
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Silencing of the long non-coding RNA LINC00265 triggers autophagy and apoptosis in lung cancer by reducing protein stability of SIN3A oncogene
1 Department of Pulmonary Oncology, Affiliated Hospital of Guangdong Medical University, Zhanjiang, 524001, China
2 Department of Thoracic Surgery, Affiliated Hospital of Guangdong Medical University, Zhanjiang, 524001, China
3 Department of Orthopedics, Affiliated Hospital of Guangdong Medical University, Zhanjiang, 524001, China
4 Guangdong Provincial Key Laboratory of Autophagy and Major Chronic Non-Communicable Diseases, Affiliated Hospital of Guangdong Medical University, Zhanjiang, 524001, China
* Corresponding Authors: GUANGHUA CHEN. Email: ; WENMEI SU. Email:
Oncology Research 2024, 32(7), 1185-1195. https://doi.org/10.32604/or.2023.030771
Received 21 April 2023; Accepted 22 August 2023; Issue published 20 June 2024
A correction of this article was approved in:
Correction: Silencing of the long non-coding RNA LINC00265 triggers autophagy and apoptosis in lung cancer by reducing protein stability of SIN3A oncogene
Read correction
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: Long non-coding RNAs are important regulators in cancer biology and function either as tumor suppressors or as oncogenes. Their dysregulation has been closely associated with tumorigenesis. LINC00265 is upregulated in lung adenocarcinoma and is a prognostic biomarker of this cancer. However, the mechanism underlying its function in cancer progression remains poorly understood. Methods: Here, the regulatory role of LINC00265 in lung adenocarcinoma was examined using lung cancer cell lines, clinical samples, and xenografts. Results: We found that high levels of LINC00265 expression were associated with shorter overall survival rate of patients, whereas knockdown of LINC00265 inhibited proliferation of cancer cell lines and tumor growth in xenografts. Western blot and flow cytometry analyses indicated that silencing of LINC00265 induced autophagy and apoptosis. Moreover, we showed that LINC00265 interacted with and stabilized the transcriptional co-repressor Switch-independent 3a (SIN3A), which is a scaffold protein functioning either as a tumor repressor or as an oncogene in a context-dependent manner. Silencing of SIN3A also reduced proliferation of lung cancer cells, which was correlated with the induction of autophagy. These observations raise the possibility that LINC00265 functions to promote the oncogenic activity of SIN3A in lung adenocarcinoma. Conclusions: Our findings thus identify SIN3A as a LINC00265-associated protein and should help to understand the mechanism underlying LINC00265-mediated oncogenesis.Keywords
Supplementary Material
Supplementary Material FileLung cancer is one of the most common malignant tumors and represents the leading cause of cancer-related deaths worldwide, with non-small cell lung cancer (NSCLC) accounting for 85% of lung cancer cases [1,2]. Despite advances in diagnosis and combined treatments, the prognosis of lung cancer patients remains unsatisfied. The 5-year survival of lung cancer is about 19% [3]. Therefore, there is an urgent need to improve the understanding of this cancer and to identify early diagnostic biomarkers and new therapeutic targets.
There is increasing evidence suggesting that long non-coding RNAs (lncRNAs) play important roles in lung cancer development by acting as both tumor suppressors and oncogenes [4]. Many lncRNAs are frequently dysregulated in cancers and associated with cancer progression [5,6]. Thus, they are promising biomarkers and therapeutic targets for lung diseases [7]. LINC00265 was first identified in lung adenocarcinoma (LUAD) as an upregulated lncRNA and a prognostic biomarker of this cancer [8], suggesting a potentially important role of this lncRNA in cancer development. Indeed, LINC00265 has been shown to promote cell proliferation and viability in a number of cancers by interacting with other non-coding RNAs and key signaling pathways such as Epidermal Growth Factor (EGF) and Wnt/β-catenin. It is upregulated in colorectal cancer (CRC) and associated with poor prognosis [9–11]. Higher levels of LINC00265 were also found in the serum of patients with acute myeloid leukemia (AML), which is correlated with lower overall survival rate because LINC00265 contributes to migration and invasion of tumor cells by regulating PI3K/AKT and signal transducer and activator of transcription 3 (STAT3) signaling [12–14]. Therefore, circulating LINC00265 can be used as a biomarker for diagnosis and therapeutic monitoring of AML [15]. However, the mechanisms underlying LINC00265 function in LUAD progression remain elusive.
The transcriptional corepressor SIN3A is a member of the SIN3 family and acts as a scaffold protein in the histone deacetylase (HDAC) complex. It can function either as a tumor suppressor or as an oncogene, depending on its biochemical interaction with specific protein partners, genetic interaction with other genes or regulation of target genes [16–18]. It has been shown that SIN3A inhibits cell invasion and is downregulated in cancers, thus functioning as a tumor suppressor [19,20]. Recently, there is also evidence indicating that SIN3A promotes the oncogenic potential of STAT3 and represses the transcription of tumor suppressor genes; interfering with its expression leads to cell death of anaplastic large-cell lymphoma through transcriptional activation of tumor suppressor genes [21].
In this work, we investigated the functional role of LINC00265 in regulating LUAD progression. Using clinical samples and NSCLC cell lines, we showed that the expression of LINC00265 is upregulated in lung cancer cells. Functional analyses indicated that knockdown of LINC00265 reduces tumor development and prevents NSCLC cell migration and invasion. Interestingly, we found that LINC00265 interacts with the transcription factor SIN3A to regulate its expression. Moreover, inhibiting the function of LINC00265 and SIN3A induced autophagy and apoptosis of NSCLC cells. These results indicate that LINC00265 plays an important role in LUAD development. They provide insights into the mechanism underlying LINC00265 activity in lung tumorigenesis, and help to identify potential targets for lung cancer therapy.
Human lung cancer tissues and paired normal tissues were collected at the Department of Thoracic Surgery of the Affiliated Hospital of Guangdong Medical University (China). Primary tumor sites and the corresponding normal tissues were dissected and subjected to RNA extraction. All participants provided written informed consent. This research was approved by the Ethical Committee of the Affiliated Hospital of Guangdong Medical University (Approval Number: YS2022083).
Human lung cell lines with different metastatic potentials (H1299, PC9, H838, A549, H1650, and H1975) and normal human bronchial epithelial (HBE) cells were purchased from Kobio Biology (Nanjing, China). H1299, PC9, H838, H1650, H1975 and HBE cells were maintained in RPMI 1640 supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin-streptomycin in a humidified incubator with 5% CO2 at 37°C. For A549 cells, RPMI 1640 was replaced by DMEM (Gibco).
Small interfering RNAs (siRNAs) and small hairpin RNAs (shRNAs) were provided by Genema (Suzhou, China). The targeting sequences are listed in Suppl. Table 1. Cell transfection was performed using the Lipofectamine® RNAiMAX reagent (Invitrogen, Carlsbad, CA, USA).
Total RNAs were prepared from tissues and cells using the Trizol reagent (TaKaRa, Japan). Cytoplasmic and nuclear RNAs from lung cancer cell lines were isolated using the RNA Subcellular Isolation kit (Active Motif, Shanghai, China). After reverse transcription using the PrimeScript RT reagent kit (TaKaRa, Japan), qPCR was performed using the Roche LightCycler® 480 System and TB Green Premix Ex Taq II kit (TakaRa, Japan). Primer sequences for qPCR are listed in Suppl. Table 2. The 2-ΔΔCt method was used to estimate relative expression levels, with GAPDH as a loading control.
Proteins were extracted using RIPA buffer containing protease inhibitors and quantified by bicinchoninic acid (BCA) analysis (Beyotime, China). They were separated by 10% SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, USA). Non-specific antibody binding was blocked with 5% skimmed milk, and the membranes were incubated with primary antibodies (Suppl. Table 3) at 4°C overnight, followed by incubation with peroxidase (HRP)-conjugated secondary antibody. After several washes, protein bands were visualized using Tanon 5200 chemiluminescence imaging system (Shanghai, China) and analyzed using ImageJ Software.
Cell proliferation and transwell assays
For proliferation assay, cells were seeded in 96-well plates with each well containing 1000 cells in 100 μL of culture medium and cultured at 37°C for an appropriate period. After adding 10 μL of (water soluble tetrazolium salt-1) WST-1 solution (Beyotime Biotechnology, Shanghai, China) to each well, the plate was further incubated for 1 h in the dark. The optical density was measured at 450 and 630 nm with a microplate reader. Transwell migration and invasion assays were performed as described previously [5].
Cells were seeded into a 6-well plate at a density of 200 cells per well and cultured for 10 to 14 days. Colonies were washed twice with PBS and fixed with methanol for 15 min. They were stained with 0.1% crystal violet for 20 min and imaged for statistical analysis.
Cells were collected by treatment with free Ethylenediaminetetraacetic acid (EDTA-free) trypsin and resuspended in 100 μL of 1 × binding buffer. After staining with 5 μL Annexin V-FITC and 5 μL propidium iodide (PI)(BD Biosciences, USA) for 15 min in the dark, cells were sorted by flow cytometry and analyzed using the BD FACSDiva6.1 software (BD Biosciences).
Five-week-old female athymic BALB/c nude mice were maintained under specific pathogen free (SPF) conditions at the animal care facility of the Experimental Animal Center of Guangxi University of Traditional Chinese Medicine. This animal experiment was approved by the Ethics Committee of Affiliated Hospital of Guangdong Medical University (Approval Number: GDY2202217). For xenograft models, H1299 cells were stably transfected with shLINC00265, or with a negative control. About 2 × 106 cells in 200 μL of sterile PBS were injected subcutaneously into the axilla of each animal. Xenografts were measured by a digital caliper every 3 days and their volumes were calculated using the following equation: volume = (length × width2)/2.
RNA immunoprecipitation (RIP) assay
The interaction between LINC00265 and SIN3A was analyzed using the EZ-Magna RIP™ RNA-Binding Protein Immunoprecipitation kit (Millipore, USA) and an antibody against SIN3A. The specificity was assayed using anti-SNRNP70 antibody and control IgG (Millipore, USA). Immunoprecipitated RNAs were reverse transcribed and analyzed by qPCR.
This was performed using a tandem fluorescent-tagged LC3 construct that produces GFP-LC3 and RFP-LC3 punctae [22]. H1299 and A549 cells were first transfected with siRNAs using the Lipofectamine® RNAiMAX Reagent and cultured for 48 h. They were then washed with phosphate-buffered saline (PBS) and transfected with the RFP-GFP-LC3B plasmid (Invitrogen, Carlsbad, CA, USA). The formation of fluorescent punctae was visualized by fluorescence microscopy.
Data were analyzed using GraphPad Prism 8.0 and presented as means ± standard deviation (SD). Student’s t-test was used for statistical analyses, and p < 0.05 was considered statistically significant. All experiments were repeated at least three times.
Upregulation of LINC00265 predicts poor prognosis in human LUAD
To identify lncRNAs that may be potentially implicated in lung cancer progression, we detected lncRNA expression profifiles using available RNA-seq. The analysis indicated that LINC00265 was significantly upregulated in lung cancer tissues, compared with adjacent tissues (Fig. 1A). This observation is consistent with the clinical information from The Cancer Genome Atlas (TCGA) database. Accordingly, analysis of ROC (receiver operating characteristic) curve showed a high degree of separability in LINC00265 expression between tumor and normal tissues, with AUC (area under the ROC curve) higher than 0.82 (Fig. 1B). Furthermore, Kaplan-Meier analysis indicated that lung cancer patients with high levels of LINC00265 expression had shorter overall survival (OS), suggesting a poor prognosis (Fig. 1C). We next examined LINC00265 expression in different NSCLC cell lines and in lung cancer tissue by RT-qPCR analysis. Compared with transformed normal bronchial epithelial cells (HBE), the expression of LINC00265 was significantly upregulated in the H1299, PC9, H838, and A549 lines, but without changes in H1650 and H1975 (Fig. 1D). The increased expression of LINC00265 in lung cancers was further confirmed using different clinical samples, as shown by analyzing 6 paired lung cancer tissues and adjacent normal tissues (Fig. 1E). Additionally, analysis of LINC00265 subcellular distribution using H1299 and A549 cells indicated a predominant presence in the cytoplasm, suggesting that it should mainly function in this cellular compartment (Fig. 1F). Taken together, these results suggest that LINC00265 may play a role in the tumorigenesis of lung cancer and may represent a novel prognostic biomarker.
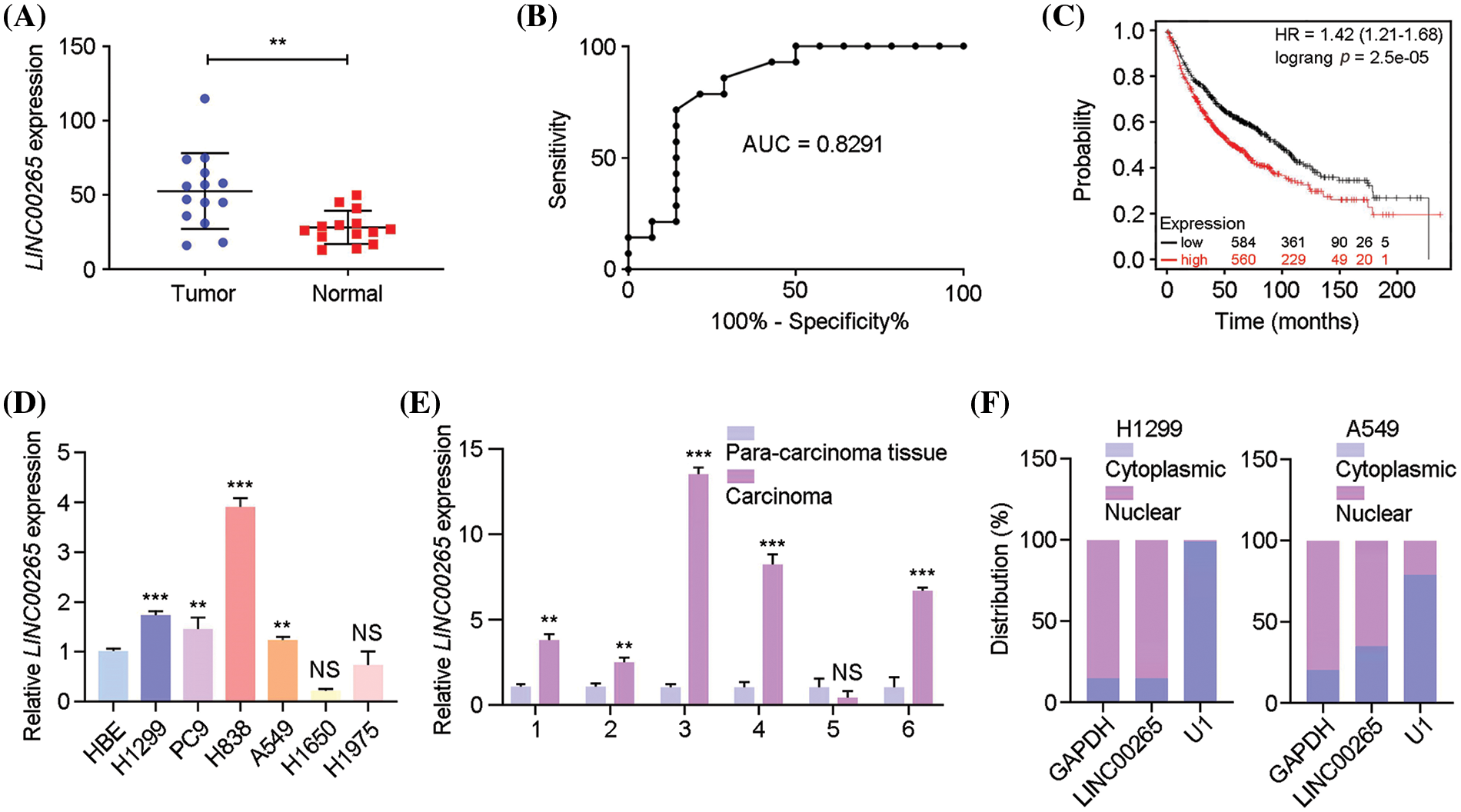
Figure 1: Upregulation of LINC00265 in lung cancers and cell lines. (A) Comparison of the expression of LINC00265 between LUAD and noncancerous lung tissues (p < 0.05). (B) ROC curve analysis shows the separability of LINC00265 expression between normal and tumor tissues (AUC = 0.8291). (C) Kaplan-Meier survival analysis shows significant association of LINC00265 expression levels with overall survival rates. (D) RT-qPCR analysis of LINC00265 expression in different NSCLC cell lines. The expression of LINC00265 in HBE cells was normalized to GAPDH and used as a reference. Data were obtained from three independent experiments (**p < 0.01; ***p < 0.001; NS, not significant). (E) The relative expression of LINC00265 was determined in 6 pairs of LUAD tissues and para-cancer tissues by qRT-PCR (**p < 0.01; ***p < 0.001; NS, not significant). (F) Analysis of cytoplasmic and nuclear accumulation of LINC00265 in H1299 and A549 cells.
Inhibition of LINC00265 reduces cell proliferation and tumor growth
The activity of LINC00265 in LUAD is poorly understood. We first examined the effects of its knockdown in NSCLC cell lines by small interfering RNA (siRNA) approach. Plasmids harbor specific siRNAs targeting LINC00265 (siLINC00265-1 and siLINC00265-2) were transfected into H1299, PC9, H838, and A549 cells. RT-qPCR analysis showed that both LINC00265-specific siRNAs significantly decreased the expression of LINC00265 in different NSCLC cell lines, when compared with a siRNA (siCtrl) that does not target any sequence (Suppl. Fig. 1). Both WST and colony formation assays indicated that LINC00265 knockdown significantly inhibited cell proliferation (Figs. 2A–2C). The tumor-inhibiting effects of LINC00265 knockdown were further confirmed using a subcutaneous xenograft model. For this purpose, we designed a short hairpin RNA (shRNA) targeting LINC00265 (shLINC00265) and a control shRNA (shCtrl). H1299 cells were infected with slow viruses to stably express the shRNAs. We found that the multiplicity of infection at 100 particles per cells was most efficient to inhibit LINC00265 expression and cell proliferation (Suppl. Fig. 2). Following implantation of H1299 cells expressing shCtrl or shLINC00265 into nude mice, tumor size in the xenografts was examined every 3 days to monitor tumor size. Consistent with the observation in NSCLC cell lines, knockdown of LINC00265 significantly reduced tumor growth from day 18 onwards (Figs. 2D–2F). In addition, the inhibition of tumor growth is correlated with a decreased LINC00265 expression level and reduced cell proliferation, as determined by immunocytochemical staining of the cell proliferation antigen Ki-67, autophagy associated indicators and apoptosis-related targets (Suppl. Fig. 3). These results indicate that inhibition of LINC00265 function can efficiently prevent the progression of lung cancers.
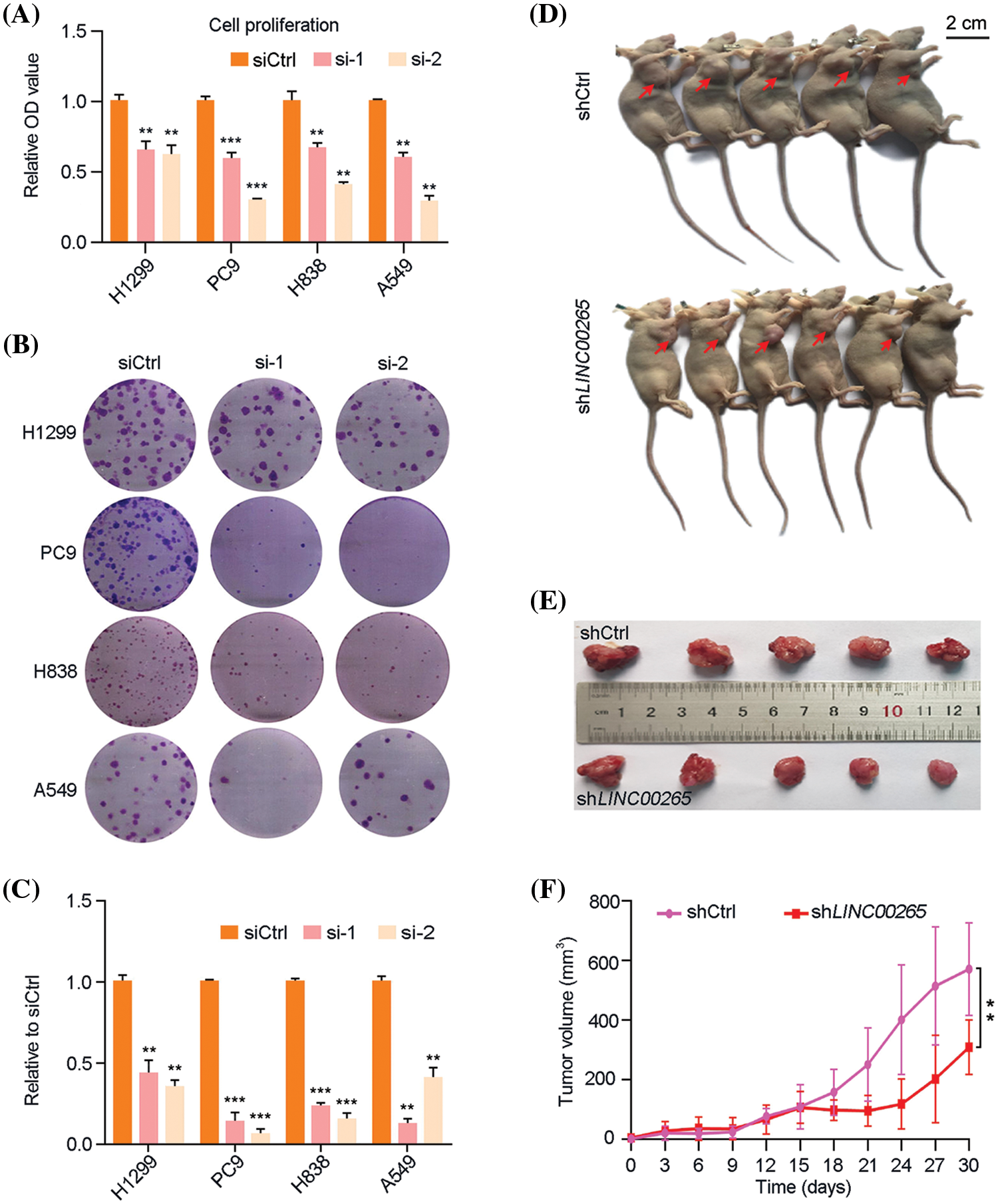
Figure 2: Knockdown of LINC00265 inhibits NSCLC cell proliferation and tumor growth. (A) WST assay shows reduced proliferation of H1299, PC9, H838 and A549 cells following LINC00265 knockdown. For each cell line, the value in siCtrl-transfected conditions is set to 1 as a reference (**p < 0.01; ***p < 0.001). (B, C) Reduced colony formation following LINC00265 knockdown in indicated NSCLC cell lines. (D, E) Representative images shows that knockdown of LINC00265 reduces tumor growth in xenografts. Images were taken at day 30 after implantation. (F) Statistical analysis of tumor growth overtime. Data were obtained using 5 animals for each time point (**p < 0.01).
Knockdown of LINC00265 inhibits migration and invasion of NSCLC cells
Transwell migration and invasion assays were used to evaluate how cell migration and invasion were affected by LINC00265-knockdown in NSCLC cell lines. We found that they were significantly decreased after LINC00265 knockdown in H1299, PC9, H838, and A549 cells (Figs. 3A–3D). This suggests that LINC00265 plays a role in the migration and invasion of NSCLC cells and that it may function as an oncogene to promote tumorigenesis or cancer progression. We then used flow cytometry to evaluate how LINC00265 knockdown affects cell viability. The results showed that inhibition of LINC00265 function led to increased apoptosis in different NSCLC cell lines (Figs. 3E–3H). Collectively, our functional analyses revealed an important role of LINC00265 in the development of lung cancer.
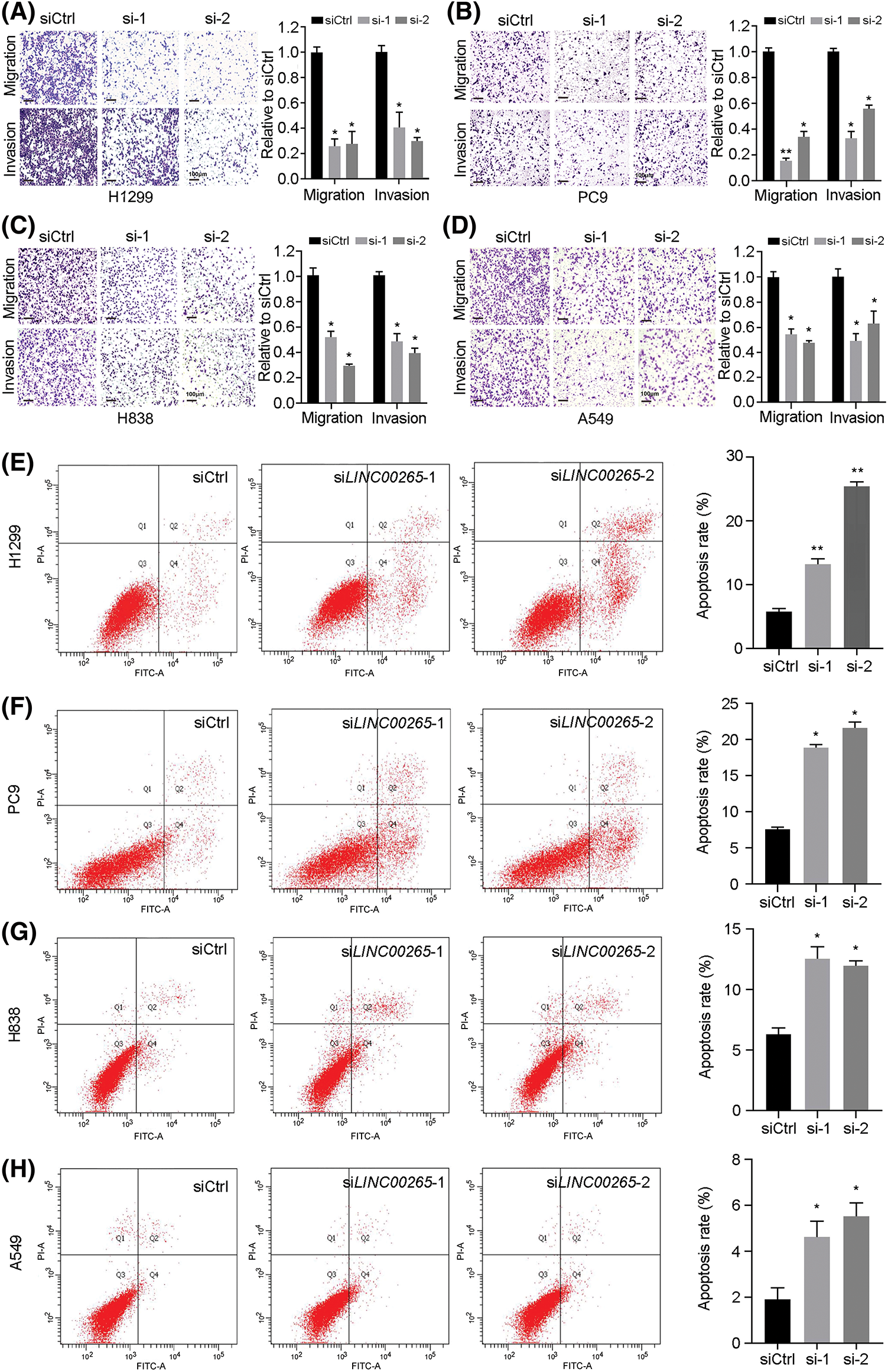
Figure 3: Silencing of LINC00265 inhibits migration and invasion of NSCLC cells and induces apoptosis. (A–D) Transwell assays show the effects of LINC00265 knockdown on NSCLC migration and invasion. The value in siCtrl-treated conditions is set to 1 as a reference. Data are the mean ± s.e.m. from three independent experiments (*p < 0.05; **p < 0.01). Scale bar, 100 μm. (E–H) Flow cytometry analyses show that knockdown of LINC00265 increases apoptosis rate in NSCLC cells (*p < 0.05; **p < 0.01).
Induction of autophagy in NSCLC cell lines following LINC00265-knockdown
Since there exist potential pathways linking apoptosis and autophagy in cancer [23], we examined how LINC00265-knockdown affects autophagy in NSCLC cell lines. The expression of p62 is constantly degraded by non-selective autophagy during the formation of autophagosomes and the maturation of autophagolysosomes [24]. Its increased level results in autophagy defects, while its decreased expression promotes autophagy [25]. During the initiation of autophagy, LC3B is recruited to autophagosomal membranes and can be used as a protein marker for the formation of autophagosomes [26]. Beclin-1 is also involved in autophagosome formation and functions to inhibit tumor growth [6]. The expression of other proteins, such as mTOR, P70 and AMPK (AMP-activated protein kinase), is also associated with autophagy in cell survival and cell death [27].
By western blot analysis of autophagy-related signature proteins, we found that expression levels of mTOR, p-mTOR, P70, p-P70 and p62 were decreased in H1299, PC9 and H838 cells at 72h after LINC00265-knockdown, concomitantly with increased expression levels of AMPK, p-AMPK, Beclin1 and LC3B (Fig. 4A, Suppl. Fig. 4). In animal models, autophagy-related proteins showed similar expression as in NSCLC cell lines (Suppl. Fig. 5). These results are reminiscent of autophagy activation, suggesting that LINC00265-knockdown can trigger autophagy in lung cancer cells. Furthermore, we examined the effects of LINC00265-knockdown on autophagy flux by fluorescence microscopy to monitor how this induces the formation of autolysosomes. H1299 and A549 cells were infected with a lentiviral vector harboring a tandem fluorescent-tagged LC3 to monitor the formation of GFP-LC3 and RFP-LC3 punctae. Since the GFP signal is quenched in acidic lysosomes, RFP punctae will be displayed in cells containing autolysosomes, which indicate autophagy termination. There were very few red and orange punctae in uninfected H1299 and A549 cells. However, silencing of LINC00265 significantly increased the number of red and orange punctae (Fig. 5A). To further confirm the induction of autophagy, we treated LINC00265-knockdwon cells with chloroquine (CQ), a known inhibitor of autophagy, because it can prevent the fusion of autophagosomes with lysosomes [28,29]. Therefore, a further increase in LC3B protein level is correlated with enhanced autophagy flux [30]. As expected, western blot analysis showed that knockdown of LINC00265 in H1299 and A549 cells followed by treatment with CQ (10 µM) further increased LC3B expression (Figs. 5B, 5C). Thus, we can conclude that inhibiting the function of LINC00265 leads to activation of autophagy in NSCLC cells.
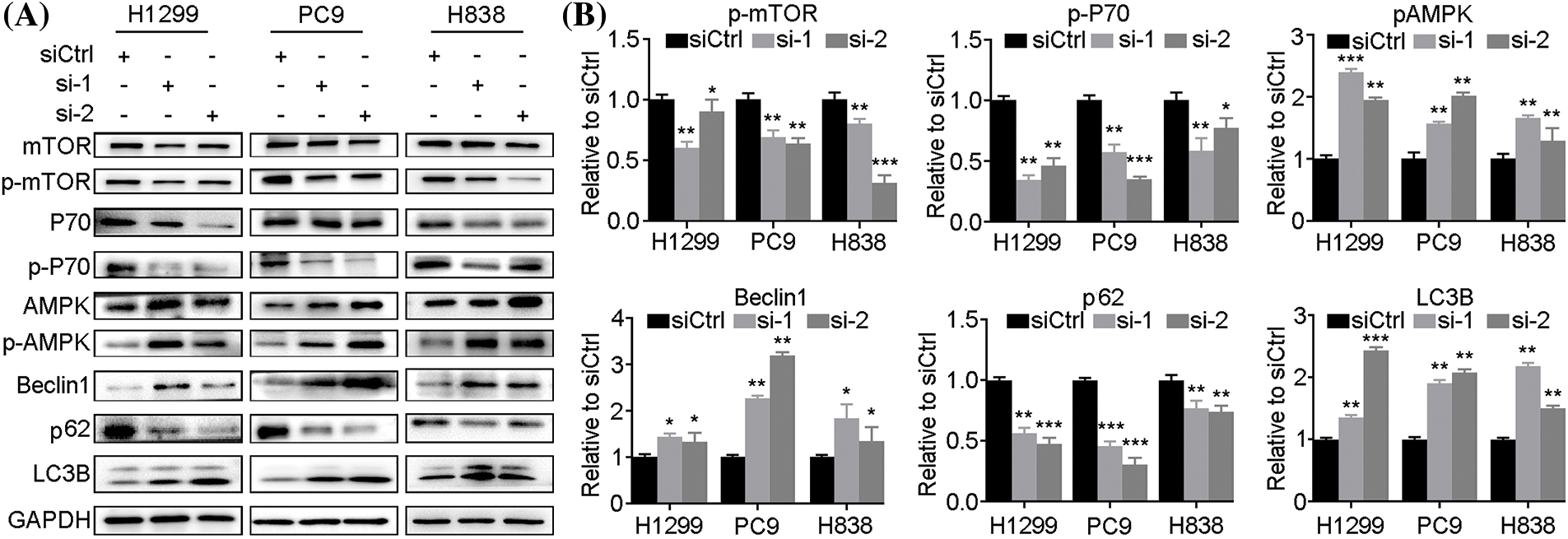
Figure 4: Knockdown of LINC00265 induces autophagy. (A) Western blot analyses of autophagy-related proteins following LINC00265 knockdown in NSCLC cell lines. (B) Quantification of western blots shows the decreased expression of p-mTOR, p-P70 and p62, but increased expression of p-AMPK, Beclin1 and LC3B, indicating autophagy activation. The value in siCtrl-treated conditions is set to 1 as a reference (*p < 0.05; **p < 0.01; ***p < 0.001).
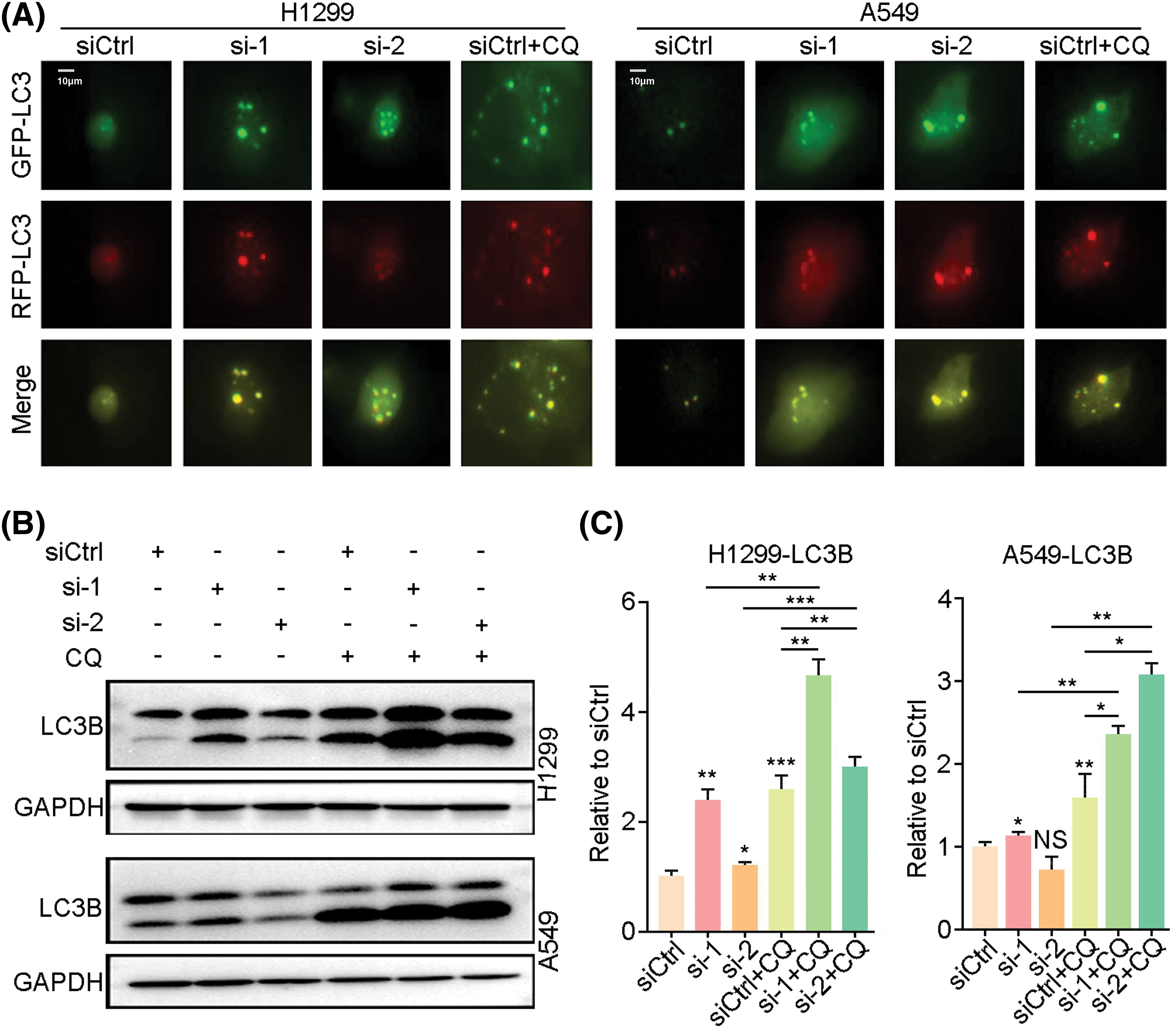
Figure 5: Knockdown of LINC00265 enhances autophagy flux. (A) Representative images show increased formation of autophagosomes after knockdown of LINC00265 in H1299 and A549 cells. Scale bar, 10 μm. (B) Western blot analyses of LC3B expression following knockdown of LINC00265 in H1299 and A549 cells treated with CQ. (C) Quantification of western blots. Increased LC3B expression in CQ-treated conditions suggests enhanced autophagy flux (*p < 0.05; **p < 0.01; ***p < 0.001; NS, not significant).
LINC00265 interacts with SIN3A protein and regulates its stability
To gain further insight into the mechanism underlying LINC00265 in LUAD, we used bioinformatics tools (GeneCards) to predict its interacting proteins. Among different possible candidates, the transcriptional co-repressor SIN3A protein was particularly interesting because its implication in tumorigenesis [21,31,32]. We further verified the physical interaction between LINC00265 and SIN3A by RIP-qPCR. The results clearly showed that LINC00265 transcripts were significantly enriched by SIN3A-specific antibody (Fig. 6A), suggesting complex formation between LINC00265 and SIN3A. To determine the biological relevance of this interaction, we analyzed SIN3A protein expression by western blot and found a decrease in LINC00265-knockdown cells (Figs. 6B, 6C). In light of the binding of LINC00265 with SIN3A protein, these results indicate that it may function in regulating the stability of SIN3A in NSCLC cells.
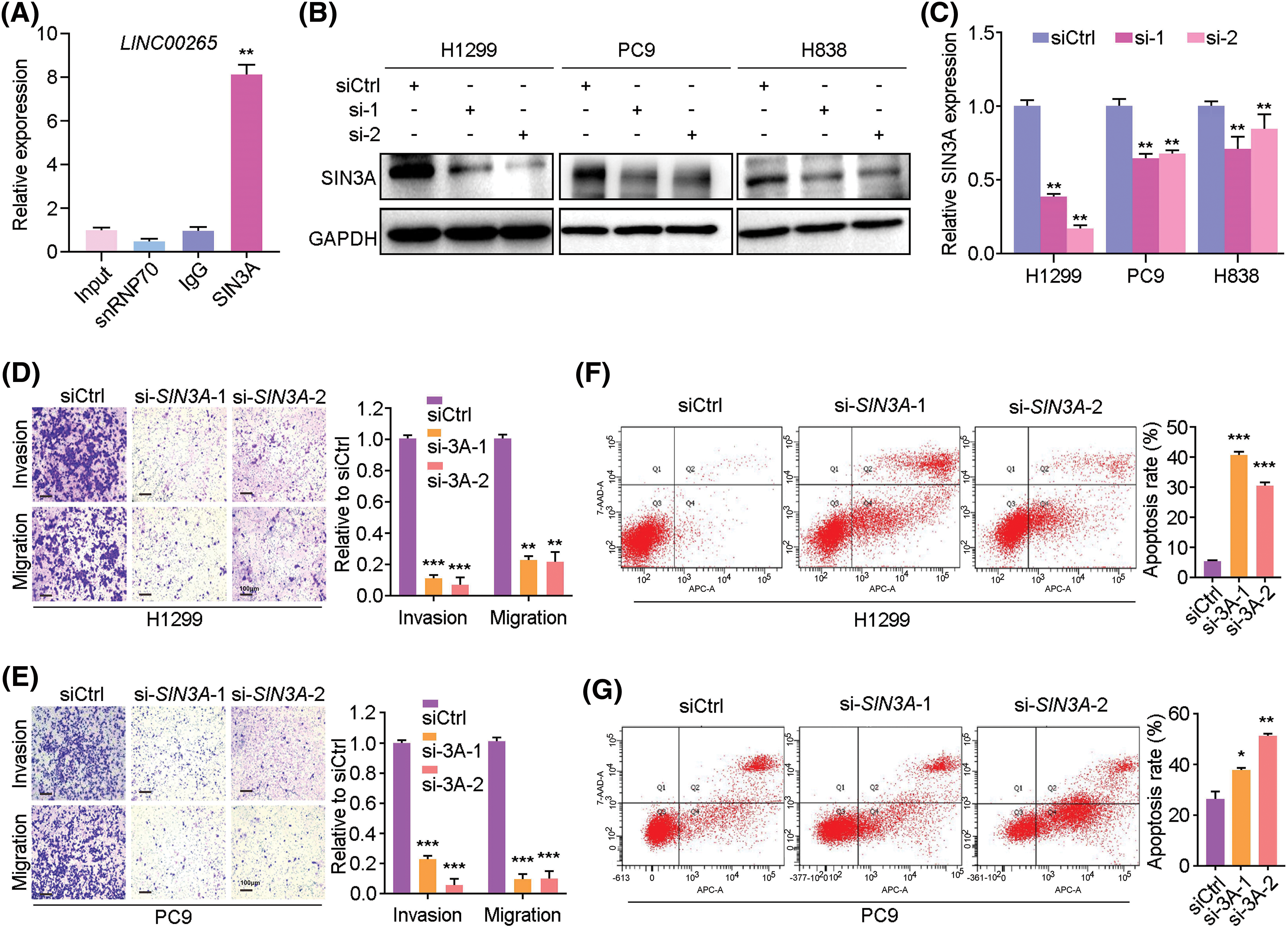
Figure 6: Interaction between LINC00265 and SIN3A protein. (A) RIP-qPCR assay shows the interaction between LINC00265 and SIN3A protein (**p < 0.01). Samples were subjected to RNA purification using anti-SIN3A antibody, anti-SNRNP70 or control IgG. The value from input sample is set to 1. (B, C) Western blot and quantification show reduced SIN3A protein levels after knockdown of LINC00265 in NSCLC cells (**p < 0.01). (D, E) Transwell assays show that knockdown of SIN3A reduces migration and invasion of H1299 and PC9 cells. Data are expressed as the mean ± s.e.m. from three independent experiments (**p < 0.01; ***p < 0.001). Scale bar, 100 μm. (F, G) Flow cytometry analysis shows that knockdown of SIN3A increases apoptosis rate in NSCLC cells. Data are the mean ± s.e.m. from three independent experiments (*p < 0.05; **p < 0.01; ***p < 0.001).
We then examined the function of SIN3A in cell proliferation using two different siRNAs. The knockdown efficiency was verified by qRT-PCR in H1299, PC9, H838 and A549 cells, as well as by western blotting and WST assay in H1299 and PC9 cells (Suppl. Fig. 6). Analysis by transwell assays indicated that silencing of SIN3A in NSCLC cells strongly reduced cell invasion and migration, when compared with the control siRNA (Figs. 6D, 6E). This was correlated with an increased apoptosis, as determined by cytometry analysis (Figs. 6F, 6G).
Knockdown of SIN3A induces autophagy in NSCLC cells
Since LINC00265 regulates autophagy, its interaction with SIN3A raises the possibility that the latter may have a similar function. We thus examined the effects of SIN3A knockdown on autophagy by western blot. The results showed that protein levels of p-AMPK, Beclin1 and LC3B were increased, whereas the expression of p-mTOR, p-P70 and p62 was reduced in H1299 and PC9 cells (Fig. 7). Therefore, this expression profile of autophagy-related proteins is similar as observed in LINC00265-knockdown cells, suggesting that LINC00265 and SIN3A may function together to regulate lung cancer development. Altogether, these observations identify LINC00265 as an interaction partner of SIN3A in LUAD. It is possible that LINC00265 stabilizes SIN3A protein to promote its oncogenic activity. This is consistent with its implication in tumorigenesis [21].
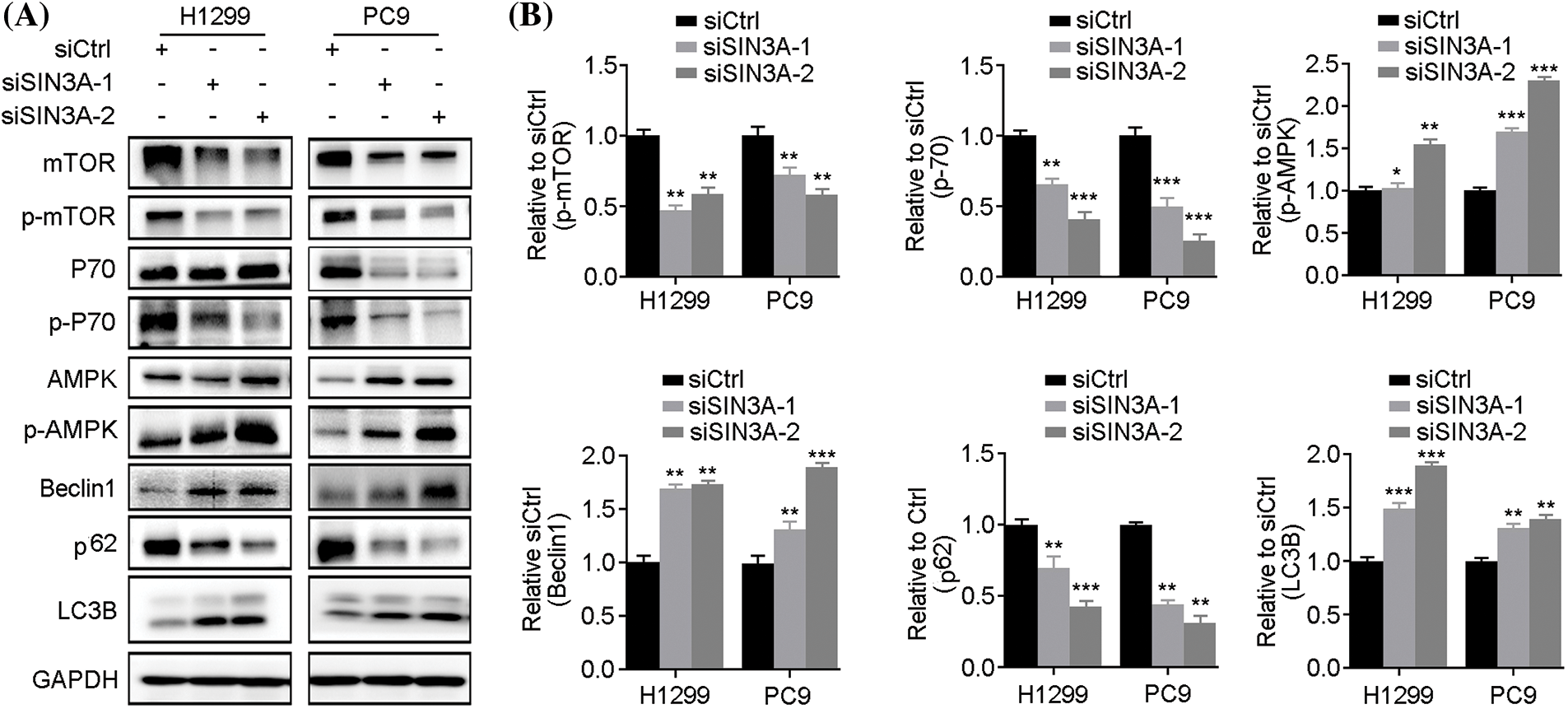
Figure 7: Knockdown of SIN3A induces autophagy. (A) Western blot analyses show the expression of autophagy-related proteins following SIN3A knockdown in NSCLC cell lines. (B) Quantification of western blots shows the decreased expression of p-mTOR, p-P70 and p62, but increased expression of p-AMPK, Beclin1 and LC3B, indicating autophagy activation (*p < 0.05; **p < 0.01; ***p < 0.001).
LncRNAs not only function in cancer immunity, metabolism and metastasis, but also play an important role in drug resistance [33–36]. Dysregulation of lncRNAs have been observed in various diseases and are critically implicated in cancer development [37,38]. LINC00265 displays oncogenic activity in several cancers, including hepatocellular carcinoma, bladder cancer, and gastric and colorectal cancers [11,39–41]. However, how LINC00265 functions in lung cancer remains elusive. In this study, we found that LINC00265 is significantly upregulated in lung cancer, which is associated with poor patient survival. Moreover, we showed that knockdown of LINC00265 reduces cancer cell proliferation and tumor growth by inducing autophagy and apoptosis. Molecularly, LINC00265 interacts with the oncogenic protein SIN3A and regulates its stability. These findings provide insights into the regulatory role of LINC00265 in LUAD progression. Thus, targeting LINC00265 could be a potential strategy for treatment of lung cancer.
It is well established that cancer cells display cuproptosis-related ferroptosis and apoptosis-linked autophagy-dependent cell death [42,43]. Silencing of LINC00265 could enhance autophagy flux because there was a further increased protein level of LC3B in the presence chloroquine. This suggests that knockdown of LINC00265 triggers autophagy and apoptosis, thereby preventing LUAD progression. On the contrary, an increased expression of LINC00265 should prevent autophagy-dependent cell death, thus promoting LUAD proliferation. Our results imply that LINC00265 should function to promote LUAD development by inhibiting autophagy-dependent cell death. There is evidence that autophagy displays stage-dependent functions in tumorigenesis. It acts as a tumor suppressor to inhibit the expression of oncogenic genes at initial stages but as a pro-tumor factor by promoting tumor cell survival at advanced stages [44]. Although the relationship between LINC00265-regulated autophagy and apoptosis remains unclear and needs further investigations, there is evidence suggesting that common pathways can regulate these processes [43]. Our results indicate that LINC00265 may function in both processes. Future works will be necessary how LINC00265 regulates autophagy-dependent or independent cell death.
SIN3A is a member of the SIN3 family and functions in the HDAC complex. It plays essential roles in various cellular processes linked to cancer pathogenesis and progression [18]. Nevertheless, how it functions in tumor biology is still subject of debate [16]. There is a possibility that SIN3A displays oncogenic or anti-tumor activity dependent on its biochemical interaction with specific protein partners, genetic interaction with other genes or regulation of target genes [17,20]. It has been shown that SIN3A could function as a tumor suppressor by regulating gene expression involved in cell invasion [17,19,20]. More recent works indicated that it interacts with and contributes to the oncogenic potential of the transcription factor STAT3 by repressing the transcription of tumor suppressor genes. Thus, interfering with its expression causes transcriptional activation tumor suppressor genes and reduces tumorigenic potential of anaplastic large-cell lymphoma by increasing tumor cell death [21]. Our present observations further support an oncogenic activity of SIN3A. They suggest that SIN3A may be implicated in regulating autophagy in NSCLC cells.
We found that LINC00265 binds to and stabilizes SIN3A protein in lung cancer cells, thus identifying a novel partner for this LncRNA. Silencing of LINC00265 results in a decreased expression of SIN3A expression. Of note, inhibiting the function of SIN3A produced similar effects as LINC00265 knockdown in NSCLC cell lines, suggesting they may function together in LUAD progression. Since LINC00265 interacts with SIN3A, its oncogenic activity may be mediated by SIN3A. There is a possibility that knockdown of SIN3A inhibits NSCLC migration and invasion at least partially by activating autophagy. Thus, our results support a pro-tumor activity of this epigenetic regulator. Consistent with this conclusion, it has been shown that the SIN3A promotes cell survival in different cancers by forming a complex with STAT3 to silence tumor repressor genes [21]. Interestingly, LINC00265 has been also shown to promote metastasis of acute lymphoblastic leukemia by regulating STAT3 signaling [14]. This raises a possibility that LINC00265 cooperates with SIN3A to enhance STAT3 signaling in LUAD. At present, it is still unclear how LINC00265 regulates the stability of SIN3A protein. However, LINC00265 has been shown to promote colorectal tumorigenesis through activation of Wnt/ß-catenin signaling. Mechanistically, LINC00265 increases the expression of ZMIZ2, which in turn recruits the deubiquitylase USP7 to stabilize β-catenin protein. Whether LINC00265 stabilizes SIN3A protein through a similar mechanism merits further investigation.
In summary, this study shows that upregulation of LINC00265 in LUAD leads to reduced overall survival rate in patients and that inhibition of LINC00265 can prevent tumor progression. Our results also indicate that LINC00265 regulates autophagy and apoptosis likely through interaction with SIN3A. These findings provide insights into the mechanism underlying LINC00265 activity in lung tumorigenesis.
Acknowledgement: We thank the surgeons and patients who participated in this study.
Funding Statement: This work was supported in part by the National Natural Science Foundation of China (NSFC) (82073388 to SWM), the Natural Outstanding Youth Fund of Guangdong Province (2022B1515020090 to SWM), Guangdong Provincial Key Laboratory of Autophagy and Major Chronic Non-Communicable Diseases (2022B1212030003 to SWM), and the Affiliated Hospital of Guangdong Medical University Clinical Research Program (LCYJ2020B005 to SWM).
Author Contributions: The authors confirm contribution to the paper as follows: SWM and YZX supervised the project. HXB, CCY and CYY designed the research and performed all experiments. ZHL, GYD and LBY completed the basic experiment part. CYH, HZ and XYL were responsible for clinical sample collection and subsequent sample delivery. SWM and CGH helped to revise the manuscript. All authors read and approved the final manuscript.
Availability of Data and Materials: All data that support the findings of this study are available from the corresponding authors upon reasonable request.
Ethics Approval: This research was approved by the Ethical Committee of the Affiliated Hospital of Guangdong Medical University (Approval Number: YS2022083). All participants provided written informed consent. This animal experiment was approved by the Ethics Committee of Affiliated Hospital of Guangdong Medical University (Approval Number: GDY2202217).
Conflicts of Interest: The authors declare that they have no conflicts interests to report regarding the present study.
Supplementary Materials: The supplementary material is available online at https://doi.org/10.32604/or.2023.030771.
References
1. Testa, U., Castelli, G., Pelosi, E. (2018). Lung cancers: Molecular characterization, clonal heterogeneity and evolution, and cancer stem cells. Cancers, 10(8), 248. https://doi.org/10.3390/cancers10080248. [Google Scholar] [PubMed] [CrossRef]
2. Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I. et al. (2021). Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians, 71(3), 209–249. [Google Scholar] [PubMed]
3. Bade, B. C., Dela Cruz, C. S. (2020). Lung cancer 2020: Epidemiology, etiology, and prevention. Clinics in Chest Medicine, 41(1), 1–24. https://doi.org/10.1016/j.ccm.2019.10.001. [Google Scholar] [PubMed] [CrossRef]
4. Ginn, L., Shi, L., Montagna, M., Garofalo, M. (2020). Lncrnas in non-small-cell lung cancer. Non-Coding RNA, 6(3), 25. https://doi.org/10.3390/ncrna6030025. [Google Scholar] [PubMed] [CrossRef]
5. Su, W., Feng, S., Chen, X., Yang, X., Mao, R. et al. (2018). Silencing of long noncoding RNA MIR22HG triggers cell survival/death signaling via oncogenes YBX1, MET, and p21 in lung cancer. Cancer Research, 78(12), 3207–3219. https://doi.org/10.1158/0008-5472.CAN-18-0222. [Google Scholar] [PubMed] [CrossRef]
6. Liang, X. H., Jackson, S., Seaman, M., Brown, K., Kempkes, B. et al. (1999). Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature, 402(6762), 672–676. https://doi.org/10.1038/45257. [Google Scholar] [PubMed] [CrossRef]
7. Vencken, S. F., Greene, C. M., McKiernan, P. J. (2015). Non-coding RNA as lung disease biomarkers. Thorax, 70(5), 501–503. https://doi.org/10.1136/thoraxjnl-2014-206193. [Google Scholar] [PubMed] [CrossRef]
8. Li, D. S., Ainiwaer, J. L., Sheyhiding, I., Zhang, Z., Zhang, L. W. (2016). Identification of key long non-coding rnas as competing endogenous RNAs for mirna-mrna in lung adenocarcinoma. European Review for Medical and Pharmacological Sciences, 20(11), 2285–2295. [Google Scholar] [PubMed]
9. Ge, H., Yan, Y., Yue, C., Liang, C., Wu, J. (2019). Long noncoding RNA linc00265 targets EGFR and promotes deterioration of colorectal cancer: A comprehensive study based on data mining and in vitro validation. OncoTargets and Therapy, 12, 10681–10692. https://doi.org/10.2147/OTT [Google Scholar] [CrossRef]
10. Zhu, Y., Gu, L., Lin, X., Cui, K., Liu, C. et al. (2020). Linc00265 promotes colorectal tumorigenesis via ZMIZ2 and USP7-mediated stabilization of β-catenin. Cell Death and Differentiation, 27(4), 1316–1327. https://doi.org/10.1038/s41418-019-0417-3. [Google Scholar] [PubMed] [CrossRef]
11. Sun, S., Li, W., Ma, X., Luan, H. (2020). Long noncoding RNA linc00265 promotes glycolysis and lactate production of colorectal cancer through regulating of mir-216b-5p/trim44 axis. Digestion, 101(4), 391–400. https://doi.org/10.1159/000500195. [Google Scholar] [PubMed] [CrossRef]
12. Ma, L., Kuai, W. X., Sun, X. Z., Lu, X. C., Yuan, Y. F. (2018). Long noncoding RNA linc00265 predicts the prognosis of acute myeloid leukemia patients and functions as a promoter by activating PI3K-AKT pathway. European Review for Medical and Pharmacological Sciences, 22(22), 7867–7876. [Google Scholar] [PubMed]
13. Zhang, F., Li, Q., Zhu, K., Zhu, J., Li, J. et al. (2020). Lncrna linc00265/mir-485-5p/irf2-mediated autophagy suppresses apoptosis in acute myeloid leukemia cells. American Journal of Translational Research, 12(6), 2451–2462. [Google Scholar] [PubMed]
14. Zhao, D., Xing, Q., Song, H., Zhao, Y., Guo, G. (2021). Linc00265/mir-4500 axis accelerates acute lymphoblastic leukemia progression by enhancing stat3 signals. Cancer Management and Research, 13, 8147–8156. https://doi.org/10.2147/CMAR.S274590. [Google Scholar] [PubMed] [CrossRef]
15. Xiao, Q., Lin, C., Peng, M., Ren, J., Jing, Y. et al. (2022). Circulating plasma exosomal long non-coding RNAs linc00265, linc00467, uca1, and snhg1 as biomarkers for diagnosis and treatment monitoring of acute myeloid leukemia. Frontiers in Oncology, 12, 1033143. https://doi.org/10.3389/fonc.2022.1033143. [Google Scholar] [PubMed] [CrossRef]
16. Kadamb, R., Mittal, S., Bansal, N., Batra, H., Saluja, D. (2013). Sin3: Insight into its transcription regulatory functions. European Journal of Cell Biology, 92(8–9), 237–246. [Google Scholar] [PubMed]
17. Kadamb, R., Leibovitch, B. A., Farias, E. F., Dahiya, N., Suryawanshi, H. et al. (2022). Invasive phenotype in triple negative breast cancer is inhibited by blocking sin3a-PF1 interaction through KLF9 mediated repression of ITGA6 and ITGB1. Translational Oncology, 16, 101320. https://doi.org/10.1016/j.tranon.2021.101320. [Google Scholar] [PubMed] [CrossRef]
18. Bansal, N., David, G., Farias, E., Waxman, S. (2016). Emerging roles of epigenetic regulator sin3 in cancer. Advances in Cancer Research, 130, 113–135. https://doi.org/10.1016/bs.acr.2016.01.006. [Google Scholar] [PubMed] [CrossRef]
19. Suzuki, H., Ouchida, M., Yamamoto, H., Yano, M., Toyooka, S. et al. (2008). Decreased expression of the sin3a gene, a candidate tumor suppressor located at the prevalent allelic loss region 15q23 in non-small cell lung cancer. Lung Cancer, 59(1), 24–31. https://doi.org/10.1016/j.lungcan.2007.08.002. [Google Scholar] [PubMed] [CrossRef]
20. Das, T. K., Sangodkar, J., Negre, N., Narla, G., Cagan, R. L. (2013). Sin3a acts through a multi-gene module to regulate invasion in drosophila and human tumors. Oncogene, 32(26), 3184–3197. https://doi.org/10.1038/onc.2012.326. [Google Scholar] [PubMed] [CrossRef]
21. Gambi, G., di Simone, E., Basso, V., Ricci, L., Wang, R. et al. (2019). The transcriptional regulator sin3a contributes to the oncogenic potential of stat3. Cancer Research, 79(12), 3076–3087. https://doi.org/10.1158/0008-5472.CAN-18-0359. [Google Scholar] [PubMed] [CrossRef]
22. Kimura, S., Noda, T., Yoshimori, T. (2007). Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged lc3. Autophagy, 3(5), 452–460. https://doi.org/10.4161/auto.4451. [Google Scholar] [PubMed] [CrossRef]
23. Su, M., Mei, Y., Sinha, S. (2013). Role of the crosstalk between autophagy and apoptosis in cancer. Journal of Oncology, 2013, 102735. [Google Scholar] [PubMed]
24. Moscat, J., Diaz-Meco, M. T. (2009). P62 at the crossroads of autophagy, apoptosis, and cancer. Cell, 137(6), 1001–1004. https://doi.org/10.1016/j.cell.2009.05.023. [Google Scholar] [PubMed] [CrossRef]
25. Mathew, R., Karp, C. M., Beaudoin, B., Vuong, N., Chen, G. et al. (2009). Autophagy suppresses tumorigenesis through elimination of p62. Cell, 137(6), 1062–1075. https://doi.org/10.1016/j.cell.2009.03.048. [Google Scholar] [PubMed] [CrossRef]
26. Tanida, I., Ueno, T., Kominami, E. (2008). Lc3 and autophagy. Methods in Molecular Biology, 445, 77–88. https://doi.org/10.1007/978-1-59745-157-4 [Google Scholar] [CrossRef]
27. Das, G., Shravage, B. V., Baehrecke, E. H. (2012). Regulation and function of autophagy during cell survival and cell death. Cold Spring Harbor Perspectives in Biology, 4(6), a008813. [Google Scholar] [PubMed]
28. Klionsky, D. J., Abdel-Aziz, A. K., Abdelfatah, S., Abdellatif, M., Abdoli, A. et al. (2021). Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy, 17(1), 1–382. https://doi.org/10.1080/15548627.2020.1797280. [Google Scholar] [PubMed] [CrossRef]
29. Mauthe, M., Orhon, I., Rocchi, C., Zhou, X., Luhr, M. et al. (2018). Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy, 14(8), 1435–1455. https://doi.org/10.1080/15548627.2018.1474314. [Google Scholar] [PubMed] [CrossRef]
30. Mizushima, N., Yoshimori, T. (2007). How to interpret LC3 immunoblotting. Autophagy, 3(6), 542–545. https://doi.org/10.4161/auto.4600. [Google Scholar] [PubMed] [CrossRef]
31. Solaimani, P., Wang, F., Hankinson, O. (2014). Sin3a, generally regarded as a transcriptional repressor, is required for induction of gene transcription by the aryl hydrocarbon receptor. The Journal of Biological Chemistry, 289(48), 33655–33662. https://doi.org/10.1074/jbc.M114.611236. [Google Scholar] [PubMed] [CrossRef]
32. Lewis, M. J., Liu, J., Libby, E. F., Lee, M., Crawford, N. P. et al. (2016). Sin3a and sin3b differentially regulate breast cancer metastasis. Oncotarget, 7(48), 78713–78725. https://doi.org/10.18632/oncotarget.v7i48 [Google Scholar] [CrossRef]
33. Jiang, M. C., Ni, J. J., Cui, W. Y., Wang, B. Y., Zhuo, W. (2019). Emerging roles of lncrna in cancer and therapeutic opportunities. American Journal of Cancer Research, 9(7), 1354–1366. [Google Scholar] [PubMed]
34. Wu, P., Mo, Y., Peng, M., Tang, T., Zhong, Y. et al. (2020). Emerging role of tumor-related functional peptides encoded by lncrna and circrna. Molecular Cancer, 19(1), 22. https://doi.org/10.1186/s12943-020-1147-3. [Google Scholar] [PubMed] [CrossRef]
35. Lin, Y. H. (2020). Crosstalk of lncrna and cellular metabolism and their regulatory mechanism in cancer. International Journal of Molecular Sciences, 21(8), 2947. https://doi.org/10.3390/ijms21082947. [Google Scholar] [PubMed] [CrossRef]
36. Shi, Q., Li, Y., Li, S., Jin, L., Lai, H. et al. (2020). Lncrna DILA1 inhibits cyclin D1 degradation and contributes to tamoxifen resistance in breast cancer. Nature Communications, 11(1), 5513. https://doi.org/10.1038/s41467-020-19349-w. [Google Scholar] [PubMed] [CrossRef]
37. Peng, W. X., Koirala, P., Mo, Y. Y. (2017). Lncrna-mediated regulation of cell signaling in cancer. Oncogene, 36(41), 5661–5667. https://doi.org/10.1038/onc.2017.184. [Google Scholar] [PubMed] [CrossRef]
38. Munschauer, M., Nguyen, C. T., Sirokman, K., Hartigan, C. R., Hogstrom, L. et al. (2018). The norad lncrna assembles a topoisomerase complex critical for genome stability. Nature, 561(7721), 132–136. https://doi.org/10.1038/s41586-018-0453-z. [Google Scholar] [PubMed] [CrossRef]
39. Yang, Z., OuYang, X., Zheng, L., Dai, L., Luo, W. (2021). Long intergenic noncoding RNA00265 promotes proliferation of gastric cancer via the microrna-144-3p/chromobox 4 axis. Bioengineered, 12(1), 1012–1025. https://doi.org/10.1080/21655979.2021.1876320. [Google Scholar] [PubMed] [CrossRef]
40. Zhi, Y., Sun, F., Cai, C., Li, H., Wang, K. et al. (2021). Linc00265 promotes the viability, proliferation, and migration of bladder cancer cells via the mir-4677-3p/fgf6 axis. Human & Experimental Toxicology, 40(12_suppl), S434–S446. https://doi.org/10.1177/09603271211043479. [Google Scholar] [PubMed] [CrossRef]
41. Ge, B., Zhang, X., Zhou, W., Mo, Y., Su, Z. H. et al. (2022). Linc00265 promotes metastasis and progression of hepatocellular carcinoma by interacting with E2F1 at the promoter of CDK2. Cell Journal, 24(6), 294–301. [Google Scholar] [PubMed]
42. Wang, T., Jiang, X., Lu, Y., Ruan, Y., Wang, J. (2023). Identification and integration analysis of a novel prognostic signature associated with cuproptosis-related ferroptosis genes and relevant lncrna regulatory axis in lung adenocarcinoma. Sedentary Life and Nutrition, 15(5), 1543–1563. [Google Scholar]
43. Liu, G., Pei, F., Yang, F., Li, L., Amin, A. D. et al. (2017). Role of autophagy and apoptosis in non-small-cell lung cancer. International Journal of Molecular Sciences, 18(2), 367. https://doi.org/10.3390/ijms18020367. [Google Scholar] [PubMed] [CrossRef]
44. Chavez-Dominguez, R., Perez-Medina, M., Lopez-Gonzalez, J. S., Galicia-Velasco, M., Aguilar-Cazares, D. (2020). The double-edge sword of autophagy in cancer: From tumor suppression to pro-tumor activity. Frontiers in Oncology, 10, 578418. https://doi.org/10.3389/fonc.2020.578418. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools