
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

GNAS mutations suppress cell invasion by activating MEG3 in growth hormone–secreting pituitary adenoma
1 Department of Neurosurgery, Affiliated Jinling Hospital, Medical School of Nanjing University, Nanjing, 210002, China
2 Department of Neurosurgery, Children’s Hospital of Nanjing Medical University, Nanjing, 210019, China
3 Affiliated Eye Hospital, Nanjing Medical University, Nanjing, 210029, China
4 Department of Neurosurgery, Jinling Hospital, Southern Medical University, Nanjing, 210002, China
5 Department of Neurosurgery, Jinling Hospital, Nanjing Medical University, Nanjing, 210002, China
* Corresponding Authors: YONG XU. Email: ; CHIYUAN MA. Email:
Oncology Research 2024, 32(6), 1079-1091. https://doi.org/10.32604/or.2024.046007
Received 14 September 2023; Accepted 08 December 2023; Issue published 23 May 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Approximately 30%–40% of growth hormone–secreting pituitary adenomas (GHPAs) harbor somatic activating mutations in GNAS (α subunit of stimulatory G protein). Mutations in GNAS are associated with clinical features of smaller and less invasive tumors. However, the role of GNAS mutations in the invasiveness of GHPAs is unclear. GNAS mutations were detected in GHPAs using a standard polymerase chain reaction (PCR) sequencing procedure. The expression of mutation-associated maternally expressed gene 3 (MEG3) was evaluated with RT-qPCR. MEG3 was manipulated in GH3 cells using a lentiviral expression system. Cell invasion ability was measured using a Transwell assay, and epithelial–mesenchymal transition (EMT)-associated proteins were quantified by immunofluorescence and western blotting. Finally, a tumor cell xenograft mouse model was used to verify the effect of MEG3 on tumor growth and invasiveness. The invasiveness of GHPAs was significantly decreased in mice with mutated GNAS compared with that in mice with wild-type GNAS. Consistently, the invasiveness of mutant GNAS-expressing GH3 cells decreased. MEG3 is uniquely expressed at high levels in GHPAs harboring mutated GNAS. Accordingly, MEG3 upregulation inhibited tumor cell invasion, and conversely, MEG3 downregulation increased tumor cell invasion. Mechanistically, GNAS mutations inhibit EMT in GHPAs. MEG3 in mutated GNAS cells prevented cell invasion through the inactivation of the Wnt/β-catenin signaling pathway, which was further validated in vivo. Our data suggest that GNAS mutations may suppress cell invasion in GHPAs by regulating EMT through the activation of the MEG3/Wnt/β-catenin signaling pathway.Keywords
Abbreviations
| GHPA | Growth hormone–secreting pituitary adenoma |
| NFPA | nonfunctioning pituitary adenoma |
| GNAS | α subunit of the stimulatory G protein |
| MEG3 | maternally expressed 3 |
| EMT | epithelial-to-mesenchymal transition |
| CCK-8 | Cell Counting Kit-8 (tradename) |
| MMP | matrix metalloproteinase |
| siRNA | small interfering RNA or short interfering RNA |
| WT | wild type |
| AC | adenylyl cyclase |
| cAMP | Cyclic 3′,5′-adenosine monophosphate |
Growth hormone–secreting pituitary adenoma (GHPA) accounts for 12.5% of pituitary neuroendocrine tumors, and excessive growth hormone results in acromegaly and systemic complications [1]. Acromegaly has been associated with a two-fold increase in mortality, mainly due to cardiovascular disease, which can be reversed by treatments for controlling hormone overproduction [2]. Approximately one-half of the patients with GHPA experienced high-risk relapse following surgical reduction owing to tumor cell infiltration of the surrounding tissues [3]. Thus, supplementary chemotherapy is warranted to control tumor recurrence.
The genome-wide analysis of epigenetic changes revealed specific subgroup molecular characteristics of pituitary adenoma [4]. For example, the hypomethylation of GH1 and SST5 promoters is associated with the overexpression of the respective genes in GHPAs, and POMC is hypomethylated in corticotroph adenomas. The somatic mutations of GNAS and USP8 combined with transcriptome analysis have been identified in GHPAs and corticotroph adenomas.
Mutations in GNAS, which encodes the α subunit of the stimulatory G protein, were detected in ~40% GHPAs [5,6]. Functional studies have suggested that GNAS mutations constitutively activate adenylyl cyclase (AC), thereby inducing the cyclic AMP (cAMP) signaling pathway in pituitary tumors [7]. Studies have reported that somatostatin and its analogs reduce GH secretion through multiple mechanisms involving AC inhibition [8]. Somatostatin binding sites are negatively coupled to AC; however, the inhibition of hormone secretion by somatostatin cannot be explained solely through AC inhibition [8]. It has been speculated that somatostatin blocks basal hormone release by a mechanism different from that of the inhibition of cAMP formation. In addition, it has been suggested that the volume of GNAS-mutant GHPA is smaller and it is less likely to be invasive [9].
Maternally expressed gene 3 (MEG3), a large non-coding RNA (lncRNA), was first identified as a tumor suppressor in the pituitary [10]. Our previous study suggested that MEG3 expression is associated with GNAS mutations and it is involved in regulation of growth hormone hypersecretion, proliferation, and invasiveness of GHPAs [11]. The cAMP response element (CRE), located in the MEG3 proximal promoter region, is critical for MEG3 expression [12]. Cyclic adenosine monophosphate‑responsive element-binding protein, as a downstream target of mutated GNAS, is also implicated in the upregulation of MEG3 by binding to the CRE site [12,13]. Some studies have suggested that MEG3 suppresses tumor cell proliferation and invasion by downregulating related proteins [14,15]. The Wnt/β-catenin pathway plays an important role in tumorigenesis, cellular proliferation, and invasion [16]. Extensive studies have shown that lncRNAs have the potential to target canonical Wnt-related components during epithelial–mesenchymal transition (EMT) process [17]. Studies have demonstrated that MEG3 negatively regulates the Wnt/β-catenin signaling pathway in tumor growth and invasion [18,19]. Recently, MEG3 was shown to induce the phosphorylation, ubiquitination, and degradation of β-catenin through GSK-3β, which in turn inactivates the Wnt pathway and ultimately inhibits the invasion and metastasis of retinoblastoma cells [20]. This study aimed to ascertain whether mutations in GNAS inhibit the invasiveness of GHPA cells mainly through MEG3-mediated inactivation of the Wnt/β-catenin pathway. The findings of this study may provide a new therapeutic approach for treating GHPA.
Patients and clinical characteristics
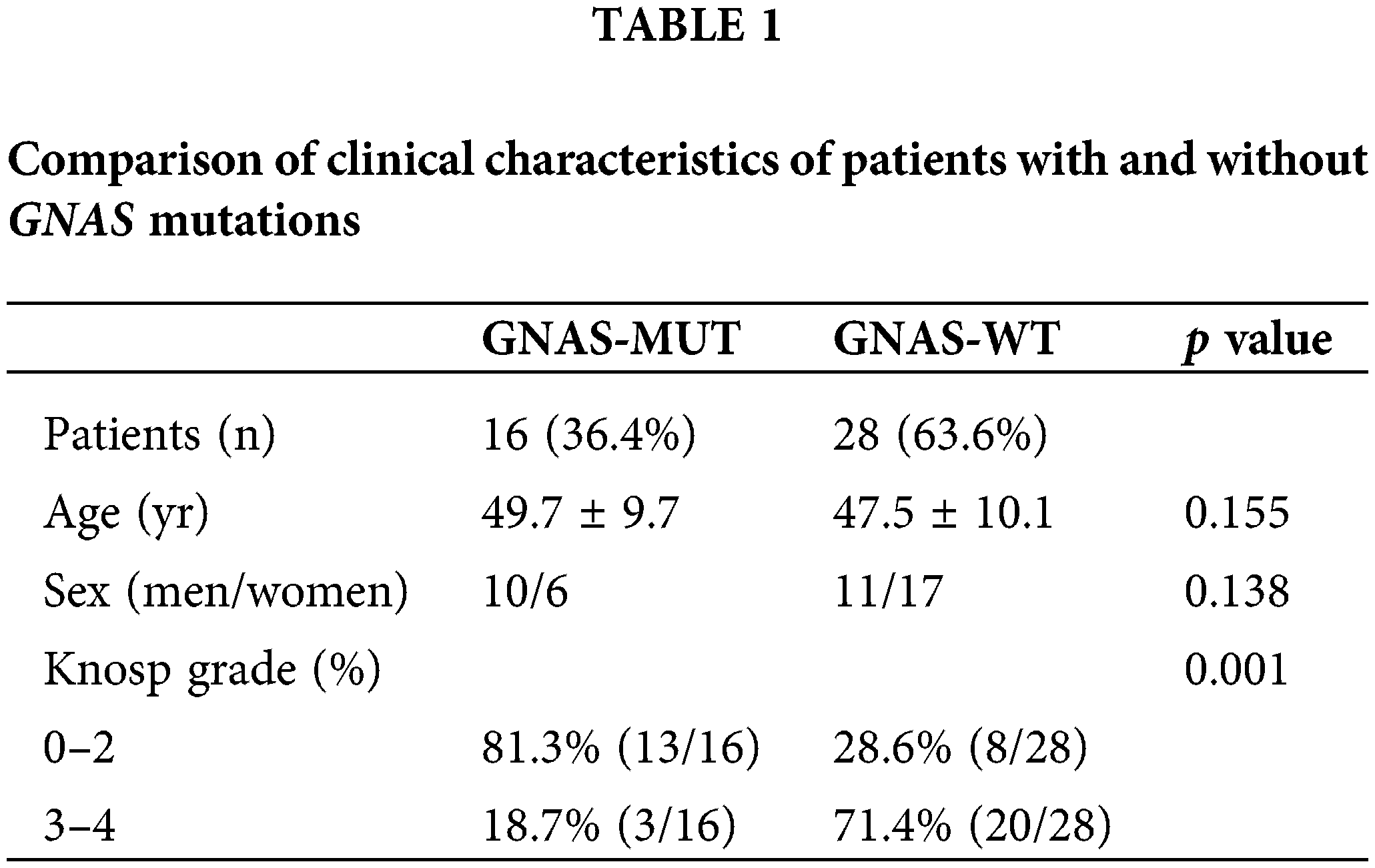
Tumor samples were collected from 44 patients (21 men and 23 women) with acromegaly who underwent endoscopic endonasal transsphenoidal surgery at the Department of Neurosurgery of Nanjing Jinling Hospital (Nanjing, China) between November 2018 and November 2019. The inclusion criteria were as follows: (i) available results of biochemical tests and magnetic resonance images (MRIs) before any treatment, (ii) pituitary adenomas that can be identified via radiological study, and (iii) histological diagnosis of GHPAs by pathologists in Nanjing Jinling Hospital. The exclusion criteria were as follows: (i) patients who have undergone radiotherapy and drug therapy before surgery and (ii) patients with residual or recurrent tumor following surgery. Overall, 10 patients with clinically nonfunctioning pituitary adenoma (NFPA) were included as negative controls. Approval for the study was obtained from the Ethical Committee of Nanjing Jinling Hospital (2018NZKY-008-02) and informed consent were obtained from all patients who participated in this study. This study was performed according to the ethical standards of the 1964 Helsinki Declaration. Pituitary adenomas were classified into invasive and noninvasive tumors according to the degree of lateral extension to the cavernous sinus space via MRI scanning [21]. Knosp grades 3 and 4 were defined as invasive pituitary adenomas, and Knosp grades 0–2 were defined as noninvasive tumors [22]. Tumor volume was determined using the formula (length × width × height × Π)/6. The clinical characteristics of the patients are described in Table 1.
Detection of mutations in GNAS
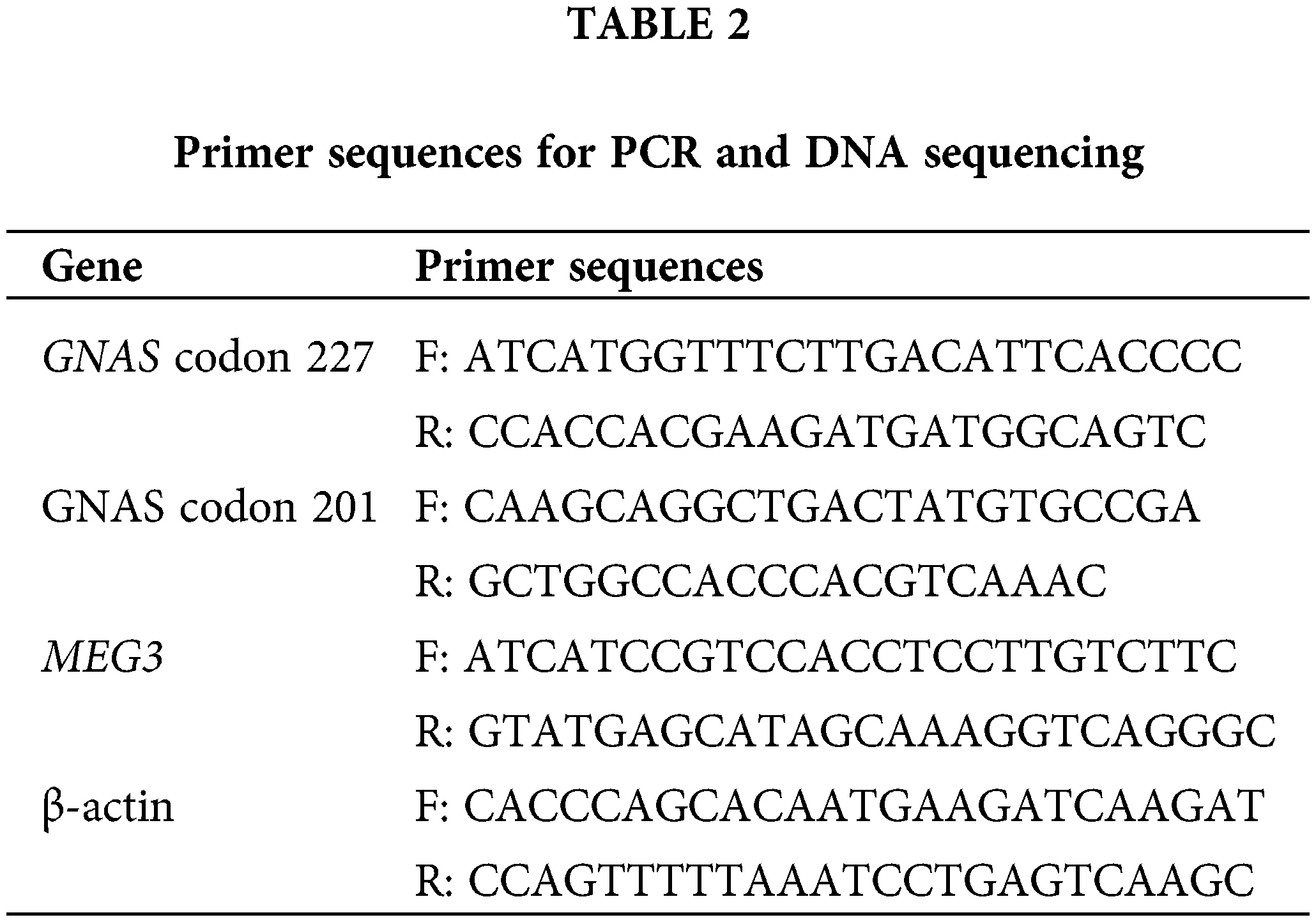
Genomic DNA was extracted from 44 GHPA and 10 NFPA tissues using a DNA miniprep kit, according to the manufacturer’s protocol (Qiagen GmbH, Hilden, Germany). Point mutations in GNAS reported in tumor specimens included the CGT-to-TGT mutation at codon 201 (Arg201Cys) and the CAG-to-CTG mutation at codon 227 (Gln227Leu) [23]. PCR amplification of codons 201 and 227 was performed using Taq DNA-Polymerase (TTH Biotools Madrid, Spain) [23]. The PCR products were purified using a PCR purification kit (Qiagen GmbH, Hilden, Germany) and sequenced using an ABI3730XL analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc., Carlsbad, CA, USA). Primer sequences used for PCR and DNA sequencing are listed in Table 2.
The GH3 cell line (rat GH-secreting pituitary tumor cell line), which produces both growth hormone and prolactin, was purchased from the Cell Culture Centre, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences (Beijing, China). GH3 cells were cultured in Ham’s F12 medium supplemented with 10% FBS and 1% streptomycin and penicillin in a humidified 5% CO2 incubator at 37°C.
A pWPT lentiviral expression vector (Genechem, Shanghai, China) was used to clone wild-type and mutant GNAS, and the generated constructs were termed pWPT-GNAS (expressing GNAS wild-type), pWPT-GNAS-Q227L (expressing GNAS mutated at Q227L), and pWPT-GNAS-R201C (expressing GNAS mutated at R201C). GH3 cells were plated in 6-well plates at 70% confluence and then injected with lentivirus to express wild-type or mutant-type GNAS. Overexpressed or knocked-down MEG3 gene was packaged by lentivirus and then transfected into GH3 cells according to the protocols (Corues Biotechnology, Nanjing, China).
RNA extraction and quantitative reverse transcription real-time quantitative PCR
Total RNA was isolated from tissues or cells using Trizol reagent and reverse transcribed into complementary DNA using the TaqMan MicroRNA Reverse Transcription Kit (TaKaRa, Dalian, China). Real-time quantitative (RT-q) PCR was performed using SYBR Green PCR Master Mix (Takara, Japan) according to the manufacturer’s instructions. The sequences of qPCR primers are listed in Table 2.
lncRNA MEG3 was normalized with β-actin and the level of MEG3 in GHPA was further normalized with its level in NFPA. The experiments were repeated at least three times.
Total RNA from GH3 cells with MEG3-overexpressed and MEG3-overexpress-vector was extracted using Trizol reagent (Thermo Fisher, Waltham, USA) according to the manufacturer’s protocol. RNA samples were digested with RNase free DNase I (Invitrogen, Carlsbad, USA) to eliminate residual genomic DNA, and the digestion products were purified using magnetic beads (Axygen, Union City, USA).
Total RNA was qualified by Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). The next-generation sequencing library was prepared according to the protocol provided by the manufacturer (NEBNext Ultra RNA Library Prep Kit for Illumina HiSeq system). The sequences and data were analyzed to identify differentially expressed genes (Genewiz, Shanghai, China).
Proteins were extracted from tumor cells and tissues in RIPA buffer (Beyotime Biotech., Shanghai, China), separated using SDS-PAGE, and transferred onto polyvinylidene fluoride membranes. The membranes were blocked with 5% fat-free milk in Tris-buffered saline containing 0.1% Tween 20 and incubated at 4°C overnight with primary antibodies against MMP-2 (#40994, rabbit mAb, 1:1000), β-catenin (#8480, rabbit mAb, 1:1000), MMP-9 (#13667, rabbit mAb, 1:1000), and β-actin (#4970, rabbit mAb, 1:1000), purchased from Cell Signaling Technology (Danvers, MA, USA). Subsequently, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (#7074, anti-rabbit IgG, HRP-linked antibody, Cell Signaling Tech.). The images were visualized using enhanced chemiluminescence (Beyotime Biotech., Shanghai, China). Pierce ECL Western Blotting Substrate (Thermo Scientific) was used to detect the chemiluminescence signals. Densitometric analysis of the western blot bands was performed using the Bio-Rad Imaging system (Bio-Rad, Hercules, California, USA). The western blotting assay was repeated three times.
Matrigel matrix diluent (300 μg/mL) was used to coat the bottom of the upper chamber of the Transwell chamber in a precooled environment at 4°C. The volume ratio of serum-free F12 medium to Matrigel in the upper chamber was 4:1, the total volume was 100 μL, and the Matrigel was frozen overnight at 4°C. The next day, 500 μL of F12 medium containing 10% FBS was added into the lower chamber. Cells (1 × 105) were seeded in the upper chamber. After incubation for 48 h, non-invading cells in the upper chamber were removed. Cells that invaded the bottom chamber were fixed with 4% paraformaldehyde and stained using 0.1% crystal violet (Beyotime Biotech., Shanghai, China). Five random fields were selected for counting under a microscope (magnification, ×200). Each experiment was performed three times.
Tumor tissue sections were prepared from the surgical resection of GHPA. Tissue slides were fixed with 4% formaldehyde, blocked with 10% normal goat serum, and incubated overnight at 4°C with primary antibodies against β-catenin, E-cadherin, N-cadherin, and vimentin (diluted at 1:100 in PBS). All antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA). The slides were removed from the incubation chamber, washed three times with PBST, and incubated for 40 min in the dark in a humidified chamber at room temperature with goat polyclonal secondary antibodies against rabbit IgG (1:300; Abcam Biotechnology, Cambridge, MA, USA) reconstituted in PBS. Sections were washed three times with PBST. After counterstaining was complete, the sections were treated with glycerol/PBS (2:1) for 10 min in the dark at room temperature. Five randomly chosen fields were selected for counting using an Axiovert 200 fluorescent microscope.
Subcutaneous xenografts in nude mice
Animal experiments were approved by the Animal Experimentation Ethics Committee of the Jinling Hospital of Nanjing University (protocol number: 2021DZGKJDWSL-0081).
Four-week-old female athymic BALB/c nude mice were purchased from the Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China) and housed and maintained in laminar airflow chambers under specific pathogen-free conditions. MEG3-overexpressing GH3 cells (107 cells/0.1 mL) were subcutaneously injected into the right back side of the mice. Following the tumor formation, a week after injection, lithium chloride (Sigma-Aldrich, St. Louis, MO, USA; 60 mg/kg/d in 100 μL saline), a β-catenin activator, was administered daily via intraperitoneal injection in the group with high levels of MEG3. The other groups were injected with saline alone as controls. Tumor volumes were measured using a vernier caliper twice a week and calculated as (length × width2)/2. Four weeks after the injection, the mice were euthanized via cervical dislocation, and the dissected tumors were weighed and processed to determine the expression levels of related proteins, including β-catenin, E-cadherin, N-cadherin, MMP-2, and MMP-9.
Tumor tissues from mice were fixed and antigens were exposed using heat-induced epitope retrieval with exposure to citric acid buffer (pH = 7.0), blocked in 10% normal goat serum, and incubated with 3% hydrogen peroxide. The tissue slides were incubated with primary antibodies against β-catenin (#8480, 1:200), E-cadherin (#3195, 1:200), N-cadherin (#13116, 1:200), MMP-2 (#40994, 1:200), and MMP-9 (#13667, 1:200) from Cell Signaling Technology (Danvers, MA, USA) overnight at 4°C followed by incubation with a goat anti‑rabbit secondary antibody (A0208, 1:50) from Beyotime for 15 min at 37°C. We repeated the experiments thrice. The number of positive cells was counted in each section in 10 randomly selected fields (magnification, ×200).
Data were statistically analyzed using SPSS 19.0. Student’s unpaired t-test and Fisher’s exact test were used for intergroup analysis. One‑way analysis of variance test was used to compare tumor volume and weight among the three groups of in vivo experiments when the mice were euthanized. The results are presented as mean ± standard deviation. Correlation was analyzed using Spearman’s correlation. p < 0.05 was considered statistically significant.
In total, 44 patients with GHPA and 10 patients with NFPA were enrolled in this study. Mutations in GNAS were scanned by directly sequencing genomic PCR products amplified from the tumor samples of patients using specific primer sets. The results revealed that 16/44 patients carried GNAS mutations, including 8 cases of codons 227 and 201 each. Conversely, no GNAS mutation was detected in the 10 patients with NFPA. Subsequently, the patients were divided into two groups (GNAS-WT, n = 28; GNAS-MUT, n = 16). The percentage of invasive tumors (Knosp grades 3 and 4) in GNAS-MUT tumors remarkably decreased (18.7% vs. 71.4%; p = 0.001; Table 1). There were no statistical differences in terms of age or gender between the two groups.
Mutant GNAS leads to the inhibition of GH3 cell invasion
The clinical analysis results predicted that GNAS mutations are associated with the invasiveness of GHPA. No GNAS mutations were detected in GH3 cells. Accordingly, GH3 cells expressing the wild- or mutant-type GNAS were constructed. The cells were injected with lentivirus carrying Flag-tagged Q227L (pWPT-GNAS-Q227L) or R201C (pWPT-GNAS-R201C) as well as GNAS-WT control (pWPT-GNAS) (Fig. 1A). The cell invasion capacity was significantly decreased in GH3-Q227L (p = 0.008) and GH3-R201C (p = 0.002) cells compared with GH3-GNAS-WT cells (Fig. 1B). In addition, the expression levels of MMP-2 (2-fold, p = 0.0015; 4-fold, p = 0.0003) and MMP-9 (2.3-fold, p = 0.0025; 1.9-fold, p = 0.0036) in GH3-Q227L and GH3-R201C cells were reduced (Fig. 1C).
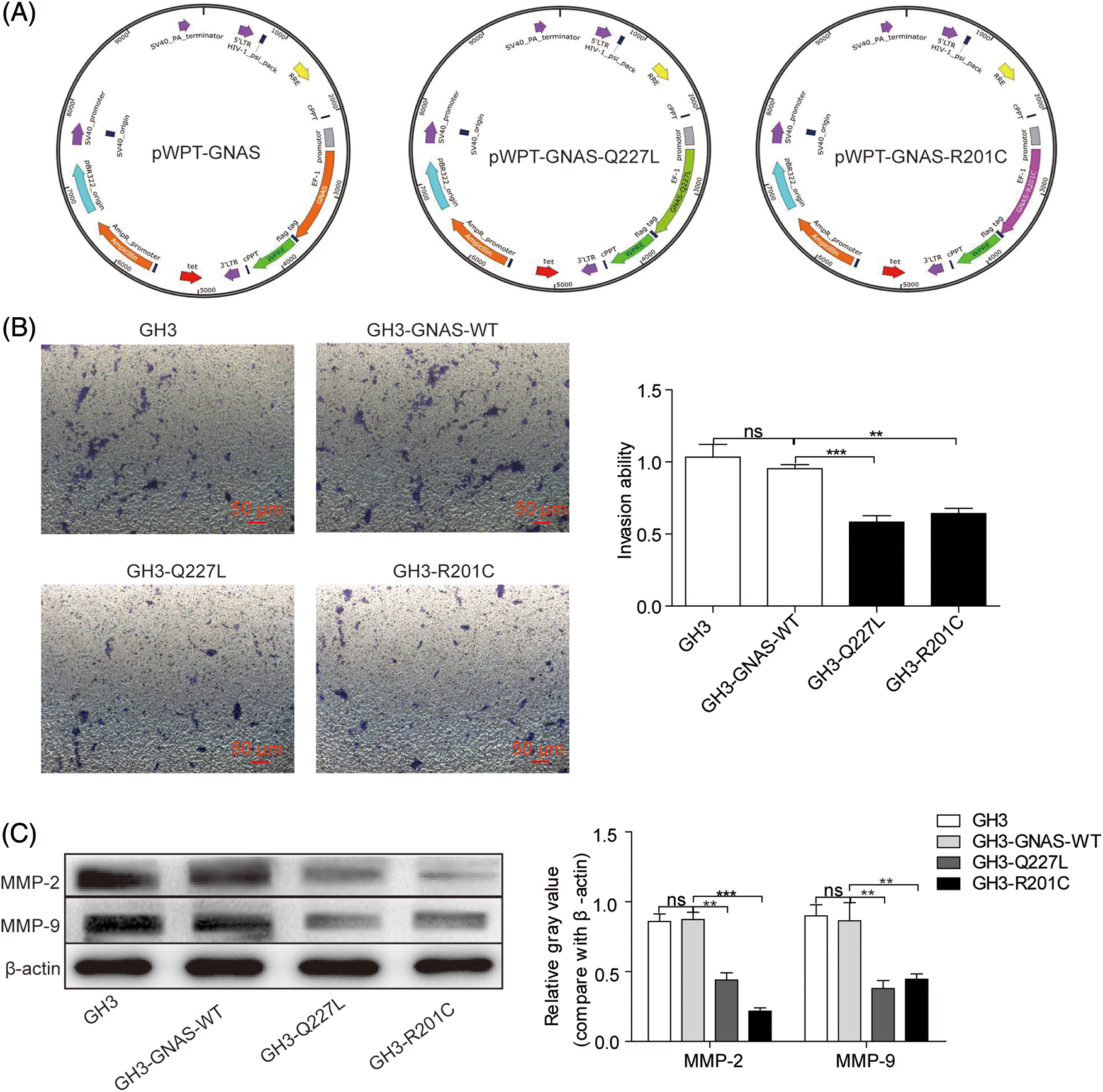
Figure 1: GNAS mutations inhibit GH3 cell invasion. A: Wild-type and mutant GNAS were expressed in GH3 cells by transduction with lentiviral vectors: pWPT-GNAS-Q227L, pWPT-GNAS-R201C, and pWPT-GNAS. B: Cell invasion was measured using the Transwell assay (magnification, ×200). C: The levels of MMP-2 and MMP-9 were quantified via western blotting. **p < 0.01, ***p < 0.001 show statistical significance between the two groups as indicated, and “ns” shows no significance. GH3-GNAS-WT: GH3 cells containing wild-type GNAS; GH3-Q227L: GH3 cells expressing the mutant GNAS at Q227L; GH3-R201C: GH3 cells expressing the mutant GNAS at R201C. Data represent the mean ± s.d. of three independent experiments
Mutant GNAS upregulates MEG3 expression
Because GNAS mutations are speculated to participate in upregulating MEG3 expression, the expression levels of MEG3 in NFPA and GHPA tumor tissues were quantified via RT-qPCR. The levels of MEG3 considerably increased in 44 GHPA tumor tissues compared with the 10 NFPA tumor tissues (p = 0.0003) (Fig. 2A). Intriguingly, MEG3 expression further increased in the 16 GHPA tumor tissues with mutant GNAS compared with the 28 wild-type tumors (p = 0.006; Fig. 2B). Consistently, the high levels of MEG3 were verified in GHPA tumor tissues with single mutant sites at Q227L and R201C (4.7-fold, p = 0.0004; 4.4-fold, p = 0.0033; Fig. 2C). Importantly, tumor invasiveness significantly declined in the group expressing high levels of MEG3 compared with the group expressing low levels of MEG3 (p = 0.0014; Fig. 2D). We suggest that MEG3 negatively correlates with tumor invasion in GHPA.
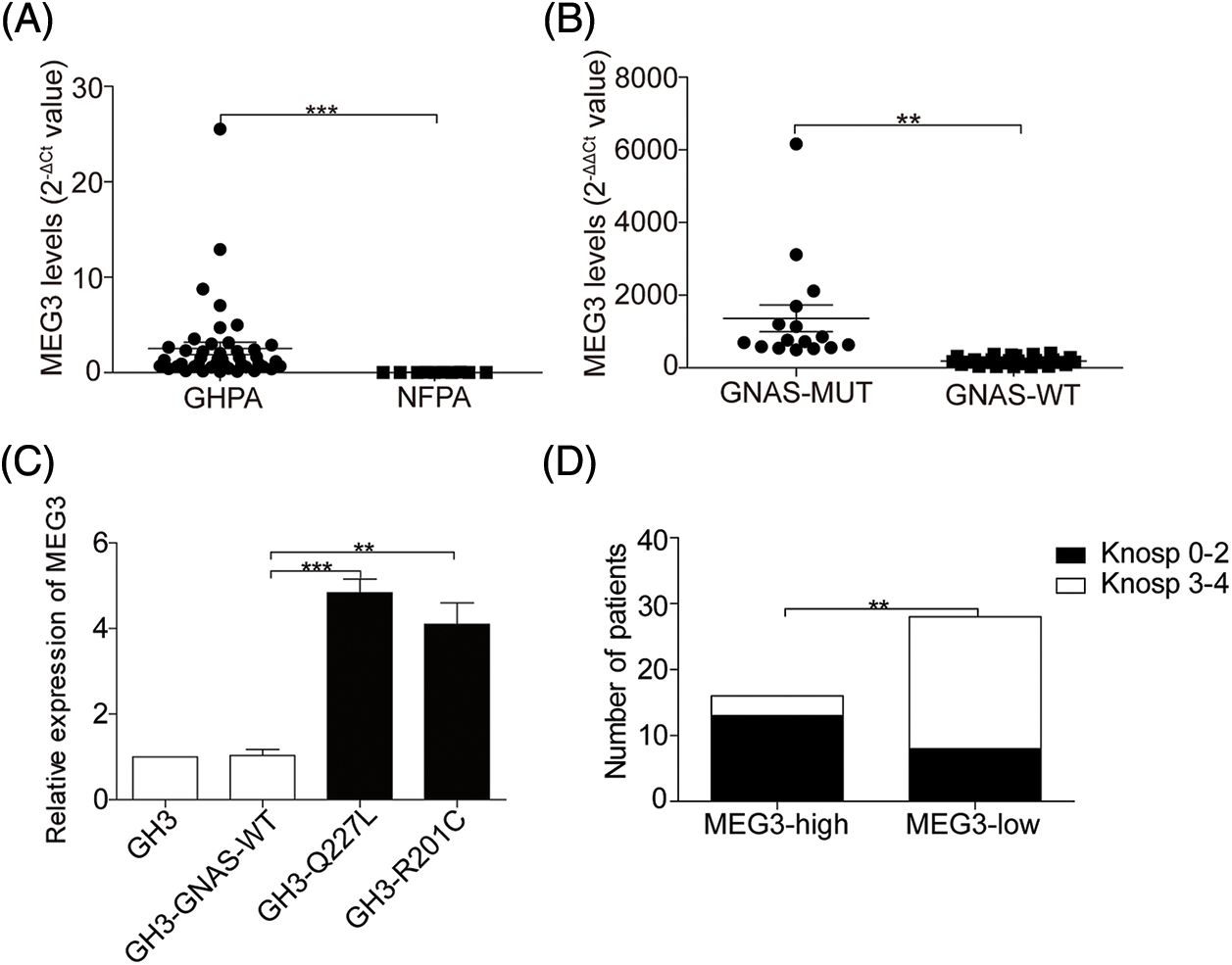
Figure 2: GNAS mutations upregulate MEG3 expression. A: The expression of MEG3 in GHPA and NFPA was quantified via RT-qPCR analysis. B and C: Correlation between MEG3 expression and GNAS mutations was determined via RT-qPCR. D: Correlation between MEG3 expression and percentage of invasive tumors was analyzed. **p < 0.01, ***p < 0.001 show statistical significances between the two groups as indicated. GHPA: growth hormone–secreting pituitary adenoma; NFPA: non‑functioning pituitary adenoma; GNAS-MUT: growth hormone–secreting pituitary adenoma possessing GNAS mutations; GNAS-WT: growth hormone–secreting pituitary adenoma possessing wild-type GNAS; MEG3-high and MEG3-low: GHPAs were divided into the MEG3 high expression group and low expression group according to the MEG3 expression level
MEG3 inhibits the invasiveness of GH3 cells
To verify whether MEG3 can inhibit rat GH3 cell invasion, MEG3 was manipulated using lentiviral expression systems for ectopic expression or knock down in GH3 cells. After lentivirus infection, MEG3 was quantified using immunofluorescence and RT-qPCR. MEG3 level increased >8-fold in MEG3-overexpressing cells compared with MEG3-overexpressing empty vector cells (p = 0.0001); MEG3 level decreased >2-fold in MEG3-si cells (p = 0.005) (Fig. 3A). Compared to the lentiviral vector control, MEG3 overexpression decreased cell invasion (p = 0.0008). By contrast, silencing MEG3 increased cell invasion (p = 0.0044) (Fig. 3B). Consistently, protein expression of MMP-2 (p < 0.05) and MMP-9 (p < 0.05) decreased in MEG3-overexpressing cells and increased in cells with MEG3 silenced (p < 0.001, p < 0.001) (Fig. 3C). To evaluate the role of MEG3 in mediating the effects of the GNAS mutation on tumor invasion, we examined the effects of MEG3 knockdown in GH3 cells expressing mutant GNAS. Cell invasion ability and protein levels of MMP-2 and MMP-9 significantly increased in GH3 cells with GNAS mutations and MEG3 knockdown (Figs. 3D and 3E). These results suggested that GNAS mutations inhibit invasiveness of rat GHPA cells by activating MEG3.
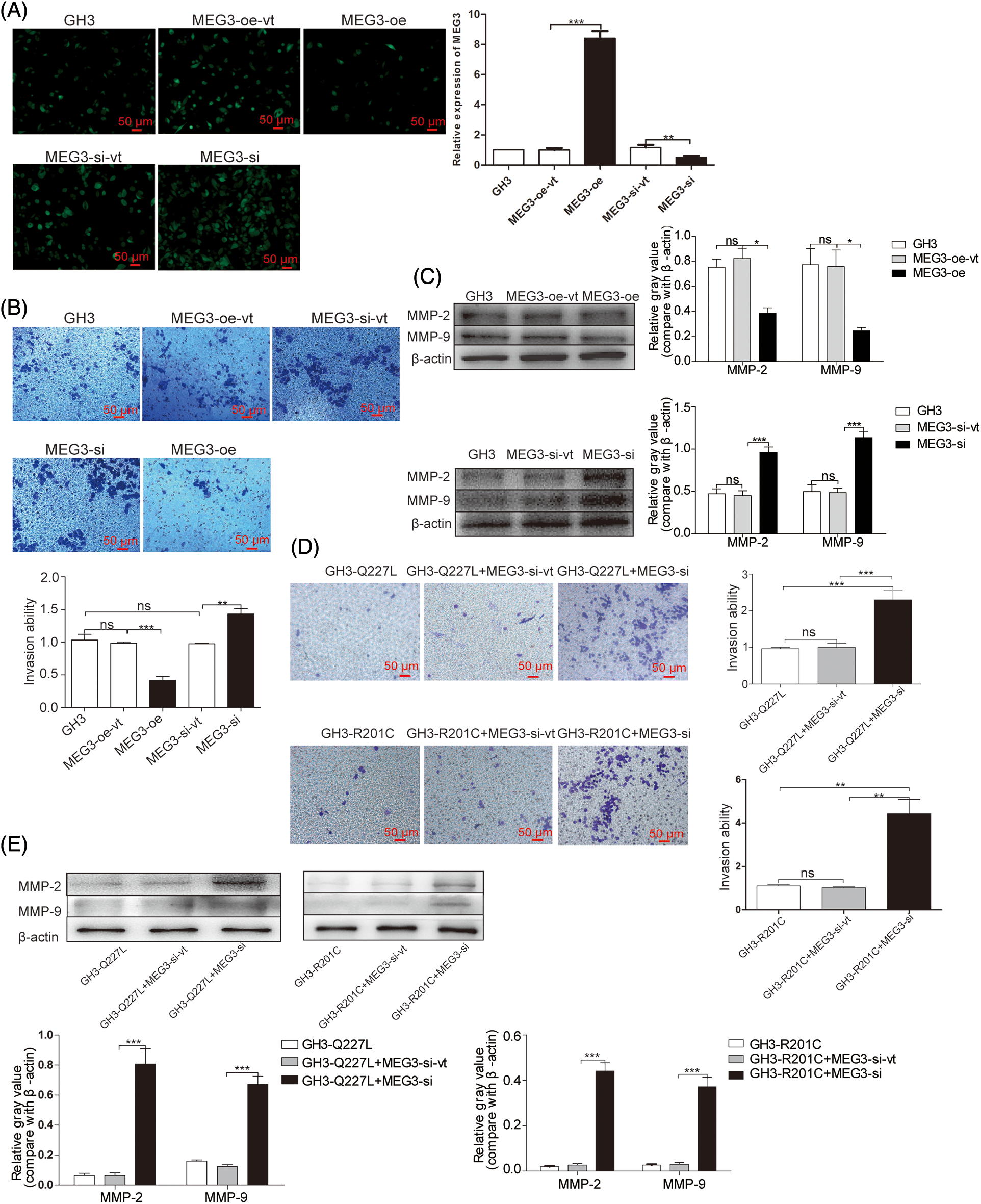
Figure 3: MEG3 inhibits the invasiveness of GHPA cells. A: MEG3 was overexpressed or knocked down in GH3 cells. Relative levels of MEG3 were quantified via immunofluorescence assay and confirmed via RT-qPCR (magnification, ×200). B: Cell invasion was analyzed using the Transwell assay (magnification, ×200). C: Expression levels of MMP-2 and MMP-9 in MEG3-overexpressing cells were measured via western blotting. D: Cell invasion of MEG3 knockdown in GH3 cells expressing mutant GNAS was detected using the Transwell assay (magnification, ×200). E: Expression levels of MMP-2 and MMP-9 of MEG3 knockdown in GH3 cells expressing mutant GNAS were measured via western blotting. *p < 0.05, **p < 0.01, and ***p < 0.001 show the statistical significance between the two groups as indicated, and “ns” shows no significance. MEG3-oe-vt: GH3 cells expressing empty vector of overexpressed MEG3; MEG3-oe: GH3 cells expressing overexpressed MEG3; MEG3-si-vt: GH3 cells containing MEG3 siRNA expressing empty vector; MEG3-si: GH3 cells containing MEG3 siRNA expressing vector
MEG3 inhibits cell invasion by inactivating the Wnt/β-catenin signaling pathway
To investigate the mechanism by which MEG3 inhibits GHPA cell invasion, MEG3 was overexpressed in GH3 cells and the RNA expression profile was analyzed by RNA-Seq. The results revealed that the Wnt/β-catenin signaling pathway could be involved in regulating cell invasion (Fig. 4A). Consistently, the mRNA level of β-catenin significantly decreased in GH3 cells with GNAS mutations (GH3-Q227L: p = 0.0047, GH3-R201C: p = 0.005) (Fig. 4B). Ectopic expression of MEG3 in GH3 cells resulted in reduced β-catenin expression (p < 0.01); conversely, silencing of MEG3 in GH3 cells increased the levels of β-catenin (p < 0.05) (Fig. 4B and C). Significant negative relationships were found between the expression levels of MEG3 and β-catenin (p < 0.05) (Fig. 4D). Consistently, the level of β-catenin in GHPA-carrying GNAS mutations (n = 16) was lower than that in wild-type tumors (n = 28) (p = 0.001), which are associated with high levels of MEG3 (Fig. 4E). The results suggest that MEG3 negatively regulates β-catenin and promotes cell invasion, particularly in GHPA cells carrying GNAS mutations.
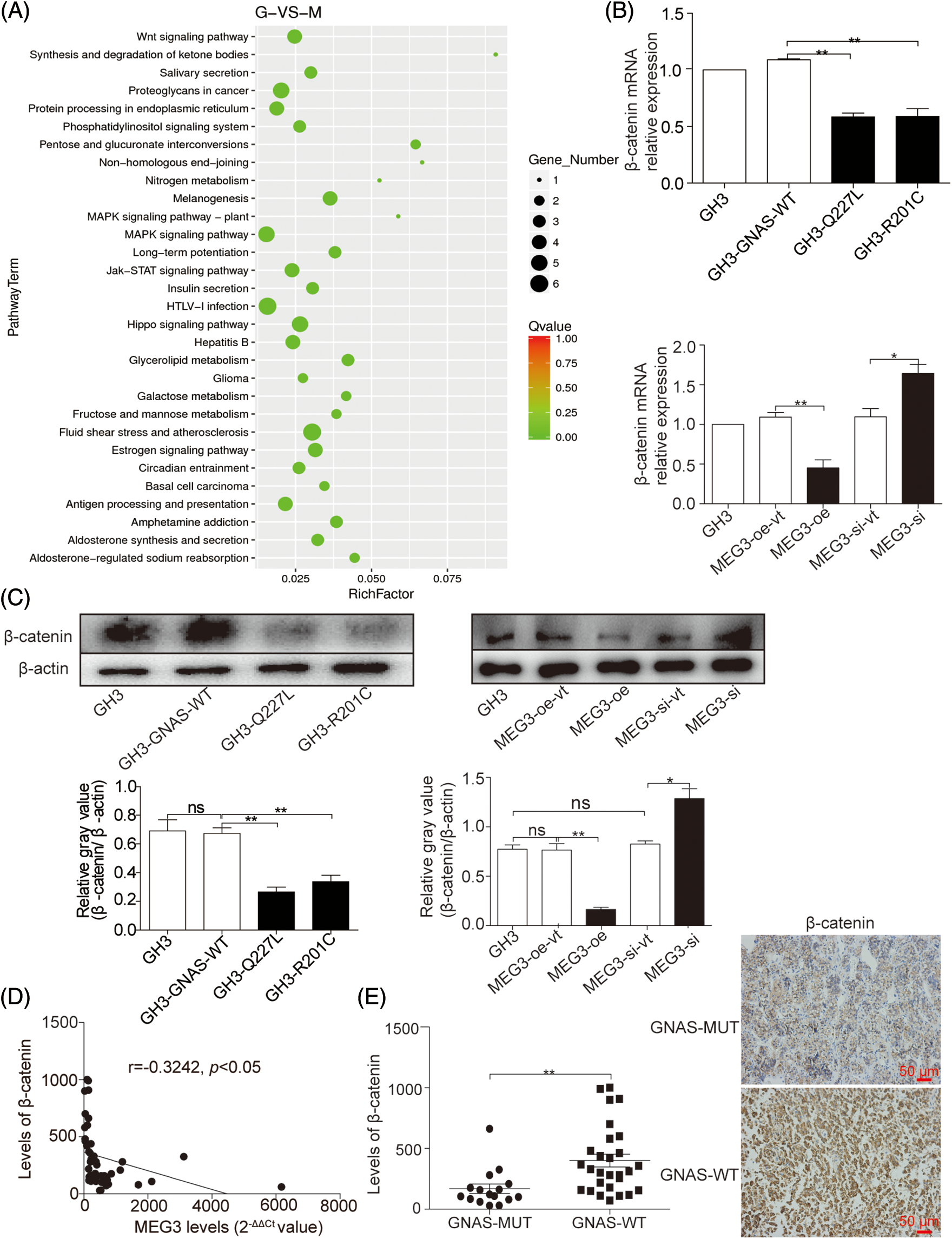
Figure 4: MEG3 inhibits the invasiveness of GHPA with GNAS mutations by inactivating Wnt/β-catenin signaling. A: RNA-Seq was used to examine the gene expression profiles in MEG3-overexpressed GH3 cells compared with vector-only controls. The altered mRNA expression profile was analyzed using Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis. B: Reduction of β-catenin mRNA expression in GHPA tumors with the GNAS mutations and GH3 cells with a high level of MEG3 was confirmed via RT-qPCR. C: Relative β-catenin levels were measured via western blotting. D: Significant negative relationships were found between the expression levels of MEG3 and β-catenin. E: β-catenin levels between GNAS-mutant and wild-type tumors were analyzed using IHC (magnification, ×200). *p < 0.05 and **p < 0.01 show the statistical significance between the two groups as indicated, and “ns” shows no significance
Inactivation of β-catenin suppresses GHPA cell invasion
LiCl is an activator and Dickkopf1 (Dkk1) is a suppressor of β-catenin [24,25]. Accordingly, MEG3-overexpressing cells were treated with 20 mM LiCl [26] and MEG3-si cells were treated with 150 ng/mL Dkk1 [27]. β-catenin expression increased in LiCl-treated cells (4.4-fold, p < 0.01) but decreased in Dkk1-treated (Sigma-Aldrich) cells (1.4-fold, p < 0.05; Fig. 5A). LiCl treatment enhanced cell invasion (1.8-fold, p < 0.05); however, Dkk1 treatment inhibited cell invasion (1.6-fold, p < 0.05; Fig. 5B). LiCl could upregulate the expression of MMP-2 (p < 0.01) and MMP-9 (p < 0.05). Conversely, Dkk1 suppressed MMP-2 (p < 0.05) and MMP-9 (p < 0.05) expression (Fig. 5C). These results suggest that MEG3 suppresses GHPA cell invasion by inhibiting activated β-catenin.
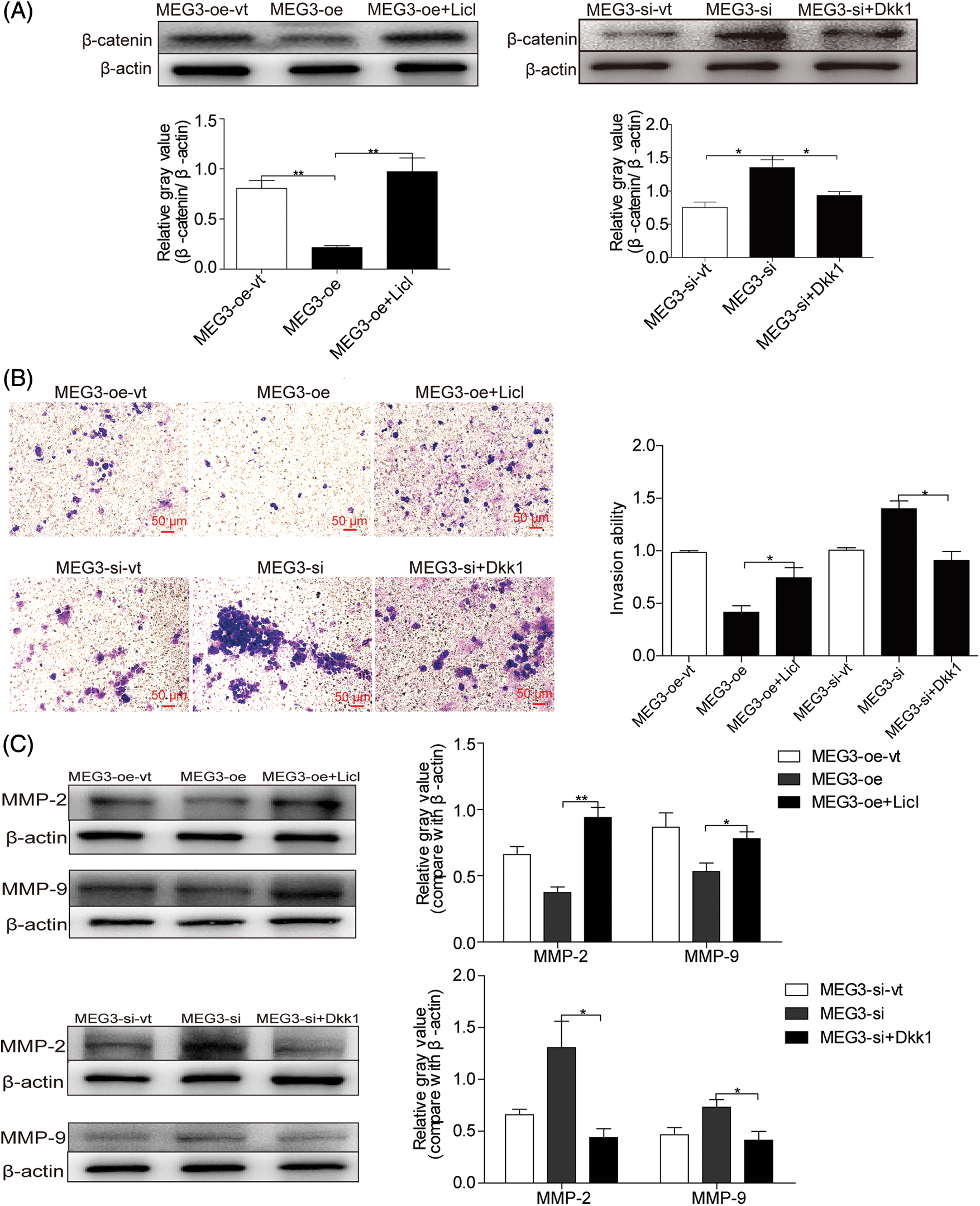
Figure 5: MEG3-mediated downregulation of β-catenin inhibits GH3 cell invasion. A: MEG3 was manipulated by expressing or silencing in GH3 cells. The expression of β-catenin was induced by LiCl (20 mM) or repressed by Dkk1 (150 ng/mL) and measured using western blotting. B: Relative cell invasion was quantified by the Transwell assay (magnification, ×200). C: Expression of MMP-2 and MMP-9 was measured using western blotting. *p < 0.05, **p < 0.01 show statistical significance between the two groups as indicated
EMT plays a fundamental role in promoting cell mobility and tumor metastasis, and Wnt/β-catenin signaling is a key mechanism in EMT [28,29]. To examine the effects of GNAS mutations on EMT, we quantified the expression of EMT-associated proteins regulated by β-catenin, such as E-cadherin (cell adhesion marker), as well as N-cadherin and vimentin (mesenchymal markers), using immunofluorescence in GHPA tissues. Compared with GHPA tissue with wild-type GNAS, GHPA tissue with GNAS mutations exhibited increased E-cadherin (p < 0.0001), decreased N-cadherin (p < 0.01) and vimentin (p < 0.05), and high constitutive levels of MEG3 (Fig. 6A and B). Thus, our findings suggest that GNAS mutations inhibit the invasiveness of GHPA by downregulating EMT (Fig. 6C).
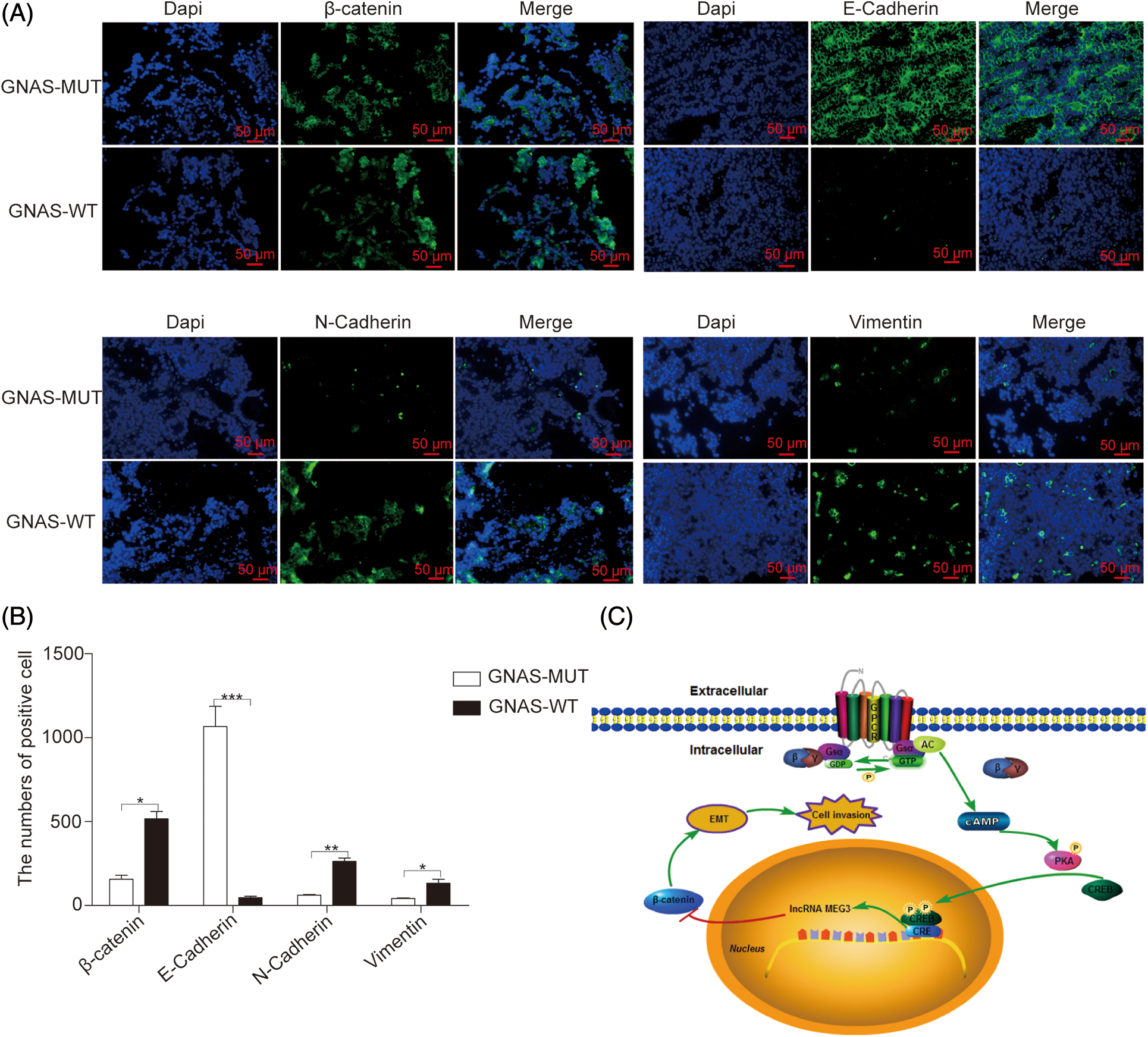
Figure 6: GNAS mutations inhibit EMT in GHPA tumors. A and B: The expression of EMT-related proteins in GHPA tumor tissues was quantified via immunofluorescence (magnification, ×200). C: Mechanism by which GNAS mutations inhibit the invasiveness of GHPA. *p < 0.05, **p < 0.01, and ***p < 0.001 show statistical significance between the two groups as indicated. GPCR: G protein coupled receptor; Gsα: α subunit of Gs protein; GDP: guanosine diphosphate; GTP: guanosine triphosphate; AC: adenylyl cyclase; cAMP: cyclic adenosine monophosphate; PKA: protein kinase A; CREB: cAMP response element-binding protein; CRE: cAMP response element; EMT: epithelial–mesenchymal transition
In vivo validation of MEG3-mediated inhibition of cell invasion
To further verify that MEG3 suppresses cell invasion in GHPA by inhibiting β-catenin-regulated EMT, β-catenin was manipulated by overexpressing MEG3 in GH3 cells. The cells were subcutaneously injected into nude mice for tumor formation (Fig. 7A). The sustained efficiency of MEG3 upregulation was verified by detecting the expression level of MEG3 in the xenograft tumor of each mouse via RT-qPCR. Notably, MEG3 level increased >four-fold in the MEG3-overexpressing group compared with the MEG3-oe-vt group and was unaffected by LiCl (Fig. 7B). Consistent with the regulated levels of β-catenin in the formed tumors, E-cadherin increased upon overexpressing MEG3 and further decreased following LiCl treatment. Conversely, the expression levels of N-cadherin, β-catenin, MMP-2, and MMP-9 decreased by elevating MEG3 and increased via LiCl-mediated induction (Figs. 7C and 7D). The results confirmed that MEG3 negatively regulates EMT by downregulating β-catenin.
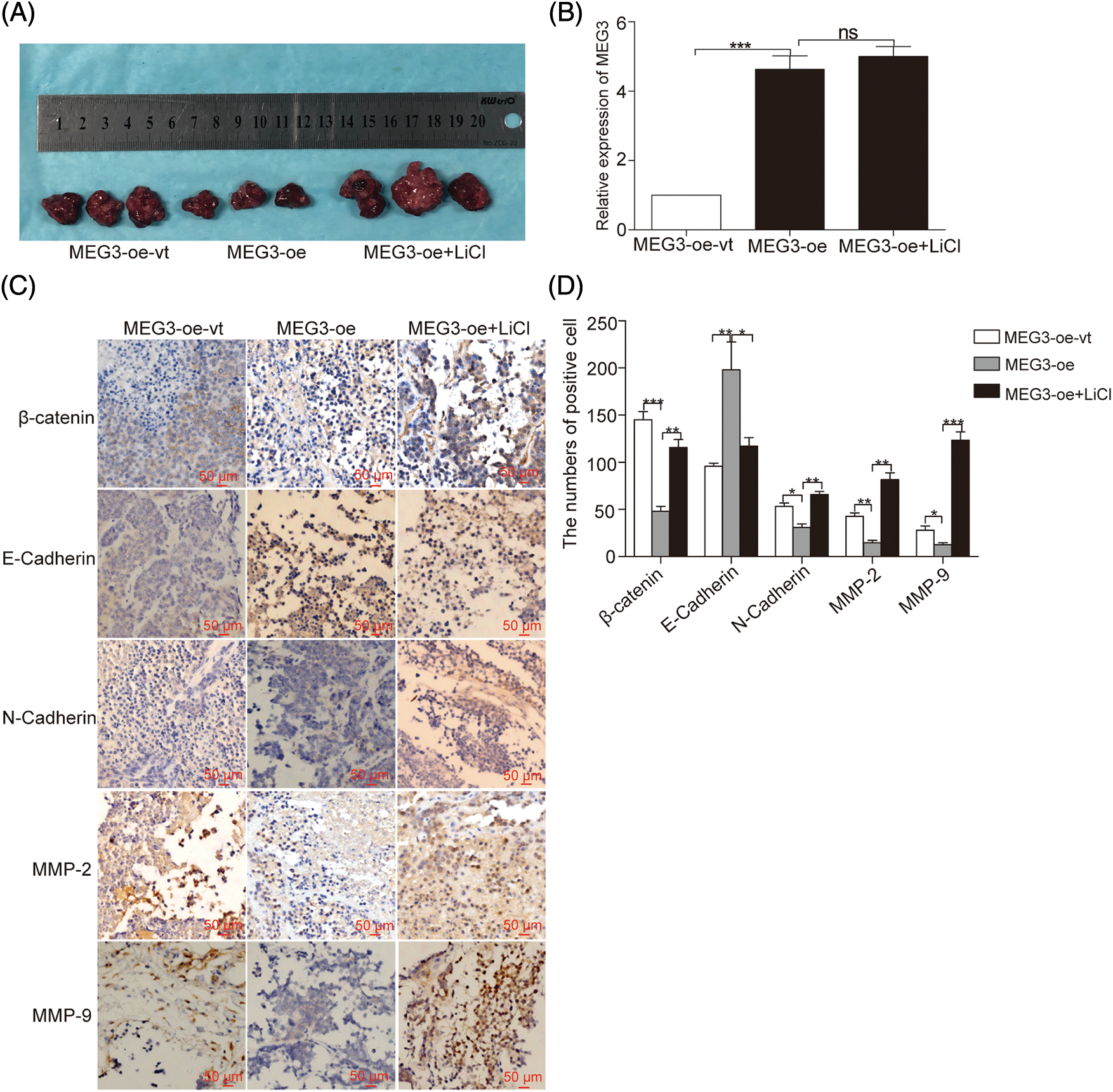
Figure 7: MEG3 inhibits GHPA cell invasion in vivo. A: GH3 cells expressing different levels of MEG3 were subcutaneously injected into null mice followed by LiCl treatment. B: Sustained efficiency of MEG3 upregulation was verified by detecting the expression level of MEG3 in the xenograft tumor of each group via RT-qPCR. C, D: Levels of β-catenin, E-cadherin, N-cadherin, MMP-2, and MMP-9 in tumor tissues were quantified via immunohistochemistry (IHC) (magnification, ×200). *p < 0.05, **p < 0.01, and ***p < 0.001 show statistical significance among the three groups as indicated, and “ns” shows no significance
GHPA is a typically benign tumor with a high incidence and large economic burden and often manifests with invasive growth [30]. Numerous studies have indicated that the presence of point mutations in GNAS strongly reflects the biological characteristics of GHPAs, such as tendency for densely granulated tumors and smaller tumor size [31–33]. A meta-analysis has suggested that GNAS mutations can be used as a prognostic factor for treatment response to somatostatin analogs [34]. A study has suggested that Gsα protein encoded by GNAS is important for activating the cAMP-dependent pathway in pituitary cells for differentiation and proliferation [35]. GNAS mutations were proposed to be involved in the constitutive activation of cAMP, which plays a causal role in pituitary adenomas [36]. However, the poor growth rate of GHPAs naturally expressing GNAS mutations strongly suggest counteraction of the putative growth advantage conferred by cAMP. Consequently, this study aimed to elucidate the effect of GNAS mutations on the GHPA phenotype, especially on tumor invasion, and the mechanism involved.
Interestingly, MEG3 was identified as a tumor suppressor that is a downstream target of cAMP [12,37,38]. Studies have reported that the level of MEG3 is uniquely high in GHPA but not in NFPAs [39]. Thus, we speculate that GNAS mutations suppress the invasiveness of GHPA mainly by activating MEG3. As expected, the high levels of MEG3 were only detected in GHPA-carrying GNAS mutations. MEG3 levels were significantly increased in GH3 cells expressing GNAS mutations compared with GH3 cells expressing wild-type GNAS, suggesting that GNAS mutations inhibit GHPA cell invasion through MEG3 activation. To ascertain the effect of MEG3 on cell invasion, we manipulated MEG3 in GH3 cells. Ectopic expression of MEG3 reduced cell invasion, and conversely, silencing MEG3 enhanced invasiveness. Altogether, our results suggest that MEG3 plays an important role in promoting GHPA invasiveness.
The canonical Wnt/β-catenin signaling pathway is a key regulator of EMT and tumor progression [40]. Upon activation of the Wnt pathway, β-catenin accumulates in nuclei and leads to EMT [17]. In this study, we showed that GNAS mutations lead to increased MEG3 expression but decreased β-catenin expression. The activation of β-catenin by LiCl enhanced cell invasion, and the inactivation of β-catenin by Dkk1 inhibited cell invasion. Subsequently, elevated MEG3 expression led to the downregulation of β-catenin in GHPA cells. In parallel, the silencing of MEG3 upregulated β-catenin expression, suggesting that MEG3 inhibits the invasiveness of GHPA cells by inactivating the Wnt/β-catenin pathway. However, in this study, the mechanism by which MEG3 regulates β-catenin is unclear. Recent studies have shown that MEG3 inhibits pituitary tumor invasiveness through the MIR-376B-3P/HMGA2 axis [41]. Other studies have shown that MEG3 negatively regulates the proliferation, migration, and apoptosis of osteosarcoma through the Wnt/β-catenin pathway by targeting miR-184 [42]. MEG3 suppressed liver cancer cell growth by inhibiting β-catenin through the activation of PKM2 and inactivation of PTEN [15]. The mechanism by which MEG3 regulates β-catenin-mediated transcriptional activation in GHPA must be investigated.
Anterior pituitary with an epithelial phenotype expresses multiple cadherin proteins, such as E-cadherin, that function in cell attachment [43]. EMT is vital for tumor cell invasion and metastasis. The loss of E-cadherin and/or the increase of N-cadherin are hallmarks of EMT. In various tumor types, the Wnt/β-catenin signaling pathway is constitutively active to promote EMT [44]. This study uncovered that β-catenin and EMT-related functional proteins are altered by MEG3 in GHPA with GNAS mutations. Accordingly, overexpressing MEG3 in GHPA cells led to the upregulation of E-cadherin but downregulation of N-cadherin and vimentin by altering the transcriptional regulation of β-catenin.
MMPs—important proteolytic enzymes in the degradation of extracellular matrix and basement membrane—are crucial for tumor cell invasion [45]. MMP-2 and MMP-9 play vital roles in tumor invasion due to their potent ability to degrade collagen type IV [46]. In particular, they play important roles in the invasiveness of GHPA [47]. Consistently, in this study, the levels of MMP-2 and MMP-9 decreased in GHPA tumors with GNAS mutations; additionally, the increase in MEG3 expression in GHPA cells resulted in a decrease in the expression of MMP-2 and MMP-9.
Of note, our findings identify a possible mechanism to explain several known GHPAs phenotypes. Our observations that MEG3 activation in somatotroph cells due to an activating GNAS mutation, concomitantly the inactivation of Wnt/β-catenin pathway and inhibition of EMT may explain distinctive somatotroph adenoma features. However, evidence from other studies suggested that somatotroph cAMP has a dual role in promoting growth hormone secretion while causing DNA damage [35]. These findings may help explain the smaller, less invasive size of GHPAs containing GNAS mutations, both of which secrete higher levels of growth hormone. Likewise, in our present study, we speculate that cAMP also plays a dual role in GNAS mutation–positive adenomas. The different mechanism is that MEG3 may also play an important role in inhibiting tumor proliferation and invasion in addition to DNA damage.
This study revealed that GNAS mutations are associated with the invasiveness of GHPA tumors, possibly by increasing the level of MEG3. MEG3 upregulation in rat GH3 cells suppress cell invasion by inhibiting Wnt/β-catenin signaling. Silencing MEG3 upregulates β-catenin and enhances EMT. Owing to the lack of growth advantage observed in GHPAs with mutant GNAS, the possible mechanisms in this study were elucidated. Moreover, the findings suggest that MEG3 serves as a biomarker for detecting the GHPA phenotype and inhibition of Wnt/β-catenin signaling can be a useful approach for treating GHPA.
Acknowledgement: We thank Dr. Xun Zhang, the Neuroendocrine Unit, Massachusetts General Hospital and Harvard Medical School, Boston, MA, USA for his advice on the structure of this article.
Funding Statement: This work was supported by the Applied Basic Research Programs of Science and Technology Commission Foundation of Jiangsu Province (No. BE2015684).
Author Contributions: The authors confirm contribution to the paper as follows: Chiyuan Ma: Conceptualization, Methodology. Chao Tang: Data curation, Writing-original draft preparation. Chunyu Zhong: Data curation, Investigation. Junhao Zhu: Investigation. Feng Yuan: Investigation, Formal analysis. Jin Yang: Software, Validation. Yong Xu: Writing-reviewing and Editing. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The datasets during and analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval: Approval for the study was obtained from the Ethical Committee of Nanjing Jinling Hospital (2018NZKY-008-02) and informed consent were obtained from all patients who participated in this study. This study was performed according to the ethical standards of the 1964 Helsinki Declaration. The animal experiment protocols were approved by the Institutional Animal Committee of Jinling Hospital.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Melmed, S. (2020). Pituitary-tumor endocrinopathies. The New England Journal of Medicine, 382, 937–950. https://doi.org/10.1056/NEJMra1810772 [Google Scholar] [PubMed] [CrossRef]
2. Fleseriu, M., Langlois, F., Lim, D. S. T., Varlamov, E. V., Melmed, S. (2022). Acromegaly: Pathogenesis, diagnosis, and management. The Lancet Diabetes & Endocrinology, 10, 804–826. https://doi.org/10.1016/S2213-8587(22)00244-3 [Google Scholar] [PubMed] [CrossRef]
3. Park, H. H., Kim, E. H., Ku, C. R., Lee, E. J., Kim, S. H. (2018). Outcomes of aggressive surgical resection in growth hormone-secreting pituitary adenomas with cavernous sinus invasion. World Neurosurgery, 117, e280−e9. [Google Scholar]
4. Melmed, S., Kaiser, U. B., Lopes, M. B., Bertherat, J., Syro, L. V. et al. (2022). Clinical biology of the pituitary adenoma. Endocrine Reviews, 43, 1003–1037. https://doi.org/10.1210/endrev/bnac010 [Google Scholar] [PubMed] [CrossRef]
5. Ershadinia, N., Tritos, N. A. (2022). Diagnosis and treatment of acromegaly: An update. Mayo Clinic Proceedings, 97, 333–346. https://doi.org/10.1016/j.mayocp.2021.11.007 [Google Scholar] [PubMed] [CrossRef]
6. Jung, H., Kim, K., Kim, D., Moon, J. H., Kim, E. H. et al. (2021). Associations of GNAS mutations with surgical outcomes in patients with growth hormone-secreting pituitary adenoma. Endocrinology and Metabolism, 36, 342–350. https://doi.org/10.3803/EnM.2020.875 [Google Scholar] [PubMed] [CrossRef]
7. Hernandez-Ramirez, L. C., Trivellin, G., Stratakis, C. A. (2018). Cyclic 3′,5′-adenosine monophosphate (cAMP) signaling in the anterior pituitary gland in health and disease. Molecular and Cellular Endocrinology, 463, 72–86. https://doi.org/10.1016/j.mce.2017.08.006 [Google Scholar] [PubMed] [CrossRef]
8. Epelbaum, J., Enjalbert, A., Krantic, S., Musset, F., Bertrand, P. et al. (1987). Somatostatin receptors on pituitary somatotrophs, thyrotrophs, and lactotrophs: Pharmacological evidence for loose coupling to adenylate cyclase. Endocrinology, 121, 2177–2185. https://doi.org/10.1210/endo-121-6-2177 [Google Scholar] [PubMed] [CrossRef]
9. Boguslawska, A., Korbonits, M. (2021). Genetics of acromegaly and gigantism. Journal of Clinical Medicine, 10, 1377. https://doi.org/10.3390/jcm10071377 [Google Scholar] [PubMed] [CrossRef]
10. Zhang, X., Zhou, Y., Mehta, K. R., Danila, D. C., Scolavino, S. et al. (2003). A pituitary-derived MEG3 isoform functions as a growth suppressor in tumor cells. The Journal of Clinical Endocrinology and Metabolism, 88, 5119–5126. https://doi.org/10.1210/jc.2003-030222 [Google Scholar] [PubMed] [CrossRef]
11. Tang, C., Zhong, C., Cong, Z., Yang, J., Wen, G. et al. (2019). MEG3 is associated with gsp oncogene regulation of growth hormone hypersecretion, proliferation and invasiveness of human GH-secreting adenomas. Oncology Letters, 17, 3495–3502 [Google Scholar] [PubMed]
12. Zhao, J., Zhang, X., Zhou, Y., Ansell, P. J., Klibanski, A. (2006). Cyclic AMP stimulates MEG3 gene expression in cells through a cAMP-response element (CRE) in the MEG3 proximal promoter region. The International Journal of Biochemistry & Cell Biology, 38, 1808–1820. https://doi.org/10.1016/j.biocel.2006.05.004 [Google Scholar] [PubMed] [CrossRef]
13. Yamamoto, K. K., Gonzalez, G. A., Menzel, P., Rivier, J., Montminy, M. R. (1990). Characterization of a bipartite activator domain in transcription factor CREB. Cell, 60, 611–617. https://doi.org/10.1016/0092-8674(90)90664-Z [Google Scholar] [PubMed] [CrossRef]
14. Li, J., Jiang, X., Li, C., Liu, Y., Kang, P. et al. (2019). LncRNA-MEG3 inhibits cell proliferation and invasion by modulating Bmi1/RNF2 in cholangiocarcinoma. Journal of Cellular Physiology, 234, 22947–22959. https://doi.org/10.1002/jcp.v234.12. [Google Scholar] [CrossRef]
15. Wang, J., Xu, W., He, Y., Xia, Q., Liu, S. (2018). LncRNA MEG3 impacts proliferation, invasion, and migration of ovarian cancer cells through regulating PTEN. Inflammation Research, 67, 927–936. https://doi.org/10.1007/s00011-018-1186-z [Google Scholar] [PubMed] [CrossRef]
16. Nusse, R., Clevers, H. (2017). Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell, 169, 985–999. https://doi.org/10.1016/j.cell.2017.05.016 [Google Scholar] [PubMed] [CrossRef]
17. Alsaab, H. O. (2023). Pathological role of long non-coding (lnc) RNA in the regulation of Wnt/β-catenin signaling pathway during epithelial-mesenchymal transition (EMT). Pathology, Research and Practice, 248, 154566. https://doi.org/10.1016/j.prp.2023.154566 [Google Scholar] [PubMed] [CrossRef]
18. Zheng, Q., Lin, Z., Xu, J., Lu, Y., Meng, Q. et al. (2018). Long noncoding RNA MEG3 suppresses liver cancer cells growth through inhibiting β-catenin by activating PKM2 and inactivating PTEN. Cell Death & Disease, 9, 253. https://doi.org/10.1038/s41419-018-0305-7 [Google Scholar] [PubMed] [CrossRef]
19. Li, P., Gao, Y., Li, J., Zhou, Y., Yuan, J. et al. (2018). LncRNA MEG3 repressed malignant melanoma progression via inactivating Wnt signaling pathway. Journal of Cellular Biochemistry, 119, 7498–7505. https://doi.org/10.1002/jcb.v119.9. [Google Scholar] [CrossRef]
20. Gao, Y., Chen, X., Zhang, J. (2022). LncRNA MEG3 inhibits retinoblastoma invasion and metastasis by inducing β-catenin degradation. American Journal of Cancer Research, 12, 3111–3127 [Google Scholar] [PubMed]
21. Cottier, J. P., Destrieux, C., Brunereau, L., Bertrand, P., Moreau, L. et al. (2000). Cavernous sinus invasion by pituitary adenoma: MR imaging. Radiology, 215, 463–469. https://doi.org/10.1148/radiology.215.2.r00ap18463 [Google Scholar] [PubMed] [CrossRef]
22. Knosp, E., Steiner, E., Kitz, K., Matula, C. (1993). Pituitary adenomas with invasion of the cavernous sinus space: A magnetic resonance imaging classification compared with surgical findings. Neurosurgery, 33, 610–617 [Google Scholar] [PubMed]
23. Goto, Y., Kinoshita, M., Oshino, S., Arita, H., Kitamura, T. et al. (2014). Gsp mutation in acromegaly and its influence on TRH-induced paradoxical GH response. Clinical Endocrinology, 80, 714–719. https://doi.org/10.1111/cen.2014.80.issue-5. [Google Scholar] [CrossRef]
24. Niehrs, C. (2006). Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene, 25, 7469–7481. https://doi.org/10.1038/sj.onc.1210054 [Google Scholar] [PubMed] [CrossRef]
25. Gupta, A., Schulze, T. G., Nagarajan, V., Akula, N., Corona, W. et al. (2012). Interaction networks of lithium and valproate molecular targets reveal a striking enrichment of apoptosis functional clusters and neurotrophin signaling. The Pharmacogenomics Journal, 12, 328–341. https://doi.org/10.1038/tpj.2011.9 [Google Scholar] [PubMed] [CrossRef]
26. Zhang, J., He, L., Yang, Z., Li, L., Cai, W. (2019). Lithium chloride promotes proliferation of neural stem cells in vitro, possibly by triggering the Wnt signaling pathway. Animal Cells and Systems, 23, 32–41. https://doi.org/10.1080/19768354.2018.1487334 [Google Scholar] [PubMed] [CrossRef]
27. Zhuang, X., Zhang, H., Li, X., Li, X., Cong, M. et al. (2017). Differential effects on lung and bone metastasis of breast cancer by Wnt signalling inhibitor DKK1. Nature Cell Biology, 19, 1274–1285. https://doi.org/10.1038/ncb3613 [Google Scholar] [PubMed] [CrossRef]
28. Li, Q., Lai, Q., He, C., Fang, Y., Yan, Q. et al. (2019). RUNX1 promotes tumour metastasis by activating the Wnt/β-catenin signalling pathway and EMT in colorectal cancer. Journal of Experimental & Clinical Cancer Research, 38, 334. https://doi.org/10.1186/s13046-019-1330-9 [Google Scholar] [PubMed] [CrossRef]
29. Ghahhari, N. M., Babashah, S. (2015). Interplay between microRNAs and WNT/β-catenin signalling pathway regulates epithelial-mesenchymal transition in cancer. European Journal of Cancer, 51, 1638–1649. https://doi.org/10.1016/j.ejca.2015.04.021 [Google Scholar] [PubMed] [CrossRef]
30. Dhaneshwar, S., Shandily, S., Tiwari, V. (2023). Growth hormone excess: Implications and management. Endocrine, Metabolic & Immune Disorders Drug Targets, 23, 748–763. https://doi.org/10.2174/1871530322666221012155533 [Google Scholar] [PubMed] [CrossRef]
31. Landis, C. A., Harsh, G., Lyons, J., Davis, R. L., McCormick, F. et al. (1990). Clinical characteristics of acromegalic patients whose pituitary tumors contain mutant Gs protein. The Journal of Clinical Endocrinology and Metabolism, 71, 1416–1420. https://doi.org/10.1210/jcem-71-6-1416 [Google Scholar] [PubMed] [CrossRef]
32. Spada, A., Arosio, M., Bochicchio, D., Bazzoni, N., Vallar, L. et al. (1990). Clinical, biochemical, and morphological correlates in patients bearing growth hormone-secreting pituitary tumors with or without constitutively active adenylyl cyclase. The Journal of Clinical Endocrinology and Metabolism, 71, 1421–1426. https://doi.org/10.1210/jcem-71-6-1421 [Google Scholar] [PubMed] [CrossRef]
33. Spada, A., Arosio, M., Bassetti, M., Vallar, L., Clementi, E. et al. (1991). Mutations in the alpha subunit of the stimulatory regulatory protein of adenylyl cyclase (Gs) in human GH-secreting pituitary adenomas. Biochemical, clinical, and morphological aspects. Pathology, Research and Practice, 187, 567–570. https://doi.org/10.1016/S0344-0338(11)80145-7 [Google Scholar] [PubMed] [CrossRef]
34. Efstathiadou, Z. A., Bargiota, A., Chrisoulidou, A., Kanakis, G., Papanastasiou, L. et al. (2015). Impact of gsp mutations in somatotroph pituitary adenomas on growth hormone response to somatostatin analogs: A meta-analysis. Pituitary, 18, 861–867. https://doi.org/10.1007/s11102-015-0662-5 [Google Scholar] [PubMed] [CrossRef]
35. Ben-Shlomo, A., Deng, N., Ding, E., Yamamoto, M., Mamelak, A. et al. (2020). DNA damage and growth hormone hypersecretion in pituitary somatotroph adenomas. The Journal of Clinical Investigation, 130, 5738–5755. https://doi.org/10.1172/JCI138540 [Google Scholar] [PubMed] [CrossRef]
36. Mantovani, G., Lania, A. G., Spada, A. (2010). GNAS imprinting and pituitary tumors. Molecular and Cellular Endocrinology, 326, 15–18. https://doi.org/10.1016/j.mce.2010.04.009 [Google Scholar] [PubMed] [CrossRef]
37. Zhang, X., Gejman, R., Mahta, A., Zhong, Y., Rice, K. A. et al. (2010). Maternally expressed gene 3, an imprinted noncoding RNA gene, is associated with meningioma pathogenesis and progression. Cancer Research, 70, 2350–2358. https://doi.org/10.1158/0008-5472.CAN-09-3885 [Google Scholar] [PubMed] [CrossRef]
38. Ma, J., Li, T. F., Han, X. W., Yuan, H. F. (2019). Downregulated MEG3 contributes to tumour progression and poor prognosis in oesophagal squamous cell carcinoma by interacting with miR-4261, downregulating DKK2 and activating the Wnt/beta-catenin signalling. Artificial Cells, Nanomedicine, and Biotechnology, 47, 1513–1523. https://doi.org/10.1080/21691401.2019.1602538 [Google Scholar] [PubMed] [CrossRef]
39. Gejman, R., Batista, D. L., Zhong, Y., Zhou, Y., Zhang, X. et al. (2008). Selective loss of MEG3 expression and intergenic differentially methylated region hypermethylation in the MEG3/DLK1 locus in human clinically nonfunctioning pituitary adenomas. The Journal of Clinical Endocrinology and Metabolism, 93, 4119–4125. https://doi.org/10.1210/jc.2007-2633 [Google Scholar] [PubMed] [CrossRef]
40. Krishnamurthy, N., Kurzrock, R. (2018). Targeting the Wnt/β-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treatment Reviews, 62, 50–60. https://doi.org/10.1016/j.ctrv.2017.11.002 [Google Scholar] [PubMed] [CrossRef]
41. Zhu, D., Xiao, Z., Wang, Z., Hu, B., Duan, C. et al. (2020). MEG3/MIR-376B-3P/HMGA2 axis is involved in pituitary tumor invasiveness. Journal of Neurosurgery, 134, 499–511. [Google Scholar]
42. Li, L., Pei, S., Sun, N. (2020). MEG3 targets miR-184 and Wnt/beta-catenin and modulates properties of osteosarcoma. Frontiers in Bioscience, 25, 1901–1912. https://doi.org/10.2741/4884 [Google Scholar] [PubMed] [CrossRef]
43. Fougner, S. L., Lekva, T., Borota, O. C., Hald, J. K., Bollerslev, J. et al. (2010). The expression of E-cadherin in somatotroph pituitary adenomas is related to tumor size, invasiveness, and somatostatin analog response. The Journal of Clinical Endocrinology and Metabolism, 95, 2334–2342. https://doi.org/10.1210/jc.2009-2197 [Google Scholar] [PubMed] [CrossRef]
44. Liang, Z., Lu, L., Mao, J., Li, X., Qian, H. et al. (2017). Curcumin reversed chronic tobacco smoke exposure induced urocystic EMT and acquisition of cancer stem cells properties via Wnt/β-catenin. Cell Death & Disease, 8, e3066. https://doi.org/10.1038/cddis.2017.452 [Google Scholar] [PubMed] [CrossRef]
45. di Nezza, L. A., Misajon, A., Zhang, J., Jobling, T., Quinn, M. A. et al. (2002). Presence of active gelatinases in endometrial carcinoma and correlation of matrix metalloproteinase expression with increasing tumor grade and invasion. Cancer, 94, 1466–1475. https://doi.org/10.1002/cncr.v94:5. [Google Scholar] [CrossRef]
46. Scheau, C., Badarau, I. A., Costache, R., Caruntu, C., Mihai, G. L. et al. (2019). The role of matrix metalloproteinases in the epithelial-mesenchymal transition of hepatocellular carcinoma. Analytical Cellular Pathology, 2019, 9423907. [Google Scholar]
47. Yang, Q., Li, X. (2019). Molecular network basis of invasive pituitary adenoma: A review. Frontiers in Endocrinology, 10, 7. https://doi.org/10.3389/fendo.2019.00007 [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools