
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW
The role of cholesterol metabolism in lung cancer
1 Department of Thoracic Oncology and State Key Laboratory of Biotherapy, Cancer Center, West China Hospital, Sichuan University, Chengdu, 610041, China
2 West China School of Medicine, Sichuan University, Chengdu, 610041, China
3 Department of Postgraduate Students, West China School of Medicine, Chengdu, 610041, China
4 Department of Biotherapy, Cancer Center, West China Hospital, Sichuan University, Chengdu, 610041, China
* Corresponding Authors: QIN ZHANG. Email: ; HUASHAN SHI. Email:
# Contributed equally to this study
Oncology Research 2024, 32(10), 1613-1621. https://doi.org/10.32604/or.2024.047933
Received 22 November 2023; Accepted 04 March 2024; Issue published 18 September 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Elevated serum cholesterol metabolism is associated with a reduced risk of lung cancer. Disrupted cholesterol metabolism is evident in both lung cancer patients and tumor cells. Inhibiting tumor cell cholesterol uptake or biosynthesis pathways, through the modulation of receptors and enzymes such as liver X receptor and sterol-regulatory element binding protein 2, effectively restrains lung tumor growth. Similarly, promoting cholesterol excretion yields comparable effects. Cholesterol metabolites, including oxysterols and isoprenoids, play a crucial role in regulating cholesterol metabolism within tumor cells, consequently impacting cancer progression. In lung cancer patients, both the cholesterol levels in the tumor microenvironment and within tumor cells significantly influence cell growth, proliferation, and metastasis. The effects of cholesterol metabolism are further mediated by the reprogramming of immune cells such as T cells, B cells, macrophages, myeloid-derived suppressor cells, among others. Ongoing research is investigating drugs targeting cholesterol metabolism for clinical treatments. Statins, targeting the cholesterol biosynthesis pathway, are widely employed in lung cancer treatment, either as standalone agents or in combination with other drugs. Additionally, drugs focusing on cholesterol transportation have shown promise as effective therapies for lung cancer. In this review, we summarized current research regarding the rule of cholesterol metabolism and therapeutic advances in lung cancer.Keywords
In recent years, a growing number of studies [1] have highlighted the significant impact of body metabolism on the initiation and progression of tumors. Tumor cells and immune cells exhibit rapid reprogramming in the tumor microenvironment (TME), influenced by the levels of metabolic substances such as carbohydrates, proteins, and lipids [2]. Furthermore, there has been increasing attention to the role of cholesterol metabolism in cancer. A previous study has demonstrated a correlation between serum cholesterol levels and progression, treatment effect and prognosis of cancer. Another study conducted in Australia suggested that higher total serum cholesterol was associated with a reduced risk of total cancer in men (hazard ratio [HR] = 0.94; 95% confidence interval, 95% [CI]: 0.88-1.00) [3]. This association may be attributed to the impact of preclinical cancer effect on cholesterol metabolism. Cancers in preclinical stages have the potential to impede the metabolism of serum cholesterol, leading to a decrease in serum cholesterol levels. For instance, chronic liver disease, a precursor to liver cancer, can impede cholesterol evacuation from tumor cells due to hepatic inadequacy, resulting in a reduction of serum cholesterol levels.
Lung cancer is one of the types with the highest occurrence and death rate, contributing to a 20% death rate associated with cancer [4]. In recent years, there has been significant attention on the relationship between cholesterol metabolism and lung cancer. Findings from a Korean cohort study indicated a negative correlation between serum cholesterol level and incidence rate of lung cancer in men (HR, 0.89, 95% CI, 0.82-0.96; p < 0.001), though no such correlation was observed in women, which may due to different lifestyle factors including smoking and eating habits [5]. Data from this large prospective study revealed that total cholesterol levels exhibit varying associations with cancer, depending on the specific cancer site. In patients with lung cancer, total cholesterol, low-density lipoprotein-cholesterol (LDL-C) and high-density lipoprotein-cholesterol (HDL-C) were significantly lower compared to the non-cancer group [6]. However, the underlying mechanisms were not elucidated in these two studies. Notably, in patients treated with immune checkpoint inhibitor therapy, an increase in total cholesterol was linked to longer overall survival (OS) (p = 0.036) [7]. Another study indicated that individuals with high serum cholesterol levels (>200 mg/dL) experienced longer OS (19.4 months vs. 5.5 months, p = 0.001) [8]. Cholesterol plays an important role in immunomodulation, functioning as both an energy resource and a component of cell membranes. It is indispensable for the activation of immune cells, including T cells, natural killer, macrophages, and others. Reduced cholesterol levels inhibit the proliferation and activation of T cells. The aim of this review is to uncover the role of cholesterol metabolism in lung cancer, encompassing the effects of cholesterol metabolic and transport pathways on lung cancer, the roles of cholesterol metabolites in lung cancer, the effects of cholesterol metabolism on immune cells associated with lung cancer, and potential strategies targeting cholesterol metabolism for lung cancer treatment.
Overview of cholesterol metabolism in tumor cells
Lipids play a crucial role in cellular functions. Among them, cholesterol, a 27-carbon-lipid, is essential for cellular metabolism and tumor cells. Cholesterol serves as a vital component of the cell membrane, influencing material transport, regulating membrane fluidity and stability, and participating in signal transduction. The intricate process of cholesterol metabolism involves exogenous uptake, biosynthesis, excretion, and storage [9]. Cholesterol is derived from both exogenous uptake and biosynthesis. To minimize energy consumption in cholesterol biosynthesis, cells express various receptors to facilitate the transport of exogenous cholesterol. In the human body, cholesterol is primarily transported by low-density lipoprotein (LDL) and high-density lipoprotein (HDL). Following absorption in the gastrointestinal tract, cholesterol from food is transported as chylomicrons to the liver and then distributed throughout the body via LDL. Inside cells, LDL is transported by LDL receptors (LDLR) and undergoes hydrolysis into fatty acids and cholesterol by lysosomal enzymes, such as lysosomal acid lipase (LAL) [10]. Evidence indicates that elevated cholesterol levels in colon cancer cells may promote cancer progression [11]. In TME, HDL can reduce cholesterol levels in tumor cells, thereby reversing tumor immune escape and inhibiting angiogenesis [12]. The elimination of LAL suppresses immune rejection and permits the growth of human lung cancer cells in LAL-deficient mice, highlighting the essential role of LAL homeostasis in maintaining antitumor immunity [13]. Endogenous cholesterol synthesis initiates with acetyl CoA, progressing to the formation of mevalonic acid through the action of 3-hydroxy-3-methylglutarate monoacyl CoA (HMG-CoA), and eventually results in cholesterol through more than 20 enzymes. The pivotal enzyme in this process is 3-Hydroxy-3-methylglutary coenzyme A reductase (HMGCR), serving as the first-rate limiting enzyme in cholesterol biosynthesis. Statin drugs exert their effects on the growth of tumor cells by dysregulating the activity of this enzyme [14]. The key regulator of cholesterol metabolism, SREBP2, upregulates the expression of HMGCR and other enzymes in response to low cellular cholesterol levels, thereby enhancing endogenous cholesterol uptake. Cholesterol metabolism also involves excretion and storage. Under the influence of Acyl coenzyme A-cholesterol acyltransferase (ACAT1), excess cholesterol can be esterified into cholesterol esters and then stored within cells as lipid droplets [15].
Conversely, intracellular cholesterol accumulation, particularly hydroxysteroid, can activate LXR [16]. This activation facilitates the removal of excess cholesterol from cells via ATP-binding cassette transporter A1 (ABCA1), and G1 (ABCG1). Subsequently, cholesterol is transported to the liver by HDL, where it is either converted into bile acid or directly discharged from the intestine through bile [17]. The removal of LXR can lead to cholesterol accumulation in tumor cells, contributing to a subtype of non-small cell lung carcinoma (NSCLC) [18].
In conclusion, there are four metabolic pathways of cholesterol in both normal cells and tumor cells. However, mechanisms have been discovered to enhance cholesterol uptake and biosynthesis in tumor cells, while cholesterol removal is weakened leading to an accumulation of cholesterol in tumor cells, including lung cancer, compared to normal tissue. The detailed mechanisms are illustrated below.
The impact of cholesterol metabolism and transport pathways on lung cancer
The cholesterol levels in tumor cells are generally recognized to be higher than in other tissues [19], indicating that factors involved in cholesterol metabolism and transport pathways can impact the progression of cancer. Favorably, factors that promote cholesterol uptake, biosynthesis and accumulation in tumor cells tend to support cancer progression. Conversely, components involved in mediating cholesterol efflux can inhibit cancer development. LXRs serve as key transcriptional regulators in cholesterol homeostasis [19]. Indeed, cholesterol accumulation in tumor cells can activate LXRs, thereby promoting cholesterol efflux via ABCA1 and ABCG1 [16]. Studies have demonstrated that LXR stimulation disrupts the AKT survival pathway, inducing apoptosis of tumor cells [20]. Interestingly, this apoptotic effect can be reversed by the addition of exogenous cholesterol. Conversely, inhibiting LXRs in tumor cells has been shown to decrease the expression of glycolytic and lipogenic genes, leading to apoptosis of tumor cells [21]. Inactivation of LXR genes results in cholesterol accumulation in lung cells, contributing to lung squamous cell carcinoma occurrence after 14 months [22]. This underscores the pivotal role of LXRs in preventing epithelial hyperplasia. Moreover, LXRαβ-/-mice develop a subtype of NSCLC at 18 months, characterized by a high proliferation rate of tumor cells [18]. A clinical study showed that higher LXRα levels correlate with significantly increased 5-year tumor-related survival rates in NSCLC patients (54% vs. 32%) [23]. ABCG1 promotes cholesterol efflux from tumor cells, thereby regulating intracellular cholesterol homeostasis [24], which is also a vital mediator of LXRs effects (Fig. 1). The upregulation of ABCG1 in lung cancer is associated with increased cancer cell proliferation, migration, and invasion, while inhibition by betulinic acid leads to G1 phase arrest, decreased proliferation, and migration potential of lung cancer cells [25,26].
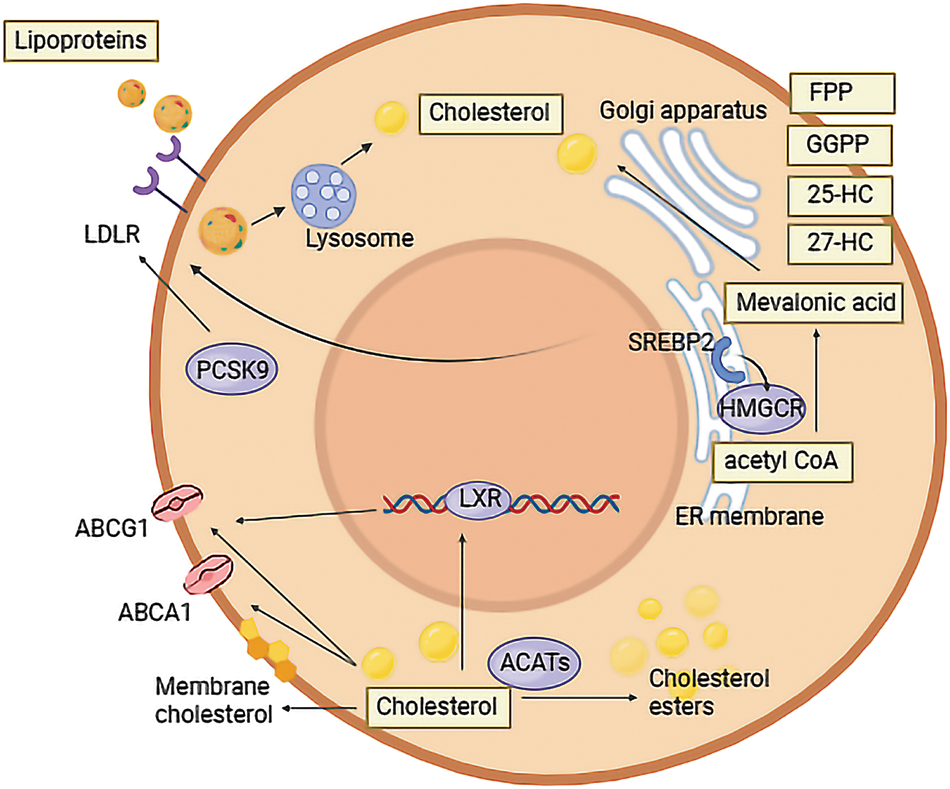
Figure 1: Cholesterol metabolism in tumor cells. In the form of LDL, cholesterol can be transported into tumor cells by LDLR, and then hydrolyzed into cholesterol. PCSK9 in tumor cells is a regulator of LDLR. Biosynthesis of cholesterol in tumor cells begins with acetyl CoA. It can be transformed into cholesterol by various enzymes, of which HMGCR is the most important. 25-HC, 27-HC, FPP and GGPP are metabolites of cholesterol biosynthesis. SREP2 can regulate HMGCR and LDLR expression in tumor cells. Apart from being esterified by ACATs and stored in cells, cholesterol can also be excreted by ABCG1 and ABCA1, under the regulation of LXR when cholesterol accumulates in tumor cells.
Additionally, LDL plays a crucial role in facilitating cholesterol uptake by tumor cells, serving as a transporter of cholesterol from the liver to various tissues, including tumors. When cholesterol levels in tumor cells are low, SPEBP2 not only stimulates cholesterol biosynthesis but also promotes LDLR expression, thereby increasing cholesterol uptake. Research suggests that lower serum LDL cholesterol levels (<70 mg/dL) are associated with a significantly higher risk of lung cancer [27] and serum LDL levels are markedly lower in lung cancer patients compared to the control group. The pro-protein convertase subtilisin/kexin 9 (PCSK9) is a key regulator of cholesterol transport via LDL [28]. As a member of the pro-protein convertase family, PCSK9 can bind to LDLR, accelerating its degradation by lysosomes and consequently decreasing cholesterol uptake [29]. Research indicates that PCSK9 inhibitors can control the progression of non-cardiovascular diseases, including cancer [19]. This therapeutic effect may arise from the removal of the inhibitory impact on cholesterol uptake by tumor cells. In NSCLC, lower serum PCSK9 levels (<95 ng/mL) are associated with higher OS [30]. Moreover, NSCLC patients with a lower baseline serum PCSK9 exhibit longer progression-free survival and a better response to ICIs [31]. In lung adenocarcinoma (LAC) cells, transfection with PCSK9 small interfering (si) RNA induces mitochondrial dysfunction and apoptosis in tumor cells [32].
Roles of cholesterol metabolites in lung cancer
Cholesterol derivatives, such as 25-hydroxy cholesterol (25-HC), 22-hydroxy cholesterol, 24-hydroxy cholesterol, and 27-hydroxy cholesterol (27-HC) have been identified as influential factors in cancer development [33]. Specifically, 25-HC and 27-HC are oxysterols generated as byproducts of cholesterol metabolism in various organs, including the lung [34]. Their significance in the progression of lung cancer has been established. Oxysterols contribute to the pro-cancerous effects of lung cancer through mechanisms such as inflammation or interactions with oxysterol-binding proteins [35]. Among oxysterols, 27-HC is the most prevalent type [36] and has been associated with the progression and metastasis of various tumors, including lung cancer [37]. In LAC, 27-HC is linked to elevated cholesterol levels and metastasis [38]. Mechanistically, 27-HC maintains cholesterol balance by promoting cholesterol execration via LXR and suppressing cholesterol production through the Insulin-induced Genes (INSIGs) [39]. Additionally, 27-HC can enhance the growth of end-stage cancers, potentially due to an imbalance in pro-survival and pro-inflammatory signals. Its effects involved the regulation of NFκB/PPIB axis and the secretion of FGF2 and IL-6. Research has shown that 27-HC can enhance osteoclast differentiation in conditioned media from LAC cells co-cultured with macrophages. This effect is induced by inhibiting miR-139 expression and activating the STAT3/c-Fos/NFATc1 pathway, ultimately leading to the inhibition of bone metastasis in LAC [40]. Additionally, 27-HC can inhibit cholesterol synthesis by suppressing HMGCR [41] and SREBP2 [42], consequently reducing cholesterol levels in tumor cells. 27-HC has been reported to have the ability to inhibit lung cancer progression, however, contrasting findings suggest that 25-HC can enhance it. 25-HC can decrease the sensitivity of tumor cells to 5-fluorouracil (5-FU), resulting in an increase in cell proliferation and decreased cell apoptosis when 5-FU is introduced [43]. Additionally, 25-HC has been shown to promote distant lung metastasis of tumor cells in vivo. As a ligand of LXR, 25-HC may impact the progression of lung cancer [44]. 25-HC was proven to promote the progression of LAC cells [45]. In the monoculture system, 25-HC promoted LAC cell migration and invasion in an LXR-dependent manner without affecting cell proliferation. However, in the co-culture system, 25-HC could stimulate interleukin-1β (IL-1β) secretion and increase the expression of LXR and Snail, enhancing the effect of 25-HC in an LXR-independent manner.
Isoprenoids, such as GGPP and farnesyl diphosphate (FPP), are also metabolites of cholesterol metabolism [46]. Cellular dysregulation in isoprenoid metabolism can be a contributing factor to cancer [47]. Geranylgeranyl diphosphate synthase (GGPPS) is the enzyme responsible for synthesizing GGPP from FPP, impacting the relative levels of FPP and GGPP in tumor cells [48]. Research has demonstrated a significantly higher expression of GGPPS in LAC tissues compared to adjacent normal tissues [49]. Moreover, the knockdown of GGPPS has been shown to inhibit the migration and invasion of LAC through the regulation of EMT due to the depletion of GGPP, while it does not affect cell proliferation and apoptosis [50].
The effects of cholesterol metabolism on immune cells associated with lung cancer
During the progression of lung cancer, tumor cells continuously interact with surrounding stromal cells, including various immune cells, to form TME, exhibiting unique metabolic characteristics [51]. Tumor-infiltrating immune cells (TIICs) are key participants in TME, regulating the growth of tumor cells and displaying either anti-tumor or tumor-promoting functions [52]. Key components of TIICs include T cells, B cells, tumor-associated macrophages (TAMs), dendritic cells (DC), myeloid-derived suppressor cells (MDSCs), neutrophils and natural killer (NK) cells [53]. Cholesterol metabolism in the TME not only affects the functions of tumor cells, but can also reprogram the activities of TIICs, influencing immune functions.
MDSCs are heterogeneous, immature cell populations derived from the bone marrow that exert a negative effect on immune responses in autoimmune diseases, cancer, and chronic infections. They also play a crucial regulatory role in the invasion and metastasis of tumor cells, as well as in the generation of tumor blood vessels [54]. In the context of lung cancer, MDSCs, through interactions with inherent defense mechanisms, can cause indirect inflammatory damage to promote the progression of lung cancer. They can also restrain the anti-tumor ability of T cells. Research has shown that high serum cholesterol levels induce the generation and accumulation of MDSCs, promoting the rapid growth and spontaneous metastasis of primary tumors, including lung cancer, by inhibiting the activation of CD8+ T cells [55]. TAMs are an important class of natural immune cells in the TME. In lung cancer, TAMs can be divided into classic activation (M1) and alternative activation (M2) macrophages, exhibiting anti-tumor or tumor-promoting characteristics. TAMs can promote lung cancer progression through mechanisms such as promoting tumor angiogenesis, tumor cell metastasis, and immune escape [56]. In a study of LAC, there was a significant accumulation of cholesterol within tumor tissue. Interestingly, the cholesterol levels were found to be reduced in TAMs isolated from LAC tissues compared to adjacent normal tissues [57]. This indicates an opposite dysregulation of cholesterol homeostasis between tumor cells and TAMs. The research further suggests that cholesterol depletion in TAMs can have a distinct impact on macrophage gene expression, promoting angiogenesis, growth, and metastasis [57].
Cholesterol metabolism can affect other immune cells in lung cancer. Research indicates that a high serum cholesterol level can impair the anti-tumor ability of CD8+ T cells, and cholesterol accumulation in TME can induce cholesterol depletion in CD8+ T cells, compromising their anti-tumor functions [55]. In contrast, suppressing cholesterol esterification in T cells by inhibition of ACAT1 can increase the cholesterol level in the plasma membrane of CD8+ T cells. Consequently, this enhancement can promote the proliferation and signaling of CD8+ T cells, thereby controlling the progression of lung cancer [58]. Regulatory B cells restrict immune and inflammatory response in lung cancer. Research has shown that the synthesis of cholesterol metabolic intermediate GGPP is crucial for IL-10 production, which mediates the function of regulatory B cells [59]. NK cells, being cytotoxic lymphocytes of the innate immune system, possess the ability to eliminate tumor cells. Elevated serum cholesterol levels have been linked to an increase in the numbers of NK cells and the expression of NK cell-activating receptors, resulting in fewer tumor metastases in the lung [15]. The interplay between cholesterol levels and the immune response involving regulatory B cells and NK cells highlights the complexity of the immune landscape in the context of lung cancer.
Targeting cholesterol metabolism for lung cancer strategies
It is well-established that alterations in cholesterol metabolism within tumor cells are considered one of the emerging hallmarks of cancer [60]. In recent years, there has been growing research into targeting cholesterol synthesis and transport processes to inhibit the occurrence and progression of various cancers, including lung cancer. These strategies primarily focus on the cholesterol biosynthesis pathway, particularly through HMGCR, such as statin drugs, and the transportation or delivery of cholesterol-modifying drugs.
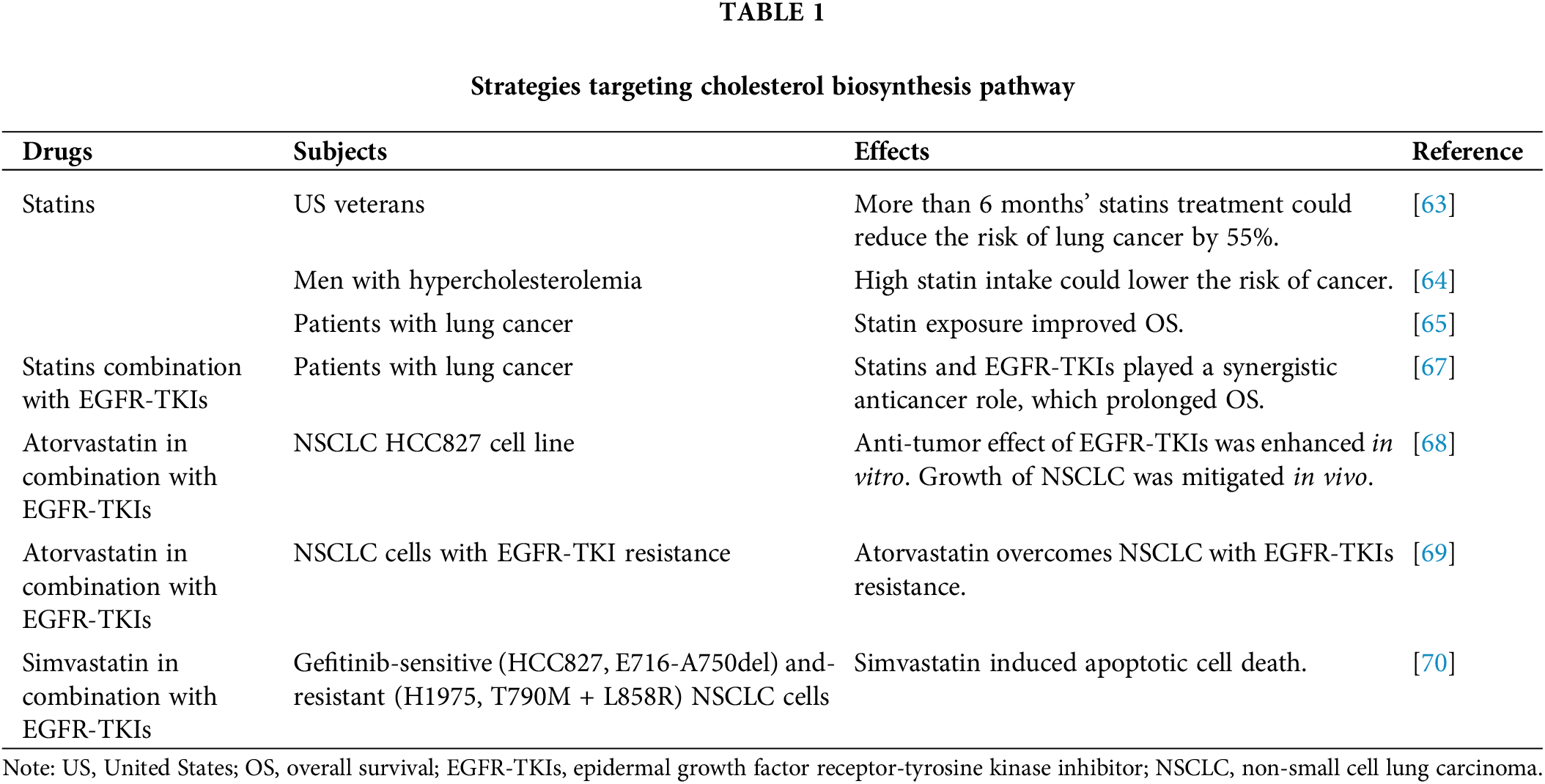
Statins have been widely recognized as a safe and effective clinical anti-tumor approach. These drugs can inhibit HMGCR, a key enzyme involved in cholesterol biosynthesis. Consequently, they can lower LDL-C levels and elevate HDL-C levels. Originally identified as a fungal metabolite [61], statins gained prominence due to their early use as cholesterol-lowering drugs with minimal adverse effects. This led to their swift adoption in clinical oncology, including strategies for managing lung cancer [62]. In the realm of clinical therapy, research has shown that individuals receiving statins for more than 6 months before a lung cancer diagnosis had a 55% reduced risk of developing lung cancer [63]. In men with hypercholesterolemia, a high intake of statins is associated with a lower risk of lung cancer [64]. Furthermore, There is evidence that statin exposure is associated with significantly improved OS among patients with lung cancer [65]. Apart from the impact of cholesterol metabolism in tumor cells, statins were also found effective in regulating lung cancer TME in order to inhibit cancer progress and enhance treatment effects. Statins could reverse immuo-cold (tumors with limited tumor-infiltrating) TME to immuno-hot via cholesterol synthesis pathways in NSCLC, which significantly enhanced ICB efficacy [65].
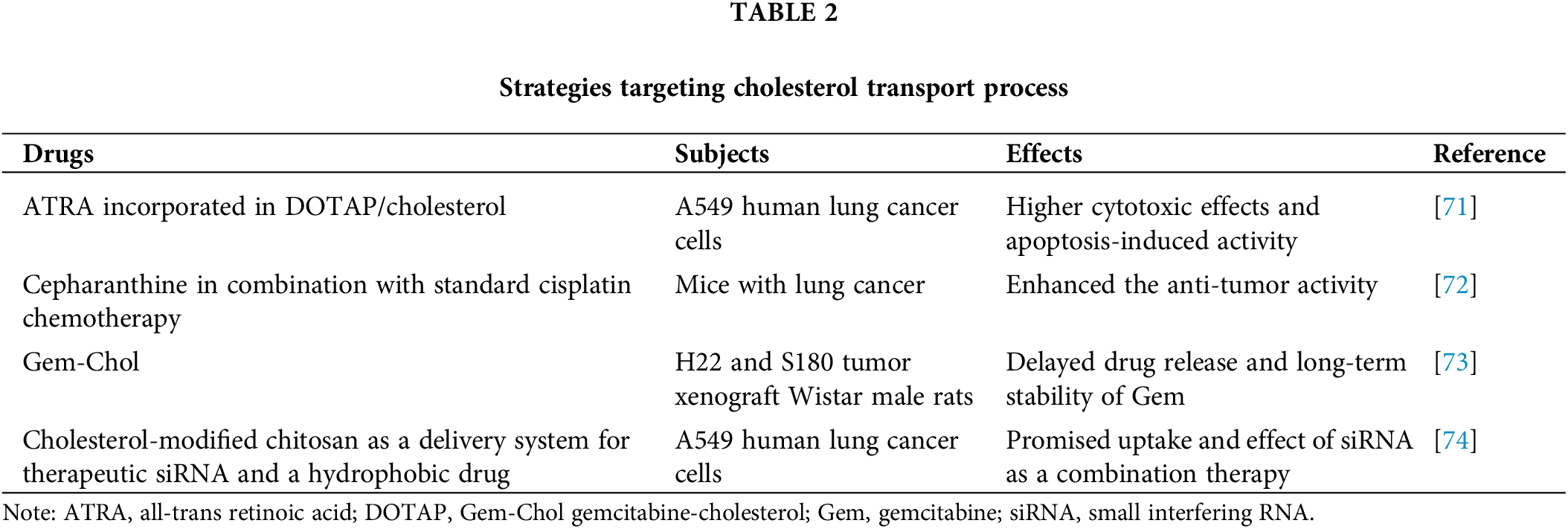
The potential of combining statins with other anti-cancer therapies has also been explored [66], and statins have been found to be effective in overcoming resistance to epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs). The combination of statins with EGFR-TKIs therapy has been associated with prolonged OS in patients with lung cancer, indicating a synergistic anti-cancer effect [67]. Moreover, the combination of atorvastatin with EGFR-TKIs in the treatment of NSCLC demonstrated an enhanced anti-tumor effect in vitro, along with a mitigated growth of NSCLC in vivo [68]. Another study has shown that atorvastatin exhibits anti-tumor properties in NSCLC and can overcome resistance to EGFR-TKIs [69]. In NSCLC cells harboring an EGFR-resistant mutation, simvastatin could induce apoptotic cell death [70] (Table 1). Various drugs targeting the cholesterol transport process have shown promising results. For instance, when incorporated into cationic liposomes composed of DOTAP/cholesterol, all-trans retinoic acid (ATRA) exhibited significantly higher cytotoxic effects and apoptosis-induced activity [71]. Cepharanthine has been identified as an inhibitor of endolysosomal trafficking of cholesterol and LDL, enhancing the anti-tumor activity of the standard chemotherapy cisplatin in lung cancer [72]. Cholesterol can also serve as a medium for delivering drugs to lung cancer cells to promote anti-tumor effects. When a cholesterol-modified derivative of gemcitabine (Gem) is synthesized (Gem-Chol), it delays drug release and provides long-term stability, thereby improving the anti-tumor effect of Gem in the treatment of NSCLC [73]. In another study, a cholesterol-modified low molecular weight chitosan was employed as a delivery system for therapeutic small interfering RNA (siRNA) and a hydrophobic drug. This approach efficiently facilitated the uptake of promising siRNA by lung cancer as part of combination cancer therapy [74] (Table 2).
In recent years, there has been a growing recognition of the significance of cholesterol metabolism in the context of lung cancer investigations. The pathways involving the uptake, biosynthesis, excretion, and storage of cholesterol play a pivotal role in the initiation and progression of lung cancer. Despite a positive correlation between higher serum cholesterol levels and a lower incidence rate of lung cancer, the cholesterol levels within lung tumor cells have been found to be significantly elevated compared to adjacent normal cells. Strategies aimed at reducing cholesterol levels in tumor cells, such as inhibiting cholesterol uptake or biosynthesis and promoting excretion, have demonstrated the potential to decrease the incidence and progression of lung cancer. Additionally, the modulation of molecules along cholesterol metabolism pathways and metabolites can exert an influence on the growth and proliferation of lung cancer cells. In the systemic environment of lung cancer, cholesterol metabolism can also impact the functionality of immune cells in TME. While there is abundant evidence concerning the impact of cholesterol metabolism on lung cancer, details of the exact function of these effects and clinical applications are not fully demonstrated. Based on these findings, researchers are encouraged to further investigate more cholesterol metabolic targets to dysregulate the lipid balance in lung cancer cells, as well as the preclinical and clinical utility of these components in lung cancer. A growing body of evidence suggests that drugs targeting the cholesterol metabolism process have the capability to inhibit the progress of lung cancer. It is possible to develop more effective drugs based on cholesterol metabolism in tumor cells, and their clinical applications could be advantageous as lung cancer treatments. Moreover, as tumor cells constantly interact with TME, further investigations on cholesterol levels in TME, and targeted therapies, such as limiting cholesterol accumulation in TME, could be clinically beneficial.
Collectively, a deeper understanding of cholesterol metabolism and its effects on both tumor cells and immune cells within the TME could pave the way for innovative metabolic therapies to combat cancer.
Acknowledgement: Some data were obtained from the clinical database of palliative care for advanced lung cancer. We would like to thank the Cancer Psychology and Health Management Committee of the Sichuan Cancer Society for their guidance.
Funding Statement: No funding.
Author Contributions: Weigang Xiu designed the study. Weigang Xiu, Xingyu Liu, Kaixin Hu, Qin Zhang, Huashan Shi conducted the experiments and analyzed the data. Xingyu Liu wrote the manuscript. All authors contributed to the article and approved the submitted version.
Availability of Data and Materials: The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Al-Zoughbi, W., Al-Zhoughbi, W., Huang, J., Paramasivan, G. S., Till, H. et al. (2014). Tumor macroenvironment and metabolism. Seminars in Oncology, 41(2), 281–295. https://doi.org/10.1053/j.seminoncol.2014.02.005. [Google Scholar] [PubMed] [CrossRef]
2. Sung, H., Siegel, R. L., Torre, L. A., Pearson-Stuttard, J., Islami, F. et al. (2019). Global patterns in excess body weight and the associated cancer burden. CA: A Cancer Journal for Clinicians, 69(2), 88–112. [Google Scholar] [PubMed]
3. Strohmaier, S., Edlinger, M., Manjer, J., Stocks, T., Bjørge, T. et al. (2013). Total serum cholesterol and cancer incidence in the metabolic syndrome and cancer project (Me-Can). PLoS One, 8(1), e54242. https://doi.org/10.1371/journal.pone.0054242. [Google Scholar] [PubMed] [CrossRef]
4. Roointan, A., Ahmad Mir, T., Ibrahim Wani, S., Mati Ur, R., Hussain, K. K. et al. (2019). Early detection of lung cancer biomarkers through biosensor technology: A review. Journal of Pharmaceutical and Biomedical Analysis, 164, 93–103. https://doi.org/10.1016/j.jpba.2018.10.017. [Google Scholar] [PubMed] [CrossRef]
5. Kitahara, C. M., Berrington de González, A., Freedman, N. D., Huxley, R., Mok, Y. et al. (2011). Total cholesterol and cancer risk in a large prospective study in Korea. Journal of Clinical Oncology, 29(12), 1592–1598. https://doi.org/10.1200/JCO.2010.31.5200. [Google Scholar] [PubMed] [CrossRef]
6. Fiorenza, A. M., Branchi, A., Sommariva, D. (2000). Serum lipoprotein profile in patients with cancer. A comparison with non-cancer subjects. International Journal of Clinical & Labratory Research, 30(3), 141–145. https://doi.org/10.1007/s005990070013. [Google Scholar] [PubMed] [CrossRef]
7. Karayama, M., Inui, N., Inoue, Y., Yoshimura, K., Mori, K. et al. (2022). Increased serum cholesterol and long-chain fatty acid levels are associated with the efficacy of nivolumab in patients with non-small cell lung cancer. Cancer Immunology, Immunotherapy, 71(1), 203–217. https://doi.org/10.1007/s00262-021-02979-4. [Google Scholar] [PubMed] [CrossRef]
8. Perrone, F., Minari, R., Bersanelli, M., Bordi, P., Tiseo, M. et al. (2020). The prognostic role of high blood cholesterol in advanced cancer patients treated with immune checkpoint inhibitors. Journal for Immunotherapu, 43(6), 196–203. https://doi.org/10.1097/CJI.0000000000000321. [Google Scholar] [PubMed] [CrossRef]
9. Ding, X., Zhang, W., Li, S., Yang, H. (2019). The role of cholesterol metabolism in cancer. American Journal of Cancer Research, 9(2), 219–227. [Google Scholar] [PubMed]
10. Lee, I. K., Song, H., Kim, H., Kim, I. S., Tran, N. L. et al. (2020). RORα regulates cholesterol metabolism of CD8+ T cells for anticancer immunity. Cancers, 12(7), 1733. https://doi.org/10.3390/cancers12071733. [Google Scholar] [PubMed] [CrossRef]
11. Mayengbam, S. S., Singh, A., Yaduvanshi, H., Bhati, F. K., Deshmukh, B. et al. (2023). Cholesterol reprograms glucose and lipid metabolism to promote proliferation in colon cancer cells. Cancer & Metabolism, 11(1), 15. https://doi.org/10.1186/s40170-023-00315-1. [Google Scholar] [PubMed] [CrossRef]
12. Ossoli, A., Wolska, A., Remaley, A. T., Gomaraschi, M. (2022). High-density lipoproteins: A promising tool against cancer. Biochimica et Biophysica Acta-Molecular and Cell Biology of Lipids, 1867(1), 159068. https://doi.org/10.1016/j.bbalip.2021.159068. [Google Scholar] [PubMed] [CrossRef]
13. Ding, X., Zhao, T., Lee, C. C., Yan, C., Du, H. (2021). Lysosomal acid lipase deficiency controls T-and B-regulatory cell homeostasis in the lymph nodes of mice with human cancer xenotransplants. American Journal of Pathology, 191(2), 353–367. https://doi.org/10.1016/j.ajpath.2020.10.007. [Google Scholar] [PubMed] [CrossRef]
14. Aguilar-Ballester, M., Herrero-Cervera, A., Vinué, Á., Martínez-Hervás, S., González-Navarro, H. (2020). Impact of cholesterol metabolism in immune cell function and atherosclerosis. Nutrients, 12(7), 2021. https://doi.org/10.3390/nu12072021. [Google Scholar] [PubMed] [CrossRef]
15. Qin, W. H., Yang, Z. S., Li, M., Chen, Y., Zhao, X. F. et al. (2020). High serum levels of cholesterol increase antitumor functions of nature killer cells and reduce growth of liver tumors in mice. Gastroenterology, 158(6), 1713–1727. https://doi.org/10.1053/j.gastro.2020.01.028. [Google Scholar] [PubMed] [CrossRef]
16. Luo, J., Yang, H., Song, B. L. (2020). Mechanisms and regulation of cholesterol homeostasis. Nature Reviews Molecular Cell Biology, 21(4), 225–245. https://doi.org/10.1038/s41580-019-0190-7. [Google Scholar] [PubMed] [CrossRef]
17. Otocka-Kmiecik, A., Mikhailidis, D. P., Nicholls, S. J., Davidson, M., Rysz, J. et al. (2012). Dysfunctional HDL: A novel important diagnostic and therapeutic target in cardiovascular disease? Progress in Lipid Research, 51(4), 314–324. https://doi.org/10.1016/j.plipres.2012.03.003. [Google Scholar] [PubMed] [CrossRef]
18. Wu, W., Sarhadi, M., Song, X., Xue, J., Dai, Y. et al. (2023). Liver X receptors and estrogen receptor β, two players in a rare subtype of NSCLC. International Journal of Biological Sciences, 19(9), 2848–2859. https://doi.org/10.7150/ijbs.85164. [Google Scholar] [PubMed] [CrossRef]
19. Huang, B., Song, B. L., Xu, C. (2020). Cholesterol metabolism in cancer: Mechanisms and therapeutic opportunities. Nature Metabolism, 2(2), 132–141. https://doi.org/10.1038/s42255-020-0174-0. [Google Scholar] [PubMed] [CrossRef]
20. Pommier, A. J., Alves, G., Viennois, E., Bernard, S., Communal, Y. et al. (2010). Liver X receptor activation downregulates AKT survival signaling in lipid rafts and induces apoptosis of prostate cancer cells. Oncogene, 29(18), 2712–2723. https://doi.org/10.1038/onc.2010.30. [Google Scholar] [PubMed] [CrossRef]
21. Flaveny, C. A., Griffett, K., El-Gendy Bel, D., Kazantzis, M., Sengupta, M. et al. (2015). Broad anti-tumor activity of a small molecule that selectively targets the warburg effect and lipogenesis. Cancer Cell, 28(1), 42–56. https://doi.org/10.1016/j.ccell.2015.05.007. [Google Scholar] [PubMed] [CrossRef]
22. Dai, Y. B., Miao, Y. F., Wu, W. F., Li, Y., D’Errico, F. et al. (2016). Ablation of liver X receptors α and β leads to spontaneous peripheral squamous cell lung cancer in mice. Proceedings of the National Academy of Sciences of the United States of America, 113(27), 7614–7619. [Google Scholar] [PubMed]
23. Melloni, G., Muriana, P., Bandiera, A., Fontana, R., Maggioni, D. et al. (2018). Prognostic role of liver X receptor-alpha in resected stage II and III non-small-cell lung cancer. The Clinical Respiratory Journal, 12(1), 241–246. https://doi.org/10.1111/crj.2018.12.issue-1 [Google Scholar] [CrossRef]
24. Sag, D., Cekic, C., Wu, R., Linden, J., Hedrick, C. C. (2015). The cholesterol transporter ABCG1 links cholesterol homeostasis and tumour immunity. Nature Communications, 6, 6354. https://doi.org/10.1038/ncomms7354. [Google Scholar] [PubMed] [CrossRef]
25. Tian, C., Huang, D., Yu, Y., Zhang, J., Fang, Q. et al. (2017). ABCG1 as a potential oncogene in lung cancer. Experimental and Therapeutic Medicine, 13(6), 3189–3194. https://doi.org/10.3892/etm.2017.4393. [Google Scholar] [PubMed] [CrossRef]
26. Zhao, H., Mu, X., Zhang, X., You, Q. (2020). Lung cancer inhibition by betulinic acid nanoparticles via adenosine 5′-triphosphate (ATP)-binding cassette transporter G1 gene downregulation. Medical Science Monitor, 26, e922092. [Google Scholar] [PubMed]
27. Benn, M., Tybjærg-Hansen, A., Stender, S., Frikke-Schmidt, R., Nordestgaard, B. G. (2011). Low-density lipoprotein cholesterol and the risk of cancer: A mendelian randomization study. Journal of the National Cancer Institute, 103(6), 508–519. https://doi.org/10.1093/jnci/djr008. [Google Scholar] [PubMed] [CrossRef]
28. Hazen, S. L. (2012). New lipid and lipoprotein targets for the treatment of cardiometabolic diseases. Journal of Lipid Research, 53(9), 1719–1721. https://doi.org/10.1194/jlr.E030205. [Google Scholar] [PubMed] [CrossRef]
29. Li, Z., Liu, Q. (2018). Proprotein convertase subtilisin/kexin type 9 inhibits hepatitis C virus replication through interacting with NS5A. Journal of General Virology, 99(1), 44–61. https://doi.org/10.1099/jgv.0.000987. [Google Scholar] [PubMed] [CrossRef]
30. Bonaventura, A., Grossi, F., Carbone, F., Vecchié, A., Minetti, S. et al. (2019). Serum PCSK9 levels at the second nivolumab cycle predict overall survival in elderly patients with NSCLC: A pilot study. Cancer Immunology and Immunotherapy, 68(8), 1351–1358. https://doi.org/10.1007/s00262-019-02367-z. [Google Scholar] [PubMed] [CrossRef]
31. Xie, M., Yu, X., Chu, X., Xie, H., Zhou, J. et al. (2022). Low baseline plasma PCSK9 level is associated with good clinical outcomes of immune checkpoint inhibitors in advanced non-small cell lung cancer. Thoracic Cancer, 13(3), 353–360. https://doi.org/10.1111/tca.v13.3 [Google Scholar] [CrossRef]
32. Xu, X., Cui, Y., Cao, L., Zhang, Y., Yin, Y. et al. (2017). PCSK9 regulates apoptosis in human lung adenocarcinoma A549 cells via endoplasmic reticulum stress and mitochondrial signaling pathways. Experimental and Therapeutic Medicine, 13(5), 1993–1999. https://doi.org/10.3892/etm.2017.4218. [Google Scholar] [PubMed] [CrossRef]
33. Giacomini, I., Gianfanti, F., Desbats, M. A., Orso, G., Berretta, M. et al. (2021). Cholesterol metabolic reprogramming in cancer and its pharmacological modulation as therapeutic strategy. Frontiers in Oncology, 11, 682911. https://doi.org/10.3389/fonc.2021.682911. [Google Scholar] [PubMed] [CrossRef]
34. Blanc, M., Hsieh, W. Y., Robertson, K. A., Kropp, K. A., Forster, T. et al. (2013). The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity, 38(1), 106–118. https://doi.org/10.1016/j.immuni.2012.11.004. [Google Scholar] [PubMed] [CrossRef]
35. Kang, K. A., Chae, S., Lee, K. H., Park, M. T., Lee, S. J. et al. (2005). Cytotoxic effect of 7β-hydroxycholesterol on human NCI-H460 lung cancer cells. Biological and Pharmaceutical Bulletin, 28(8), 1377–1380. https://doi.org/10.1248/bpb.28.1377. [Google Scholar] [PubMed] [CrossRef]
36. Pataj, Z., Liebisch, G., Schmitz, G., Matysik, S. (2016). Quantification of oxysterols in human plasma and red blood cells by liquid chromatography high-resolution tandem mass spectrometry. Journal of Chromatography A, 1439, 82–88. https://doi.org/10.1016/j.chroma.2015.11.015. [Google Scholar] [PubMed] [CrossRef]
37. Chimento, A., Casaburi, I., Avena, P., Trotta, F., de Luca, A. et al. (2018). Cholesterol and its metabolites in tumor growth: Therapeutic potential of statins in cancer treatment. Frontiers in Endocrinology, 9, 807. [Google Scholar] [PubMed]
38. Li, X., Chen, H., Zhang, L., Chen, L., Wei, W. et al. (2022). 27-hydroxycholesterol linked high cholesterol diet to lung adenocarcinoma metastasis. Oncogene, 41(19), 2685–2695. https://doi.org/10.1038/s41388-022-02285-y. [Google Scholar] [PubMed] [CrossRef]
39. Abdalkareem Jasim, S., Kzar, H. H., Haider Hamad, M., Ahmad, I., Al-Gazally, M. E. et al. (2022). The emerging role of 27-hydroxycholesterol in cancer development and progression: An update. International Immunopharmacology, 110, 109074. https://doi.org/10.1016/j.intimp.2022.109074. [Google Scholar] [PubMed] [CrossRef]
40. Zhang, L., Liu, M., Liu, J., Li, X., Yang, M. et al. (2019). 27-hydroxycholesterol enhanced osteoclastogenesis in lung adenocarcinoma microenvironment. Journal of Cellular Physiology, 234(8), 12692–12700. https://doi.org/10.1002/jcp.v234.8 [Google Scholar] [CrossRef]
41. SchroepferJr, G. J. (2000). Oxysterols: Modulators of cholesterol metabolism and other processes. Physiological Reviews, 80(1), 361–554. https://doi.org/10.1152/physrev.2000.80.1.361. [Google Scholar] [PubMed] [CrossRef]
42. Adams, C. M., Reitz, J., de Brabander, J. K., Feramisco, J. D., Li, L. et al. (2004). Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. Journal of Biology Chemistry, 279(50), 52772–52780. https://doi.org/10.1074/jbc.M410302200. [Google Scholar] [PubMed] [CrossRef]
43. Wang, S., Yao, Y., Rao, C., Zheng, G., Chen, W. (2019). 25-HC decreases the sensitivity of human gastric cancer cells to 5-fluorouracil and promotes cells invasion via the TLR2/NF-κB signaling pathway. International Journal of Oncology, 54(3), 966–980. [Google Scholar] [PubMed]
44. Koarai, A., Yanagisawa, S., Sugiura, H., Ichikawa, T., Kikuchi, T. et al. (2012). 25-Hydroxycholesterol enhances cytokine release and Toll-like receptor 3 response in airway epithelial cells. Respiratory Research, 13(1), 63. https://doi.org/10.1186/1465-9921-13-63. [Google Scholar] [PubMed] [CrossRef]
45. Chen, L., Zhang, L., Xian, G., Lv, Y., Lin, Y. et al. (2017). 25-Hydroxycholesterol promotes migration and invasion of lung adenocarcinoma cells. Biochemical and Biophysical Research Communications, 484(4), 857–863. https://doi.org/10.1016/j.bbrc.2017.02.003. [Google Scholar] [PubMed] [CrossRef]
46. Reid, T. S., Terry, K. L., Casey, P. J., Beese, L. S. (2004). Crystallographic analysis of CaaX prenyltransferases complexed with substrates defines rules of protein substrate selectivity. Journal of Molecular Biology, 343(2), 417–433. https://doi.org/10.1016/j.jmb.2004.08.056. [Google Scholar] [PubMed] [CrossRef]
47. Xu, N., Shen, N., Wang, X., Jiang, S., Xue, B. et al. (2015). Protein prenylation and human diseases: A balance of protein farnesylation and geranylgeranylation. Science China Life Sciences, 58(4), 328–335. https://doi.org/10.1007/s11427-015-4836-1. [Google Scholar] [PubMed] [CrossRef]
48. Perez-Sala, D. (2007). Protein isoprenylation in biology and disease: General overview and perspectives from studies with genetically engineered animals. Frontiers in Biosciences, 12, 4456–4472. https://doi.org/10.2741/2401. [Google Scholar] [PubMed] [CrossRef]
49. Okayama, H., Kohno, T., Ishii, Y., Shimada, Y., Shiraishi, K. et al. (2012). Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Research, 72(1), 100–111. https://doi.org/10.1158/0008-5472.CAN-11-1403. [Google Scholar] [PubMed] [CrossRef]
50. Wang, X., Xu, W., Zhan, P., Xu, T., Jin, J. et al. (2018). Overexpression of geranylgeranyl diphosphate synthase contributes to tumour metastasis and correlates with poor prognosis of lung adenocarcinoma. Journal of Cellular and Molecular Medicine, 22(4), 2177–2189. https://doi.org/10.1111/jcmm.2018.22.issue-4 [Google Scholar] [CrossRef]
51. Deng, M., Gui, X., Kim, J., Xie, L., Chen, W. et al. (2018). LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature, 562(7728), 605–609. https://doi.org/10.1038/s41586-018-0615-z. [Google Scholar] [PubMed] [CrossRef]
52. Gonzalez, H., Hagerling, C., Werb, Z. (2018). Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes & Development, 32(19–20), 1267–1284. [Google Scholar]
53. Zhao, L., Li, J., Liu, Y., Kang, L., Chen, H. et al. (2018). Cholesterol esterification enzyme inhibition enhances antitumor effects of human chimeric antigen receptors modified T cells. Journal of Immunotherapy, 41(2), 45–52. https://doi.org/10.1097/CJI.0000000000000207. [Google Scholar] [PubMed] [CrossRef]
54. Condamine, T., Mastio, J., Gabrilovich, D. I. (2015). Transcriptional regulation of myeloid-derived suppressor cells. Journal of Leukocyte Biology, 98(6), 913–922. https://doi.org/10.1189/jlb.4RI0515-204R. [Google Scholar] [PubMed] [CrossRef]
55. Vuong, J. T., Stein-Merlob, A. F., Nayeri, A., Sallam, T., Neilan, T. G. et al. (2022). Immune checkpoint therapies and atherosclerosis: Mechanisms and clinical implications: JACC state-of-the-art review. Journal of the American College of Cardiology, 79(6), 577–593. https://doi.org/10.1016/j.jacc.2021.11.048. [Google Scholar] [PubMed] [CrossRef]
56. Kimura, Y., Sumiyoshi, M., Baba, K. (2016). Antitumor and antimetastatic activity of synthetic hydroxystilbenes through inhibition of lymphangiogenesis and M2 macrophage differentiation of tumor-associated macrophages. Anticancer Research, 36(1), 137–148. [Google Scholar] [PubMed]
57. Hoppstädter, J., Dembek, A., Höring, M., Schymik, H. S., Dahlem, C. et al. (2021). Dysregulation of cholesterol homeostasis in human lung cancer tissue and tumour-associated macrophages. EBioMedicine, 72, 103578. https://doi.org/10.1016/j.ebiom.2021.103578. [Google Scholar] [PubMed] [CrossRef]
58. Yang, W., Bai, Y., Xiong, Y., Zhang, J., Chen, S. et al. (2016). Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature, 531(7596), 651–655. https://doi.org/10.1038/nature17412. [Google Scholar] [PubMed] [CrossRef]
59. Bibby, J. A., Purvis, H. A., Hayday, T., Chandra, A., Okkenhaug, K. et al. (2020). Cholesterol metabolism drives regulatory B cell IL-10 through provision of geranylgeranyl pyrophosphate. Nature Communications, 11(1), 3412. https://doi.org/10.1038/s41467-020-17179-4. [Google Scholar] [PubMed] [CrossRef]
60. Pavlova, N. N., Zhu, J., Thompson, C. B. (2022). The hallmarks of cancer metabolism: Still emerging. Cell Metabolism, 34(3), 355–377. https://doi.org/10.1016/j.cmet.2022.01.007. [Google Scholar] [PubMed] [CrossRef]
61. Ahmadi, M., Amiri, S., Pecic, S., Machaj, F., Rosik, J. et al. (2020). Pleiotropic effects of statins: A focus on cancer. Biochimica et Biophysica Acta-Molecular Basis Disease, 1866(12), 165968. https://doi.org/10.1016/j.bbadis.2020.165968. [Google Scholar] [PubMed] [CrossRef]
62. Fatehi Hassanabad, A., McBride, S. A. (2019). Statins as potential therapeutics for lung cancer: Molecular mechanisms and clinical outcomes. American Journal of Clinical Oncology, 42(9), 732–736. https://doi.org/10.1097/COC.0000000000000579. [Google Scholar] [PubMed] [CrossRef]
63. Khurana, V., Bejjanki, H. R., Caldito, G., Owens, M. W. (2007). Statins reduce the risk of lung cancer in humans: A large case-control study of US veterans. Chest, 131(5), 1282–1288. https://doi.org/10.1378/chest.06-0931. [Google Scholar] [PubMed] [CrossRef]
64. Kwon, Y. J., You, N. Y., Lee, J. W., Kim, J., Kang, H. T. (2019). High receipt of statins reduces the risk of lung cancer in current smokers with hypercholesterolemia: The national health insurance service-health screening cohort. Clinical Lung Cancer, 20(2), e177–e185. https://doi.org/10.1016/j.cllc.2018.11.009. [Google Scholar] [PubMed] [CrossRef]
65. Chen, Y., Li, X., Zhang, R., Xia, Y., Shao, Z. et al. (2019). Effects of statin exposure and lung cancer survival: A meta-analysis of observational studies. Pharmacological Research, 141, 357–365. https://doi.org/10.1016/j.phrs.2019.01.016. [Google Scholar] [PubMed] [CrossRef]
66. Longo, J., Van Leeuwen, J. E., Elbaz, M., Branchard, E., Penn, L. Z. (2020). Statins as anticancer agents in the era of precision medicine. Clinical Cancer Research, 26(22), 5791–5800. https://doi.org/10.1158/1078-0432.CCR-20-1967. [Google Scholar] [PubMed] [CrossRef]
67. Nguyen, P. A., Chang, C. C., Galvin, C. J., Wang, Y. C., An, S. Y. et al. (2020). Statins use and its impact in EGFR-TKIs resistance to prolong the survival of lung cancer patients: A cancer registry cohort study in Taiwan. Cancer Science, 111(8), 2965–2973. https://doi.org/10.1111/cas.v111.8 [Google Scholar] [CrossRef]
68. Luo, Y., Yang, Y., Peng, P., Zhan, J., Wang, Z. et al. (2021). Cholesterol synthesis disruption combined with a molecule-targeted drug is a promising metabolic therapy for EGFR mutant non-small cell lung cancer. Translational Lung Cancer Research, 10(1), 128–142. https://doi.org/10.21037/tlcr [Google Scholar] [CrossRef]
69. Ali, A., Levantini, E., Fhu, C. W., Teo, J. T., Clohessy, J. G. et al. (2019). CAV1-GLUT3 signaling is important for cellular energy and can be targeted by atorvastatin in non-small cell lung cancer. Theranostics, 9(21), 6157–6174. https://doi.org/10.7150/thno.35805. [Google Scholar] [PubMed] [CrossRef]
70. Lee, H. Y., Kim, I. K., Lee, H. I., Mo, J. Y., Yeo, C. D. et al. (2016). The apoptotic effect of simvastatin via the upregulation of BIM in nonsmall cell lung cancer cells. Experimental Lung Research, 42(1), 14–23. https://doi.org/10.3109/01902148.2015.1125970. [Google Scholar] [PubMed] [CrossRef]
71. Kawakami, S., Suzuki, S., Yamashita, F., Hashida, M. (2006). Induction of apoptosis in A549 human lung cancer cells by all-trans retinoic acid incorporated in DOTAP/cholesterol liposomes. Journal of Controlled Release, 110(3), 514–521. https://doi.org/10.1016/j.jconrel.2005.10.030. [Google Scholar] [PubMed] [CrossRef]
72. Lyu, J., Yang, E. J., Head, S. A., Ai, N., Zhang, B. et al. (2017). Pharmacological blockade of cholesterol trafficking by cepharanthine in endothelial cells suppresses angiogenesis and tumor growth. Cancer Letters, 409, 91–103. https://doi.org/10.1016/j.canlet.2017.09.009. [Google Scholar] [PubMed] [CrossRef]
73. Li, T., Chen, L., Deng, Y., Liu, X., Zhao, X. et al. (2017). Cholesterol derivative-based liposomes for gemcitabine delivery: Preparation, in vitro, and in vivo characterization. Drug Development and Industrial Pharmacy, 43(12), 2016–2025. https://doi.org/10.1080/03639045.2017.1361965. [Google Scholar] [PubMed] [CrossRef]
74. Muddineti, O. S., Shah, A., Rompicharla, S. V. K., Ghosh, B., Biswas, S. (2018). Cholesterol-grafted chitosan micelles as a nanocarrier system for drug-siRNA co-delivery to the lung cancer cells. International Journal of Biological Macromolecules, 118, 857–863. https://doi.org/10.1016/j.ijbiomac.2018.06.114. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools