, are involved in the orientation of PGA1. Modification of AKR1B10 by biotinylated PGA1 resulted in loss of enzyme activity. Further, PGA1 induced the accumulation of doxorubicin, an AKR substrate in lung cancer cells with consequent cell cycle arrest. These findings characterize PGA1 as an AKR inhibitor that can effectively counter cancer chemoresistance [174]. Other endogenous substances that inhibit AKRs, particularly, AKR1B10 include steroid hormones, bile acids, isolithocholic acid, and androst-4-ene-3,6-dione [175,176].
NSAIDs and other potential drug candidates
Endo et al. [177] demonstrated selective inhibition of AKR1B10 by NSAIDS, N-phenylanthranilic acids, and glycyrrhetic acid. Molecular docking studies revealed that the side chain of Val301 and hydrogen bonds between Val301, Gln114, and Ser304 are crucial for the selective inhibitory potency of the NSAIDs to target AKR1B10 and function as anticancer agents. Cemtirestat (3-mercapto-5H-[1,2,4]-triazino[5,6-b]indole-5-acetic acid), a highly efficient ARI and potent antioxidant was designed and patented. The cytotoxicity of cemtirestat was significantly lower than that of epalrestat. Based on in silico predictions, in vitro and in vivo assays, cemtirestat was found to be a safe drug with therapeutic potential that can progress to clinical trials [178].
Trans-(±)-kusunokinin ((±)KU), a synthetic antiproliferative agent that inhibits the growth of several malignant tumor cells both in vitro and in vivo, was shown to be a selective inhibitor of AKR1B1 [179–181]. (±)KU exerted more potent cytotoxic effects on triple-negative breast and ovarian cancer cells compared to epalrestat [182]. (±)KU was shown to bind to AKR1B1 and inhibit oxidative stress as well as the migration, EMT, and invasion of breast cancer cells.
AKR inhibitors of plant origin
Although many synthetic AKR inhibitors have been developed, many of these have been withdrawn following clinical trials due to low efficacy and safety concerns. The discovery and development of AKR inhibitors from natural sources have therefore gained the focus of research attention. Veeresham et al. [182] classified AKR inhibitors of plant origin into different groups based on their chemical structure, which include flavonoids, tannins, alkaloids, terpenoids, coumarins, and miscellaneous compounds.
A variety of flavonoids abundantly present in fruits, vegetables, herbs, and spices are known to inhibit AKR. Varma et al. [183] demonstrated that quercetin, quercetrin, and myrcitrin are effective inhibitors of AKR. Rutin is one of the commonly found dietary flavonols with AKR-inhibiting activity [184]. Xanthohumol, isoxanthohumol, and 8-prenylnaringenin flavonones found in hops (Humulus lupulus) that exhibit anticancer activity are potent inhibitors of both AKR1B1 and AKR1B10 but do not affect the activity of AKR1A1 [185]. Zemanova et al. [186] examined 40 different phenolics and alkaloids for their potential to inhibit AKR1B10. They identified flavones such as apigenin, luteolin, and 7-hydroxyflavone as potent inhibitors of AKR1B10 based on IC50, selectivity, and mechanism of action. These compounds were found to significantly inhibit AKR1B10-catalysed reduction of the chemotherapeutic agent daunorubicin without causing cytotoxicity.
Among the various tannins investigated, beta-glucogallin, a major component of gooseberry (Emblica officinalis) and precursor of ellagic acid in strawberry and raspberry, is a potent AKR inhibitor [187]. Recently, Suyanto et al. [188] provided evidence to show that β-glucogallin prevents the proliferation and migration of cholangiocarcinoma cells. A hydrolyzable tannin pentagalloyl glucose isolated from the bark of Rhus verniciflua displayed potent inhibition of human recombinant AR [189]. Alkaloids are secondary metabolites produced by plants that offer immense scope for the development of various drugs including anticancer agents [190]. The alkaloid dehydrocorydaline isolated from methanolic extract of the tuber of Corydalis turtschaninovii that prevents metastasis of NSCLC via suppression of MMPs and BCL2 is also an AKR inhibitor [191,192]. Berberine, another plant alkaloid that exhibits remarkable anticancer properties [193] inhibits AR activity in vivo [194].
Terpenoids also known as isoprenoids that exhibit a wide array of pharmacological effects including antiproliferative activity, are potent AKR inhibitors [185,195]. The triterpenoids or limonoids from the neem tree (Azadirachta indica) display potent anticancer properties [196]. Two neem limonoids, gedunin and nimbolide have been demonstrated to abrogate oncogenic signaling pathways and prevent the acquisition of cancer hallmarks by inhibiting AKR. Administration of gedunin, a neem limonoid, prevented angiogenesis in hamster oral tumors via the inactivation of AR. The mechanism involved the blockade of PI3K/Akt and NF-κB pathways and the downregulation of the expression of the oncomiR, miR-21, HIF-1α, and VEGF [197]. In SCC131 oral cancer cells, gedunin alone and in combination with epalrestat prevented cancer hallmarks presumably by inhibiting AR-mediated ROS signaling and co-inactivation of Akt, Erk, and IKK/NF-κB signaling, with the combination being more effective than the single agents. Cotreatment with gedunin and epalrestat induced G1/S phase cell cycle arrest, and cell death initially by autophagy and subsequently by apoptosis. Furthermore, the combination inhibited angiogenesis in both SCC131 cells as well as in Eahy926 endothelial cells [198]. Nimbolide, another neem limonoid was demonstrated to inhibit angiogenesis in breast cancer cells as well as in a xenografted nude mouse model of breast cancer by AR inhibition with the consequent abrogation of the insulin-like growth factor-1 (IGF-1)/PI3K/Akt and HIF-1α/VEGF signaling. Further, nimbolide was shown to regulate AR and IGF-1/PI3K/Akt signaling by epigenetic modulation. Most importantly, co-administration of nimbolide with metformin or the chemotherapeutic drugs tamoxifen/cisplatin was found to be more efficacious than single agents in abrogating IGF-1/PI3K/Akt/AR signaling suggesting that AKR inhibitors show promise in chemosensitization [199].
Coumarins that contain a benzopyran-2-one or chromen-2-one ring system, are found in various natural products. Kawanishi et al. [200] listed 41 coumarins that exhibit AR inhibitory activity. Angular dihydropyranocoumarins isolated from the flowers of Peucedanum japonicum (Umbelliferae) exhibited significant AKR1C1 inhibitory activity in A549 human non-small-cell lung cancer cells [201]. Nodakenin and scopoletin are naturally occurring coumarins that are reported to exert anticancer effects as well as AKR inhibitory activity [202,203]. A wide variety of dietary compounds identified as AKR inhibitors are also anticancer agents. These include spinach, cinnamon, lemon, basil, fenugreek, and curcumin among several others [154,204].
Aldo-keto reductases are a superfamily of enzymes that play crucial roles in various cellular processes. AKRs play a significant role in cancer biology, contributing to metabolic reprogramming, drug resistance, and tumorigenesis. Targeting AKRs through various strategies including the use of synthetic or natural ARIs holds promise for the development of novel cancer therapies. Further research is needed to elucidate the specific roles of individual members of the AKR family in different cancer types, identify biomarkers of AKR activity, and optimize therapeutic approaches to effectively target AKRs in cancer treatment.
In general, downregulation of AKRs is less common in malignancies compared to overexpression. While AKR1B1 is significantly elevated in liver and lung cancer, it is more widely overexpressed in a range of malignancies and is associated with worse outcomes. Given the significant association between diabetes and increased cancer risk, AKR1B1 due to its involvement in the polyol pathway and oncogenic signaling could presumably emerge as a vital link in the context of coincident disease. On the other hand, AKR1B10 is implicated in cell proliferation and chemoresistance and is less widely distributed in normal tissues relative to AKR1B1. These characteristics render AKR1B10 as a promising tumor marker and therapeutic target in specific tumors.
Despite convincing evidence implicating the role of AKRs in cancer, more intensive clinical studies are warranted on a broader range of malignant tumors, especially concerning the different isoforms. Clinical research has largely focused on AKR1B1 and AKR1B10 with very little emphasis on other members that could shed light on specific pathways and cellular processes that are specific to cancer type and tissue of origin.
Although many preclinical studies have demonstrated the efficacy of AKR inhibitors as anticancer agents, only a few have entered clinical trials. It is important to identify AKR inhibitors that exhibit high potency, safety, and isoform selectivity. A major bottleneck in the development of selective AKR inhibitors is the structural similarity among AKR family proteins. It is important to unravel subtle structural differences that can prevent cross-inhibition and facilitate selective inhibition. Natural products with their rich repertoire of chemical entities offer promise in the design and development of novel AKR inhibitors.
Acknowledgement: The authors thank Krishna K Kalahasti for his help in the preparation of the manuscript.
Funding Statement: SN and GBR are supported by grants from the Science and Engineering Research Board, Government of India (EMR/2016/001984), and Indian Council of Medical Research.
Author Contributions: SN, GBR conceptualized; SN, GBR, PRK, and KKT prepared the manuscript; SN, GBR, and PRK reviewed and edited the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: All authors declare no conflicts of interest whatsoever.
Supplementary Materials: The supplementary material is available online at https://doi.org/10.32604/or.2024.049918.
References
1. Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I. et al. (2021). Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians, 71(3), 209–249. [Google Scholar] [PubMed]
2. Sanchez-Vega, F., Mina, M., Armenia, J., Chatila, W. K., Luna, A. et al. (2018). Oncogenic signaling pathways in the cancer genome atlas. Cell, 173(2), 321–337. https://doi.org/10.1016/j.cell.2018.03.035. [Google Scholar] [PubMed] [CrossRef]
3. Hanahan, D. (2022). Hallmarks of cancer: New dimensions. Cancer Discovery, 12(1), 31–46. https://doi.org/10.1158/2159-8290.CD-21-1059. [Google Scholar] [PubMed] [CrossRef]
4. Paskeh, M. D. A., Entezari, M., Mirzaei, S., Zabolian, A., Saleki, H. et al. (2022). Emerging role of exosomes in cancer progression and tumor microenvironment remodeling. Journal of Hematology and Oncology, 15(1), 83. https://doi.org/10.1186/s13045-022-01305-4. [Google Scholar] [PubMed] [CrossRef]
5. Qin, Y., Ashrafizadeh, M., Mongiardini, V., Grimaldi, B., Crea, F. et al. (2023). Autophagy and cancer drug resistance in dialogue: Pre-clinical and clinical evidence. Cancer Letters, 570, 216307. https://doi.org/10.1016/j.canlet.2023.216307. [Google Scholar] [PubMed] [CrossRef]
6. Ramos, A., Sadeghi, S., Tabatabaeian, H. (2021). Battling chemoresistance in cancer: Root causes and trategies to uproot them. International Journal of Molecular Sciences, 22(17), 9451. https://doi.org/10.3390/ijms22179451. [Google Scholar] [PubMed] [CrossRef]
7. Zhao, M., Ma, J., Li, M., Zhang, Y., Jiang, B. et al. (2021). Cytochrome P450 enzymes and drug metabolism in humans. International Journal of Molecular Sciences, 22(23), 12808. https://doi.org/10.3390/ijms222312808. [Google Scholar] [PubMed] [CrossRef]
8. Li, Y., Steppi, A., Zhou, Y., Mao, F., Miller, P. C. et al. (2017). Tumoral expression of drug and xenobiotic metabolizing enzymes in breast cancer patients of different ethnicities with implications to personalized medicine. Scientific Reports, 7(1), 4747. https://doi.org/10.1038/s41598-017-04250-2. [Google Scholar] [PubMed] [CrossRef]
9. Rekka, E. A., Kourounakis, P. N., Pantelidou, M. (2019). Xenobiotic metabolising enzymes: Impact on pathologic conditions, drug interactions and drug design. Current Topics in Medicinal Chemistry, 19(4), 276–291. https://doi.org/10.2174/1568026619666190129122727. [Google Scholar] [PubMed] [CrossRef]
10. Manthalkar, L., Ajazuddin, Bhattacharya, S. (2022). Evidence-based capacity of natural cytochrome enzyme inhibitors to increase the effectivity of antineoplastic drugs. Discover Oncology, 13(1), 142. https://doi.org/10.1007/s12672-022-00605-y. [Google Scholar] [PubMed] [CrossRef]
11. Penning, T. M. (2015). The Aldo-keto reductases (AKRsOverview. Chemico Biological Interactions, 234, 236–246. https://doi.org/10.1016/j.cbi.2014.09.024. [Google Scholar] [PubMed] [CrossRef]
12. Hyndman, D., Bauman, D. R., Heredia, V. V., Penning, T. M. (2003). The aldo-keto reductase superfamily homepage. Chemico Biological Interactions, 143, 621–631. [Google Scholar] [PubMed]
13. Shen, Y., Ma, J., Yan, R., Ling, H., Li, X. et al. (2015). Impaired self-renewal and increased colitis and dysplastic lesions in colonic mucosa of AKR1B8-deficient mice. Clinical Cancer Research, 21(6), 1466–1476. https://doi.org/10.1158/1078-0432.CCR-14-2072. [Google Scholar] [PubMed] [CrossRef]
14. Luo, D. X., Huang, M. C., Ma, J., Gao, Z., Liao, D. F et al. (2011). Aldo-keto reductase family 1, member B10 is secreted through a lysosome-mediated non-classical pathway. The Biochemical Journal, 438(1), 71–80. https://doi.org/10.1042/BJ20110111. [Google Scholar] [PubMed] [CrossRef]
15. Banerjee, S. (2021). Aldo keto reductases AKR1B1 and AKR1B10 in cancer: Molecular mechanisms and signaling networks. Advances in Experimental Medicine and Biology, 1347, 65–82. https://doi.org/10.1007/978-3-030-80492-3. [Google Scholar] [CrossRef]
16. Giménez-Dejoz, J., Kolář, M. H., Ruiz, F. X., Crespo, I., Cousido-Siah, A. et al. (2015). Substrate specificity, inhibitor selectivity and structure-function relationships of Aldo-Keto Reductase 1B15: A novel human retinaldehyde reductase. PLoS One, 10(7), e0134506. https://doi.org/10.1371/journal.pone.0134506. [Google Scholar] [PubMed] [CrossRef]
17. Hers, H. G. (1956). The mechanism of the transformation of glucose in fructose in the seminal vesicles. Biochimica et Biophysica Acta, 22(1), 202–203. https://doi.org/10.1016/0006-3002(56)90247-5. [Google Scholar] [PubMed] [CrossRef]
18. Oatesvan, H. R. (1959). Formation of polyols by the lens of the rat with ‘sugar’ cataract. Nature, 184, 194–195. https://doi.org/10.1038/184194b0. [Google Scholar] [CrossRef]
19. Kinoshita, J. H., Kador, P., Catiles, M. (1981). Aldose reductase in diabetic cataracts. The Journal of the American Medical Association, 246, 257–261. https://doi.org/10.1001/jama.1981.03320030049032. [Google Scholar] [CrossRef]
20. Oates, P. J. (2008). Aldose reductase, still a compelling target for diabetic neuropathy. Current Drug Targets, 9(1), 14–36. https://doi.org/10.2174/138945008783431781. [Google Scholar] [PubMed] [CrossRef]
21. Srivastava, S. K., Ramana, K. V., Bhatnagar, A. (2005). Role of aldose reductase and oxidative damage in diabetes and the consequent potential for therapeutic options. Endocrine Reviews, 26(3), 380–392. https://doi.org/10.1210/er.2004-0028. [Google Scholar] [PubMed] [CrossRef]
22. Ramana, K. V., Srivastava, S. K. (2010). Aldose reductase: A novel therapeutic target for inflammatory pathologies. The International Journal of Biochemistry Cell Biology, 42(1), 17–20. https://doi.org/10.1016/j.biocel.2009.09.009. [Google Scholar] [PubMed] [CrossRef]
23. Srivastava, S. K., Yadav, U. C. S., Reddy, A. B., Saxena, A., Tammali, R. et al. (2011). Aldose reductase inhibition suppresses oxidative stress-induced inflammatory disorders. Chemico Biological Interactions, 191(1–3), 330–338. [Google Scholar] [PubMed]
24. Liu, J., Wen, G., Cao, D. (2009). Aldo-keto reductase family 1 member B1 inhibitors: Old drugs with new perspectives. Recent Patents on Anti-Cancer Drug Discovery, 4(3), 246–253. https://doi.org/10.2174/157489209789206931. [Google Scholar] [PubMed] [CrossRef]
25. Singh, M., Kapoor, A., Bhatnagar, A. (2021). Physiological and pathological roles of aldose reductase. Metabolites, 11(10), 655. https://doi.org/10.3390/metabo11100655. [Google Scholar] [PubMed] [CrossRef]
26. Khayami, R., Hashemi, S. R., Kerachian, M. A. (2020). Role of aldo-keto reductase family 1 member B1 (AKR1B1) in the cancer process and its therapeutic potential. Journal of Cellular and Molecular Medicine, 24(16), 8890–8902. https://doi.org/10.1111/jcmm.v24.16. [Google Scholar] [CrossRef]
27. Penning, T. M., Jonnalagadda, S., Trippier, P. C., Rižner, T. L. (2021). Aldo-keto reductases and cancer drug resistance. Pharmacological Reviews, 73(3), 1150–1171. https://doi.org/10.1124/pharmrev.120.000122. [Google Scholar] [PubMed] [CrossRef]
28. Fang, C. Y., Lin, Y. H., Chen, C. L. (2019). Overexpression of AKR1B10 predicts tumor recurrence and short survival in oral squamous cell carcinoma patients. Journal of Oral Pathology Medicine, 48(8), 712–719. https://doi.org/10.1111/jop.v48.8. [Google Scholar] [CrossRef]
29. Zhou, Y., Lin, Y., Li, W., Liu, Q., Gong, H. et al. (2023). Expression of AKRs superfamily and prognostic in human gastric cancer. Medicine, 102(8), e33041. https://doi.org/10.1097/MD.0000000000033041. [Google Scholar] [PubMed] [CrossRef]
30. Wu, X., Li, X., Fu, Q., Cao, Q., Chen, X. et al. (2017). AKR1B1 promotes basal-like breast cancer progression by a positive feedback loop that activates the EMT program. The Journal of Experimental Medicine, 214(4), 1065–1079. https://doi.org/10.1084/jem.20160903. [Google Scholar] [PubMed] [CrossRef]
31. Ma, J., Luo, D. X., Huang, C., Shen, Y., Bu, Y. et al. (2012). AKR1B10 overexpression in breast cancer: Association with tumor size, lymph node metastasis and patient survival and its potential as a novel serum marker. International Journal of Cancer, 131(6), E862–871. [Google Scholar] [PubMed]
32. Chang, K. C., Petrash, J. M. (2018). Aldo-keto reductases: Multifunctional proteins as therapeutic argets in diabetes and inflammatory disease. Advances in Experimental Medicine and Biology, 1032, 173–202. https://doi.org/10.1007/978-3-319-98788-0. [Google Scholar] [CrossRef]
33. Maccari, R., Ottanà, R. (2015). Targeting aldose reductase for the treatment of diabetes complications and inflammatory diseases: New insights and future directions. Journal of Medicinal Chemistry, 58(5), 2047–2067. https://doi.org/10.1021/jm500907a. [Google Scholar] [PubMed] [CrossRef]
34. Quattrini, L., La Motta, C. (2019). Aldose reductase inhibitors: 2013-present. Expert Opinion on Therapeutic Patents, 29, 199–213. https://doi.org/10.1080/13543776.2019.1582646. [Google Scholar] [PubMed] [CrossRef]
35. Sonowal, H., Ramana, K. V. (2021). Development of aldose reductase inhibitors for the treatment of inflammatory disorders and cancer: Current drug design strategies and future directions. Current Medicinal Chemistry, 28(19), 3683–3712. [Google Scholar] [PubMed]
36. Graham, A., Brown, L., Hedge, P. J., Gammack, A. J., Markham, A. F. (1991). Structure of the human aldose reductase gene. The Journal of Biological Chemistry, 266(11), 6872–6877. https://doi.org/10.1016/S0021-9258(20)89582-9. [Google Scholar] [CrossRef]
37. Liu, Z., Zhong, L., Krishack, P. A., Robbins, S., Cao, J. X. et al. (2009). Structure and promoter characterization of aldo-keto reductase family 1 B10 gene. Gene, 437(1–2), 39–44. [Google Scholar] [PubMed]
38. Nishinaka, T., Miura, T., Shimizu, K., Terada, T. (2017). Identification and characterization of functional antioxidant response elements in the promoter of the aldo-keto reductase AKR1B10 gene. Chemico Biological Interactions, 276, 160–166. https://doi.org/10.1016/j.cbi.2017.02.008. [Google Scholar] [PubMed] [CrossRef]
39. Mindnich, R. D., Penning, T. M. (2009). Aldo-keto reductase (AKR) superfamily: Genomics and annotation. Human Genomics, 3(4), 362–370. https://doi.org/10.1186/1479-7364-3-4-362. [Google Scholar] [PubMed] [CrossRef]
40. Petrash, J. M., Harter, T. M., Devine, C. S., Olins, P. O., Bhatnagar, A. et al. (1992). Involvement of cysteine residues in catalysis and inhibition of human aldose reductase. site-directed mutagenesis of Cys-80, −298, and −303. Journal of Biological Chemistry, 267(34), 24833–24840. https://doi.org/10.1016/S0021-9258(18)35839-3. [Google Scholar] [CrossRef]
41. Fournier, B., Bendeif, E. E., Guillot, B., Podjarny, A., Lecomte et al. (2009). Charge density and electrostatic interactions of fidarestat, an inhibitor of human aldose reductase. Journal of the American Chemical Society, 131(31), 10929–10941. https://doi.org/10.1021/ja8095015. [Google Scholar] [PubMed] [CrossRef]
42. Srivastava, S., Watowich, S. J., Petrash, J. M., Srivastava, S. K., Bhatnagar, A. (1999). Structural and kinetic determinants of aldehyde reduction by aldose reductase. Biochemistry, 38(1), 42–54. https://doi.org/10.1021/bi981794l. [Google Scholar] [PubMed] [CrossRef]
43. PeCousido-Siah, A., Ruiz, F. X., Mitschler, A., Porté, S., de Lera, Á. R., et al. (2014). Identification of a novel polyfluorinated compound as a lead to inhibit the human enzymes aldose reductase and AKR1B10: Structure determination of both ternary complexes and implications for drug design. Acta Crystallographica Section D, Biological Crystallography, 70(3), 889–903. https://doi.org/10.1107/S1399004713033452. [Google Scholar] [PubMed] [CrossRef]
44. Penning, T. M., Wangtrakuldee, P., Auchus, R. J. (2019). Structural and functional biology of aldo-keto reductase steroid-transforming enzymes. Endocrine Reviews, 40(2), 447–475. https://doi.org/10.1210/er.2018-00089. [Google Scholar] [PubMed] [CrossRef]
45. Thakur, S., Gupta, S. K., Ali, V., Singh, P., Verma, M. (2021). Aldose reductase: A cause and a potential target for the treatment of diabetic complications. Archives of Pharmacal Research, 44(7), 655–667. https://doi.org/10.1007/s12272-021-01343-5. [Google Scholar] [PubMed] [CrossRef]
46. Singh, M., Kapoor, A., Bhatnagar, A. (2015). Oxidative and reductive metabolism of lipid-peroxidation derived carbonyls. Chemico Biological Interactions, 234, 261–273. https://doi.org/10.1016/j.cbi.2014.12.028. [Google Scholar] [PubMed] [CrossRef]
47. Gelboin, H. V. (1980). Benzo[α]pyrene metabolism, activation and carcinogenesis: Role and regulation of mixed-function oxidases and related enzymes. Physiological Reviews, 60(4), 1107–1166. https://doi.org/10.1152/physrev.1980.60.4.1107. [Google Scholar] [PubMed] [CrossRef]
48. Guengerich, F. P., Johnson, W. W. (1999). Kinetics of hydrolysis and reaction of aflatoxin B1 exo-8,9-epoxide and relevance to toxicity and detoxication. Drug Metabolism Reviews, 31(1), 141–158. https://doi.org/10.1081/DMR-100101911. [Google Scholar] [PubMed] [CrossRef]
49. Hecht, S. S. (1998). Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chemical Research in Toxicology, 11(6), 559–603. https://doi.org/10.1021/tx980005y. [Google Scholar] [PubMed] [CrossRef]
50. Upadhyaya, P., Kenney, P. M., Hochalter, J. B., Wang, M., Hecht, S. S. (1999). Tumorigenicity and metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol enantiomers and metabolites in the A/J mouse. Carcinogenesis, 20(8), 1577–1582. https://doi.org/10.1093/carcin/20.8.1577. [Google Scholar] [PubMed] [CrossRef]
51. Endo, S., Matsunaga, T., Nishinaka, T. (2021). The role of AKR1B10 in physiology and pathophysiology. Metabolites, 11(6), 332. https://doi.org/10.3390/metabo11060332. [Google Scholar] [PubMed] [CrossRef]
52. Quinn, A. M., Harvey, R. G., Penning, T. M. (2008). Oxidation of PAH trans-dihydrodiols by human aldo-keto reductase AKR1B10. Chemical Research in Toxicology, 21(11), 2207–2215. https://doi.org/10.1021/tx8002005. [Google Scholar] [PubMed] [CrossRef]
53. Giménez-Dejoz, J., Weber, S., Fernández-Pardo, Á., Möller, G., Adamski, J. et al. (2019). Engineering aldo-keto reductase 1B10 to mimic the distinct 1B15 topology and specificity towards inhibitors and substrates, including retinoids and steroids. Chemico-Biological Interactions, 307, 186–194. https://doi.org/10.1016/j.cbi.2019.04.030. [Google Scholar] [PubMed] [CrossRef]
54. Penning, T. M., Burczynski, M. E., Hung, C. F., McCoull, K. D. et al. (1999). Dihydrodiol dehydrogenases and polycyclic aromatic hydrocarbon activation: Generation of reactive and redox active o-quinones. Chemical Research in Toxicology, 12(1), 1–18. https://doi.org/10.1021/tx980143n. [Google Scholar] [PubMed] [CrossRef]
55. Jiang, H., Shen, Y. M., Quinn, A. M., Penning, T. M. (2005). Competing roles of cytochrome P450 1A1/1B1 and aldo-keto reductase 1A1 in the metabolic activation of (±)-7,8-dihydroxy-7,8-dihydro-benzo[α]pyrene in human bronchoalveolar cell extracts. Chemical Research in Toxicology, 18(2), 365–374. https://doi.org/10.1021/tx0497245. [Google Scholar] [PubMed] [CrossRef]
56. Ko, B. C., Ruepp, B., Bohren, K. M., Gabbay, K. H., Chung, S. S. (1997). Identification and characterization of multiple osmotic response sequences in the human aldose reductase gene. The Journal of Biological Chemistry, 272(26), 16431–16437. https://doi.org/10.1074/jbc.272.26.16431. [Google Scholar] [PubMed] [CrossRef]
57. Nguyen, T., Rushmore, T. H., Pickett, C. B. (1994). Transcriptional regulation of a rat liver glutathione S-transferase Ya subunit gene. Analysis of the antioxidant response element and its activation by the phorbol ester 12-O-tetradecanoylphorbol-13-acetate. The Journal of Biological Chemistry, 269(18), 13656–13662. https://doi.org/10.1016/S0021-9258(17)36880-1. [Google Scholar] [CrossRef]
58. Rushmore, T. H., Morton, M. R., Pickett, C. B. (1991). The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. The Journal of Biological Chemistry, 266(18), 11632–11639. https://doi.org/10.1016/S0021-9258(18)99004-6. [Google Scholar] [CrossRef]
59. Penning, T. M., Drury, J. E. (2007). Human aldo-keto reductases: Function, gene regulation, and single nucleotide polymorphisms. Archives of Biochemistry and Biophysics, 464(2), 241–250. https://doi.org/10.1016/j.abb.2007.04.024. [Google Scholar] [PubMed] [CrossRef]
60. Rizner, T. L., Smuc, T., Rupreht, R., Sinkovec, J., Penning, T. M. (2006). AKR1C1 and AKR1C3 may determine progesterone and estrogen ratios in endometrial cancer. Molecular and Cellular Endocrinology, 248(1–2), 126–135. [Google Scholar] [PubMed]
61. Penning, T. M., Steckelbroeck, S., Bauman, D. R., Miller, M. W., Jin, Y. et al. (2006). Aldo-keto reductase (AKR) 1C3: Role in prostate disease and the development of specific inhibitors. Molecular and Cellular Endocrinology, 248(1–2), 182–191. [Google Scholar] [PubMed]
62. Steckelbroeck, S., Jin, Y., Gopishetty, S., Oyesanmi, B., Penning, T. M. (2004). Human cytosolic 3alpha-hydroxysteroid dehydrogenases of the aldo-keto reductase superfamily display significant 3beta-hydroxysteroid dehydrogenase activity: Implications for steroid hormone metabolism and action. The Journal of Biological Chemistry, 279(11), 10784–10795. https://doi.org/10.1074/jbc.M313308200. [Google Scholar] [PubMed] [CrossRef]
63. Lefrançois-Martinez, A. M., Bertherat, J., Val, P., Tournaire, C., Gallo-Payet, N. et al. (2004). Decreased expression of cyclic adenosine monophosphate-regulated aldose reductase (AKR1B1) is associated with malignancy in human sporadic adrenocortical tumors. The Journal of Clinical Endocrinology and Metabolism, 89(6), 3010–3019. https://doi.org/10.1210/jc.2003-031830. [Google Scholar] [PubMed] [CrossRef]
64. Reddy, K. A., Kumar, P. U., Srinivasulu, M., Triveni, B., Sharada, K. et al. (2017). Overexpression and enhanced specific activity of aldoketo reductases (AKR1B1 & AKR1B10) in human breast cancers. Breast, 31, 137–143. https://doi.org/10.1016/j.breast.2016.11.003. [Google Scholar] [PubMed] [CrossRef]
65. Ji, J., Xu, M. X., Qian, T. Y., Zhu, S. Z., Jiang, F. et al. (2020). The AKR1B1 inhibitor epalrestat suppresses the progression of cervical cancer. Molecular Biology Reports, 47(8), 6091–6103. https://doi.org/10.1007/s11033-020-05685-z. [Google Scholar] [PubMed] [CrossRef]
66. Matsunaga, T., Okumura, N., Saito, H., Morikawa, Y., Suenami, K. et al. (2020). Significance of aldo-keto reductase 1C3 and ATP-binding cassette transporter B1 in gain of irinotecan resistance in colon cancer cells. Chemico Biological Interactions, 332, 109295. https://doi.org/10.1016/j.cbi.2020.109295. [Google Scholar] [PubMed] [CrossRef]
67. Kropotova, E. S., Tychko, R. A., Zinov'eva, O. L., Zyrianova, A. F., Khankin, S. L. et al. (2010). Downregulation of AKR1B10 gene expression in colorectal cancer. Molekuliarnaia Biologiia, 44(2), 243–250. [Google Scholar] [PubMed]
68. Nakarai, C., Osawa, K., Akiyama, M., Matsubara, N., Ikeuchi, H. et al. (2015). Expression of AKR1C3 and CNN3 as markers for detection of lymph node metastases in colorectal cancer. Clinical and Experimental Medicine, 15(3), 333–341. https://doi.org/10.1007/s10238-014-0298-1. [Google Scholar] [PubMed] [CrossRef]
69. Xie, C., Ye, X., Zeng, L., Zeng, X., Cao, D. (2023). Serum AKR1B10 as an indicator of unfavorable survival of hepatocellular carcinoma. Journal of Gastroenterology, 58(10), 1030–1042. https://doi.org/10.1007/s00535-023-02011-9. [Google Scholar] [PubMed] [CrossRef]
70. Endo, S., Xia, S., Suyama, M., Morikawa, Y., Oguri, H. et al. (2017). Synthesis of potent and selective inhibitors of aldo-keto reductase 1B10 and their efficacy against proliferation, metastasis, and cisplatin resistance of lung cancer cells. Journal of Medicinal Chemistry, 60(20), 8441–8455. https://doi.org/10.1021/acs.jmedchem.7b00830. [Google Scholar] [PubMed] [CrossRef]
71. Liu, C., Li, Y., Wei, M., Zhao, L., Yu, Y. et al. (2019). Identification of a novel glycolysis-related gene signature that can predict the survival of patients with lung adenocarcinoma. Cell Cycle, 18(5), 568–579. https://doi.org/10.1080/15384101.2019.1578146. [Google Scholar] [PubMed] [CrossRef]
72. Zhang, W., Li, H., Yang, Y., Liao, J., Yang, G. Y. (2014). Knockdown or inhibition of aldo-keto reductase 1B10 inhibits pancreatic carcinoma growth via modulating Kras-E-cadherin pathway. Cancer Letters, 355(2), 273–280. https://doi.org/10.1016/j.canlet.2014.09.031. [Google Scholar] [PubMed] [CrossRef]
73. Meng, M., Yang, L., Zhou, H., Cheng, Q., Peng, R. et al. (2023). LINC00978 regulates metabolic rewiring to promote the malignancy of glioblastoma through AKR1B1. Cancer Letters, 567, 216277. https://doi.org/10.1016/j.canlet.2023.216277. [Google Scholar] [PubMed] [CrossRef]
74. Laffin, B., Petrash, M. (2012). Expression of the aldo-ketoreductases AKR1B1 and AKR1B10 in human cancers. Frontiers in Pharmacology, 3, 104. [Google Scholar] [PubMed]
75. Saraswat, M., Mrudula, T., Kumar, P. U., Suneetha, A., Rao, T. S. et al. (2006). Overexpression of aldose reductase in human cancer tissues. Medical Science Monitor, 12(12), CR525–529. [Google Scholar] [PubMed]
76. Jin, J., Liao, W., Yao, W., Zhu, R., Li, Y. et al. (2016). Aldo-keto reductase family 1 member B 10 mediates liver cancer cell proliferation through sphingosine-1-phosphate. Scientific Reports, 6, 22746. https://doi.org/10.1038/srep22746. [Google Scholar] [PubMed] [CrossRef]
77. van Weverwijk, A., Koundouros, N., Iravani, M., Ashenden, M., Gao, Q. et al. (2019). Metabolic adaptability in metastatic breast cancer by AKR1B10-dependent balancing of glycolysis and fatty acid oxidation. Nature Communications, 10(1), 2698. https://doi.org/10.1038/s41467-019-10592-4. [Google Scholar] [PubMed] [CrossRef]
78. Chung, Y. T., Matkowskyj, K. A., Li, H., Bai, H., Zhang, W. et al. (2012). Overexpression and oncogenic function of aldo-keto reductase family 1B10 (AKR1B10) in pancreatic carcinoma. Modern Pathology, 25(5), 758–766. https://doi.org/10.1038/modpathol.2011.191. [Google Scholar] [PubMed] [CrossRef]
79. Hung, J. J., Yeh, Y. C., Hsu, W. H. (2018). Prognostic significance of AKR1B10 in patients with resected lung adenocarcinoma. Thoracic Cancer, 9(11), 1492–1499. https://doi.org/10.1111/tca.2018.9.issue-11. [Google Scholar] [CrossRef]
80. Taskoparan, B., Seza, E. G., Demirkol, S., Tuncer, S., Stefek, M. et al. (2017). Opposing roles of the aldo-keto reductases AKR1B1 and AKR1B10 in colorectal cancer. Cellular Oncology, 40(6), 563–578. https://doi.org/10.1007/s13402-017-0351-7. [Google Scholar] [PubMed] [CrossRef]
81. Constantinescu, S., Hecht, K., Sobotzki, N., Erzinger, M. M., Bovet, C. et al. (2014). Transcriptomic responses of cancerous and noncancerous human colon cells to sulforaphane and selenium. Chemical Research in Toxicology, 27(3), 377–386. https://doi.org/10.1021/tx400427t. [Google Scholar] [PubMed] [CrossRef]
82. Yuan, C., Yuan, M., Chen, M., Ouyang, J., Tan, W. et al. (2021). Prognostic implication of a novel metabolism-related gene signature in hepatocellular carcinoma. Frontiers in Oncology, 11, 666199. https://doi.org/10.3389/fonc.2021.666199. [Google Scholar] [PubMed] [CrossRef]
83. DiStefano, J. K., Davis, B. (2019). Diagnostic and prognostic potential of AKR1B10 in human hepatocellular carcinoma. Cancers, 11(4), 486. https://doi.org/10.3390/cancers11040486. [Google Scholar] [PubMed] [CrossRef]
84. Ha, S. Y., Song, D. H., Lee, J. J., Lee, H. W., Cho, S. Y. et al. (2014). High expression of aldo-keto reductase 1B10 is an independent predictor of favorable prognosis in patients with hepatocellular carcinoma. Gut and Liver, 8(6), 648–654. https://doi.org/10.5009/gnl13406. [Google Scholar] [PubMed] [CrossRef]
85. Han, C., Gao, L., Zhao, L., Sheng, Q., Zhang, C. et al. (2018). Immunohistochemistry detects increased expression of aldo-keto reductase family 1 member B10 (AKR1B10) in early-stage hepatocellular carcinoma. Medical Science Monitor, 24, 7414–7423. https://doi.org/10.12659/MSM.910738. [Google Scholar] [PubMed] [CrossRef]
86. Liu, T. A., Jan, Y. J., Ko, B. S., Wu, Y. J., Lu, Y. J. et al. (2015). Regulation of aldo-keto-reductase family 1 B10 by 14-3-3ε and their prognostic impact of hepatocellular carcinoma. Oncotarget, 6(36), 38967–38982. https://doi.org/10.18632/oncotarget.v6i36. [Google Scholar] [CrossRef]
87. Wang, Z., Pei, Y., Li, W., Zhang, J., Liu, J. (2022). Clinical value of AKR1B10 in hepatocellular carcinoma: A systematic review and meta-analysis. PLoS One, 17(12), e0279591. https://doi.org/10.1371/journal.pone.0279591. [Google Scholar] [PubMed] [CrossRef]
88. Guengerich, F. P., Arneson, K. O., Williams, K. M., Deng, Z., Harris, T. M. (2002). Reaction of aflatoxin B(1) oxidation products with lysine. Chemical Research in Toxicology, 15(6), 780–792. https://doi.org/10.1021/tx010156s. [Google Scholar] [PubMed] [CrossRef]
89. Schwab, A., Siddiqui, A., Vazakidou, M. E., Napoli, F., Böttcher, M. et al. (2018). Polyol pathway links glucose metabolism to the aggressiveness of cancer cells. Cancer Research, 78(7), 1604–1618. https://doi.org/10.1158/0008-5472.CAN-17-2834. [Google Scholar] [PubMed] [CrossRef]
90. Fukumoto, S., Yamauchi, N., Moriguchi, H., Hippo, Y., Watanabe, A. et al. (2005). Overexpression of the aldo-keto reductase family protein AKR1B10 is highly correlated with smokers' non-small cell lung carcinomas. Clinical Cancer Research, 11, 1776–1785. https://doi.org/10.1158/1078-0432.CCR-04-1238. [Google Scholar] [PubMed] [CrossRef]
91. Kang, M. W., Lee, E. S., Yoon, S. Y., Jo, J., Lee, J. et al. (2011). AKR1B10 is associated with smoking and smoking-related non-small-cell lung cancer. The Journal of International Medical Research, 39(1), 78–85. https://doi.org/10.1177/147323001103900110. [Google Scholar] [PubMed] [CrossRef]
92. Cubillos-Angulo, J. M., Fukutani, E. R., Cruz, L. A. B., Arriaga, M. B., Lima, J. V. et al. (2020). Systems biology analysis of publicly available transcriptomic data reveals a critical link between AKR1B10 gene expression, smoking and occurrence of lung cancer. PLoS One, 15(2), e0222552. https://doi.org/10.1371/journal.pone.0222552. [Google Scholar] [PubMed] [CrossRef]
93. Liu, W., Song, J., Du, X., Zhou, Y., Li, Y. et al. (2019). AKR1B10 (Aldo-keto reductase family 1 B10) promotes brain metastasis of lung cancer cells in a multi-organ microfluidic chip model. Acta Biomaterialia, 91, 195–208. https://doi.org/10.1016/j.actbio.2019.04.053. [Google Scholar] [PubMed] [CrossRef]
94. Jin, J., Krishack, P. A., Cao, D. (2006). Role of aldo-keto reductases in development of prostate and breast cancer. Frontiers in Bioscience, 1(11), 2767–2773. [Google Scholar]
95. Hofland, J., van Weerden, W. M., Dits, N. F. J., Steenbergen, J., Van Leenders, G. J. L. H., et al. (2010). Evidence of limited contributions for intratumoral steroidogenesis in prostate cancer. Cancer Research, 70(3), 1256–1264. https://doi.org/10.1158/0008-5472.CAN-09-2092. [Google Scholar] [PubMed] [CrossRef]
96. Hamid, A. R. A. H., Pfeiffer, M. J., Verhaegh, G. W., Schaafsma, E., Brandt, A. et al. (2013). Aldo-keto reductase family 1 member C3 (AKR1C3) is a biomarker and therapeutic target for castration-resistant prostate cancer. Molecular Medicine, 18(1), 1449–1455. [Google Scholar] [PubMed]
97. Mitsiades, N., Sung, C. C., Schultz, N., Danila, D. C., He, B. et al. (2012). Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. Cancer Research, 72(23), 6142–6152. https://doi.org/10.1158/0008-5472.CAN-12-1335. [Google Scholar] [PubMed] [CrossRef]
98. Penning, T. M., Byrns, M. C. (2009). Steroid hormone transforming aldo-keto reductases and cancer. Annals of the New York Academy of Sciences, 1155, 33–42. https://doi.org/10.1111/nyas.2009.1155.issue-1. [Google Scholar] [CrossRef]
99. Byrns, M. C., Duan, L., Lee, S. H., Blair, I. A., Penning, T. M. (2010). Aldo-keto reductase 1C3 expression in MCF-7 cells reveals roles in steroid hormone and prostaglandin metabolism that may explain its over-expression in breast cancer. The Journal of Steroid Biochemistry and Molecular Biology, 118(3), 177–187. https://doi.org/10.1016/j.jsbmb.2009.12.009. [Google Scholar] [PubMed] [CrossRef]
100. McNamara, K. M., Sasano, H. (2019). The role of 17βHSDs in breast tissue and breast cancers. Molecular and Cellular Endocrinology, 489, 32–44. https://doi.org/10.1016/j.mce.2018.10.019. [Google Scholar] [PubMed] [CrossRef]
101. Penning, T. M., Burczynski, M. E., Jez, J. M., Hung, C. F., Lin, H. K. et al. (2000). Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: Functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. The Biochemical Journal, 351(1), 67–77. https://doi.org/10.1042/bj3510067. [Google Scholar] [CrossRef]
102. Li, Z., He, X., Xing, S., Ni, J., Zhang, W. et al. (2013). Overexpression of Aldo-keto reductase family 1 B10 protein in ductal carcinoma in situ of the breast correlates with HER2 positivity. Cancer Biomarkers, 13(3), 181–192. https://doi.org/10.3233/CBM-130337. [Google Scholar] [PubMed] [CrossRef]
103. Yoshitake, H., Takahashi, M., Ishikawa, H., Nojima, M., Iwanari, H. et al. (2007). Aldo-keto reductase family 1, member B10 in uterine carcinomas: A potential risk factor of recurrence after surgical therapy in cervical cancer. International Journal of Gynecological Cancer, 17(6), 1300–1306. https://doi.org/10.1111/j.1525-1438.2007.00932.x. [Google Scholar] [PubMed] [CrossRef]
104. Li, X., Yang, J., Gu, X., Xu, J., Li, H. et al. (2020). The expression and clinical significance of aldo-keto reductase 1 member B1 in gastric carcinoma. DNA and Cell Biology, 39(7), 1322–1327. https://doi.org/10.1089/dna.2020.5550. [Google Scholar] [PubMed] [CrossRef]
105. Liu, L., Zhu, L., Cheng, Z., Sun, Y., Zhou, Y. et al. (2023). Aberrant expression of AKR1B1 indicates poor prognosis and promotes gastric cancer progression by regulating the AKT-mTOR pathway. Aging, 15(18), 9661–9675. https://doi.org/10.18632/aging.v15i18. [Google Scholar] [CrossRef]
106. Kang, Y. L., Kim, J., Kwak, S. B., Kim, Y. S., Huh, J. et al. (2024). The polyol pathway and nuclear ketohexokinase a signaling drive hyperglycemia-induced metastasis of gastric cancer. Experimental and Molecular Medicine, 56(1), 220–234. https://doi.org/10.1038/s12276-023-01153-3. [Google Scholar] [PubMed] [CrossRef]
107. Zhao, Z., Hao, Z., Zhang, Z., Zhan, X. (2023). Bioinformatics analysis reveals the vital role of AKR1B1 in immune infiltration and clinical outcomes of gastric cancer. DNA and Cell Biology, 42(7), 372–389. https://doi.org/10.1089/dna.2022.0644. [Google Scholar] [PubMed] [CrossRef]
108. Yao, H. B., Xu, Y., Chen, L. G., Guan, T. P., Ma, Y. Y. et al. (2014). AKR1B10, a good prognostic indicator in gastric cancer. European Journal of Surgical Oncology, 40(3), 318–324. https://doi.org/10.1016/j.ejso.2013.12.014. [Google Scholar] [PubMed] [CrossRef]
109. Syamprasad, N. P., Rajdev, B., Jain, S., Panda, S. R., Puppala, E. R. et al. (2023). Pivotal role of AKR1B1 in pathogenesis of colitis associated colorectal carcinogenesis. International Immunopharmacology, 119, 110145. https://doi.org/10.1016/j.intimp.2023.110145. [Google Scholar] [PubMed] [CrossRef]
110. Uzozie, A., Nanni, P., Staiano, T., Grossmann, J., Barkow-Oesterreicher, S. et al. (2014). Sorbitol dehydrogenase overexpression and other aspects of dysregulated protein expression in human precancerous colorectal neoplasms: A quantitative proteomics study. Molecular and Cellular Proteomics, 13(5), 1198–1218. https://doi.org/10.1074/mcp.M113.035105. [Google Scholar] [PubMed] [CrossRef]
111. Ohashi, T., Idogawa, M., Sasaki, Y., Suzuki, H., Tokino, T. (2013). AKR1B10, a transcriptional target of p53, is downregulated in colorectal cancers associated with poor prognosis. Molecular Cancer Research, 11(12), 1554–1563. https://doi.org/10.1158/1541-7786.MCR-13-0330-T. [Google Scholar] [PubMed] [CrossRef]
112. Ko, H. H., Cheng, S. L., Lee, J. J., Chen, H. M., Kuo, M. Y. P. et al. (2017). Expression of AKR1B10 as an independent marker for poor prognosis in human oral squamous cell carcinoma. Head and Neck, 39(7), 1327–1332. https://doi.org/10.1002/hed.v39.7. [Google Scholar] [CrossRef]
113. Ko, H. H., Peng, H. H., Cheng, S. J., Kuo, M. Y. P. (2018). Increased salivary AKR1B10 level: Association with progression and poor prognosis of oral squamous cell carcinoma. Head and Neck, 40(12), 2642–2647. https://doi.org/10.1002/hed.v40.12. [Google Scholar] [CrossRef]
114. Nagaraj, N. S., Beckers, S., Mensah, J. K., Waigel, S., Vigneswaran, N. et al. (2006). Cigarette smoke condensate induces cytochromes P450 and aldo-keto reductases in oral cancer cells. Toxicology Letters, 165(2), 182–194. https://doi.org/10.1016/j.toxlet.2006.03.008. [Google Scholar] [PubMed] [CrossRef]
115. Breton, J., Gage, M. C., Hay, A. W., Keen, J. N., Wild, C. P. et al. (2008). Proteomic screening of a cell line model of esophageal carcinogenesis identifies cathepsin D and aldo-keto reductase 1C2 and 1B10 dysregulation in Barrett's esophagus and esophageal adenocarcinoma. Journal of Proteome Research, 7(5), 1953–1962. https://doi.org/10.1021/pr7007835. [Google Scholar] [PubMed] [CrossRef]
116. Luo, L., Chen, C., He, H., Cai, M., Ling, C. (2020). Silencing of long non-coding RNA (LncRNA) non-coding RNA activated by DNA damage (NORAD) inhibits proliferation, invasion, migration, and promotes apoptosis of glioma cells via downregulating the expression of AKR1B1. Medical Science Monitor, 26, e922659. [Google Scholar] [PubMed]
117. Bacolod, M. D., Lin, S. M., Johnson, S. P., Bullock, N. S., Colvin, M. et al. (2008). The gene expression profiles of medulloblastoma cell lines resistant to preactivated cyclophosphamide. Current Cancer Drug Targets, 8(3), 172–179. https://doi.org/10.2174/156800908784293631. [Google Scholar] [PubMed] [CrossRef]
118. Himura, R., Kawano, S., Nagata, Y., Kawai, M., Ota, A. et al. (2024). Inhibition of aldo-keto reductase 1C3 overcomes gemcitabine/cisplatin resistance in bladder cancer. Chemico Biological Interactions, 388, 110840. https://doi.org/10.1016/j.cbi.2023.110840. [Google Scholar] [PubMed] [CrossRef]
119. Feitelson, M. A., Arzumanyan, A., Kulathinal, R. J., Blain, S. W., Holcombe, R. F. et al. (2015). Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Seminars in Cancer Biology, 35, S25–S54. https://doi.org/10.1016/j.semcancer.2015.02.006. [Google Scholar] [PubMed] [CrossRef]
120. Tammali, R., Reddy, A. B. M., Ramana, K. V., Petrash, J. M., Srivastava, S. K. (2009). Aldose reductase deficiency in mice prevents azoxymethane-induced colonic preneoplastic aberrant crypt foci formation. Carcinogenesis, 30(5), 799–807. https://doi.org/10.1093/carcin/bgn246. [Google Scholar] [PubMed] [CrossRef]
121. Ramana, K. V., Tammali, R., Srivastava, S. K. (2010). Inhibition of aldose reductase prevents growth factor-induced G1-S phase transition through the AKT/phosphoinositide 3-kinase/E2F-1 pathway in human colon cancer cells. Molecular Cancer Therapeutics, 9(4), 813–824. https://doi.org/10.1158/1535-7163.MCT-09-0795. [Google Scholar] [PubMed] [CrossRef]
122. Tammali, R., Reddy, A. B. M., Srivastava, S. K., Ramana, K. V. (2011). Inhibition of aldose reductase prevents angiogenesis in vitro and in vivo. Angiogenesis, 14(2), 209–221. https://doi.org/10.1007/s10456-011-9206-4. [Google Scholar] [PubMed] [CrossRef]
123. Wang, J., Zhou, Y., Fei, X., Chen, X., Chen, Y. (2018). Biostatistics mining associated method identifies AKR1B10 enhancing hepatocellular carcinoma cell growth and degenerated by miR-383-5p. Scientific Reports, 8(1), 11094. https://doi.org/10.1038/s41598-018-29271-3. [Google Scholar] [PubMed] [CrossRef]
124. Matkowskyj, K. A., Bai, H., Liao, J., Zhang, W., Li, H. et al. (2014). Aldo-Ketoreductase family 1 B10 (AKR1B10) as a biomarker to distinguish hepatocellular carcinoma from benign liver lesions. Human Pathology, 45(4), 834–843. https://doi.org/10.1016/j.humpath.2013.12.002. [Google Scholar] [PubMed] [CrossRef]
125. Shoeb, M., Ramana, K. V., Srivastava, S. K. (2013). Aldose reductase inhibition enhances TRAIL-induced human colon cancer cell apoptosis through AKT/FOXO3a-dependent upregulation of death receptors. Free Radical Biology and Medicine, 63, 280–290. https://doi.org/10.1016/j.freeradbiomed.2013.05.039. [Google Scholar] [PubMed] [CrossRef]
126. Burada, F., Nicoli, E. R., Ciurea, M. E., Uscatu, D. C., Ioana, M. et al. (2015). Autophagy in colorectal cancer: An important switch from physiology to pathology. World Journal of Gastrointestinal Oncology, 7(11), 271–284. https://doi.org/10.4251/wjgo.v7.i11.271. [Google Scholar] [PubMed] [CrossRef]
127. Li, W., Liu, C., Huang, Z., Shi, L., Zhong, C. et al. (2021). AKR1B10 negatively regulates autophagy through reducing GAPDH upon glucose starvation in colon cancer. Journal of Cell Science, 134(8), jcs255273. https://doi.org/10.1242/jcs.255273. [Google Scholar] [PubMed] [CrossRef]
128. Abou Khouzam, R., Brodaczewska, K., Filipiak, A., Zeinelabdin, N. A., Buart, S. et al. (2020). Tumor hypoxia regulates immune escape/invasion: Influence on angiogenesis and potential impact of hypoxic biomarkers on cancer therapies. Frontiers in Immunology, 11, 613114. [Google Scholar] [PubMed]
129. Tammali, R., Saxena, A., Srivastava, S. K., Ramana, K. V. (2011). Aldose reductase inhibition prevents hypoxia-induced increase in hypoxia-inducible factor-1α (HIF-1α) and vascular endothelial growth factor (VEGF) by regulating 26 S proteasome-mediated protein degradation in human colon cancer cells. The Journal of Biological Chemistry, 286(27), 24089–24100. https://doi.org/10.1074/jbc.M111.219733. [Google Scholar] [PubMed] [CrossRef]
130. Tammali, R., Reddy, A. B. M., Saxena, A., Rychahou, P. G., Evers, B. M. et al. (2011). Inhibition of aldose reductase prevents colon cancer metastasis. Carcinogenesis, 32(8), 1259–1267. https://doi.org/10.1093/carcin/bgr102. [Google Scholar] [PubMed] [CrossRef]
131. Tian, K., Deng, Y., Li, Z., Zhou, H., Yao, H. (2023). AKR1B10 inhibits the proliferation and metastasis of hepatocellular carcinoma cells by regulating the PI3K/AKT pathway. Oncology Letters, 27(1), 18. https://doi.org/10.3892/ol. [Google Scholar] [CrossRef]
132. Cheng, Y., Ni, Y. J., Tang, L. M. (2023). ZNF521/EBF1 axis regulates AKR1B1 to promote the proliferation, migration, and invasion of gastric cancer cells. The Kaohsiung Journal of Medical Sciences, 39(3), 244–253. https://doi.org/10.1002/kjm2.v39.3. [Google Scholar] [CrossRef]
133. Zhong, H., Yin, H. (2015). Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. Redox Biology, 4, 193–199. https://doi.org/10.1016/j.redox.2014.12.011. [Google Scholar] [PubMed] [CrossRef]
134. Tammali, R., Srivastava, S. K., Ramana, K. V. (2011). Targeting aldose reductase for the treatment of cancer. Current Cancer Drug Targets, 11(5), 560–571. https://doi.org/10.2174/156800911795655958. [Google Scholar] [PubMed] [CrossRef]
135. Patel, M., Horgan, P. G., McMillan, D. C., Edwards, J. (2018). NF-κB pathways in the development and progression of colorectal cancer. Translational Research, 197, 43–56. https://doi.org/10.1016/j.trsl.2018.02.002. [Google Scholar] [PubMed] [CrossRef]
136. Taniguchi, K., Matsuoka, A., Kizuka, F., Lee, L., Tamura, I. et al. (2010). Prostaglandin F2α (PGF2α) stimulates PTGS2 expression and PGF2α synthesis through NFKB activation via reactive oxygen species in the corpus luteum of pseudopregnant rats. Reproduction, 140(6), 885–892. https://doi.org/10.1530/REP-10-0240. [Google Scholar] [PubMed] [CrossRef]
137. Koundouros, N., Poulogiannis, G. (2018). Phosphoinositide 3-Kinase/Akt signaling and redox metabolism in cancer. Frontiers in Oncology, 8, 160. https://doi.org/10.3389/fonc.2018.00160. [Google Scholar] [PubMed] [CrossRef]
138. Zhao, J. X., Yuan, Y. W., Cai, C. F., Shen, D. Y., Chen, M. L. et al. (2017). Aldose reductase interacts with AKT1 to augment hepatic AKT/mTOR signaling and promote hepatocarcinogenesis. Oncotarget, 8(40), 66987–67000. https://doi.org/10.18632/oncotarget.v8i40. [Google Scholar] [CrossRef]
139. Penning, T. M. (2017). Aldo-Keto reductase regulation by the nrf2 system: Implications for stress response, chemotherapy drug resistance, and carcinogenesis. Chemical Research in Toxicology, 30(1), 162–176. https://doi.org/10.1021/acs.chemrestox.6b00319. [Google Scholar] [PubMed] [CrossRef]
140. MacLeod, A. K., Acosta-Jimenez, L., Coates, P. J., McMahon, M., Carey, F. A. et al. (2016). Aldo-keto reductases are biomarkers of NRF2 activity and are co-ordinately overexpressed in non-small cell lung cancer. British Journal of Cancer, 115(12), 1530–1539. https://doi.org/10.1038/bjc.2016.363. [Google Scholar] [PubMed] [CrossRef]
141. Zuo, J., Zhang, Z., Li, M., Yang, Y., Zheng, B. et al. (2022). The crosstalk between reactive oxygen species and noncoding RNAs: From cancer code to drug role. Molecular Cancer, 21(1), 30. https://doi.org/10.1186/s12943-021-01488-3. [Google Scholar] [PubMed] [CrossRef]
142. Saxena, A., Shoeb, M., Ramana, K. V., Srivastava, S. K. (2013). Aldose reductase inhibition suppresses colon cancer cell viability by modulating microRNA-21 mediated programmed cell death 4 (PDCD4) expression. European Journal of Cancer, 49(15), 3311–3319. https://doi.org/10.1016/j.ejca.2013.05.031. [Google Scholar] [PubMed] [CrossRef]
143. Saxena, A., Tammali, R., Ramana, K. V., Srivastava, S. K. (2013). Aldose reductase inhibition prevents colon cancer growth by restoring phosphatase and tensin homolog through modulation of miR-21 and FOXO3a. Antioxidants & Redox Signaling, 18(11), 1249–1262. https://doi.org/10.1089/ars.2012.4643. [Google Scholar] [PubMed] [CrossRef]
144. Man, Q., Zhang, G., Chen, X., Na, S. R., Bai, S. et al. (2024). EPS15-AS1 inhibits AKR1B1 expression to enhance ferroptosis in hepatocellular carcinoma cells. Journal of Cancer, 15(4), 1030–1040. https://doi.org/10.7150/jca.89993. [Google Scholar] [PubMed] [CrossRef]
145. Bailly, C. (2022). Moving toward a new horizon for the aldose reductase inhibitor epalrestat to treat drug-resistant cancer. European Journal of Pharmacology, 931, 175191. https://doi.org/10.1016/j.ejphar.2022.175191. [Google Scholar] [PubMed] [CrossRef]
146. Scher, H. I., Beer, T. M., Higano, C. S., Anand, A., Taplin, M. E. et al. (2010). Antitumour activity of MDV3100 in castration-resistant prostate cancer: A phase 1–2 study. Lancet, 375(9724), 1437–1446. https://doi.org/10.1016/S0140-6736(10)60172-9. [Google Scholar] [PubMed] [CrossRef]
147. Fizazi, K., Scher, H. I., Molina, A., Logothetis, C. J., Chi, K. N. et al. (2012). Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: Final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. The Lancet Oncology, 13(10), 983–992. https://doi.org/10.1016/S1470-2045(12)70379-0. [Google Scholar] [PubMed] [CrossRef]
148. Byrns, M. C., Steckelbroeck, S., Penning, T. M. (2008). An indomethacin analogue, N-(4-chlorobenzoyl)-melatonin, is a selective inhibitor of aldo-keto reductase 1C3 (type 2 3alpha-HSD, type 5 17beta-HSD, and prostaglandin F synthasea potential target for the treatment of hormone dependent and hormone independent malignancies. Biochemical Pharmacology, 75(2), 484–493. [Google Scholar] [PubMed]
149. El-Kabbani, O., Dhagat, U., Hara, A. (2011). Inhibitors of human 20α-hydroxysteroid dehydrogenase (AKR1C1). The Journal of Steroid Biochemistry and Molecular Biology, 125(1–2), 105–111. [Google Scholar] [PubMed]
150. El-Kabbani, O., Scammells, P. J., Day, T., Dhagat, U., Endo, S. et al. (2010). Structure-based optimization and biological evaluation of human 20α-hydroxysteroid dehydrogenase (AKR1C1) salicylic acid-based inhibitors. European Journal of Medicinal Chemistry, 45(11), 5309–5317. https://doi.org/10.1016/j.ejmech.2010.08.052. [Google Scholar] [PubMed] [CrossRef]
151. Chatzopoulou, M., Pegklidou, K., Papastavrou, N., Demopoulos, V. J. (2013). Development of aldose reductase inhibitors for the treatment of inflammatory disorders. Expert Opinion on Drug Discovery, 8(11), 1365–1380. https://doi.org/10.1517/17460441.2013.843524. [Google Scholar] [PubMed] [CrossRef]
152. Hijazi, M. A., Gessner, A., El-Najjar, N. (2023). Repurposing of chronically used drugs in cancer therapy: A chance to grasp. Cancers, 15(12), 3199. https://doi.org/10.3390/cancers15123199. [Google Scholar] [PubMed] [CrossRef]
153. Ruiz, F. X., Parés, X., Farrés, J. (2021). Perspective on the structural basis for human aldo-keto reductase 1B10 inhibition. Metabolites, 11(12), 865. https://doi.org/10.3390/metabo11120865. [Google Scholar] [PubMed] [CrossRef]
154. Grewal, A. S., Bhardwaj, S., Pandita, D., Lather, V., Sekhon, B. S. (2016). Updates on aldose reductase inhibitors for management of diabetic complications and non-diabetic diseases. Mini Reviews in Medicinal Chemistry, 16(2), 120–162. [Google Scholar] [PubMed]
155. Rafiullah, M., Siddiqui, K. (2022). Pharmacological treatment of diabetic peripheral neuropathy: An update. CNS Neurological Disorders Drug Targets, 21(10), 884–900. https://doi.org/10.2174/1871527320666210303111939. [Google Scholar] [PubMed] [CrossRef]
156. Zhang, L., Zhang, H., Zhao, Y., Li, Z., Chen, S. et al. (2013). Inhibitor selectivity between aldo-keto reductase superfamily members AKR1B10 and AKR1B1: Role of Trp112 (Trp111). FEBS Letters, 587(22), 3681–3686. https://doi.org/10.1016/j.febslet.2013.09.031. [Google Scholar] [PubMed] [CrossRef]
157. Zhang, K. R., Zhang, Y. F., Lei, H. M., Tang, Y. B., Ma, C. S. et al. (2021). Targeting AKR1B1 inhibits glutathione de novo synthesis to overcome acquired resistance to EGFR-targeted therapy in lung cancer. Science Translational Medicine, 13(614), eabg6428. https://doi.org/10.1126/scitranslmed.abg6428. [Google Scholar] [PubMed] [CrossRef]
158. Zhang, J., Wang, N., Li, Q., Zhou, Y., Luan, Y. (2021). A two-pronged photodynamic nanodrug to prevent metastasis of basal-like breast cancer. Chemical Communications, 57(18), 2305–2308. https://doi.org/10.1039/D0CC08162K. [Google Scholar] [PubMed] [CrossRef]
159. Banala, V. T., Urandur, S., Sharma, S., Sharma, M., Shukla, R. P. et al. (2019). Targeted co-delivery of the aldose reductase inhibitor epalrestat and chemotherapeutic doxorubicin via a redox-sensitive prodrug approach promotes synergistic tumor suppression. Biomaterials Science, 7(7), 2889–2906. https://doi.org/10.1039/C9BM00221A. [Google Scholar] [PubMed] [CrossRef]
160. Shukla, K., Sonowal, H., Saxena, A., Ramana, K. V., Srivastava, S. K. (2017). Aldose reductase inhibitor, fidarestat regulates mitochondrial biogenesis via Nrf2/HO-1/AMPK pathway in colon cancer cells. Cancer Letters, 411, 57–63. https://doi.org/10.1016/j.canlet.2017.09.031. [Google Scholar] [PubMed] [CrossRef]
161. Saxena, A., Shoeb, M., Tammali, R., Ramana, K. V., Srivastava, S. K. (2014). Aldose reductase inhibition suppresses azoxymethane-induced colonic premalignant lesions in C57BL/KsJ-db/db mice. Cancer Letters, 355(1), 141–147. https://doi.org/10.1016/j.canlet.2014.09.006. [Google Scholar] [PubMed] [CrossRef]
162. Pandey, S. (2015). Aldose reductase inhibitor fidarestat as a promising drug targeting autophagy in colorectal carcinoma: A pilot study. Asian Pacific Journal of Cancer Prevention, 16(12), 4981–4985. https://doi.org/10.7314/APJCP.2015.16.12.4981. [Google Scholar] [PubMed] [CrossRef]
163. Tammali, R., Ramana, K. V., Singhal, S. S., Awasthi, S., Srivastava, S. K. (2006). Aldose reductase regulates growth factor-induced cyclooxygenase-2 expression and prostaglandin E2 production in human colon cancer cells. Cancer Research, 66(19), 9705–9713. https://doi.org/10.1158/0008-5472.CAN-06-2105. [Google Scholar] [PubMed] [CrossRef]
164. Kawamura, I., Lacey, E., Yamamoto, N., Sakai, F., Takeshita, S. et al. (1999). Ponalrestat, an aldose reductase inhibitor, inhibits cachexia syndrome induced by colon26 adenocarcinoma in mice. Anticancer Research, 19(5B), 4105–4111. [Google Scholar] [PubMed]
165. Cao, Z., Zhou, B., Chen, X., Huang, D., Zhang, X. et al. (2014). Statil suppresses cancer cell growth and proliferation by the inhibition of tumor marker AKR1B10. Anti-Cancer Drugs, 25(8), 930–937. https://doi.org/10.1097/CAD.0000000000000121. [Google Scholar] [PubMed] [CrossRef]
166. Kavitha, C. V., Nambiar, M., Ananda Kumar, C. S., Choudhary, B., Muniyappa, K. et al. (2009). Novel derivatives of spirohydantoin induce growth inhibition followed by apoptosis in leukemia cells. Biochemical Pharmacology, 77(3), 348–363. https://doi.org/10.1016/j.bcp.2008.10.018. [Google Scholar] [PubMed] [CrossRef]
167. Tokalı, F. S., Demir, Y., Demircioğlu, İ.H., Türkeş, C., Kalay, E. et al. (2022). Synthesis, biological evaluation, and in silico study of novel library sulfonates containing quinazolin-4(3H)-one derivatives as potential aldose reductase inhibitors. Drug Development Research, 83(3), 586–604. [Google Scholar] [PubMed]
168. Altıntop, M. D., Demir, Y., Türkeş, C., Öztürk, R. B., Cantürk, Z. et al. (2023). A new series of hydrazones as small-molecule aldose reductase inhibitors. Archiv der Pharmazie, 356(4), e2200570. https://doi.org/10.1002/ardp.v356.4. [Google Scholar] [CrossRef]
169. Han, Z., Li, J., Xu, Z., Su, Y., Wang, Y. et al. (2023). Design and synthesis of novel quinazolin-4(1H)-one derivatives as potent and selective inhibitors targeting AKR1B1. Archiv der Pharmazie, 356(4), e2200577. https://doi.org/10.1002/ardp.v356.4. [Google Scholar] [CrossRef]
170. Han, Z., Qi, G., Zhu, J., Zhang, Y., Xu, Y. et al. (2020). Novel 3,4-dihydroquinolin-2(1H)-one derivatives as dual inhibitor targeting AKR1B1/ROS for treatment of diabetic complications: Design, synthesis, and biological evaluation. Bioorganic Chemistry, 105, 104428. https://doi.org/10.1016/j.bioorg.2020.104428. [Google Scholar] [PubMed] [CrossRef]
171. Hamdi, A., Yaseen, M., Ewes, W. A., Bhat, M. A., Ziedan, N. I. et al. (2023). Development of new thiazolidine-2,4-dione hybrids as aldose reductase inhibitors endowed with antihyperglycaemic activity: Design, synthesis, biological investigations, and in silico insights. Journal of Enzyme Inhibition and Medicinal Chemistry, 38(1), 2231170. https://doi.org/10.1080/14756366.2023.2231170. [Google Scholar] [PubMed] [CrossRef]
172. Chen, W., Chen, X., Zhou, S., Zhang, H., Wang, L. et al. (2016). Design and synthesis of polyhydroxy steroids as selective inhibitors against AKR1B10 and molecular docking. Steroids, 110, 1–8. https://doi.org/10.1016/j.steroids.2016.03.004. [Google Scholar] [PubMed] [CrossRef]
173. Cousido-Siah, A., Ruiz, F. X., Fanfrlík, J., Giménez-Dejoz, J., Mitschler, A. et al. (2016). IDD388 polyhalogenated derivatives as probes for an improved structure-based selectivity of AKR1B10 inhibitors. ACS Chemical Biology, 11(10), 2693–2705. https://doi.org/10.1021/acschembio.6b00382. [Google Scholar] [PubMed] [CrossRef]
174. Díez-Dacal, B., Gayarre, J., Gharbi, S., Timms, J. F., Coderch, C. et al. (2011). Identification of aldo-keto reductase AKR1B10 as a selective target for modification and inhibition by prostaglandin A(1Implications for antitumoral activity. Cancer Research, 71(12), 4161–4171. https://doi.org/10.1158/0008-5472.CAN-10-3816. [Google Scholar] [PubMed] [CrossRef]
175. Endo, S., Matsunaga, T., Mamiya, H., Ohta, C., Soda, M. et al. (2009). Kinetic studies of AKR1B10, human aldose reductase-like protein: Endogenous substrates and inhibition by steroids. Archives of Biochemistry and Biophysics, 487(1), 1–9. https://doi.org/10.1016/j.abb.2009.05.009. [Google Scholar] [PubMed] [CrossRef]
176. Huang, L., He, R., Luo, W., Zhu, Y. S., Li, J. et al. (2016). Aldo-eto reductase family 1 member B10 inhibitors: Potential drugs for cancer treatment. Recent Patents on Anti-Cancer Drug Discovery, 11(2), 184–196. https://doi.org/10.2174/1574892811888160304113346. [Google Scholar] [PubMed] [CrossRef]
177. Endo, S., Matsunaga, T., Soda, M., Tajima, K., Zhao, H. -T. et al. (2010). Selective inhibition of the tumor marker AKR1B10 by antiinflammatory N-phenylanthranilic acids and glycyrrhetic acid. Biological and Pharmaceutical Bulletin, 33(5), 886–890. https://doi.org/10.1248/bpb.33.886. [Google Scholar] [PubMed] [CrossRef]
178. Prnová, M. Š., Račková, L., Kováčiková, L., Balleková, J., Viskupičová, J. et al. (2019). General toxicity assessment of the novel aldose reductase inhibitor cemtirestat. Interdisciplinary Toxicology, 12(3), 120–128. https://doi.org/10.2478/intox-2019-0014. [Google Scholar] [PubMed] [CrossRef]
179. Rattanaburee, T., Thongpanchang, T., Wongma, K., Tedasen., A., Sukpondma, Y. et al. (2019). Anticancer activity of synthetic (±)-kusunokinin and its derivative (±)-bursehernin on human cancer cell lines. Biomedicine and Pharmacotherapy, 117, 109115. https://doi.org/10.1016/j.biopha.2019.109115. [Google Scholar] [PubMed] [CrossRef]
180. Tedasen, A., Dokduang, S., Sukpondma, Y., Lailerd, N., Madla, S. et al. (2020). (-)-Kusunokinin inhibits breast cancer in N-nitrosomethylurea-induced mammary tumor rats. European Journal of Pharmacology, 882, 173311. https://doi.org/10.1016/j.ejphar.2020.173311. [Google Scholar] [PubMed] [CrossRef]
181. Tanawattanasuntorn, T., Rattanaburee, T., Thongpanchang, T., Graidist, P. (2022). Trans-(±)-Kusunokinin binding to AKR1B1 inhibits oxidative stress and proteins involved in migration in aggressive breast cancer. Antioxidants, 11(12), 2347. https://doi.org/10.3390/antiox11122347. [Google Scholar] [PubMed] [CrossRef]
182. Veeresham, C., Rama Rao, A., Asres, K. (2014). Aldose reductase inhibitors of plant origin. Phytotherapy Research, 28(3), 317–333. https://doi.org/10.1002/ptr.v28.3. [Google Scholar] [CrossRef]
183. Varma, S. D., Mikuni, I., Kinoshita, J. H. (1975). Flavonoids as inhibitors of lens aldose reductase. Science, 188(4194), 1215–1216. https://doi.org/10.1126/science.1145193. [Google Scholar] [PubMed] [CrossRef]
184. Reddy, G., Puppala, M., Akileshwari, C., Saraswat, M., Petrash, J. (2011). Inhibition of aldose reductase and sorbitol accumulation by dietary rutin. Current Science, 101, 1191–1197. [Google Scholar]
185. Seliger, J. M., Misuri, L., Maser, E., Hintzpeter, J. (2018). The hop-derived compounds xanthohumol, isoxanthohumol and 8-prenylnaringenin are tight-binding inhibitors of human aldo-keto reductases 1B1 and 1B10. Journal of Enzyme Inhibition and Medicinal Chemistry, 33(1), 607–614. https://doi.org/10.1080/14756366.2018.1437728. [Google Scholar] [PubMed] [CrossRef]
186. Zemanova, L., Hofman, J., Novotna, E., Musilek, K., Lundova, T. et al. (2015). Flavones inhibit the activity of AKR1B10, a promising therapeutic target for cancer treatment. Journal of Natural Products, 78(11), 2666–2674. https://doi.org/10.1021/acs.jnatprod.5b00616. [Google Scholar] [PubMed] [CrossRef]
187. Puppala, M., Ponder, J., Suryanarayana, P., Reddy, G. B., Petrash, J. M. et al. (2012). The isolation and characterization of β-glucogallin as a novel aldose reductase inhibitor from emblica officinalis. PLoS One, 7(4), e31399. https://doi.org/10.1371/journal.pone.0031399. [Google Scholar] [PubMed] [CrossRef]
188. Suyanto, E., Gorantla, J. N., Santi, M., Fatchiyah, F., Ketudat-Cairns, M. et al. (2024). Enzymatic synthesis of phenolic acid glucosyl esters to test activities on cholangiocarcinoma cells. Applied Microbiology and Biotechnology, 108(1), 69. https://doi.org/10.1007/s00253-023-12895-5. [Google Scholar] [PubMed] [CrossRef]
189. Lee, E. H., Song, D. G., Lee, J. Y., Pan, C. H., Um, B. H. et al. (2008). Inhibitory effect of the compounds isolated from Rhus verniciflua on aldose reductase and advanced glycation endproducts. Biological and Pharmaceutical Bulletin, 31(8), 1626–1630. https://doi.org/10.1248/bpb.31.1626. [Google Scholar] [PubMed] [CrossRef]
190. Qiu, S., Sun, H., Zhang, A. H., Xu, H. Y., Yan, G. L. et al. (2014). Natural alkaloids: Basic aspects, biological roles, and future perspectives. Chinese Journal of Natural Medicines, 12(6), 401–406. https://doi.org/10.1016/S1875-5364(14)60063-7. [Google Scholar] [PubMed] [CrossRef]
191. Kubo, M., Matsuda, H., Tokuoka, K., Kobayashi, Y., Ma, S. et al. (1994). Studies of anti-cataract drugs from natural sources. I. effects of a methanolic extract and the alkaloidal components from Corydalis tuber on in vitro aldose reductase activity. Biological and Pharmaceutical Bulletin,17(3), 458–459. https://doi.org/10.1248/bpb.17.458. [Google Scholar] [PubMed] [CrossRef]
192. Lee, J., Sohn, E. J., Yoon, S. W., Kim, C. G., Lee, S. et al. (2017). Anti-metastatic effect of dehydrocorydaline on H1299 non-small cell lung carcinoma cells via inhibition of matrix metalloproteinases and B cell lymphoma 2. Phytotherapy Research, 31(3), 441–448. https://doi.org/10.1002/ptr.v31.3. [Google Scholar] [CrossRef]
193. Goel, A. (2023). Current understanding and future prospects on berberine for anticancer therapy. Chemical Biology and Drug Design, 102(1), 177–200. https://doi.org/10.1111/cbdd.v102.1. [Google Scholar] [CrossRef]
194. Zych, M., Wojnar, W., Kielanowska, M., Folwarczna, J., Kaczmarczyk, S. I. (2020). Effect of berberine on glycation, aldose reductase activity, and oxidative stress in the lenses of streptozotocin-induced diabetic rats in vivo–A preliminary study. International Journal of Molecular Sciences, 21(12), 4278. https://doi.org/10.3390/ijms21124278. [Google Scholar] [PubMed] [CrossRef]
195. Kamran, S., Sinniah, A., Abdulghani, M. A. M., Alshawsh, M. A. (2022). Therapeutic potential of certain terpenoids as anticancer agents: A scoping review. Cancers, 14(5), 1100. https://doi.org/10.3390/cancers14051100. [Google Scholar] [PubMed] [CrossRef]
196. Patel, S. M., Nagulapalli Venkata, K. C., Bhattacharyya, P., Sethi, G., Bishayee, A. (2016). Potential of neem (Azadirachta indica L.) for prevention and treatment of oncologic diseases. Seminars in Cancer Biology, 40–41, 100–115. [Google Scholar] [PubMed]
197. Kishore, T. K. K., Ganugula, R., Gade, D. R., Reddy, G. B., Nagini, S. (2016). Gedunin abrogates aldose reductase, PI3K/Akt/mToR, and NF-κB signaling pathways to inhibit angiogenesis in a hamster model of oral carcinogenesis. Tumour Biology, 37(2), 2083–2093. https://doi.org/10.1007/s13277-015-4003-0. [Google Scholar] [PubMed] [CrossRef]
198. Tanagala, K. K. K., Baba, A. B., Kowshik, J., Reddy, G. B., Nagini, S. (2018). Gedunin, a neem limonoid in combination with epalrestat inhibits cancer hallmarks by attenuating aldose reductase-driven oncogenic signaling in SCC131 oral cancer cells. Anti-Cancer Agents in Medicinal Chemistry, 18(14), 2042–2052. [Google Scholar] [PubMed]
199. Nivetha, R., Arvindh, S., Baba, A. B., Gade, D. R., Gopal, G. et al. (2022). Nimbolide, a neem limonoid, inhibits angiogenesis in breast cancer by abrogating aldose reductase mediated IGF-1/PI3K/Akt signalling. Anti-Cancer Agents in Medicinal Chemistry, 22(14), 2619–2636. https://doi.org/10.2174/1871520622666220204115151. [Google Scholar] [PubMed] [CrossRef]
200. Kawanishi, K., Ueda, H., Moriyasu, M. (2003). Aldose reductase inhibitors from the nature. Current Medicinal Chemistry, 10(15), 1353–1374. https://doi.org/10.2174/0929867033457304. [Google Scholar] [PubMed] [CrossRef]
201. Park, J., Ngo, T. H., Paudel, S. B., Kil, Y. S., Ryoo, G. H. et al. (2024). Angular dihydropyranocoumarins from the flowers of Peucedanum japonicum and their aldo-keto reductase inhibitory activities. Phytochemistry, 219, 113974. https://doi.org/10.1016/j.phytochem.2024.113974. [Google Scholar] [PubMed] [CrossRef]
202. Kim, T. W. (2023). Nodakenin induces ROS-dependent apoptotic cell death and ER stress in radioresistant breast cancer. Antioxidants, 12(2), 492. https://doi.org/10.3390/antiox12020492. [Google Scholar] [PubMed] [CrossRef]
203. Parama, D., Girisa, S., Khatoon, E., Kumar, A., Alqahtani, M. S. et al. (2022). An overview of the pharmacological activities of scopoletin against different chronic diseases. Pharmacological Research, 179, 106202. https://doi.org/10.1016/j.phrs.2022.106202. [Google Scholar] [PubMed] [CrossRef]
204. Muthenna, P., Suryanarayana, P., Gunda, S. K., Petrash, J. M., Reddy, G. B. (2009). Inhibition of aldose reductase by dietary antioxidant curcumin: Mechanism of inhibition, specificity, and significance. FEBS Letters, 583(22), 3637–3642. https://doi.org/10.1016/j.febslet.2009.10.042. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools