 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE
Polarized Autologous Macrophages (PAM) Can Be a Tumor Vaccine
1
The M.O.E. Key Laboratory of Laboratory Medical Diagnostics, The College of Laboratory Medicine, Chongqing Medical
University, Chongqing, 400016, China
2
CAS Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, Institute of High Energy Physics and University of
Chinese Academy of Sciences (UCAS), Chinese Academy of Sciences (CAS), Beijing, 100049, China
* Corresponding Authors: Dongqing Wang. Email: ; Yi Hu. Email:
Oncologie 2022, 24(3), 441-449. https://doi.org/10.32604/oncologie.2022.024898
Received 12 June 2022; Accepted 15 August 2022; Issue published 19 September 2022
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Immunotherapy is currently recognized as one of the most promising anticancer strategies. In the tumor microenvironment, tumor-associated macrophages are mainly M2-type macrophages with tumor-promoting effects. Therefore, the reprogramming of tumor-associated macrophages from M2 to M1 type is a potential strategy for cancer therapy. We have previously shown the anticancer effects of implantable allogeneic M1 macrophages in mice. Here, we further engineered autologous mouse bone marrow cells into M1 macrophages and then embedded them into a sodium alginate gel to prepare an implantable immunotherapeutic agent (M1@Gel). We demonstrate that M1@Gel repolarizes M2 macrophages to M1 type and activates the immune responses in mice. As a result, M1@Gel can potently inhibit the tumor recurrence in mice after the surgical tumor removal. These results suggest that the implantation of autologous M1 macrophages might be a promising strategy for preventing postoperative tumor recurrence. We envisage that the employment of polarized autologous macrophages as a tumor vaccine might be translated into clinic.Keywords
Currently, adoptive cellular immunotherapy, tumor vaccines and other cancer immunotherapy have achieved certain successes [1–4]. Among them, macrophages have been suggested as one of the important therapeutic targets in tumor development and tumor resistance [5,6]. Based on the phenotypes and functions, macrophages can be generally divided into M1 (pro-inflammatory, classically activated macrophages) and M2 (anti-inflammatory, alternately activated macrophages) types [7].
Briefly, M1 macrophages can be in vitro induced by interferon-gamma (IFN-γ), lipopolysaccharide (LPS), and granulocyte-macrophage colony-stimulating factor (GM-CSF). GM-CSF can enhance dendritic cell (DC) functions and induce the polarization of M1 macrophages to play an antitumor role [8,9]. M1 macrophages can express antigen-presenting major histocompatibility complex (MHC) to activate adaptive immune response [10,11]. Activated M1 macrophages can produce nitric oxide and reactive oxygen species [12]. In addition, M1 macrophages can secrete cytokines such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-6 and IL-12 to promote inflammatory response. M2 macrophages can be in vitro induced by IL-4, IL-13, M-CSF/CSF1, IL-10, and transforming growth factor-β (TGF-β) [13,14]. In most solid tumors, tumor-associated macrophages (TAMs) are considered as M2-like macrophages because they share most of the properties of M2 macrophages, typically including arginase 1, mannose receptors and low levels of MHC class II complex. TAMs may perform tumor-promoting functions, and therefore TAMs could be a potential target for cancer therapy [15,16]. Previous studies have suggested that the repolarization from M2 to M1 macrophages is plausible to inhibit tumors [17,18]. Moreover, macrophages can act as professional antigen-presenting cells to activate adaptive antitumor immune response [19]. Therefore, adoptively transferring autologous macrophages to cancer patients was thought to be a potential antitumor strategy [20]. However, these attempts failed in the clinic since the solid tumor microenvironment could repolarize the transferred M1 macrophages to M2 phenotype [21]. To circumvent this problem, we attempted to employ M1 macrophages as a tumor vaccine.
Herein, we evaluated the inhibitory effect of autologous M1 macrophages on tumor recurrence. We extracted autologous bone marrow cells from the mice and engineered them into autologous M1-type macrophages in vitro. Autologous M1 macrophages were embedded in the alginate hydrogel as implantable immunotherapeutic agent, named as M1@Gel. As antigen presenting cells and inflammatory cells, M1 macrophages can further improve antigen presentation, release inflammatory cytokines and regulate macrophage polarity, thereby activating adaptive immune responses and natural immune effects to prevent primary tumor recurrence (Scheme 1).
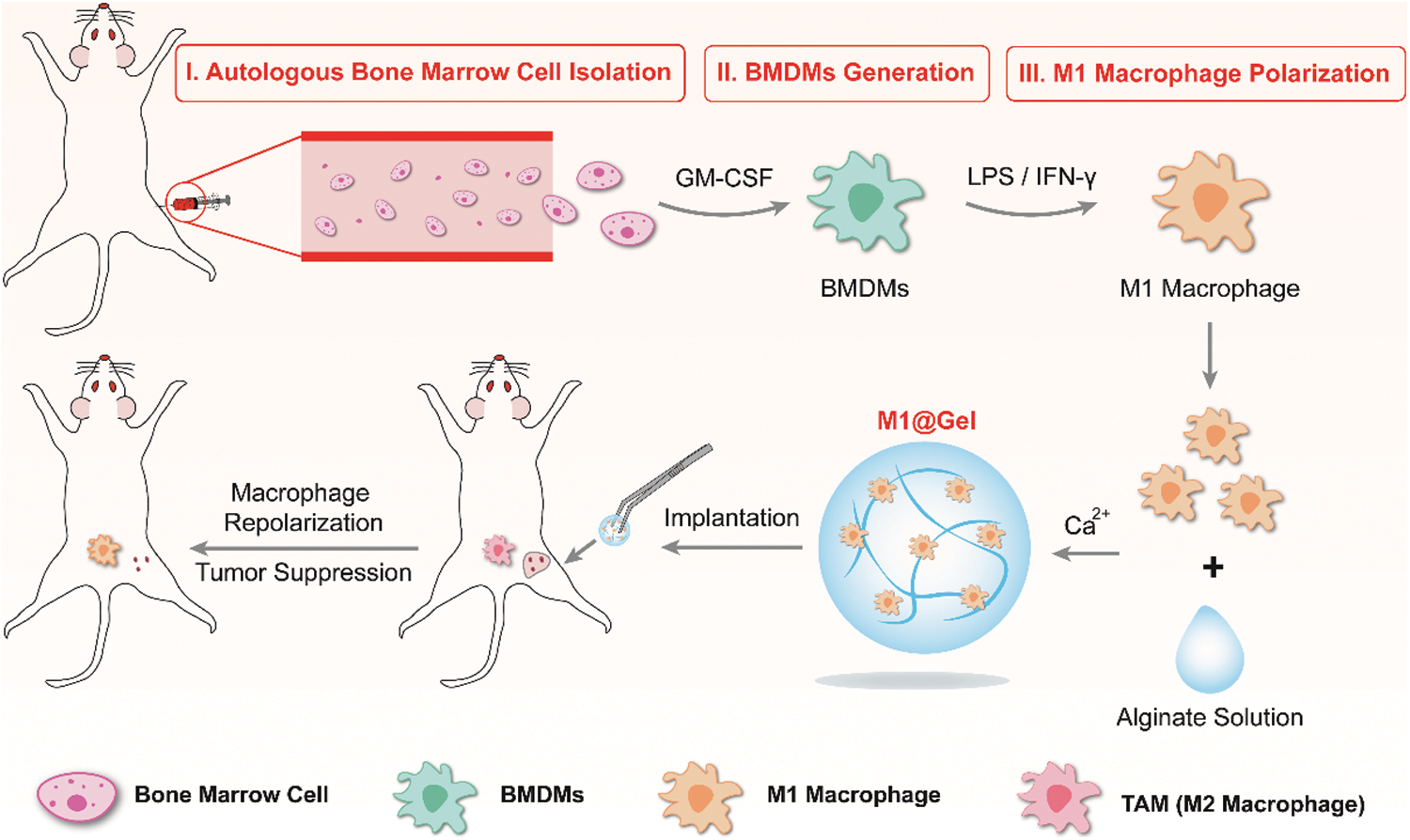
Scheme 1: Preparation of autologous M1 macrophages as a tumor vaccine
LPS was purchased from Sigma Aldrich. Mouse GM-CSF and IL-4 were obtained from Peprotech (Rocky Hill, CQ). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), RPMI 1640 medium, and penicillin-streptomycin were provided by Thermo Fisher Scientific. Red blood cells lysis buffer was provided by Beijing Solarbio Science & Technology Co., Ltd. (China). Cell counting assay kit (CCK-8) was provided by MCE (China). Anti-CD11c-Brilliant Violet 421TM (mouse, Clone: N418, Cat: 117343) and anti-mouse I-A/I-E-FITC (mouse, Clone: M1/42, Cat: 125508) were provided by Biolegend (USA). Anti-CD3e-FITC (mouse, Clone: 145–2C11, Cat: 553061), anti-CD8a-PE (mouse, Clone: 53–6.7, Cat: 553032), anti-CD4-APC (mouse, Clone: RM4–5, Cat: 553051), anti-CD45-APC-Cy7 (mouse, Clone: 30-F11, Cat: 557659), anti-CD11b-FITC (mouse, Clone: M1/70, Cat: 557396), anti-F4/80-BV421TM (mouse, Clone: T45–2342, Cat: 565411), anti-CD86-PE (mouse, Clone: GL1, Cat: 553692) and anti-CD206-Alexa Fluor 647 (mouse, Clone: MR5D3, Cat: 565250) were purchased from BD Biosciences (USA). If not specified, all the chemical reagents were provided by Sigma Aldrich (St. Louis, MO, USA).
The female BALB/c mice (5–6 weeks) were obtained from Chongqing Medical University. The animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Chongqing Medical University. 4T1 cells were purchased from ATCC (Manassas, VA). 4T1 cells were grown in RPMI 1640 containing 10% FBS and 1% penicillin-streptomycin (10000 U/mL). RAW264.7 cells were grown in DMEM containing 10% FBS and 1% penicillin-streptomycin (10000 U/mL).
2.3 Generation of Autologous M1 Macrophages
The mice were placed in the lateral position after the mice were anesthetized. After anticoagulation with heparin, a 1 mL syringe (heparin anticoagulation) was inserted into the mouse femoral and tibial joint at a 15° angle to extract 5–10 μL of bone marrow cells, which were resuspended in DMEM with 20% FBS. After the centrifugation at 1200 rpm for 5 min, the cells were cultured in DMEM with 20% FBS, 1% penicillin-streptomycin, GM-CSF (20 ng/mL), LPS (20 ng/mL) and IFN-γ (10 ng/mL) to obtain the autologous M1 macrophages. For the preparation of autologous M2 macrophages, the cells were cultured in DMEM with 20% FBS, 1% penicillin-streptomycin, GM-CSF (20 ng/mL) and IL-4 (20 ng/mL). 7 d later, autologous macrophages were collected. Anti-CD11b-FITC and anti-CD86-PE were used to verify autologous M1 macrophages. Anti-CD11b-FITC and anti-CD206-Alexa Fluor 647 were used to verify autologous M2 macrophages.
Autologous M1 macrophages (5 × 106) were dispersed in sodium alginate aqueous solution (10 mg/mL). Afterward, the solution was added dropwise into PBS buffer with 20 mM CaCl2 to prepare M1@Gel.
To examine the antitumor effects of autologous M1 macrophages, we seeded 5 × 103 macrophages in the upper transwell chamber (0.4 μm-sized) and 4T1 cells (8 × 103 cells/well) in the lower transwell chamber. After 24 h, the viability of 4T1 cells was analyzed by using the CCK-8 assay.
Female BALB/c mice (6–8 weeks) were used to establish the tumor model. 1 × 106 of 4T1 cells were subcutaneously injected into the mouse right flank. The tumors (∼90%) were excised when tumor volume was ∼200 mm3. Then the mice were implanted with hydrogel microspheres in 4 groups (n = 3): PBS, Gel, M1, M1@Gel. After the different treatments, the body weight of the mice and tumor volume were obtained every 3 d. At 21 d, the mice were sacrificed to collect the tumors and lymph nodes. The tumor single cell suspensions were labeled with anti-CD45-APC-Cy7, anti-CD11b-FITC, anti-F4/80-BV421TM, anti-CD86-PE and anti-CD206-Alexa 647 to analyze macrophage reprogramming. In addition, the tumor single cell suspensions were stained with anti-CD3e-FITC, anti-CD8a-PE and anti-CD4-APC antibodies for the analysis of infiltrating T cells. Moreover, single cell suspensions of lymph nodes were stained with Brilliant anti-CD11c-Violet 421TM, anti-MHC II-FITC and anti-CD86-PE for 30 min at 4°C to verify the DC maturation.
Macrophages are key effectors in cancer immunotherapy, and M1 macrophages can promote the antitumor immune response. In this work, mouse autologous bone marrow cells were extracted and induced into autologous M1-type macrophages, as shown in Fig. 1. Flow cytometry (Fig. 2A) verified the markers (CD11b and CD86) of autologous M1 macrophages and the markers (CD11b and CD206) of autologous M2 macrophages, indicating that autologous bone marrow cells were transformed into autologous M1 macrophages by the co-induction of GM-CSF, LPS and IFN-γ, and autologous M2 macrophages by the co-induction of GM-CSF and IL-4. Subsequently, to demonstrate the inhibitory effect of autologous macrophages on tumor cells, we obtained M1 macrophages from autologous bone marrow macrophages and a co-culture experiment (transwell chamber assays) was performed to mimic the macrophages and tumor cells in the tumor microenvironment. Autologous macrophages were grown in the upper chamber, while 4T1 cells were seeded in the lower chamber. After 24 h, CCK-8 assay was used to examine the viability of 4T1 cells. The results showed that ∼26.5% of 4T1 cells were suppressed by autologous M1 macrophages (Fig. 2B), indicating that autologous M1 macrophages possessed the antitumor potential. By contrast, autologous M2 macrophages appeared to promote the proliferation of 4T1 cells (Fig. 2B).
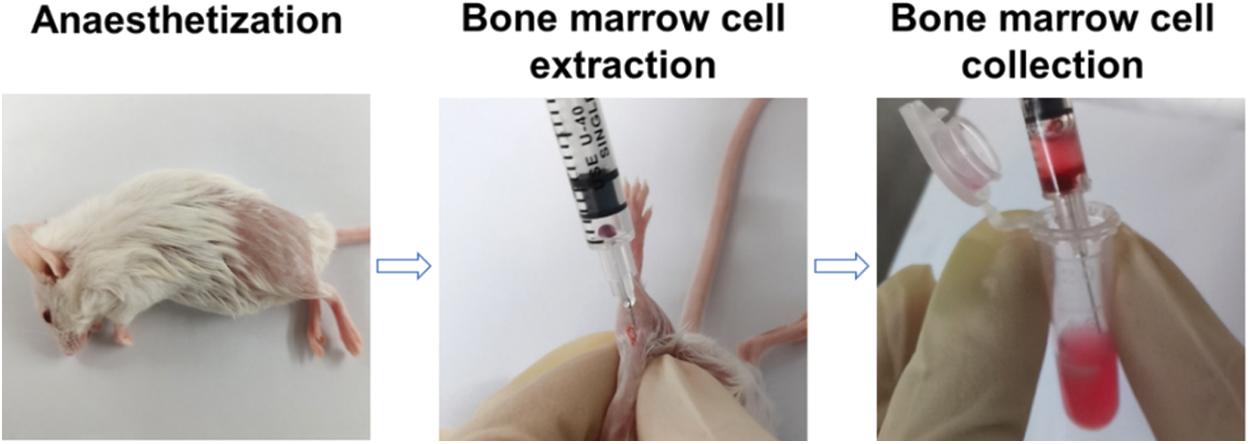
Figure 1: Extraction of autologous bone marrow cells from the mice
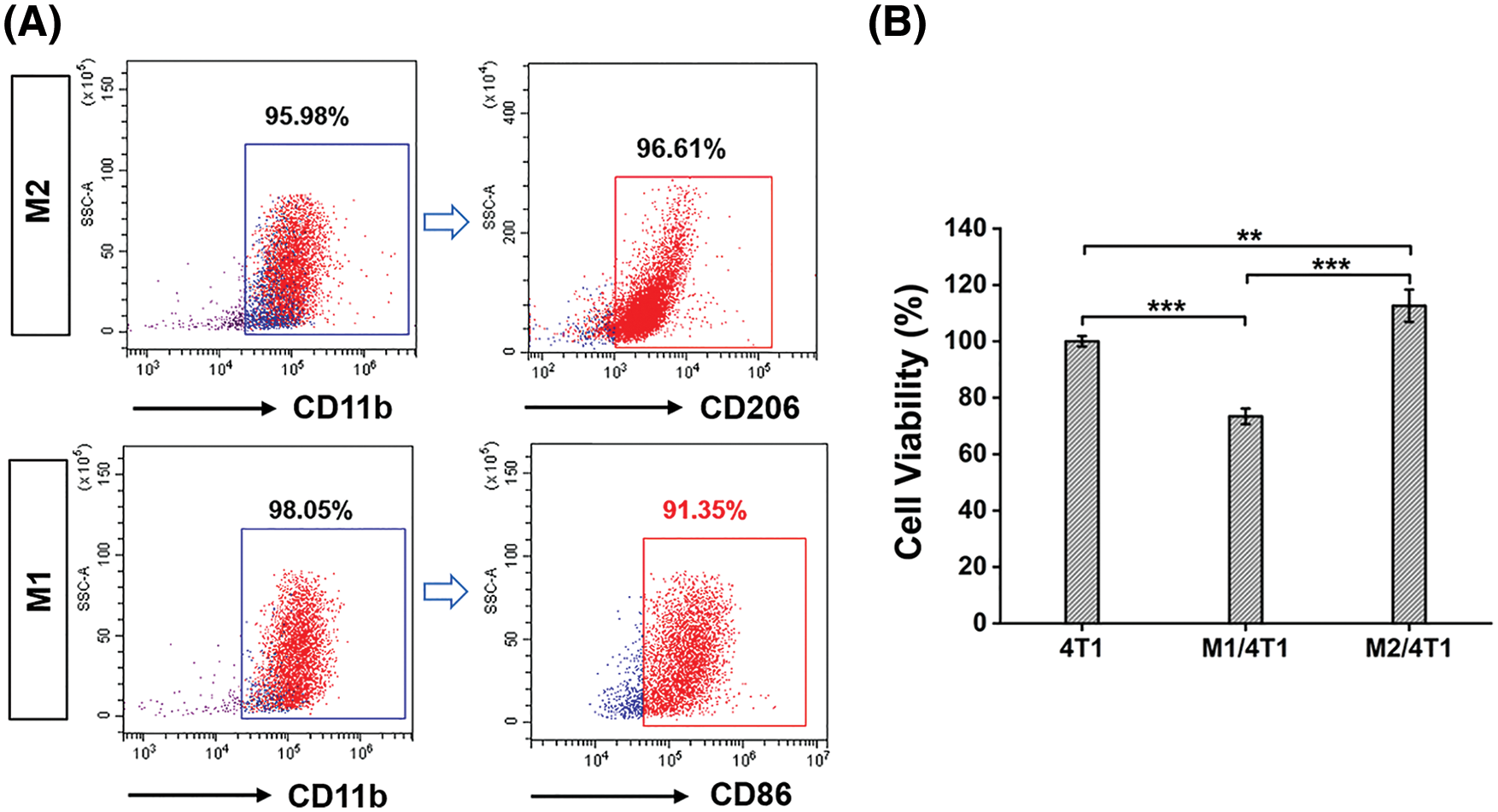
Figure 2: Preparation of autologous M1 macrophages from the mice. (A) Validation of autologous M1 macrophages (CD11b, CD86) and M2 macrophages (CD11b, CD206) by flow cytometry analysis. (B) Transwell assays verified the antitumor effects of autologous macrophages (M1). Significant differences are indicated. **, P < 0.01; ***, P < 0.001
3.2 Antitumor Effect of M1@Gel in Mice
M1 macrophages as professional antigen-presenting cells can promote adaptive antitumor immune responses and inhibit tumor growth by phagocytosing tumor debris or secreting inflammatory cytokines [19]. The strategy of adoptive transfer of autologous M1 macrophages to solid tumors has become one of the strategies for developing macrophage-based tumor therapy. The improvement of the autologous M1 macrophage retention effect and the slow-release effect might facilitate the immune regulatory effects in the tumor microenvironment. For example, hybrid macrophage membranes embedded in the alginate hydrogel had potent antitumor effects [22]. We prepared the alginate hydrogel (10 mg/mL) and embedded the autologous M1 macrophages into the hydrogel to make M1@Gel.
A subcutaneous tumor model with 4T1 cells was developed in BALB/c mice. When tumor volume was ∼200 mm3, ∼95% of tumors tissues were surgically removed. Subsequently, the mice were randomly assigned to PBS, Gel, M1 (autologous M1 macrophages) and M1@Gel (autologous M1 macrophages embedded in alginate hydrogel) for different treatments, respectively. Then, recurrent tumor volume and body weight were monitored every 3 d for 21 d. As the result shown, both PBS and Gel groups showed significant tumor recurrence after the surgery (Figs. 3A–3B). In contrast, the M1 group partially suppressed tumor recurrence at the primary site, and the M1@Gel group exhibited an excellent recurrence-suppressing effect (Figs. 3A–3B). Furthermore, the recurrent tumors were excised after the treatments. Recurrent tumor weight in M1@Gel group was lower than that in PBS and Gel groups (Fig. 3C). For all the treatments, no significant change was observed in the body weight of the mice (Fig. 3D). These results indicated that M1@Gel effectively inhibited the tumor recurrence.
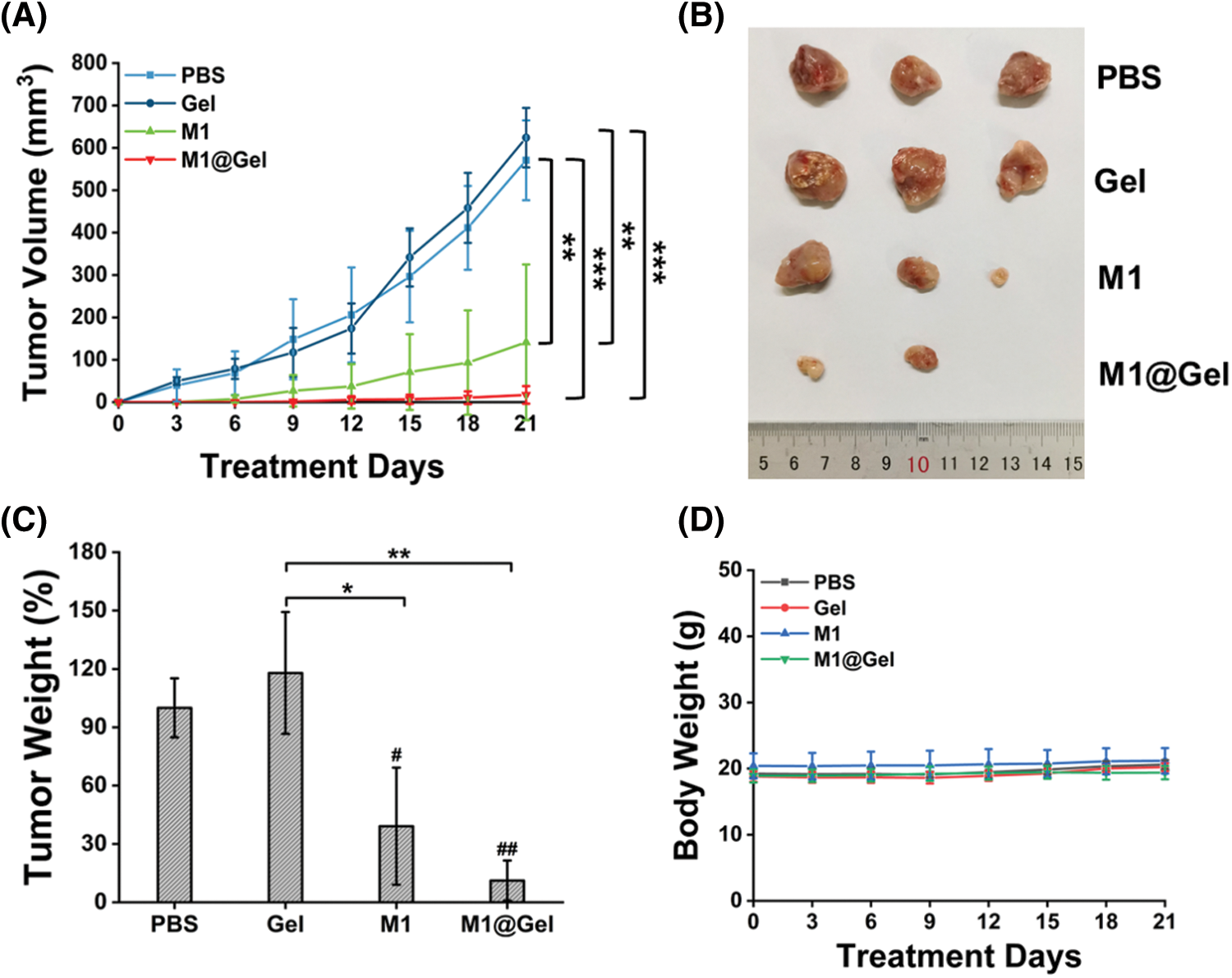
Figure 3: Antitumor effect of autologous M1 macrophages in vivo. The volume (A), image (B) and weight (C) of tumors in different treatment groups. (B) and (C) were from the mice after 21 d treatments. (D) The body weight of the mice in different treatment groups for 21 d. Significant differences are indicated. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (between groups). #, P < 0.05; ##, P < 0.01 (compared with the PBS group)
M2-like TAMs promote malignant tumor progression and thus become potential targets for cancer therapy. Fortunately, macrophage remodeling (from M2 phenotype to M1 phenotype) may suppress tumorigenesis by inducing an inflammatory microenvironment. We next explored whether M1@Gel could remodel TAMs to M1-type in mice. After the treatments with the different groups, the recurrent tumors were excised to prepare single cell suspensions for the flow cytometry analysis. M1 macrophages (CD45+CD11b+F4/80+CD86+) and M2 macrophages (CD45+CD11b+F4/80+CD206+) could be distinguished according to different markers. Compared with the PBS and Gel groups, the percentage of M1-type macrophages was relatively higher in the M1 group, and M2 macrophages were decreased in the M1 group (Figs. 4A–4B). The highest proportion of M1-type macrophages and the lowest proportion of M2-type macrophages were present in M1@Gel group (Figs. 4A–4B). As shown in Fig. 4C, the M1@Gel group had a lower M2/M1 ratio as compared with the control group, suggesting more M1-type macrophage infiltration in the tumor microenvironment. These results indicated that M1@Gel vaccine could modulate the polarity of TAMs in mice, thereby inhibiting tumor recurrence.
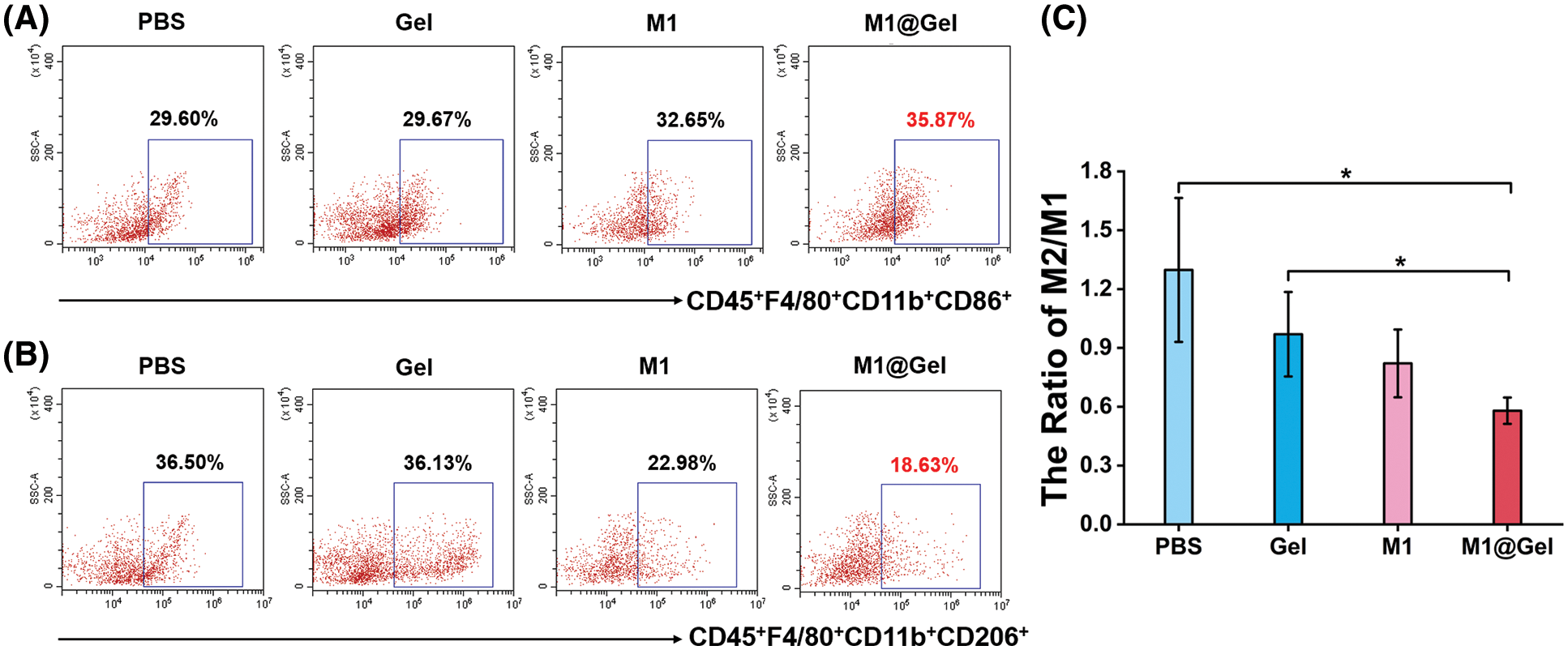
Figure 4: Immunomodulation by autologous M1 macrophages in tumor microenvironment of the mice after the treatments. Flow cytometry analysis of M1 macrophages (CD45+F4/80+CD11b+CD86+) (A) and M2 macrophages (CD45+F4/80+CD11b+CD206+) (B) after different treatments. (C) Quantitative analysis of M2/M1 ratios in mice. *, P < 0.05 (mean ± SD, n = 3)
As the antigen-presenting cells, M1 macrophages can promote the antigen-presenting effects of DCs, activate adaptive immune responses and enhance T cell activation and tumor infiltration. The maturation of DCs was enhanced in M1@Gel group after the treatment (Fig. 5A). Furthermore, CD4+ and CD8+ T cells in the mice were considerably increased by M1@Gel (Fig. 5B). We noticed that M1 group had a certain inhibitory effect on tumor recurrence compared to the control groups (Fig. 3). Results also showed that DC maturation and T cell activation were relatively enhanced in M1 group (Fig. 5).
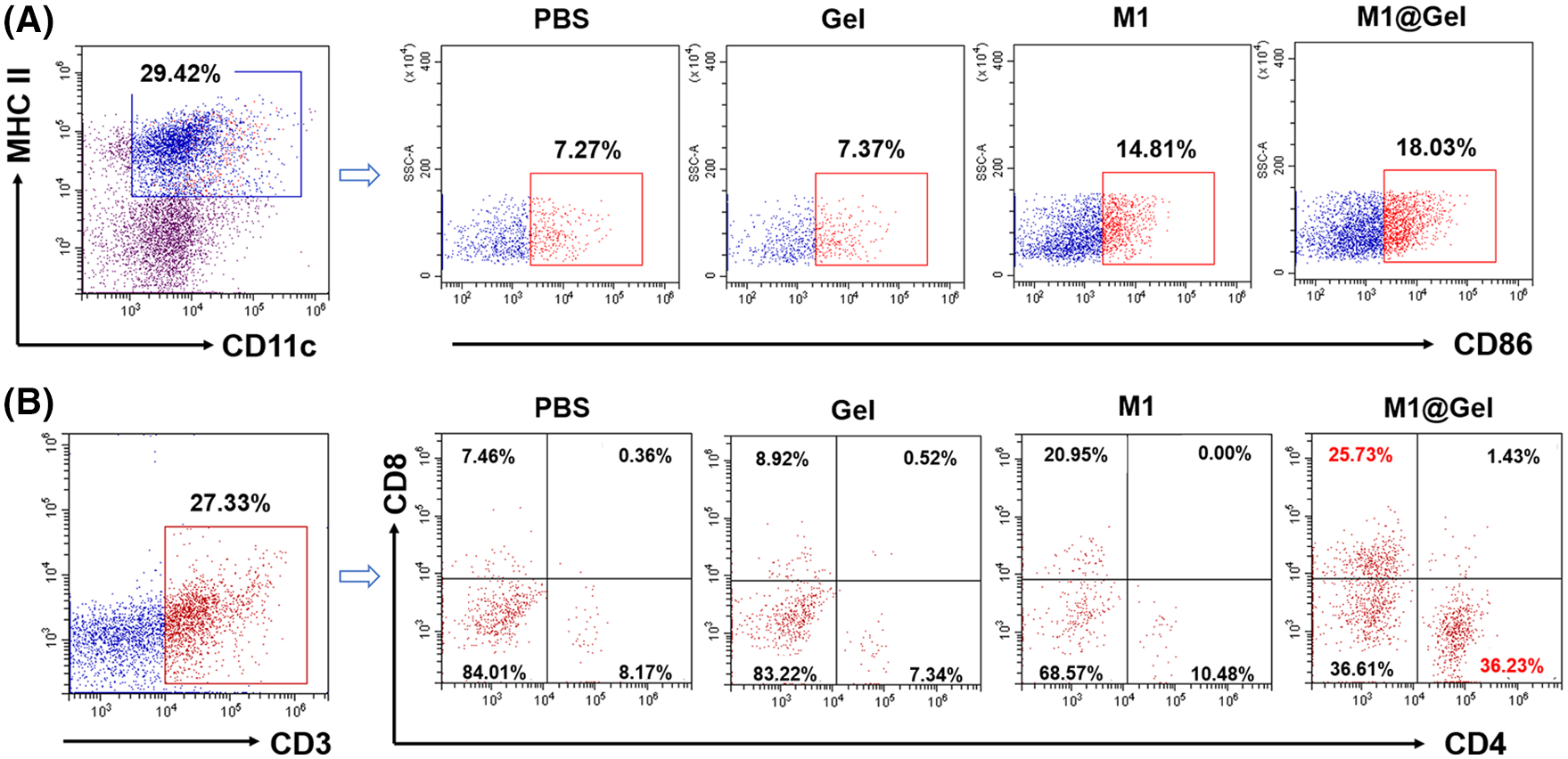
Figure 5: Activation of adaptive immune responses in mice. (A) Flow cytometry analysis of the maturation of DCs (CD11c+MHC II+CD86+) after different treatments in vivo. (B) T cell infiltration in tumor tissues after different treatments in vivo
Due to the supporting effects of alginate gel, autologous M1 macrophages were gradually released and subsequently secreted inflammatory cytokines to induce the repolarization of TAMs. In addition, autologous M1 macrophages might recruit other immune cells to synergistically reverse the immunosuppressive microenvironment of the tumor. Indeed, the potent antitumor effect induced by repolarization of macrophages in the M1@Gel group is striking.
Tumor and associated cells can produce molecules such as collagen to form a dense extracellular matrix and build barriers that block the diffusion of antitumor drugs or molecules to the tumor cells. Therefore, tumor penetration is one of the challenges for anticancer strategies, and it also accounts for the fact that CAR-T cell therapy is relatively ineffective against solid tumors [23,24]. Although primary infusion of CAR-M cells has shown antitumor effects in animal models [25,26], CAR-M may face a similar problem as CAR-T for solid tumors.
In the tumor microenvironment, TAMs are one of the main causes of tumor immunotherapy resistance. These immunosuppressive cells are powerful enough to repolarize transferred M1 macrophages to M2 phenotype. That is why M1 macrophages cannot be directly used to treat the solid tumors in the clinic. To circumvent this problem, we sought to employ M1 macrophages as a tumor vaccine after the surgery. Our data demonstrated that the controlled release of autologous M1 macrophages from the hydrogel can induce potent antitumor effects, suggesting that autologous macrophage repolarization is a promising antitumor strategy.
We developed an immunotherapeutic strategy by implanting autologous M1 macrophages to the microenvironment after surgical removal of mouse tumors. The implantable autologous M1 macrophages (M1@Gel) reprogrammed the TAMs, promoted the maturation of DCs, and activated adaptive immune responses to suppress tumor growth. The alginate hydrogel loaded with autologous M1 macrophages provided a controlled release of immune cells and achieved the prominent immune regulation effects. Therefore, the implantation of autologous M1 macrophages has been shown to be a promising strategy for cancer therapy.
Authors’ Contribution: The authors confirm contribution to the paper as follows: D. Wang and Y. Hu designed the experiments. D. Wang, H. Chen conducted in vitro and in vivo experiments. D. Wang wrote the original draft. Y. Hu revised the manuscript and coined the term “PAM”. All the authors commented on the manuscript.
Ethical Approval and Informed Consent Statement: The animal study was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Chongqing Medical University, School of Medicine. All the animal experiments were conducted under protocols approved by the IACUC of Chongqing Medical University (SYXK2018-0003).
Funding Statement: This work is financially supported by Beijing Natural Science Foundation (No. 7212212).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Restifo, N. P., Dudley, M. E., Rosenberg, S. A. (2012). Adoptive immunotherapy for cancer: Harnessing the T cell response. Nature Reviews Immunology, 12(4), 269–281. DOI 10.1038/nri3191. [Google Scholar] [CrossRef]
2. Singh, A. K., McGuirk, J. P. (2020). CAR T cells: Continuation in a revolution of immunotherapy. The Lancet Oncology, 21(3), e168–e178. DOI 10.1016/S1470-2045(19)30823-X. [Google Scholar] [CrossRef]
3. Riley, R. S., June, C. H., Langer, R., Mitchell, M. J. (2019). Delivery technologies for cancer immunotherapy. Nature Reviews Drug Discovery, 18(3), 175–196. DOI 10.1038/s41573-018-0006-z. [Google Scholar] [CrossRef]
4. Ren, W. F., Cai, X. M., Chen, J., Ruan, L. F., Lu, H. R. et al. (2021). GM-CSF-loaded nanoparticles for photothermal-assisted immunotherapy against orthotopic bladder cancer. Oncologie, 23(3), 359–371. DOI 10.32604/Oncologie.2021.018605. [Google Scholar] [CrossRef]
5. Cassetta, L., Pollard, J. W. (2018). Targeting macrophages: Therapeutic approaches in cancer. Nature Reviews Drug Discovery, 17(12), 887–904. DOI 10.1038/nrd.2018.169. [Google Scholar] [CrossRef]
6. Ruffell, B., Coussens, L. M. (2015). Macrophages and therapeutic resistance in cancer. Cancer Cell, 27(4), 462–472. DOI 10.1016/j.ccell.2015.02.015. [Google Scholar] [CrossRef]
7. DeNardo, D. G., Ruffell, B. (2019). Macrophages as regulators of tumour immunity and immunotherapy. Nature Reviews Immunology, 19(6), 369–382. DOI 10.1038/s41577-019-0127-6. [Google Scholar] [CrossRef]
8. Xia, Y., Rao, L., Yao, H., Wang, Z., Ning, P. et al. (2020). Engineering macrophages for cancer immunotherapy and drug delivery. Advanced Materials, 32(40), e2002054. DOI 10.1002/adma.202002054. [Google Scholar] [CrossRef]
9. Duan, Z., Luo, Y. (2021). Targeting macrophages in cancer immunotherapy. Signal Transduction and Targeted Therapy, 6(1), 127. DOI 10.1038/s41392-021-00506-6. [Google Scholar] [CrossRef]
10. Roche, P. A., Furuta, K. (2015). The ins and outs of MHC class II–mediated antigen processing and presentation. Nature Reviews Immunology, 15(4), 203–216. DOI 10.1038/nri3818. [Google Scholar] [CrossRef]
11. Kambayashi, T., Laufer, T. M. (2014). Atypical MHC class II-expressing antigen-presenting cells: Can anything replace a dendritic cell? Nature Reviews Immunology, 14(11), 719–730. DOI 10.1038/nri3754. [Google Scholar] [CrossRef]
12. Kapralov, A. A., Yang, Q., Dar, H. H., Tyurina, Y. Y., Anthonymuthu, T. S. et al. (2020). Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nature Chemical Biology, 16(3), 278–290. DOI 10.1038/s41589-019-0462-8. [Google Scholar] [CrossRef]
13. Mills, C. D., Lenz, L. L., Harris, R. A. (2016). A breakthrough: Macrophage-directed cancer immunotherapy. Cancer Research, 76(3), 513–516. DOI 10.1158/0008-5472.CAN-15-1737. [Google Scholar] [CrossRef]
14. Wang, D., Chen, H., Lei, L., Chen, J., Gao, J. et al. (2022). Biofabricated macrophage and fibroblast membranes synergistically promote skin wound healing. Bioengineering & Translational Medicine, e10344. DOI 10.1002/btm2.10344. [Google Scholar] [CrossRef]
15. Mantovani, A., Marchesi, F., Malesci, A., Laghi, L., Allavena, P. (2017). Tumour-associated macrophages as treatment targets in oncology. Nature Reviews Clinical Oncology, 14(7), 399–416. DOI 10.1038/nrclinonc.2016.217. [Google Scholar] [CrossRef]
16. Ngambenjawong, C., Gustafson, H. H., Pun, S. H. (2017). Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Advanced Drug Delivery Reviews, 114, 206–221. DOI 10.1016/j.addr.2017.04.010. [Google Scholar] [CrossRef]
17. Mantovani, A., Sozzani, S., Locati, M., Allavena, P., Sica, A. (2002). Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends in Immunology, 23(11), 549–555. DOI 10.1016/S1471-4906(02)02302-5. [Google Scholar] [CrossRef]
18. Chanmee, T., Ontong, P., Konno, K., Itano, N. (2014). Tumor-associated macrophages as major players in the tumor microenvironment. Cancers, 6(3), 1670–1690. DOI 10.3390/cancers6031670. [Google Scholar] [CrossRef]
19. Anderson, N. R., Minutolo, N. G., Gill, S., Klichinsky, M. (2021). Macrophage-based approaches for cancer immunotherapy. Cancer Research, 81(5), 1201–1208. DOI 10.1158/0008-5472.CAN-20-2990. [Google Scholar] [CrossRef]
20. Lee, S., Kivimae, S., Dolor, A., Szoka, F. C. (2016). Macrophage-based cell therapies: The long and winding road. Journal of Controlled Release, 240, 527–540. DOI 10.1016/j.jconrel.2016.07.018. [Google Scholar] [CrossRef]
21. Shields, C. W., Evans, M. A., Wang, L. L. W., Baugh, N., Iyer, S. et al. (2020). Cellular backpacks for macrophage immunotherapy. Science Advances, 6(18), eaaz6579. DOI 10.1126/sciadv.aaz6579. [Google Scholar] [CrossRef]
22. Wang, D., Xue, M., Chen, J., Chen, H., Liu, J. et al. (2021). Macrophage-derived implantable vaccine prevents postsurgical tumor recurrence. Biomaterials, 278, 121161. DOI 10.1016/j.biomaterials.2021.121161. [Google Scholar] [CrossRef]
23. Newick, K., O’Brien, S., Moon, E., Albelda, S. M. (2017). CAR T cell therapy for solid tumors. Annual Review of Medicine, 68, 139–152. DOI 10.1146/annurev-med-062315-120245. [Google Scholar] [CrossRef]
24. Liu, G., Rui, W., Zhao, X., Lin, X. (2021). Enhancing CAR-T cell efficacy in solid tumors by targeting the tumor microenvironment. Cellular & Molecular Immunology, 18(5), 1085–1095. DOI 10.1038/s41423-021-00655-2. [Google Scholar] [CrossRef]
25. Klichinsky, M., Ruella, M., Shestova, O., Lu, X. M., Best, A. et al. (2020). Human chimeric antigen receptor macrophages for cancer immunotherapy. Nature Biotechnology, 38(8), 947–953. DOI 10.1038/s41587-020-0462-y. [Google Scholar] [CrossRef]
26. Mukhopadhyay, M. (2020). Macrophages enter CAR immunotherapy. Nature Methods, 17(6), 561. DOI 10.1038/s41592-020-0862-4. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2022 The Author(s). Published by Tech Science Press.
Copyright © 2022 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools