| Oncologie |
DOI: 10.32604/oncologie.2022.023629
REVIEW
Delaying Emergence of Resistance to KRAS Inhibitors with Adaptive Therapy: “Treatment-to-Contain” Instead of “Treatment-to-Cure”
Pharmacotherapeutics Unit, Department of Medicine, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, Serdang, Malaysia
*Corresponding Author: Johnson Stanslas. Email: jstanslas@yahoo.co.uk
Received: 06 May 2022; Accepted: 10 June 2022
Abstract: KRAS mutations are among the most common oncogenic abnormalities in cancer. Until recently, drug discovery pursuing KRAS did not produce therapeutic benefits for patients. Specific KRAS inhibitors, such as sotorasib and adagrasib, which bind covalently to codon 12 of substituted glycine to cysteine residue of the protein (G12C), have been approved by the FDA recently for the treatment of lung cancers. Binding of these drugs to the protein inhibits the activation of the GDP-bound inactive state to the GTP-bound active state. Phase 1/2 trials have shown potential anti-tumor activity, particularly in patients with previously treated non-small cell lung cancer. Acquired resistance, on the other hand, is inevitable, and the mechanisms include new KRAS mutations such as Y96D/C and other RAS-MAPK effector protein abnormalities. “Adaptive Therapy,” an ecologically inspired concept, focuses on extending the treatment-free period in a treatment course to delay the emergence of resistance. This review focuses on acquired mechanisms of resistance to KRAS G12C inhibitors, as well as the application of adaptive therapy in the treatment of KRAS-mutated patients to maintain acquired resistance sub-clones and extend progression-free survival.
Keywords: KRAS inhibitors; adaptive therapy; drug resistance; cancer
Over the years, cancer treatment has tremendously improved with the emergence of novel treatment strategies such as selective targeted therapies that bind to oncoproteins [1]. Approximately 27% of diagnosed cancer patients are affected by the RAS family genes, namely, Kirsten rat sarcoma (KRAS), Harvey rat sarcoma (HRAS) and neuroblastoma rat sarcoma virus (NRAS). For decades, mutated KRAS was known to be undruggable due to the oncoprotein’s ability to independently hydrolyze guanosine triphosphate (GTP), thus making it constantly activated for survival and proliferation in a cancer cell [2]. In addition, about 50% of patients with KRAS-mutant-driven tumours harbour the KRAS mutation at codon 12, where substitution of glycine (G) to cysteine (C) often occurs (KRAS G12C). Additionally, point mutations occur at codon 12, with aspartic acid (G12D) or valine (G12V) replacing glycine, and these mutations are usually linked with nonsmokers [3,4]. In 2021, KRAS inhibitors such as sotorasib (AMG510) and adagrasib (MRTX849) targeting G12C mutant protein became available, which earned a breakthrough designation by the US Food and Drug Administration (FDA) and is clinically approved for treating KRAS G12C patients with metastatic lung cancer. The efficacy of sotorasib was referred to the CodeBreak-100 trial (NCT03600883). It reported an objective response rate (ORR) of 32% and a disease control rate (DCR) of 88% among lung cancer patients [5]. Aside from that, the outcome from the KRYSTAL-1 trial (NCT03785249) for adagrasib proved a promising outcome with an ORR of 45% [6]. Therefore, these drugs opened the door for many other drug discovery research aimed at KRAS oncoproteins, with some undergoing preclinical and clinical trials [7]. However, acquired resistance against targeted inhibitors has been shown to reduce therapeutic efficacy, and preclinical studies have shown the signs of different levels of sensitivity to sotorasib and/or adagrasib [8,9]. Acquired resistance to KRAS G12C inhibitors is expected, based on experience with other targeted treatments such as tyrosine kinase inhibitors for mutated epidermal growth factor receptor (EGFR). Managing the progression of drug resistance leads to the concept of “adaptive therapy”, aiming to benefit ecological (varying in the tumour volume) and evolutionary dynamics (changes in cancer cell subclones) to prolong the growth of resistant subclones and achieve progression-free survival in patients.
In this review, we focus on describing the recent reports of acquired and intrinsic resistance to KRAS inhibitors, defining adaptive therapy, and how it can be implemented in clinical settings with future KRAS inhibitors.
Drug resistance in cancer accounts for 90% of cancer-related mortalities because of poor drug treatment. Despite promising drug discoveries in cancer treatment over recent years, resistance to cytotoxic chemotherapeutic agents remains a hurdle in treating cancer in patients. They kill cancer cells by destroying their DNA, but they have several drawbacks, including relatively high toxicity and a lack of selectivity for non-cancer cells. The development of targeted therapies, such as KRAS G12C inhibitors, has progressively become a trend in recent years to precisely target and halt cancer survival. Although targeted therapy has been shown to be a highly successful treatment, the majority of patients acquire resistance throughout the course of treatment and must be switched to an alternative therapeutic strategy [10,11]. Therefore, this leads to the main reason that prevents cancer patients from achieving a complete response to a treatment. The severity of cancer acquiring resistance is responsible for around 30%–55% of non-small cell lung cancer (NSCLC) patients’ dying from the disease due to relapse [12]. In addition, 50%–70% of ovarian adenocarcinoma patients experienced relapse within 1-year post-surgery [13]. Similarly, in a multi-center study, approximately 20% of paediatric acute lymphoblastic leukaemia patients have developed recurrence due to resistance [14]. A better understanding of what and how acquired resistance arises is important in enhancing KRAS G12C inhibitors in patients.
2.1 Acquired Resistance in KRAS
Cancer cells acquire resistance by modifying the amino acid residue of the drug binding site, thus preventing further downstream inhibition. For example, this phenomenon hinders the binding site of adagrasib in the hydrophobic pocket of codon 12 and is found in nearly 50% of NSCLC patients. Concurrent point mutations in the form of G12V and G12C alleles have been documented in a NSCLC patient. Similarly, some have reported having found KRAS G12A as well as KRAS G12W [15]. Nonetheless, KRAS G12C was reported to be a common activating mutation that is not eradicated by drug exposure, as indicated by a consistent high allelic frequency in cell-free DNA, as compared to other reported acquired emerging mutations [16].
Other than mutational resistance at codon 12, novel discoveries have been found in multiple sites in the KRAS switch-II adagrasib-binding pocket, namely, R68S; H95D; Y96C [15]. These mutations in the switch-II pocket could result in disruption of the non-covalent binding interaction of adagrasib and additionally form a weaker interaction between H95 residue and sotorasib, which was confirmed by X-ray crystallography. Furthermore, in the Ba/F3 hybridoma cell line revealed concomitant mutations of KRAS G12C and novel pocket mutations such as R68S, H95D, H95Q, and Y96C that conferred resistance to adagrasib, as well as R68S and Y96C to sotorasib. In contrast, H95D, H95Q, and H95R acquired mutations remained sensitive to sotorasib [17]. As demonstrated by structural analysis [18], the Y96D residue, which corresponds to a tyrosine substitution aspartate mutation at position 96, is able to have a collective vulnerability to the various clinical KRAS G12C drugs. A report has suggested that point mutation Y96D prevents the crucial hydrogen bond for the covalent binding of adagrasib’s pyrimidine ring and position 96 tyrosine hydroxyl group. Furthermore, Y96D is believed to have the ability to make the switch-II pocket more hydrophilic for the binding of adagrasib, thus reducing the efficacy of drug interaction. It also impacts other KRAS G12C inhibitors, indicating that numerous drugs have common lability. The Y96D possesses a functional role as an acquired resistance mechanism by allowing continuous KRAS activation and resistance to KRAS G12C drugs with the evidence of preclinical ectopic insertion of KRAS G12C-addicted cancer cell lines [16,18].
2.3 Intrinsic Resistance in KRAS
In medicine, intrinsic (primary) resistance is thought to be identical with “de novo resistance,” which clinicians interpret as “from the start.” It is regarded as a poor first response to a drug in a patient who has not previously undergone therapy, resulting in decreased efficacy, such as tyrosine kinase inhibitors (TKI), over the course of treatment. In the clinic, where resistance is largely focused on when it occurs rather than how it occurs, de novo resistance contradicts acquired (secondary) resistance. Patients with colorectal cancer who were resistant to cetuximab, a monoclonal antibody that binds the extracellular domain of EGFR, had a pre-existing KRAS mutation. Misale et al. hypothesised that the emergence of a cetuximab resistant population could derive from selection of a pre-existing KRAS amplified or mutant clone, or as the result of de novo acquisition of a KRAS mutation under the pressure of cetuximab treatment. The percentage of KRAS mutant alleles detected in the resistant tumours ranged from 0.4% to 17% [19]. Aside from that, KRAS mutations have been designated to be a driver mutation in cancer, with several publications demonstrating a low allelic frequency of KRAS concomitantly existing alongside other driver mutations such as EGFR [20,21], and it was found that de novo KRAS G12C mutation was present ranging from 0.2% [22] to 1.17% of detected samples (n = 6) [23,24]. Detecting de novo KRAS mutation requires high sensitivity and specificity technology, and it was found that KRAS mutation was absent using droplet digital PCR. However, with better technology, such as next-generation sequencing, a variant allelic frequency as low as 3% was detected in EGFR-mutated patients [25]. As a result, the existence of a de novo KRAS mutation on top of another driver mutation in early diagnosis influences therapy decisions in overcoming tumour heterogeneity in cancer [26,27].
Traditionally, cancer treatment has revolved around the notion of maximal cell killing as a way of treating the disease or, at the very least, extending one’s life. The continuous dosing regimen criterion in targeted therapies focuses on “hit-hard and hit-fast,” and the goal of killing as many cancer cells as possible in the shortest amount of time may be evolutionarily naive due to natural selection of high drug-selective pressure in facilitating cancer cell resistance mutations such as R68S, H95D, and Y96C. Furthermore, one of the first-generation tyrosine kinase inhibitors, erlotinib (150 mg/daily), has a progression-free survival of approximately 13 months before the tumour progresses due to acquired resistance mutation [28]. However, a case report described a patient receiving similar drug dosage treatment who was able to safely maintain tumour burden for three years (4 weeks on/4 weeks off) before tumour progression [29]. Although the continuous treatment strategy showed promising early clinical results, the concept negates the tumour’s complex ecosystem, which contains normal cells as well as areas with poor blood flow and oxygen content. An adequate dose is necessary to affect all cancer cells, creating an environment where resistance sub-clones can emerge [30,31].
A new cancer treatment paradigm, called “adaptive therapy,” substitutes “treatment-to-cure” with “treatment-to-contain.” Sub-clonal survival is emphasised in this eco-evolutionary paradigm through competitive interactions between sensitive and resistant cells [30,31]. Besides acquiring resistant mutations, these cells downregulate their cellular fitness as a compensation. The cells possess increased rates of DNA repair or other costly metabolic activities required to pump toxic drugs across cell membranes, which results in a reduction in cellular growth rate. This phenomenon has been studied in vitro in a NSCLC cell line, PC-9, that acquired the point mutation T790M, which is shown to proliferate roughly 1.22 times slower than parental cells [32,33]. As a result, this leads to the general principle of adaptive therapy is that treatment should be used to reduce the total cancer population below a certain symptomatic threshold while sustaining a significant population of sensitive cells, and the extended treatment-free period allows both sensitive and resistant cells to proliferate competitively for survival [34–36] (Fig. 1).
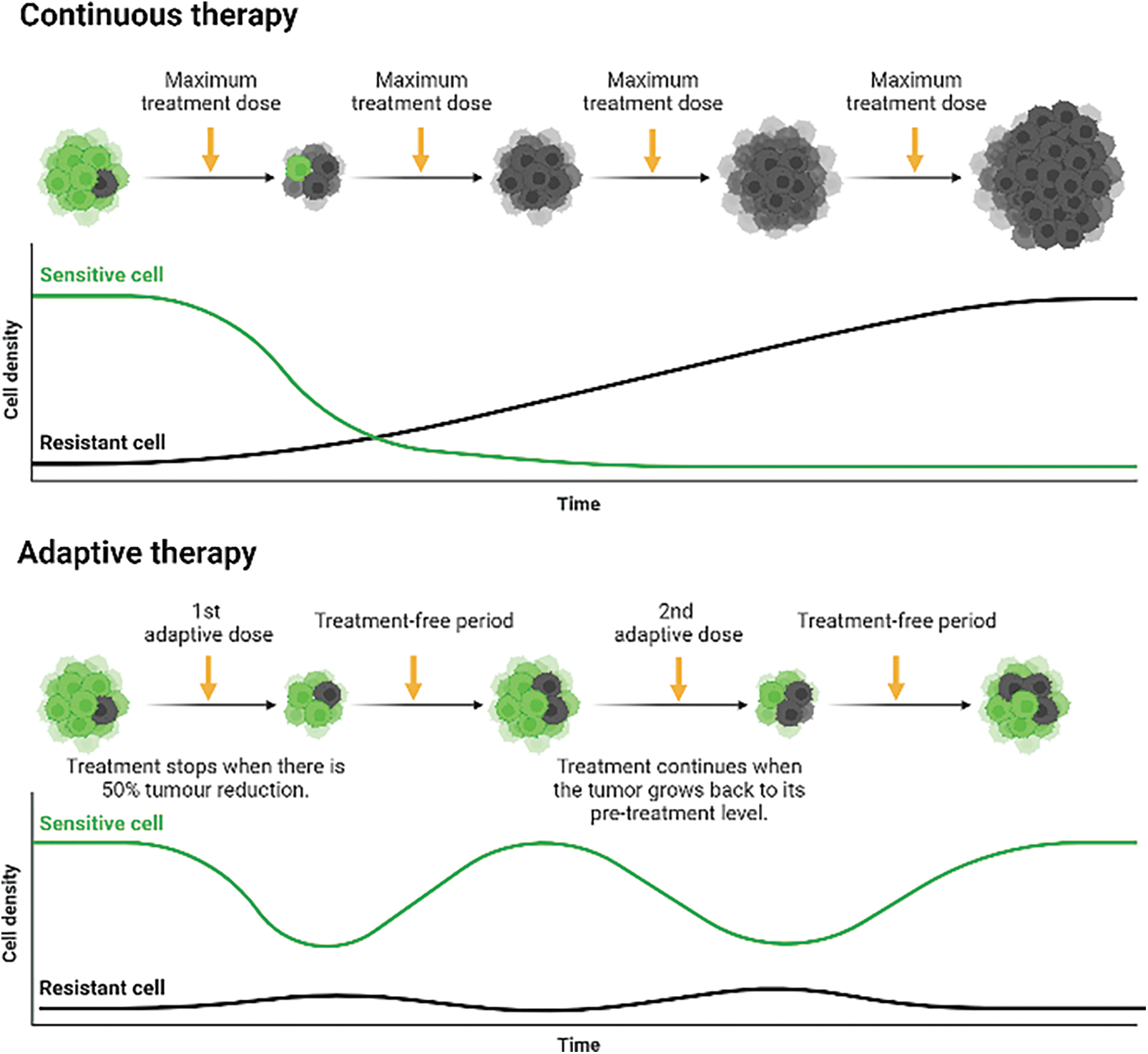
Figure 1: Continuous therapy against adaptive therapy in administering anti-cancer drugs to delay complete tumorigenic resistance. Continuous therapy: Upon diagnosis, continuous treatment will eventually eliminate all sensitive cells and select for the growth of pre-existing (de novo) and acquired resistance cells, eradicating cellular competition. Adaptive therapy: The combination of a prolonged treatment-free period and a cellular fitness difference between sensitive and resistant cells creates a cycle of cellular eradication, followed by regrowth, creating a stable tumour burden. The higher growth rate of sensitive cells outcompetes resistant cells for nutrients and space during the elongated treatment-free period, leading to the eradication of some resistant cells. Therefore, it delays the growth and emergence of both de novo resistance cells and acquired resistance cells. Created with https://BioRender.com
3.2 Clinical Evidence of Adaptive Therapy
In practise, the length of the adaptive cycle is largely consistent and manageable in each patient, although it can range between 4 and 18 months depending on the number of subpopulations, their proliferation rate, and the level of competition between subpopulations [37]. Furthermore, in a preliminary clinical study, tumour burden stability in patients with prostate cancer could be maintained with a treatment-to-failure period of at least 27 months and a drug accumulation decrease of 47% to 50% of the conventional dose [37]. This study was also supported in patients with recurrent prostate cancer [38] and metastatic castrate-resistant prostate cancer [39]. Adaptive therapy requires constant monitoring of the tumour progression that is feasible and non-invasive to the patient. In the case of prostate cancer, fluctuating levels of prostate-specific antigen (PSA) has been used as a biomarker to predict treatment response, both in early-and advanced-stage prostate cancer. It was debated whether intermittent treatment in prostate cancer with PSA monitoring was superior to continuous treatment in terms of efficacy and toxicity safety [40–43]. Brady-Nicholls et al. demonstrated that their computational model could predict clinical outcome with 81% accuracy using PSA dynamics in metastatic castrate-resistant prostate cancer [44]. This shows that the model correlates PSA dynamics with tumour burden to understand the mechanism of a patient’s evolving disease. Therefore, the correlation can provide ample time for clinicians to provide alternative treatment options or dosages instead of continuously treating the tumour to provide a better outcome with lower toxicity side effects. Designing dosage regimens to maintain a constant tumour burden rather than total eradication requires a long-term, multi-layered strategy that goes beyond the rapid therapeutic benefits of any one medication.
Adaptive therapy does not completely halt the development of resistance. In fact, it slows down the rate at which cancer cells gain resistance with a prolonged treatment-free period owing to reduced drug exposure [39–45]. In contrast, the lack of prolonged treatment-free time during the continuous administration of a drug, such as sotorasib or adagrasib, results in an increased rate of cancer cells acquiring resistance mutations and prevents patients from achieving a complete response to a drug [46,47]. In the long run, this strategy has the potential for KRAS patients to be given the same treatment for a longer period, safely maintaining a safe tumour burden that is financially feasible for patients and increasing progression-free survival [48].
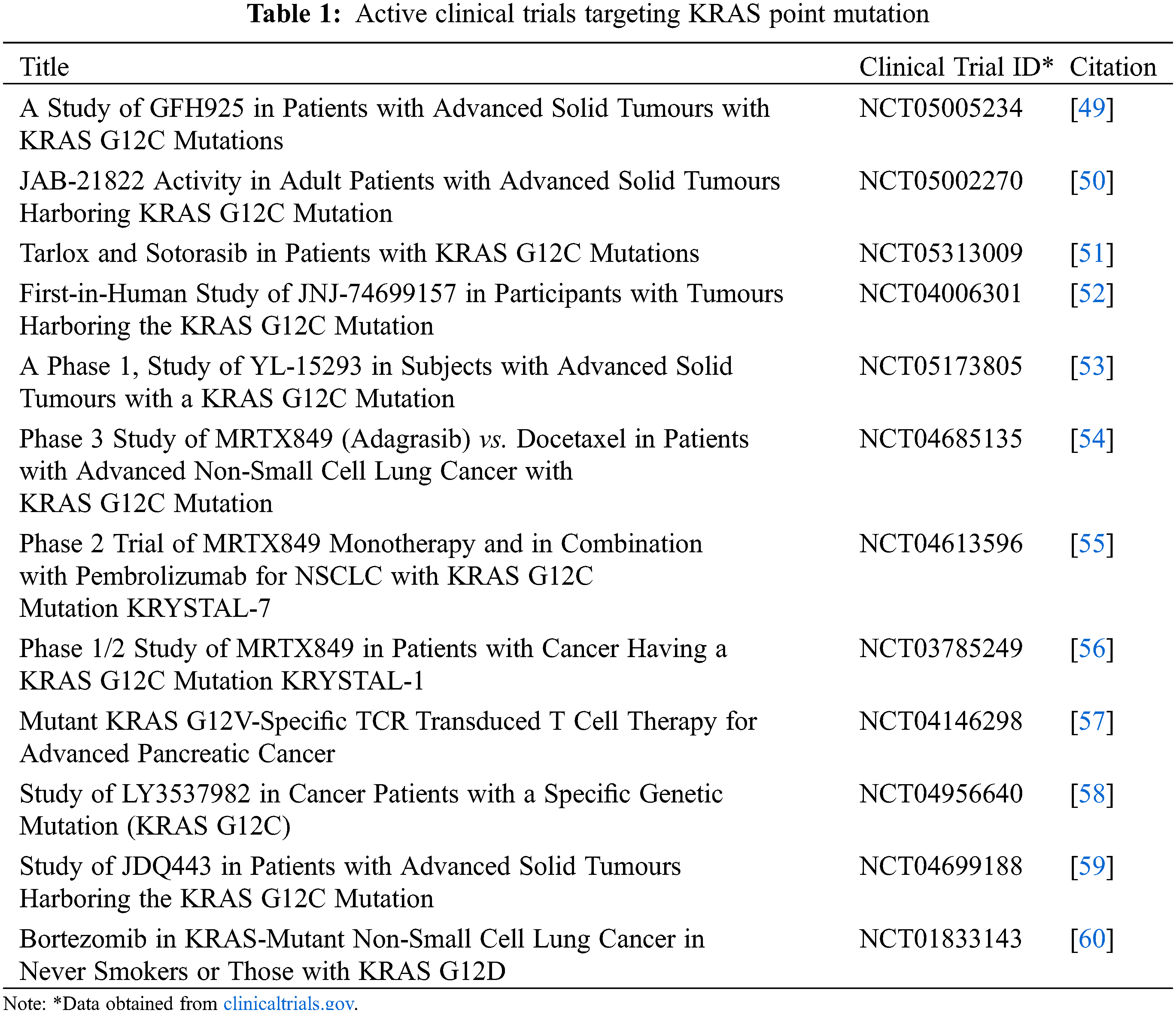
The discovery of sotorasib and adagrasib targeting KRAS G12C paves the way for much more research on KRAS (Table 1). However, drug resistance is inevitable, and resistance to sotorasib and/or adagrasib was reported a few months after FDA approval. Immediately, a new problem arises: what type of drug should be used to overcome the novel acquired resistance to KRAS inhibitors? Multiple clinical trials are in pursuit of finding drug combinations and novel drugs to overcome resistance to KRAS G12C inhibitors or finding a solution to extend progression-free survival. Even though adaptive therapy has not been applied in KRAS-mutated patients yet, the theory behind adaptive therapy is a breath of fresh air in terms of delaying the emergence of drug resistance, and by managing drug resistant sub-clones, it can be used without fear of cancer tumours acquiring resistance in a short period. Additionally, the concept does not promote a new set of drugs to do so; instead, it highlights treating a tumour with initial drug treatment with the additional feature of elongating the treatment-free period. Finally, adaptive therapy has been shown to be capable of preparing for the emergence of drug resistance and potentially leading to years of progression-free survival, including being financially feasible for patients in the coming years of KRAS inhibitor drug discovery.
Authors Contributions: The authors confirm contribution to the paper as follows: study conception and design, data collection, analysis and interpretation of data, draft manuscript preparation: Amir Imran Faisal Hamdi, Johnson Stanslas. All authors reviewed the results and approved the final version of the manuscript.
Ethics Approval and Informed Consent Statement: Not applicable.
Availability of Data and Materials: Not applicable.
Funding Statement: This work was supported by the Fundamental Research Grant Scheme [Grant No. FRGS/1/2017/SKK06/UPM/01/1 to J Stanslas].
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Herbst, R. S., Morgensztern, D., Boshoff, C. (2018). The biology and management of non-small cell lung cancer. Nature, 553(7689), 446–454. DOI 10.1038/nature25183. [Google Scholar] [CrossRef]
2. Hong, D. S., Fakih, M. G., Strickler, J. H., Desai, J., Durm, G. A. et al. (2020). KRASG12C inhibition with sotorasib in advanced solid tumors. The New England Journal of Medicine, 383(13), 1207–1217. DOI 10.1056/NEJMoa1917239. [Google Scholar] [CrossRef]
3. Govindan, R., Ding, L., Griffith, M., Subramanian, J., Dees, N. D. et al. (2012). Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell, 150(6), 1121–1134. DOI 10.1016/j.cell.2012.08.024. [Google Scholar] [CrossRef]
4. Jones, R. P., Sutton, P. A., Evans, J. P., Clifford, R., McAvoy, A. et al. (2017). Specific mutations in KRAS codon 12 are associated with worse overall survival in patients with advanced and recurrent colorectal cancer. British Journal of Cancer, 116(7), 923–929. DOI 10.1038/bjc.2017.37. [Google Scholar] [CrossRef]
5. Skoulidis, F., Li, B. T., Dy, G. K., Price, T. J., Falchook, G. S. et al. (2021). Sotorasib for lung cancers with KRAS p.G12C mutation. The New England Journal of Medicine, 384(25), 2371–2381. DOI 10.1056/NEJMoa2103695. [Google Scholar] [CrossRef]
6. Riely, G. J., Ou, S. H. I., Rybkin, I., Spira, A., Papadopoulos, K. (2021). 99O_PR KRYSTAL-1: Activity and preliminary pharmacodynamic (PD) analysis of adagrasib (MRTX849) in patients (Pts) with advanced non–small cell lung cancer (NSCLC) harboring KRASG12C mutation. Journal of Thoracic Oncology, 16(4), S751–S752. DOI 10.1016/S1556-0864(21)01941-9. [Google Scholar] [CrossRef]
7. Kidger, A. M., Sipthorp, J., Cook, S. J. (2018). ERK1/2 inhibitors: New weapons to inhibit the RAS-regulated RAF-MEK1/2-ERK1/2 pathway. Pharmacology & Therapeutics, 187, 45–60. DOI 10.1016/j.pharmthera.2018.02.007. [Google Scholar] [CrossRef]
8. Dunnett-Kane, V., Nicola, P., Blackhall, F., Lindsay, C. (2021). Mechanisms of resistance to KRASG12C inhibitors. Cancers, 13(1), 151. DOI 10.3390/cancers13010151. [Google Scholar] [CrossRef]
9. Hallin, J., Engstrom, L. D., Hargis, L., Calinisan, A., Aranda, R. (2020). The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discovery, 10(1), 54–71. DOI 10.1158/2159-8290.CD-19-1167. [Google Scholar] [CrossRef]
10. Konieczkowski, D. J., Johannessen, C. M., Garraway, L. A. (2018). A Convergence-based framework for cancer drug resistance. Cancer Cell, 33(5), 801–815. DOI 10.1016/j.ccell.2018.03.025. [Google Scholar] [CrossRef]
11. McGranahan, N., Swanton, C. (2017). Clonal heterogeneity and tumor evolution: Past, present, and the future. Cell, 168(4), 613–628. DOI 10.1016/j.cell.2017.01.018. [Google Scholar] [CrossRef]
12. Uramoto, H., Tanaka, F. (2014). Recurrence after surgery in patients with NSCLC. Translational Lung Cancer Research, 3(4), 242–249. DOI 10.3978/j.issn.2218-6751.2013.12.05. [Google Scholar] [CrossRef]
13. Castells, M., Thibault, B., Delord, J. P., Couderc, B. (2012). Implication of tumor microenvironment in chemoresistance: Tumor-associated stromal cells protect tumor cells from cell death. International Journal of Molecular Sciences, 13(8), 9545–9571. DOI 10.3390/ijms13089545. [Google Scholar] [CrossRef]
14. O’Connor, D., Sibson, K., Caswell, M., Connor, P., Cummins, M. et al. (2011). Early UK experience in the use of clofarabine in the treatment of relapsed and refractory paediatric acute lymphoblastic leukaemia. British Journal of Haematology, 154(4), 482–485. DOI 10.1111/j.1365-2141.2011.08752.x. [Google Scholar] [CrossRef]
15. Awad, M. M., Liu, S., Rybkin, I. I., Arbour, K. C., Dilly, J. et al. (2021). Acquired resistance to KRASG12C inhibition in cancer. The New England Journal of Medicine, 384(25), 2382–2393. DOI 10.1056/NEJMoa2105281. [Google Scholar] [CrossRef]
16. Pinnelli, M., Trusolino, L. (2021). The gRASs is greener: Potential New therapies in lung cancer with acquired resistance to KRASG12C inhibitors. Cancer Discovery, 11(8), 1874–1876. DOI 10.1158/2159-8290.CD-21-0609. [Google Scholar] [CrossRef]
17. Amodio, V., Yaeger, R., Arcella, P., Cancelliere, C., Lamba, S. et al. (2020). EGFR blockade reverts resistance to KRASG12C inhibition in colorectal cancer. Cancer Discovery, 10(8), 1129–1139. DOI 10.1158/2159-8290.CD-20-0187. [Google Scholar] [CrossRef]
18. Tanaka, N., Lin, J. J., Li, C., Ryan, M. B., Zhang, J. (2021). Clinical acquired resistance to KRASG12C inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS-MAPK reactivation. Cancer Discovery, 11(8), 1913–1922. DOI 10.1158/2159-8290.CD-21-0365. [Google Scholar] [CrossRef]
19. Misale, S., Yaeger, R., Hobor, S., Scala, E., Janakiraman, M. et al. (2012). Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature, 486(7404), 532–536. DOI 10.1038/nature11156. [Google Scholar] [CrossRef]
20. Li, S., Li, L., Zhu, Y., Huang, C., Qin, Y. et al. (2014). Coexistence of EGFR with KRAS, or BRAF, or PIK3CA somatic mutations in lung cancer: A comprehensive mutation profiling from 5125 Chinese cohorts. British Journal of Cancer, 110(11), 2812–2820. DOI 10.1038/bjc.2014.210. [Google Scholar] [CrossRef]
21. Guo, Y., Song, J., Wang, Y., Huang, L., Sun, L. et al. (2020). Concurrent genetic alterations and other biomarkers predict treatment efficacy of EGFR-TKIs in EGFR-mutant non-small cell lung cancer: A review. Frontiers in Oncology, 10, 610923. DOI 10.3389/fonc.2020.610923. [Google Scholar] [CrossRef]
22. Nardo, G., Carlet, J., Marra, L., Bonanno, L., Boscolo, A. et al. (2021). Detection of low-frequency KRAS mutations in cfDNA from EGFR-mutated NSCLC patients after first-line EGFR tyrosine kinase inhibitors. Frontiers in Oncology, 10, 607840. DOI 10.3389/fonc.2020.607840. [Google Scholar] [CrossRef]
23. Serna-Blasco, R., Sánchez-Herrero, E., Sanz-Moreno, S., Rodriguez-Festa, A., García-Veros, E. et al. (2021). KRAS p.G12C mutation occurs in 1% of EGFR-mutated advanced non-small-cell lung cancer patients progressing on a first-line treatment with a tyrosine kinase inhibitor. ESMO Open, 6(5), 100279. DOI 10.1016/j.esmoop.2021.100279. [Google Scholar] [CrossRef]
24. Aran, V., Zalis, M., Montella, T., de Sousa, C., Ferrari, B. L. et al. (2021). Evaluation of KRAS concomitant mutations in advanced lung adenocarcinoma patients. Medicina, 57(10), 1039. DOI 10.3390/medicina57101039. [Google Scholar] [CrossRef]
25. Rachiglio, A. M., Fenizia, F., Piccirillo, M. C., Galetta, D., Crinò, L. et al. (2019). The presence of concomitant mutations affects the activity of EGFR tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer (NSCLC) patients. Cancers, 11(3), 341. DOI 10.3390/cancers11030341. [Google Scholar] [CrossRef]
26. He, H., Xu, C., Cheng, Z., Qian, X., Zheng, L. (2019). Drug combinatorial therapies for the treatment of KRAS mutated lung cancers. Current Topics in Medicinal Chemistry, 19(23), 2128–2142. DOI 10.2174/1568026619666190902150555. [Google Scholar] [CrossRef]
27. Zarredar, H., Pashapour, S., Ansarin, K., Khalili, M., Baghban, R. et al. (2019). Combination therapy with KRAS siRNA and EGFR inhibitor AZD8931 suppresses lung cancer cell growth in vitro. Journal of Cellular Physiology, 234(2), 1560–1566. DOI 10.1002/jcp.27021. [Google Scholar] [CrossRef]
28. Becker, A., van Wijk, A., Smit, E. F., Postmus, P. E. (2010). Side-effects of long-term administration of erlotinib in patients with non-small cell lung cancer. Journal of Thoracic Oncology, 5(9), 1477–1480. DOI 10.1097/JTO.0b013e3181e981d9. [Google Scholar] [CrossRef]
29. Matsuzaki, T., Iwami, E., Sasahara, K., Kuroda, A., Nakajima, T. et al. (2019). A case report of metastatic lung adenocarcinoma with long-term survival for over 11 years. Medicine, 98(4), e14100. DOI 10.1097/MD.0000000000014100. [Google Scholar] [CrossRef]
30. Gatenby, R. A. (2009). A change of strategy in the war on cancer. Nature, 459(7246), 508–509. DOI 10.1038/459508a. [Google Scholar] [CrossRef]
31. Gatenby, R. (2012). Perspective: Finding cancer’s first principles. Nature, 491(7425), S55. DOI 10.1038/491s55a. [Google Scholar] [CrossRef]
32. Chmielecki, J., Foo, J., Oxnard, G. R., Hutchinson, K., Ohashi, K. et al. (2011). Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Science Translational Medicine, 3(90), 90ra59. DOI 10.1126/scitranslmed.3002356. [Google Scholar] [CrossRef]
33. Xie, X., Li, L., Xie, L., Liu, Z., Gao, X. et al. (2021). A new single-cell level R-index for EGFR-TKI resistance and survival prediction in LUAD. bioRxiv. DOI 10.1101/2021.07.30.454426. [Google Scholar] [CrossRef]
34. Enriquez-Navas, P. M., Kam, Y., Das, T., Hassan, S., Silva, A. et al. (2016). Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Science Translational Medicine, 8(327), DOI 10.1126/scitranslmed.aad7842. [Google Scholar] [CrossRef]
35. Grolmusz, V. K., Chen, J., Emond, R., Cosgrove, P. A., Pflieger, L. et al. (2020). Exploiting collateral sensitivity controls growth of mixed culture of sensitive and resistant cells and decreases selection for resistant cells in a cell line model. Cancer Cell International, 20, 253. DOI 10.1186/s12935-020-01337-1. [Google Scholar] [CrossRef]
36. Emond, R., Griffiths, J. I., Grolmusz, V. K., Sousa, R. S., Bild, A. H. (2021). Ecological interactions in breast cancer: Cell facilitation promotes growth and survival under drug pressure. bioRxiv. DOI 10.1101/2021.02.01.429214. [Google Scholar] [CrossRef]
37. Zhang, J., Cunningham, J. J., Brown, J. S., Gatenby, R. A. (2017). Integrating evolutionary dynamics into treatment of metastatic castrate-resistant prostate cancer. Nature Communications, 8(1), 1816. DOI 10.1038/s41467-017-01968-5. [Google Scholar] [CrossRef]
38. Brady-Nicholls, R., Nagy, J. D., Gerke, T. A., Zhang, T., Wang, A. Z. et al. (2020). Prostate-specific antigen dynamics predict individual responses to intermittent androgen deprivation. Nature Communications, 11(1), 1750. DOI 10.1038/s41467-020-15424-4. [Google Scholar] [CrossRef]
39. Cunningham, J., Thuijsman, F., Peeters, R., Viossat, Y., Brown, J. et al. (2020). Optimal control to reach eco-evolutionary stability in metastatic castrate-resistant prostate cancer. PLoS One, 15(12), e0243386. DOI 10.1371/journal.pone.0243386. [Google Scholar] [CrossRef]
40. Klotz, L. (2013). Intermittent versus continuous androgen deprivation therapy in advanced prostate cancer. Current Urology Reports, 14(3), 159–167. DOI 10.1007/s11934-013-0325-x. [Google Scholar] [CrossRef]
41. Hussain, M., Tangen, C. M., Berry, D. L., Higano, C. S., Crawford, E. D. et al. (2013). Intermittent versus continuous androgen deprivation in prostate cancer. The New England Journal of Medicine, 368(14), 1314–1325. DOI 10.1056/NEJMoa1212299. [Google Scholar] [CrossRef]
42. Higano, C. S. (2014). Intermittent versus continuous androgen deprivation therapy. Journal of the National Comprehensive Cancer Network : JNCCN, 12(5), 727–733. DOI 10.6004/jnccn.2014.0074. [Google Scholar] [CrossRef]
43. Botrel, T. E., Clark, O., dos Reis, R. B., Pompeo, A. C., Ferreira, U. et al. (2014). Intermittent versus continuous androgen deprivation for locally advanced, recurrent or metastatic prostate cancer: A systematic review and meta-analysis. BMC Urology, 14, 9. DOI 10.1186/1471-2490-14-9. [Google Scholar] [CrossRef]
44. Brady-Nicholls, R., Zhang, J., Zhang, T., Wang, A. Z., Butler, R. et al. (2021). Predicting patient-specific response to adaptive therapy in metastatic castration-resistant prostate cancer using prostate-specific antigen dynamics. Neoplasia, 23(9), 851–858. DOI 10.1016/j.neo.2021.06.013. [Google Scholar] [CrossRef]
45. Cunningham, J. J. (2019). A call for integrated metastatic management. Nature Ecology & Evolution, 3(7), 996–998. DOI 10.1038/s41559-019-0927-x. [Google Scholar] [CrossRef]
46. Gallaher, J. A., Enriquez-Navas, P. M., Luddy, K. A., Gatenby, R. A., Anderson, A. (2018). Spatial heterogeneity and evolutionary dynamics modulate time to recurrence in continuous and adaptive cancer therapies. Cancer Research, 78(8), 2127–2139. DOI 10.1158/0008-5472.CAN-17-2649. [Google Scholar] [CrossRef]
47. Strobl, M., Gallaher, J., West, J., Robertson-Tessi, M., Maini, P. K. et al. (2022). Spatial structure impacts adaptive therapy by shaping intra-tumoral competition. Communications Medicine, 2, 46. DOI 10.1038/s43856-022-00110-x. [Google Scholar] [CrossRef]
48. Thomas, F., Donnadieu, E., Charriere, G. M., Jacqueline, C., Tasiemski, A. et al. (2018). Is adaptive therapy natural? PLoS Biology, 16(10), e2007066. DOI 10.1371/journal.pbio.2007066. [Google Scholar] [CrossRef]
49. Zhejiang Genfleet Therapeutics Co., Ltd. (2022). An open-label, multi-center phase I/II clinical study evaluating the safety/Tolerability, pharmacokinetics, and effectiveness of GFH925 in patients with advanced solid tumors with KRAS G12C mutations. https://clinicaltrials.gov/ct2/show/NCT05005234. [Google Scholar]
50. Jacobio Pharmaceuticals Co., Ltd. (2022). A phase 1/2, multi-center, open-label study to evaluate the safety, tolerability, pharmacokinetics, and preliminary evidence of antitumor activity of JAB-21822 monotherapy and combination therapy in adult patients with advanced solid tumors harboring KRAS G12C mutation. https://clinicaltrials.gov/ct2/show/NCT05002270. [Google Scholar]
51. Medical University of South Carolina. (2022). A phase Ib/II trial of tarloxotinib and sotorasib in patients with KRAS G12C mutations. https://clinicaltrials.gov/ct2/show/NCT05313009. [Google Scholar]
52. Janssen Research & Development, LLC (2022). A First-in-human study of the safety, pharmacokinetics, pharmacodynamics, and preliminary antitumor activity of JNJ-74699157 in participants with advanced solid tumors harboring the KRAS G12C mutation. https://clinicaltrials.gov/ct2/show/NCT04006301. [Google Scholar]
53. Shanghai YingLi Pharmaceutical Co, Ltd. (2022). A phase 1/2, study of YL-15293 in subjects with advanced solid tumors with a KRAS G12C mutation. https://clinicaltrials.gov/ct2/show/NCT05173805. [Google Scholar]
54. Mirati Therapeutics Inc. (2022). A randomized phase 3 study of MRTX849 versus docetaxel in patients with previously treated non-small cell lung cancer with KRAS G12C mutation. https://clinicaltrials.gov/ct2/show/NCT04685135. [Google Scholar]
55. Mirati Therapeutics Inc. (2022). A phase 2 trial of MRTX849 monotherapy and in combination with pembrolizumab in patients with advanced non-small cell lung cancer with KRAS G12C mutation. https://clinicaltrials.gov/ct2/show/NCT04613596. [Google Scholar]
56. Mirati Therapeutics Inc. (2022). A phase 1/2 multiple expansion cohort trial of MRTX849 in patients with advanced solid tumors with KRAS G12C mutation KRYSTAL-1. [Google Scholar]
57. Guo, S. W. (2022). Clinical trial evaluating the safety and activity of mutant KRAS G12 V-specific TCR transduced T cell therapy for advanced pancreatic cancer. https://clinicaltrials.gov/ct2/show/NCT04146298. [Google Scholar]
58. Eli Lilly and Company (2022). A phase 1a/1b study of LY3537982 in patients with KRAS G12C-mutant advanced solid tumors. https://clinicaltrials.gov/ct2/show/NCT04956640. [Google Scholar]
59. Novartis Pharmaceuticals (2022). A phase Ib/II open-label, multi-center dose escalation study of JDQ443 in patients with advanced solid tumors harboring the KRAS G12C mutation. https://clinicaltrials.gov/ct2/show/NCT04699188. [Google Scholar]
60. Memorial Sloan Kettering Cancer Center (2022). A phase 2 trial of bortezomib in KRAS-mutant non-small cell lung cancer in never smokers or those with KRAS G12D. https://clinicaltrials.gov/ct2/show/NCT01833143. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |