| Oncologie |
DOI: 10.32604/oncologie.2022.021490
REVIEW
Leucémie à Tricholeucocytes et Autres Proliférations à Cellules Chevelues: Diagnostic et Traitement
Hairy Cell Leukemia and HCL-Like Disorders: Diagnosis and Treatment
Hématologie, CHU Côte de Nacre, Caen Cedex, 14033, France
*Corresponding Author: Xavier Troussard. Email: troussard-x@chu-caen.fr
Received: 17 January 2022; Accepted: 04 March 2022
Résumé: La leucémie à tricholeucocytes (LT) représente 2% de l’ensemble des leucémies. Le diagnostic repose sur la présence dans le sang et/ou la moelle de tricholeucocytes: cellules lymphoïdes B au cytoplasme chevelu exprimant le CD103, CD123, CD11c et CD25. La mutation BRAFV600E, marqueur moléculaire de la maladie, est présente dans plus de 80% des cas. La LT doit être distinguée des autres syndromes lymphoprolifératifs chroniques B, notamment des autres proliférations à cellules chevelues, forme variante de la leucémie à tricholeucocytes (LT-V) et lymphome splénique diffus de la pulpe rouge (LSDPR). Des progrès thérapeutiques ont été récemment réalisés. Les analogues des purines (PNAs) en monothérapie: désoxycoformycine (DCF) ou 2-chloro-désoxyadénosine (CDA), restent le traitement de référence de la première ligne. Ils peuvent être associés aux anticorps monoclonaux anti-CD20 (rituximab, obinutuzumab) dès la première ligne, l’immunochimiothérapie permettant l’obtention de rémissions complètes (RC) prolongées. Chez les patients avec une maladie en rechute/réfractaire (R/R), de nouvelles options thérapeutiques ont émergé: immunotoxines, inhibiteurs de BRAF (BRAFi) ou de BTK. Ces traitements sont à discuter en réunion de concertation pluridisciplinaire (RCP). Chez les patients avec une LT-V, l’immunochimiothérapie est maintenant le traitement de référence en première ligne. Dans tous les cas, la surveillance hématologique prolongée est nécessaire, en raison du risque augmenté de cancer secondaire et notamment celui d’hémopathie maligne.
Abstract: Hairy cell leukemia (LT) accounts for 2% of all leukemias. The diagnosis is based on the presence in the blood and/or the marrow of hairy cells expressing CD103, CD123, CD11c and CD25. The BRAFV600E mutation, a molecular marker of the disease, is present in more than 80% of cases. LT should be distinguished from other chronic B-cell lymphoproliferative disorders, including the variant form of hairy cell leukemia (HCL-V) and diffuse splenic red pulp lymphoma (DSRPL). Progress has recently been made in the management of patients. The purine analogues (PNAs) in monotherapy, deoxycoformycin (DCF) or 2-chloro-deoxyadenosine (CDA), remain the first-line reference treatment. PNAs can be associated with anti-CD20 monoclonal antibodies (rituximab, obinutuzumab) and are now being introduced from the first line, with immunochemotherapy making it possible to obtain prolonged complete remissions (CR). In patients with relapsed/refractory (R/R) hairy cell leukemia, new therapeutic options emerged: immunotoxins, BRAF (BRAFi) or BTK inhibitors. The different options have to be discussed in a multidisciplinary consultation meeting. In patients with HCL-V, immunochemotherapy is the first-line standard of treatment. In all cases, prolonged haematological monitoring is necessary, because of the increased risk of secondary cancer, particularly the risk of hematologic malignancies.
Mots-clés: Leucémie à tricholeucocytes; HCL, mutation BRAFV600E; forme variante de la leucémie à; tricholeucocytes; lymphome diffus de la pulpe rouge de la rate; Analogues des purines; Anticorps monoclonaux anti-CD20; Immunotoxines; Inhibiteurs de BRAF
Keywords: Hairy cell leukemia; HCL; BRAFV600E mutation; variant form of hairy cell leukemia; splenic diffuse red pulp lymphoma; Purine nucleoside analogs; anti-CD20 monoclonal antibodies; immunotoxins; BRAF inhibitors
Parmi les hémopathies lymphoïdes B matures de la classification WHO 2008 des tumeurs hématopoïétiques et des tissus lymphoïdes, nous étudierons dans cet article les proliférations à cellules chevelues. Elles comprennent: 1) deux entités reconnues: la leucémie à tricholeucocytes (LT) et le lymphome splénique de la zone marginale (LSZM) avec cellules villeuses circulantes, 2) deux entités provisoires classées dans les lymphomes/leucémies spléniques inclassables; la forme variante de la LT (LT-V) et le lymphome diffus de la pulpe rouge de la rate (LDPRR). Le pronostic de ces hémopathies malignes s’est amélioré ces dernières années avec notamment l’introduction de nouvelles drogues, qui ont modifié le traitement de ces maladies.
2 Leucémie à Tricholeucocytes (LT)
La leucémie à tricholeucocytes (LT) appelée aussi HCL (Hairy Cell Leukemia) représente moins de 1% de l’ensemble des hémopathies malignes (HM) et 2% de l’ensemble des leucémies [1–4]. Le nombre de cas incidents est de 1,500 en Europe [5], 300 en France [4] et de 1,100 aux Etats-Unis [6]. L’âge médian au diagnostic est de 63 ans chez l’homme et de 59 ans chez la femme. Dans huit cas sur dix, c’est l’homme qui est atteint. Le taux d’incidence standardisé ajusté sur la population mondiale reste stable en France: il est de 0, 39/100,000 personnes-années chez l’homme et 0, 09 chez la femme [4]. La survie nette standardisée est de 95% à 5 ans, 97% à 40 ans et 92% à 80 ans. Elle s’améliore avec le temps en Europe et aux Etats-Unis [7,8] pour être à 5 ans superposable à celle de la population générale [9]. La cause de la LT reste inconnue. Des formes familiales sont observées [10–12] et plusieurs variants génétiques non récurrents ont pu être identifiés [13]. Les études suédoises ont montré un risque plus élevé de LT chez les membres de la famille de patients atteints de leucémie lymphoïde chronique (LLC) [14]. Le métier d’agriculteur (culture de fourrage, expositions aux insecticides organophosphorés) [15–17] pourrait constituer un facteur de risque professionnel ainsi qu’une exposition au benzène [18]. Un lien négatif avec la consommation de tabac a été démontré. L’incidence de la maladie est plus basse chez les afro-américains [6,19] et la survie inférieure à celle observée chez les Blancs [20]. La LT peut être associée à de nombreuses autres HM, notamment à d’autres lymphomes non-hodgkiniens à petites cellules ou à la LLC [21,22].
2.2 Le Diagnostic Repose sur Trois Examens
Les circonstances du diagnostic sont très variées. Une splénomégalie sans adénopathies superficielles, des infections bactériennes, une tuberculose [23], de la fièvre peuvent révéler la maladie. Des manifestations hémorragiques ou des signes en rapport avec l’anémie peuvent être aussi révélateurs. Un hémogramme demandé de façon systématique peut faire découvrir une neutropénie, une anémie, une thrombopénie et/ou une monocytopénie. Des localisations inhabituelles [24] osseuses [25], cutanées [26] ou des pertes brutales d’audition ont été rapportées [27].
Il montre une pancytopénie, parfois une neutropénie, une monocytopénie, une thrombopénie ou une anémie discrètement macrocytaire. Les automates identifient les tricholeucocytes comme des monocytes, masquant la monocytopénie caractéristique du diagnostic. L’examen du frottis sanguin détecte la présence de tricholeucocytes (hairy cells). Parfois en nombre réduit, les cellules sont de taille moyenne et présentent un cytoplasme étendu, faiblement et irrégulièrement basophile avec de fines projections cytoplasmiques (Fig. 1). La présence de ces projections serait liée à des modifications d’expression de la β actine et du transcrit spécifique leucocytaire 1 (LST-1). Les inclusions cytoplasmiques granulolamellaires avec l’aspect de bâtonnets discrètement basophiles à zone centrale claire sont parfois observées. Le rapport nucléocytoplasmique est bas. Excentré, ovale ou arrondi, le noyau peut être parfois réniforme. La chromatine nucléaire a un aspect finement dispersé et le nucléole peu ou pas visible est de petite taille et souvent unique. La fibrose réticulinique rend l’aspiration médullaire difficile. La biopsie médullaire évalue le degré de l’infiltration tumorale, avec sur coupe, des cellules avec un noyau ovalaire ou réniforme et une zone claire, qui sépare chaque noyau conséquence de la taille des cytoplasmes peu visibles et rétractés. L’identification des tricholeucocytes peut être facilitée par la mise en évidence d’une activité phosphatase acide tartrate résistante (TRAP) de moins en moins recherchée, non spécifique mais néanmoins caractéristique [28] ou par immunohistochimie. La splénectomie est inutile au diagnostic: l’histologie montrerait une infiltration de la pulpe rouge, un effacement de la pulpe blanche et la formation de pseudosinus spléniques avec élargissement des cordons pulpaires.
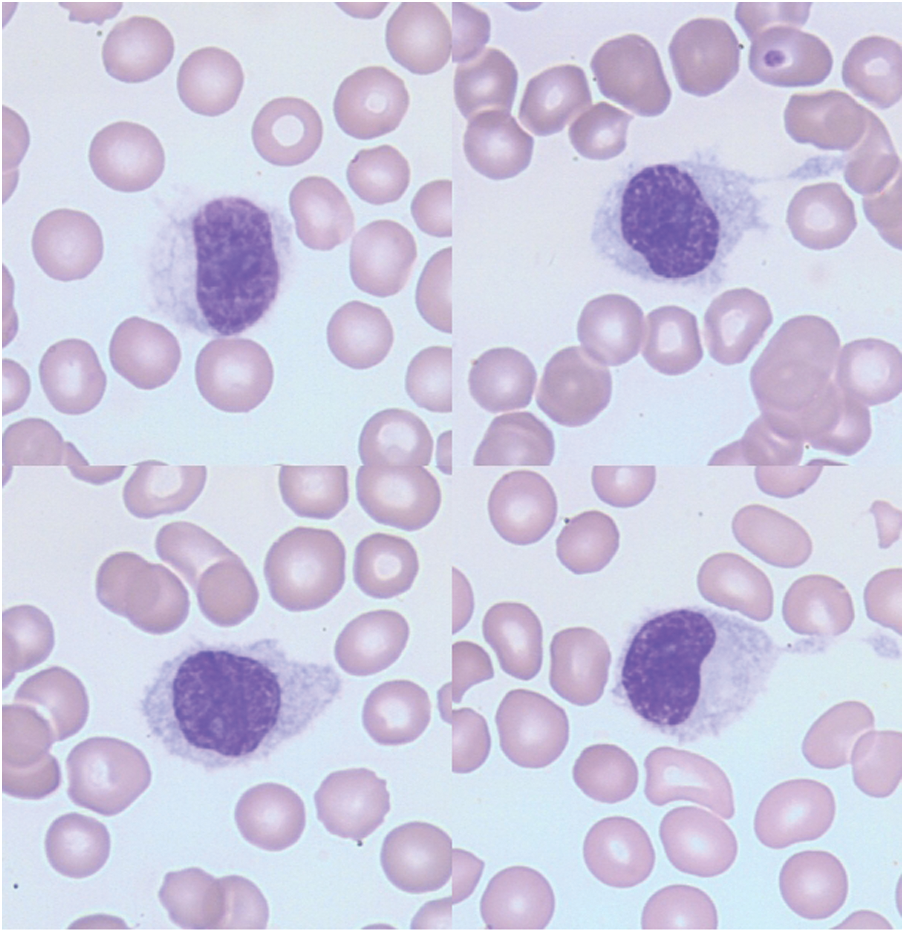
Figure 1: Aspects cytologiques de la LT (Laboratoire Hématologie, CHU de Caen, France)
2.2.2 Cytométrie en Flux (CMF) sur le Sang et/ou la Moelle
Les tricholeucocytes expriment les marqueurs de la lignée B: CD19, CD20 (expression modérée à forte), CD22 (expression forte) et CD200 (expression forte) sans expression du CD5, CD10, CD23 ou du CD27, un marqueur des cellules B mémoires. L’expression du CD11c est forte et celle du CD25 modérée à intense [29]. Les cellules tumorales expriment le CD123 [30,31] et le CD103. L’expression du CD76 (DBA44) [32,33] n’est pas spécifique et peut être observée dans 80% des cas de lymphome splénique de la zone marginale (LSZM). L’annexine A1, exprimée aussi sur les cellules myéloïdes, est un marqueur caractéristique des tricholeucocytes [34]. Un score immunologique a été développé pour faciliter le diagnostic de la LT: il est basé sur l’expression de quatre marqueurs: CD103, CD123, CD25, CD11c. Un point est attribué pour une expression positive et une absence de point pour une expression négative. Le score est de 3 ou 4 dans 98% des cas de LT, contrairement à la LT variante (LT-V) et au LSZM où le score est habituellement de 0 ou 1 [35]. Des phénotypes inhabituels ont été décrits: perte du CD123 [36] notamment en cas d’infection à SARS-Cov-2 associée, expression du CD5 [37] ou du CD10 [38]. De rares cas de LT biclonales ont été rapportés [39,40]. Pour distinguer la HCL de la HCL-V, la médiane d’intensité de fluorescence (MFI) du CD43, CD200 et du CD79b peut être utilisée: elle est plus élevée dans la HCL, mais celle du CD81 est plus basse comparée à celle observée dans la HCL-V [41].
Identifiée en 2011 sur l’exon 15 du gène BRAF (7q34), la mutation BRAFV600E correspond à un remplacement en position 600 de la valine (V) par l’acide glutamique (E). Véritable marqueur moléculaire de la maladie [42], elle a été Initialement décrite chez tous les patients avec une LT et les études ultérieures ont montré son absence dans environ 10% des cas [43–45]. Des mutations alternatives identifiées dans l’exon 11 doivent être recherchées [46], particulièrement chez les patients utilisant le VH4-34 ou chez les patients avec une LT-V. Les patients BRAFWT pourraient avoir un plus mauvais pronostic. La mutation est aussi présente dans les mélanomes (80%), les histiocytoses à cellules de Langerhans et la maladie d’Erdheim Chester (MEC) (50%), les cancers papillaires de la thyroïde (40%), plus rarement les cancers colorectaux, les cancers pulmonaires, les glioblastomes et certaines HM, dont le myélome multiple (MM) et la leucémie lymphoïde chronique (LLC) [47,48]. La mutation active la voie de signalisation MAPK/ERK, permettant la transcription des gènes de la famille ETS, FOS et MYC mais aussi de gènes inhibiteurs, notamment les DUSP (Dual Specificity Phosphatases) [49]. Des mutations autres que les mutations des gènes de la voie MAP-kinases (BRAF, MAP2K1) ont été identifiées: mutations des gènes du cycle cellulaire (CDKN1B dans 10–25%, beaucoup plus rarement TP53), mutations des gènes de la voie NF-kB (KLF2), mutations des gènes du spliceosome (U2AF1) ou des mutations des gènes régulateurs de l’épigénétique (KMT2C, EZH2, ARID1A, ARID1B, CREBP, KDM6A) [50–54]. Le rôle de ces différentes mutations dans la progression de la maladie reste à préciser [55].
2.3 Patients à Risque élevé de Rechute
Des facteurs de mauvais pronostic ont été décrits: splénomégalie > 3 centimètres, leucocytose élevée, présence dans le sang de plus de 5 × 109/L tricholeucocytes, B2 microglobuline élevée (>2 N) [56] et LDH augmentées [57]. Le séquençage des gènes de la partie variable de la chaîne lourde des immunoglobulines (IGHV) montre un profil IGHV muté dans environ 85% des cas avec une utilisation du VH3 dans 66% des cas, du VH4 dans 23% et du VH1, VH2, VH5 ou VH7 dans 3% des cas, le VH6 étant peu utilisé. Le profil non muté (moins de 20% des cas) est associé à une masse tumorale élevée, une absence de mutation BRAFV600E, une survie globale plus courte et une moins bonne réponse au traitement par les analogues des purines (PNA) [58,59]. Les patients utilisant VH4-34 (40% des cas de LT-V et 10% des cas de LT) ont aussi un mauvais pronostic. Une hétérogénéité intraclonale a été observée dans la majorité des cas testés.
L’expression de 57 protéines médullaires après traitement par CDA et par Digital Special Profiling (DSP) a montré chez 9 patients une réduction significative de marqueurs tumoraux B (CD20, CD11c, CD45, BCL2), une augmentation d’expression de marqueurs myéloïdes (CD14, CD68) sans modification des marqueurs T CD3, CD4, CD8 ou NK CD56 et une meilleure réponse au traitement en cas de diminution d’expression du CD20 et du CD44 et en cas d’augmentation d’expression du CD3 et du CD8 (absence de corrélation avec le CD4) [60].
Des recommandations nationales et internationales ont été établies pour harmoniser les pratiques: associées aux algorithmes, elles permettent de guider les décisions thérapeutiques [61–66], en période normale et pendant les périodes de crise sanitaire [67].
2.4.1 En cas de Maladie Symptomatique, le Traitement Doit être Initié
Le traitement est à débuter en cas de splénomégalie, d’anémie inférieure à 10 g/dL, une neutropénie inférieure à 1 G/L, une thrombopénie inférieure à 100 G/L ou des infections sévères et répétées [63].
2.4.2 Critères de Réponse au Traitement
La réponse complète (RC) hématologique (RCH) est définie par la disparition des signes cliniques et une correction des anomalies de l’hémogramme (hémoglobine > 11 g/dL (sans transfusion), plaquettes > 100G/L, neutrophiles > 1, 5 G/L). Chez le sujet jeune, l’objectif du traitement est d’obtenir la RC associant une RCH et l’absence de tricholeucocytes à l’examen de la biopsie ostéomédullaire [63] à réaliser 4 à 6 mois après la fin du traitement par les analogues des purines (PNAs). Il peut être important de préciser notamment chez le sujet jeune si la RC est associée à une maladie résiduelle (MRD) indétectable (MRDu). La MRD peut être étudiée par de nombreuses techniques: immunohistochimie [68,69], PCR allèle spécifique [70], PCR digitale [71,72] ou CMF [73]. Des efforts d’harmonisation sont nécessaires pour permettre la comparaison des résultats entre les différents laboratoires [74]: pour les techniques de biologie moléculaire, la recherche de MRD doit être précédée d’un séquençage IGHV qui n’est pas forcément réalisé en pratique.
2.4.3 Traitement de Première Ligne
Les analogues des purines (PNAs) sont le traitement de référence
-Monothérapie. La supériorité de la désoxycoformycine (DCF) comparée au traitement par interférons-alpha (IFNs) a été démontrée en 1995 dans une étude randomisée chez 356 patients [75]. Cette étude a montré la supériorité des PNAs, considérés comme le traitement de référence de la maladie dans sa phase précoce. Contrairement à la DCF et la 2-chloro-désoxyadénosine (CDA), l’efficacité de la fludarabine n’a pas été mise en évidence [76,77]. Le choix entre la DCF et la CDA dépend de l’existence ou non d’une altération de la fonction rénale, la DCF étant contre-indiquée en cas d’insuffisance rénale.
La DCF a été introduite en 1987 chez 27 patients à la dose de 5 mg/m2 pendant deux jours consécutifs tous les 14 jours [78] pendant 8 à 10 cycles. Une RC a été obtenue chez 16 patients (59%) en trois mois: elle est durable et persiste pendant une médiane de 228 jours. Une rémission partielle (RP) est obtenue dans 37% des cas. Un échec du traitement est observé chez un seul patient. Des taux de RC de 80% ont été observés dans des séries ultérieures avec un nombre plus élevé de patients [79–81]. Malgré l’obtention d’une RC, les cellules tumorales résiduelles persistent habituellement [81] expliquant la survenue de rechutes dans environ 20% des cas.
La CDA a été introduite trois ans plus tard en 1990 chez deux patients à la dose de 0, 1 mg/kg par jour en perfusion continue pendant sept jours. La RC identifiée chez un patient a persisté plus d’un an [82]. Les RC sont observées de façon très fréquente dans les séries ultérieures avec un recul prolongé [83–92]. En France, le schéma habituel est une dose de 0, 14 mg/kg par jour par voie sous-cutanée pendant cinq jours. Aux États-Unis, la perfusion est continue pendant sept jours ou les schémas discontinus sont utilisés: perfusion de deux heures par jour pendant cinq jours ou de trois heures une fois par semaine pendant six semaines [93]. Malgré des réponses initiales durables, 40% des patients rechutent après un traitement de première ligne. Les patients à nouveau traités en monothérapie deviennent de moins en moins sensibles aux traitements et la durée de la réponse est de plus en plus courte [90,91]. L’intérêt de changer de PNA lors d’une rechute n’a pas été clairement démontré [85,87].
Les PNAs sont myélotoxiques et immunosuppresseurs. Avec la DCF, la réduction du nombre de lymphocytes CD4+ se corrige en deux ans [93]. Chez 349 patients traités par CDA, 71% ont développé une neutropénie sévère de grade 4 et 42% une fièvre avec une infection documentée dans 13% des cas. L’introduction conjointe de filgrastim n’a pas réduit le risque infectieux [94]. Après une diminution initiale des cellules CD8+, l’augmentation est observée trois mois environ après le début du traitement. La normalisation du nombre des cellules CD20 et CD4 est plus tardive (un à deux ans), expliquant un rapport CD4/CD8 inférieur à 1 de façon prolongée [95,96]. La diminution des cellules CD4 correspond à une réduction des cellules naïves CD4+ CD45RA+ [97]. Des infections opportunistes ont été rapportées [98] justifiant une prophylaxie par le triméthoxazole et le valaciclovir à débuter une semaine après le début du traitement et à poursuivre jusqu’à récupération d’un taux de CD4 suffisant. D’autres effets secondaires ont été décrits: hyperéosinophilies régressives une semaine après l’arrêt du traitement [99] et survenue de syndromes myélodysplasiques ou de cancers secondaires [100].
-Association aux anticorps monoclonaux anti-CD20. L’immunochimiothérapie a été introduite en 2006 chez 13 patients, dont 11 patients en première ligne. La CDA, IV en 2 heures à la dose de 5, 6 mg/m2 pendant 5 jours, est associée à du rituximab (R) en consolidation un mois après la fin du PNA à la dose de 375 mg/m2 une fois par semaine pendant 8 semaines [101]. La RC est obtenue dans tous les cas, avec une MRDu par PCR ou CMF dans 92% des cas (12/13 patients). Ces excellents résultats sont confirmés cinq ans plus tard chez 36 patients (31 patients avec une LT, 5 patients avec une LT-V) [102]: la RC est obtenue chez tous les patients. Après un suivi médian de 25 mois, un seul patient avec une LT-V a rechuté. Une MRDu évaluée par CMF est obtenue dans 79% des cas (22/28 patients) et par PCR dans 70% des cas (19/27 patients). L’étude a été étendue chez 80 patients (59 en première ligne, 14 en seconde ligne, 7 patients avec une LT-V) [103]: la RC est obtenue chez tous les patients avec une LT et dans 86% des cas de LT-V. La MRDu évaluée par CMF est obtenue dans 94% des cas (75 patients évaluables): elle devient détectable chez 6 patients dont 2 patients avec une LT-V. Une autre étude randomisée a comparé chez 68 patients en première ligne le schéma CDA-R associant CDA (0, 15 mg/kg IV pendant 5 jours) et R introduit en même temps que la CDA à la dose de 375 mg/m2 toutes les semaines pendant 8 semaines au schéma CDA combinant CDA et R en consolidation débuté 6 mois après la CDA [104]. Cette étude montre une supériorité du schéma CDA-R, avec un taux de RC dans 100% des cas et une MRDu obtenue 6 mois après le début du traitement par CDA dans 97% des cas alors qu’elle est de seulement 24% avec le schéma CDA. Après un suivi médian de 7, 7 ans, un seul patient a reçu du R dans le groupe CDA-R et 21 patients dans le groupe CDA.
-Cas particuliers. Les traitements par IFNs, introduits en 1984 [105], parfois à faible dose de façon prolongée, peuvent être indiqués si les traitements précédents ne peuvent pas être initiés [65]. L’accès à ces produits devient de plus en plus difficile, compte tenu de l’arrêt de leur production. Ils peuvent être, dans une version pégylée, une solution alternative chez la femme enceinte ou chez les patients avec une neutropénie sévère fébrile. Le traitement par vemurafénib en monothérapie peut-être recommandé chez les patients avec une infection non contrôlée avant l’utilisation des PNAs [106] ou en cas d’infection à SARS Cov2 [67]. La splénectomie reste indiquée chez les patients dont le diagnostic est incertain ou chez les patients avec une LT réfractaire.
Patients avec une HCL en rechute/réfractaire (R/R)
Il est nécessaire d’inclure, si possible, le patient dans un essai clinique.
-Inhibiteurs de BRAF (BRAFi)
Monothérapie. Le vemurafénib est transitoirement efficace en monothérapie. 54 patients (28 patients dans la série italienne, 26 patients dans la série américaine) antérieurement traités (médiane de ligne de traitements: 3 ou réfractaires aux PNAs dans la moitié des cas), ont été traités à la dose de 960 mg deux fois par jour [107] pendant une durée limitée de 16 et 18 semaines. Une réponse a été obtenue dans respectivement 96% et 100% des cas, dont 35% et 42% de RC. L’efficacité du traitement est de courte durée, la survie sans rechute étant de 9 mois: 19 mois en cas de RC et 6 mois en cas de RP. La survie sans traitement est de 21, 5 mois: elle est identique chez les patients en RC (25 mois) ou en RP (18 mois). Des doses plus faibles de vemurafénib (240 mg deux fois par jour) ont été utilisées chez 17 patients: la RC est obtenue dans 40% des cas. En cas de rechute, un nouveau traitement par BRAFi demeure efficace, permettant d’envisager des stratégies de stop and go [108,109]. Les effets indésirables sont marqués par une toxicité cutanée (éruption, hyperkératose palmoplantaire, photosensibilité et kérato-canthomes), oculaire (rétinopathie grave centrale et occlusion veineuse rétinienne), cardiaque (allongement de l’intervalle QTc) ou hépatique (élévation des aminotransférases et de la bilirubine).
Un autre BRAFi, le dabrafénib, a été utilisé chez 10 patients à la dose de 150 mg deux fois par jour pendant une période limitée de 8 à 12 semaines. Les patients ont reçu une médiane de traitement antérieur de 3, 5. Une RC est observée chez 3 patients avec une MRD détectable dans tous les cas, une RP chez 5 patients et un effet clinique bénéfique chez les deux derniers patients avec une réponse mineure (RM) (régression de la splénomégalie chez un des patients [110].
Les BRAFi sont non myélotoxiques et non immunosuppresseurs: ils peuvent être proposés en cas de neutropénie fébrile [106,111,112] ou en cas de risque élevé d’infection à SARS-Cov2 [67]. Des maladies hématologiques préexistantes sont susceptibles de progresser lors de l’utilisation de ces médicaments: progression par un mécanisme Ras dépendant: leucémie myélomonocytaire chronique (mutation NRAS G12R) [113], lymphocytose B monoclonale (mutation KRASG12D) [114], érythroleucémie (mutation PI3KE54K connue pour activer RAS) [108] ou non: LLC en absence de mutations RAS [115].
Association aux anti-CD20. L’association vemurafenib (960 mg deux fois par jour pendant 2 cycles de 4 semaines) plus R (8 injections au total ; J1 et J15 du cycle 1 et 2 plus 4 doses de R de consolidation tous les 15 jours) a été proposée chez 31 patients en R/R. Le taux de RC obtenu à la fin du traitement est l’objectif principal de l’étude: il est de 87% (26/30 pts), un taux deux fois plus élevé que celui obtenu avec le vemurafenib en monothérapie [116]. La RC est associée à une MRDu sanguine et médullaire dans 60% des cas (18/30 pts). Elle est durable avec une survie sans rechute (RFS) à 34 mois de 85% (13–50), comparée à 4 mois en cas d’utilisation du vemurafenib en monothérapie. La RFS n’est pas modifiée par une préalable exposition au R mais elle est plus courte en cas d’exposition antérieure aux BRAFi (57% chez les 7 pts avec RC exposés aux BRAFi vs. 95% chez les autres) et de MRD détectable (56% chez les pts avec une MRD détectable vs. 100% en cas de MRDu). L’association a été aussi utilisée avec une bonne tolérance chez les patients traités antérieurement par le Moxetumomab pasudotox [117].
Association aux inhibiteurs de MEK (MEKi). L’association du dabrafénib (150 mg deux fois par jour) à un inhibiteur de MEK (MEKi), le tramétinib (2 mg une fois par jour) est en cours d’évaluation chez les patients en R/R [118].
-Moxetumomab pasudotox (Moxe). C’est une immunotoxine constituée du domaine variable de l’immunoglobuline murine anti-CD22 fusionné à une forme tronquée de PE38, une exotoxine de Pseudomonas. Le CD22, fortement exprimé à la surface des tricholeucocytes, intervient dans la survie, la prolifération, l’activation et la signalisation des lymphocytes B. Après liaison au domaine extracellulaire du CD22, le Moxe est internalisé permettant la libération de l’exotoxine PE38 à l’intérieur de la cellule. Ce processus inhibe la synthèse protéique cellulaire et provoque la mort de la cellule. Le Moxe a été autorisé par la Food and Drug Administration (FDA) en septembre 2018 chez les patients en R/R après au moins deux lignes de traitement incluant une ligne de PNA. L’autorisation donnée se base sur une étude de phase 1 [119] et sur l’essai pivotal à un bras multicentrique [120,121]. L’objectif principal de cette dernière étude est l’obtention d’une RC prolongée se maintenant pendant 180 jours et évaluée par un comité indépendant. Quatre-vingt patients, avec un âge médian de 60 ans (34–84) ont été inclus avec un suivi médian de 24, 6 mois. Le nombre médian de lignes de traitement antérieurement reçu est de 3 (2–11) ; 60 patients (75%) ont reçu du R et 14 (17.5%) un BRAFi. Le Moxe a été donné à la dose de 40 mg/kg par voie IV en 30 min les jours 1, 3 et 5 de chaque cycle de 28 jours. La durée du traitement est de 6 cycles au maximum. 33 patients (41%) présentent une RC et 29 patients (36%) une RC durable de plus de 180 jours. Une MRDu est obtenue chez 27/33 patients avec RC. Parmi les 29 (90%) patients avec RC durable, 26 ont une MRDu. Les effets indésirables de grade 3 ou 4 sont les infections chez 13 patients (16%, 3%) dont une infection liée au traitement chez deux patients (2%, 5%), un syndrome de fuite capillaire chez 2 patients (2%, 5%) et un syndrome hémolytique et urémique chez 4 patients (5%). Les deux dernières complications ont été dans tous les cas réversibles à l’arrêt du traitement. Trois patients ont reçu un second traitement par Moxe après rechute d’un traitement antérieur par Moxe: chez deux patients les cytopénies se sont corrigées [122]. Le Moxe est devenu inaccessible en Europe compte tenu de l’arrêt de production par le laboratoire de ce médicament.
-Inhibiteurs de BTK (BTKi). Trente-sept patients, 28 patients avec une HCL et 9 patients avec une HCL-V, ont reçu un traitement par ibrutinib à la dose de 420 mg/jour (24 pts) ou 840 mg/jour (13 patients) jusqu’à progression ou toxicité inacceptable [123]. Le nombre de traitement reçu antérieurement est de 4. L’objectif principal de l’étude est l’ORR à 32 semaines. Il est de 24% (1 pt avec RC, 8 pts avec PR) et augmente à 36% à 48 semaines. Le meilleur taux d’ORR obtenu est de 54%, dont 7 RC (19%) (3 MRDu) et 13 RP (35%). L’efficacité du traitement est prolongée, la médiane de survie sans progression (PFS) étant non atteinte et estimée à 36 mois à 73%. La médiane d’OS est de 69 mois, avec une OS estimée à 36 mois de 85%. Les effets indésirables observés sont identiques à ceux observés dans la LLC. La fibrillation atriale est observée chez 6 patients (16%), toujours de grade 1 ou 2, le flutter atrial chez 2 patients (5%), de grade 1 ou 2 dans un cas et de grade > ou égal à 3 chez l’autre patient. Une baisse des plaquettes est observée chez 15 patients (41%), de grade 1 ou 2 chez 7 patients (19%) ou >ou égal à 3 chez 8 patients (22%). Il n’existe pas de différence entre la LT. et la LT-V en terme d’ORR, de PFS ou d’OS. Le mécanisme d’action de l’ibrutinib pourrait être différent de celui observé dans la LLC: 1) Il n’existe pas d’augmentation dans le sang du nombre de cellules leucémiques tumorales, 2) chez 4 patients avec progression (LT: 2 pts, HCL-V: 2 pts) aucune mutation de BTK ou de PLCγ2 n’a pu être identifiée, 3) enfin, l’ibrutinib maintient son efficacité en cas de phosphorylation d’ERK.
-Inhibiteurs de Bcl2 (Bcl-2i). Le venetoclax est un inhibiteur sélectif de BCL-2, qui restaure l’apoptose dans les cellules cancéreuses. Le traitement par venetoclax est autorisé dans la LLC et la LAM (AML) chez les patients adultes inéligibles à un traitement intensif. Il pourrait être aussi efficace dans la LT et la LT-V. Des données récentes montrent que des concentrations de venetoclax (0.1 et 1 μM) pourraient induire in vitro l’apoptose des cellules primaires. Une diminution de la cytotoxicité des cellules HCL exposées pendant 48 heures à différents stimuli (activated T lymphocytes, stromal cells, TLR-9 agonist CpG, TLR-2 agonist PAM3) a été aussi montrée, laissant suggérer que le venetoclax associé à des cibles du microenvironnement pourrait aussi améliorer son efficacité [124]. L’intérêt du venetoclax chez les patients avec une LT en R/R reste à évaluer et l’association venetoclax plus ibrutinib, utilisée dans la LLC pourrait être utile chez certains patients avec une LT [40].
En première ligne, les PNAs associés au R sont challengés par la combinaison vemurafenib (960 mg deux fois par jour pendant 4 mois) plus obinutuzumab (1000 mg J1, J8 et J15 du cycle 2 et 1000 mg à J1 des cycles 3 et 4). Dans une phase II, l’association vemurafenib plus obinutuzumab a été évaluée chez 30 patients en première ligne. La RC à la fin du traitement est obtenue chez tous les 27 patients évaluables et une MRDu est observée dans 92% des cas (24/26 patients). Après un suivi médian de 19, 7 mois (2, 1–42), aucune rechute n’est observée [125]. Un essai est actuellement en cours comparant le schéma CDA-R au traitement vemurafebnib obinutuzumab.
Chez les patients en R/R, le R pourrait amplifier l’efficacité des PNAs, des BRAFi et du Moxe. Un essai de phase I associant le Moxe avec le R est en cours chez des patients ayant reçu deux lignes de traitement antérieur ou une rechute précoce inférieure à 2 ans. L’association ibrutinib et venetoclax a montré une efficacité dans la LLC et pourrait être utilisée dans la LT. L’association vemurafenib plus Moxe pourrait être aussi une combinaison d’intérêt. La possibilité d’utiliser des anticorps monoclonaux dirigés contre le CD38 [126] ou le CD123 [31] reste aussi à investiguer.
Le risque de cancers secondaires est augmenté chez les patients avec une LT. Le mécanisme reste débattu en lien avec la maladie elle-même ou être secondaire aux traitements utilisés [127,128]. Le risque de tumeurs hématopoïétiques est multiplié par 40 [129]. Chez 487 patients, il a été observé une histoire familiale de cancers dans 18% des cas et un SIR (ratio standardisé d’incidence) de 1, 86 (intervalle de confiance [IC] 95%: 1, 34–2, 51) pour les cancers secondaires et de 5, 32 (IC 95%: 2, 90–8, 92) pour les hémopathies secondaires. Ce risque impose une surveillance hématologique prolongée chez tous les patients.
3 Autres Proliférations à Cellules Chevelues
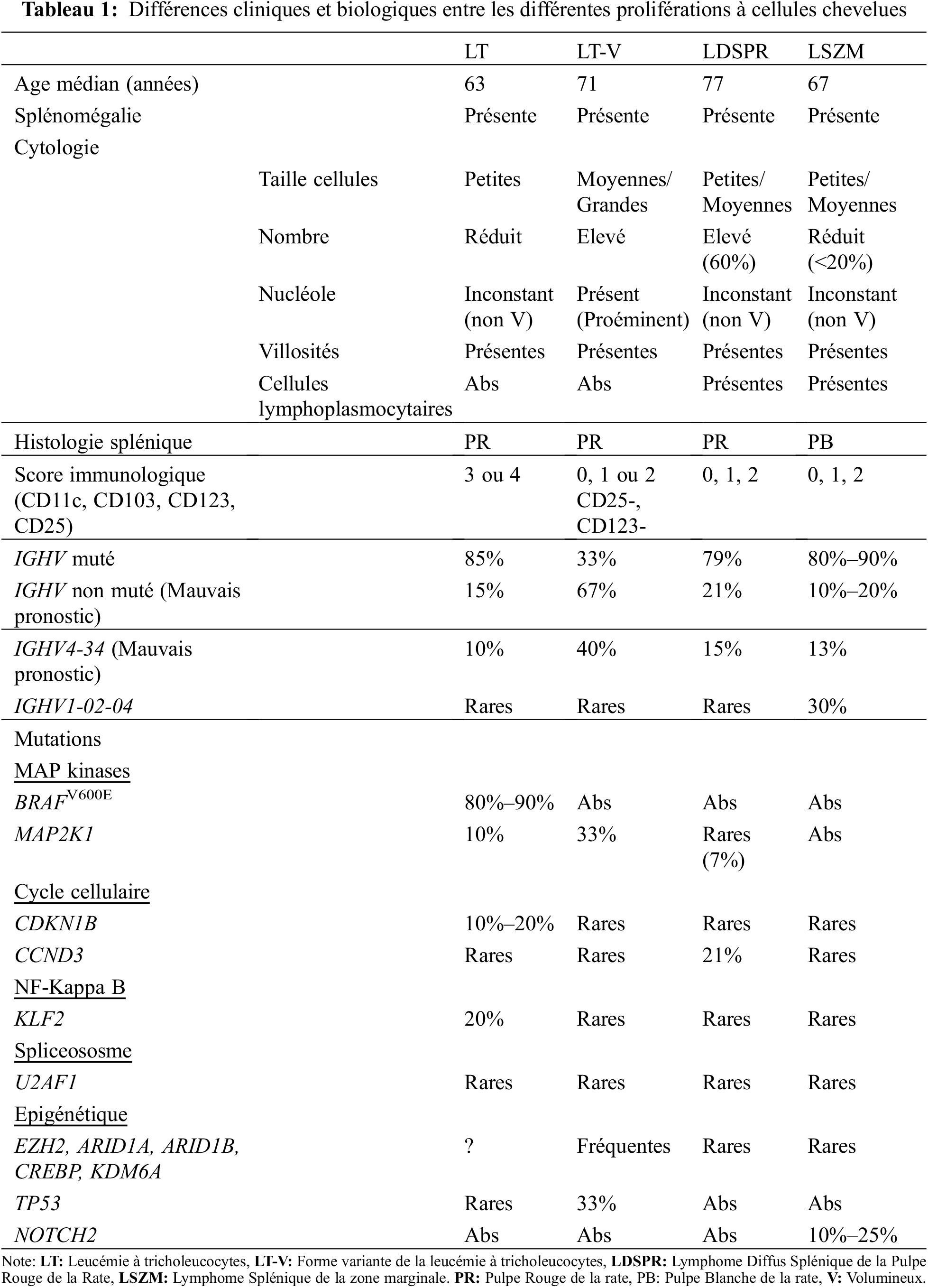
Il est important de distinguer les autres proliférations à cellules chevelues car leur traitement est différent. Aucune de ces entités ne présente de mutation BRAFV600E. Les principales différences entre la LT et les autres proliférations à cellules chevelues sont présentées dans le Tableau 1.
3.1 Lymphome Splénique Diffus de la Pulpe Rouge
Le lymphome splénique diffus de la pulpe rouge (LSDPR) (Splenic Diffuse Red Pulp Lymphoma) représente au sein des lymphomes spléniques non classables une entité provisoire. Décrit en 2008 chez 37 patients [130–133], les séries publiées sont limitées [134,135]. L’âge médian des patients est de 77 ans (46-91) et la prédominance masculine est moins marquée que celle observée dans la LT, le ratio hommes/femmes étant de 1, 64 : 1. La splénomégalie est présente dans 81% des cas, alors que la présence d’adénopathies est notée dans seulement 11% des cas. Un composant monoclonal est parfois identifié (6/37). L’aspect du frottis sanguin est monomorphe. Les cellules lymphoïdes anormales sont fréquentes et atteignent 60% de la lymphocytose (26%–91%) ; elles sont de taille petite à moyenne avec des villosités à distribution polaire et la présence d’un nucléole à peine visible ou invisible. La présence d’un nucléole proéminent est rarement notée, identifiée chez seulement 4 patients en progression/transformation de la maladie. A noter aussi la présence fréquente de cellules lymphoplasmocytaires. Les cellules sont B CD19 positives et expriment fortement le CD22 et le CD11c (97%), de façon plus inconstante le CD103 (38%), le CD123 (16%) et le CD25 (3%). Le profil IGHV est dans la plupart des cas muté (79%), avec un usage préférentiel d’IGHV3 (12/33) et IGHV4 (13/33) et de VH4-34 chez 5 patients et de VH3-23 chez 6 patients. La même série étendue à 48 patients a montré une utilisation d’IGHV3 chez 20 patients et d’IGHV4 chez 17 patients avec une utilisation de VH4-34 et VH3-23 chez 10 et 9 patients, respectivement. La mutation BRAFV600E n’est jamais détectée, sauf dans un cas où une mutation BRAF-K601Q est identifiée [135]. Les mutations TP53 sont absentes ou rarement présentes. Des mutations de BCOR, CCND3 et MAP2K1 ont été décrites dans 24%, 21% et 7% des cas [135–137]. Compte tenu du nombre de séries limitées, il n’existe pas de données de survie et les indications de traitement ne sont pas clairement établies. Une évolution en leucémie à prolymphocytes B (PLL-B) a été rapportée [138].
3.2 Forme Variante de la Leucémie à Tricholeucocytes
Décrite par John Cawley en 1980, [139] la forme variante de la LT (LT-V) est reconnue comme une entité provisoire depuis 2008: elle est comme le SDRPL classée parmi les lymphomes spléniques B inclassables. Elle est rare et le nombre de nouveaux cas incidents estimés aux Etats-Unis est de 810 [6]. Le pronostic de la LT-V est plus réservé que celui de la LT: la médiane de survie étant de neuf ans, avec 15% des patients survivant à plus de 17 ans [140]. L’âge médian des patients au diagnostic est de 71 ans, un âge de 10 ans plus élevé que celui observé dans la LT. La prédominance masculine est moins marquée que dans la LT et le rapport hommes/femmes est de 1.5 : 2. [140–143] Chez 52 patients, la splénomégalie souvent volumineuse est présente dans 85% des cas, alors que l’hépatomégalie et les adénopathies superficielles sont observées dans 19% et 15% des cas, respectivement [140]. L’anémie et la thrombopénie sont notées dans 29 et 43% des cas et la leucocytose est souvent augmentée, 10% des patients présentant un chiffre normal de leucocytes. Contrairement à la LT, il n’existe pas de monocytopénie, ni de neutropénie. L’aspect du frottis sanguin est monomorphe, avec des cellules de taille intermédiaire à grande, un noyau régulier avec un nucléole unique et proéminent (Fig. 2), une chromatine relativement condensée et un cytoplasme plus ou moins abondant avec des projections.
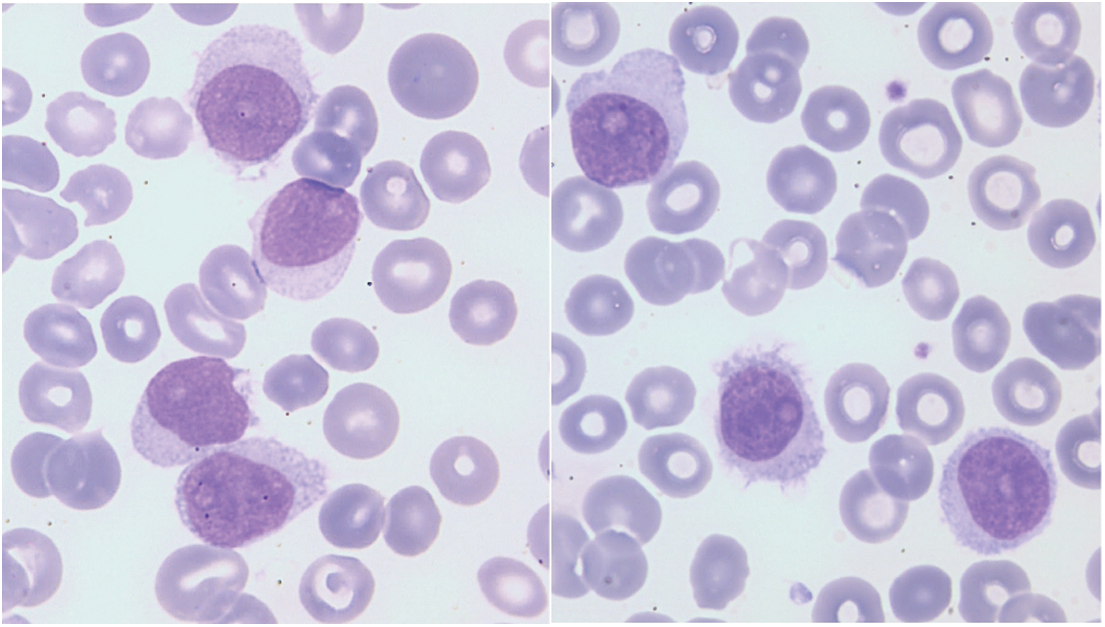
Figure 2: Aspects cytologiques de la LT-V (Laboratoire Hématologie, CHU de Caen Normandie, France)
La moelle est facile à aspirer. Une infiltration interstitielle médullaire est présente dans 75% des cas: elle est mixte, interstitielle et nodulaire dans 10% des cas. L’histologie splénique est identique à celle de la LT: il existe une hypertrophie de la pulpe rouge, avec une pulpe blanche réduite ou même absente. La présence de cellules anormales dans les sinusoïdes est souvent observée. Les cellules expriment fortement les immunoglobulines de surface (plus souvent IgG et lambda), CD11c de façon forte, habituellement CD103 (deux tiers des cas) mais n’expriment ni le CD123, ni le CD25. La moyenne de fluorescence (MFI) des marqueurs CD43, CD81 et CD200 peut être une aide à l’identification de la LT-V, la moyenne d’expression du CD81 étant plus élevée que celle observée dans la LT mais celle du CD43 et du CD200 étant moindre [41]. Le profil IGHV dans la LT-V est muté dans un tiers des cas (85% dans la LT, 80% dans le SDRPL), avec une utilisation préférentielle du VH4-34 [144–146]. Les mutations TP53, rares dans la LT et le SDRPL, sont fréquentes dans la LT-V [141]. La mutation de BRAFV600E est absente dans la LT-V, contrairement à la mutation du gène MAP2K1 (MEK1) identifiée dans plus d’un tiers des cas [52]. Des mutations de KMT2C (MLL3), de CCND3 A et de U2AF1 ont été aussi retrouvées [146]. Une évolution clonale a été observée chez un patient avec une LLC stade A au diagnostic évoluant en stade B et présentant un caryotype complexe. Ce patient, traité par bendamustine plus R présente deux plus tard une LT-V sans mutation de MAP2K1 mais avec des mutations de TP53. Le diagnostic de lymphome composite est retenu: profil non muté de la LLC (IGHV1-3*01) et non muté de la LT-V (IGHV4-34*01). Les deux clones sont identifiés au diagnostic, le clone LLC étant prédominant. Après traitement, le clone de la LT-V est devenu majoritaire avec une disparition presque complète du clone LLC. La mutation TP53 est présente au diagnostic de la LLC et au moment de l’apparition de la LT-V [147]. La prise en charge de la LT-V n’est pas standardisée. Les agents alkylants, les IFNs-α et la CDA en monothérapie sont inefficaces [148]. La splénectomie a une efficacité transitoire. Des traitements par rituximab, obinutuzumab [149] ou bendamustine plus R [150] ont été efficaces chez certains patients. L’association CDA plus R reste l’option de première ligne [151]. Une étude récente de phase II chez 20 patients, dont 8 non antérieurement traités, a montré que l’association CDA plus R permettait une RC à 4 semaines dans 70% des cas (14 patients) et une MRDu dans 45% (9 patients). Six mois après le traitement par CDA-R, la RC se maintient dans 90% des cas (18/20 patients), dont 80% (16 patients) avec une MRDu évaluée par IHC et CMF sur la moelle. La PFS à 5 ans est de 63%, 3% et l’OS à 5 ans de 73%, 9%. Les patients avec des mutations de TP53 (26% des cas: 5/19) ont une probabilité plus élevée d’avoir une MRD détectable à 6 mois et présentent une PFS plus courte (36, 4 mois versus PFS non atteinte). Dans les formes en rechute ou réfractaires, les immunotoxines et l’ibrutinib [152–154] peuvent être discutés dans les lignes ultérieures de traitement. Une réponse a été rapportée chez un patient traité par l’association ibrutinib plus venetoclax [40].
3.3 Lymphome Splénique de la Zone Marginale Avec Cellules Villeuses Circulantes
Le LSZM est une entité à part entière. Il existe dans les formes spléniques des lymphomes de la zone marginale (SMZL) des formes avec cellules circulantes, souvent en faible nombre (<20%) avec des villosités courtes et associées à un composant lymphoplasmocytaire [155]. La splénectomie, si elle était réalisée, montrerait une expansion de la pulpe blanche et une infiltration de la pulpe rouge. Les cellules B monotypiques ont un score RMH (score Matutes) < 3 le plus souvent à 0 et expriment CD180 et CD200. Il existe des phénotypes inhabituels, avec expression du CD5, CD23, CD11c, CD25 et CD103 rendant le diagnostic plus complexe. Le profil IGHV est muté, avec une utilisation préférentielle du VH1-2 et en particulier de l’allèle VH1-02*04 dans 30% des cas mais sans impact sur le pronostic, de l’IGHV3-23 dans 8% des cas et du VH4-34 dans 13% des cas. Les anomalies cytogénétiques les plus fréquentes sont la délétion 7q, la trisomie complète ou partielle du chromosome 3 ou 12 et/ou des anomalies touchant les chromosomes 8, 9p34, 12q23-24, 18q et 17p. Parmi les mutations observées, les mutations NOTCH2 dans 10–25% des cas, KLF2 dans 20 à 30% ou TP53 dans 10%–15% ainsi que des mutations dans la voie NFkB soit dans la voie canonique (MYD88, CD79/A/B, CARD11, IKBKB) ou non canonique (TRAF3, BIRC3). La survie du LSZM est bonne ; le traitement est à initier en cas de maladie symptomatique et repose sur l’immunochimiothérapie.
La connaissance de la LT s’est améliorée avec l’identification de la mutation BRAFV600E comme marqueur moléculaire de la maladie. Néanmoins, le tricholeucocyte reste une cellule bien mystérieuse. Le traitement de première ligne fait appel, en cas de maladie symptomatique, aux PNAs. En cas de rechute ou de maladie réfractaire, notamment dans les LT utilisant IGHV4–34, le traitement, à discuter avec les spécialistes en réunion de concertation pluridisciplinaire, fait appel à un inhibiteur de BRAF associé ou non aux inhibiteurs de MEK, aux immunotoxines ou aux inhibiteurs de BTK. Les progrès accomplis sont assez importants pour que nous soyons en droit d’envisager la guérison. Il est aussi nécessaire de distinguer la LT des autres proliférations à cellules chevelues, qui ont par ailleurs un pronostic et un traitement différents.
Les proliférations à cellules chevelues regroupent des entités hétérogènes: il est essentiel de distinguer la leucémie à tricholeucocytes des autres proliférations, notamment de la forme variante de la leucémie à tricholeucocytes et du lymphome splénique de la pulpe rouge de la rate. La prise en charge de ces patients est différente.
Le diagnostic de la leucémie à tricholeucocytes repose sur l’hémogramme et l’examen du frottis sanguin associé à un examen médullaire par aspiration et/ou biopsie médullaire, permettant d’identifier les tricholeucocytes. L’analyse par cytométrie en flux (CMF) identifie ces cellules comme des cellules exprimant de façon très caractéristique les molécules CD11c, CD25, CD103 et CD123. La recherche de la mutation BRAFV600E est essentielle: elle constitue un véritable marqueur moléculaire de la maladie. Le traitement de première ligne repose sur les analogues des purines utilisés en monothérapie et de plus en plus fréquemment en association avec les anticorps monoclonaux anti-CD20. L’immunochimiothérapie est de plus en plus utilisée dès la première ligne. Les nouvelles drogues, le moxetumomab pasudotox, les inhibiteurs de BRAF ou de BTK sont utiles chez les patients avec une maladie en rechute/réfractaire. Une surveillance hématologique est nécessaire, compte tenu du risque augmenté de cancers secondaires.
La forme variante de la leucémie à tricholeucocytes est rare: les cellules habituellement n’expriment pas les molécules CD123 et CD25. La mutation BRAFV600E est absente mais une mutation MAP2K1 (MEK1) serait observée dans plus d’un tiers des cas. Le lymphome splénique diffus de la pulpe rouge reste une entité encore mal identifiée et proche de la forma variante de la leucémie à tricholeucocytes: des mutations de BCOR, CCND3 et MAPK1 ont été observées.
Acknowledgement: Les auteurs remercient la Tumorothèque de Basse Normandie (TCBN) pour la contribution à la collection du matériel.
Author Contributions: XT and EM ont contribué à la conception de l’article. Préparation du matériel, collection des données, analyses des données. La première version de l’article a été réalisée par XT. XT et EM ont réalisé la version définitive et approuvé le document final.
Ethics Approval and Informed Consent Statement: Not applicable.
Funding Statement: Absence de financement.
Conflicts of Interest: Xavier Troussard. Consultant pour Innate Pharma, Astra Zeneca, Abbvie. EM. Absence de conflit d’intérêt.
1. Troussard, X. (2020). Epidemiology of hairy cell leukemia (HCL) and HCL-like disorders. Medical Research Archives, 8(10), 3–11. DOI 10.18103/mra.v8i10.2259. [Google Scholar] [CrossRef]
2. Wiber, M., Maitre, E., poncet, J. M., Duchenet, V., Damaj, G. et al. (2020). A Population-based study of hairy cell leukemia over a period of 20 years. Cancer Treatment and Research Communications, 25, 100236–100242. DOI 10.1016/j.ctarc.2020.100236. [Google Scholar] [CrossRef]
3. Bernstein, L., Newton, P., Ross, R. K. (1990). Epidemiology of hairy cell leukemia in Los Angeles County. Cancer Research, 50(12), 3605–3609. [Google Scholar]
4. Troussard, X., Maitre, E., Maynadie, M., Monnereau, A., Mounier, M. et al. (2020). Etude de la survie des personnes atteintes d’un cancer en France métropolitaine 1990–2018. leucémie à tricholeucocytes. hémopathies malignes. Étude à Partir des Registres des Cancers du Réseau Francim, 2, 87–90. [Google Scholar]
5. Gatta, G., Capocaccia, R., Botta, L., Mallone, S., De Angelis, R. et al. (2017). Burden and centralised treatment in Europe of rare tumours: Results of RARECAREnet—A population-based study. The Lancet Oncology, 18(8), 1022–1039. DOI 10.1016/S1470-2045(17)30445-X. [Google Scholar] [CrossRef]
6. Teras, L. R., DeSantis, C. E., Cerhan, J. R., Morton, L. M., Jemal, A. et al. (2016). 2016 US lymphoid malignancy statistics by world health organization subtypes: 2016 US lymphoid malignancy statistics by world health organization subtypes. CA: A Cancer Journal for Clinicians, 66(6), 443–459. DOI 10.3322/caac.21357. [Google Scholar] [CrossRef]
7. Dinmohamed, A. G., Posthuma, E. F. M., Visser, O., Kater, A. P., Raymakers, R. A. P. et al. (2018). Relative survival reaches a plateau in hairy cell leukemia: A population-based analysis in the Netherlands. Blood, 131(12), 1380–1383. DOI 10.1182/blood-2017-12-820381. [Google Scholar] [CrossRef]
8. Chandran, R., Gardiner, S. K., Smith, S. D., Spurgeon, S. E. (2013). Improved survival in hairy cell leukaemia over three decades: A SEER database analysis of prognostic factors. British Journal of Haematology, 163(3), 407–409. DOI 10.1111/bjh.12490. [Google Scholar] [CrossRef]
9. Madanat, Y. F., Rybicki, L., Radivoyevitch, T., Jagadeesh, D., Dean, R. et al. (2017). Long-term outcomes of hairy cell leukemia treated with purine analogs: A comparison with the general population. Clinical Lymphoma Myeloma and Leukemia, 17(12), 857–862. DOI 10.1016/j.clml.2017.07.003. [Google Scholar] [CrossRef]
10. Casado, L., Mouleon, P., Villarrubia, B., Toledo, M. C., Martinez-Frejo, M. C. (1998). Familial hairy cell leukemia: A HLA-linked disease or farmers-linked disease? Haematologica, 83(8), 751–752. [Google Scholar]
11. Makower, D., Marino, P., Frank, M., Wiernik, P. H. (1998). Familial hairy cell leukemia. Leukemia & Lymphoma, 29(1–2), 193–197. DOI 10.3109/10428199809058395. [Google Scholar] [CrossRef]
12. Virchis, A., Mehta, A. (1997). Familial occurrence of hairy cell leukaemia in a father and daughter-A case report. Blood, 90(10), 4115. [Google Scholar]
13. Pemov, A., Pathak, A., Jones, S. J., Dewan, R., Merberg, J. et al. (2020). In search of genetic factors predisposing to familial hairy cell leukemia (HCLExome-sequencing of four multiplex HCL pedigrees. Leukemia, 34(7), 1934–1938. DOI 10.1038/s41375-019-0702-7. [Google Scholar] [CrossRef]
14. Goldin, L. R., Björkholm, M., Kristinsson, S. Y., Turesson, I., Landgren, O. (2009). Elevated risk of chronic lymphocytic leukemia and other indolent non-hodgkin’s lymphomas among relatives of patients with chronic lymphocytic leukemia. Haematologica, 94(5), 647. DOI 10.3324/haematol.2008.003632. [Google Scholar] [CrossRef]
15. Clavel, J., Mandereau, L., Cordier, S., Goaster, C. L., Heamon, D. et al. (1995). Hairy cell leukaemia, occupation, and smoking. British Journal of Haematology, 91(1), 154–161. DOI 10.1111/j.1365-2141.1995.tb05261.x. [Google Scholar] [CrossRef]
16. Monnereau, A., Slager, S. L., Hughes, A. M., Smith, A., Glimelius, B. et al. (2014). Medical history, lifestyle, and occupational risk factors for hairy cell leukemia: The InterLymph non-hodgkin lymphoma subtypes project. JNCI Monographs, 2014(48), 115–124. DOI 10.1093/jncimonographs/lgu004. [Google Scholar] [CrossRef]
17. Orsi, L., Delabre, L., Monnereau, A., Delval, P., Berthou et al. (2009). Occupational exposure to pesticides and lymphoid neoplasms among men: Results of a French case-control study. Occupational and Environmental Medicine, 66(5), 291–298. DOI 10.1136/oem.2008.040972. [Google Scholar] [CrossRef]
18. Rana, I., Dahlberg, S., Steinmaus, C., Zhang, L. (2021). Benzene exposure and non-hodgkin lymphoma: A systematic review and meta-analysis of human studies. The Lancet Planetary Health, 5(9), e633–e643. DOI 10.1016/S2542-5196(21)00149-2. [Google Scholar] [CrossRef]
19. Morton, L. M., Turner, J. J., Cerhan, J. R., Linet, M. S., Treseler, P. A. et al. (2007). Proposed classification of lymphoid neoplasms for epidemiologic research from the pathology working group of the international lymphoma epidemiology consortium (InterLymph). Blood, 110(2), 695–708. DOI 10.1182/blood-2006-11-051672. [Google Scholar] [CrossRef]
20. Giri, S., Shrestha, R., Pathak, R., Bhatt, V. R. (2015). Racial differences in the overall survival of hairy cell leukemia in the United States: A population-based analysis of the surveillance, epidemiology, and End results database. Clinical Lymphoma, Myeloma and Leukemia, 15(8), 484–488. DOI 10.1016/j.clml.2015.03.001. [Google Scholar] [CrossRef]
21. Gine, E., Bosch, F., Villamor, N., Rozman, M., Colomer, D. et al. (2002). Simultaneous diagnosis of hairy cell leukemia and chronic lymphocytic leukemia/small lymphocytic lymphoma: A frequent association? Leukemia, 16(8), 1454–1459. DOI 10.1038/sj.leu.2402553. [Google Scholar] [CrossRef]
22. Obiorah, I. E., Francischetti, I. M. B., Wang, H. W., Ahn, I. E., Wang, W. X. et al. (2020). Concurrent chronic lymphocytic leukemia/small lymphocytic lymphoma and hairy cell leukemia: Clinical, pathologic and molecular features. Leukemia & Lymphoma, 61(13), 3177–3187. DOI 10.1080/10428194.2020.1797007. [Google Scholar] [CrossRef]
23. Marie, J., Degos, L., Flandrin, G. (1977). Hairy-cell leukemia and tuberculosis. New England Journal of Medicine, 297(24), 1354–1354. DOI 10.1056/NEJM197712152972420. [Google Scholar] [CrossRef]
24. Pattnaik, S. A., Padhi, S., Chhabra, G., Panigrahi, M. K., Das, P. K. et al. (2020). Atypical presentation of hairy cell leukemia: A report and comprehensive review. Blood Research, 55(2), 123–127. DOI 10.5045/br.2020.2020069. [Google Scholar] [CrossRef]
25. Robak, P., Jesionek-Kupnicka, D., Kupnicki, P., Polliack, A., Robak, T. (2020). Bone lesions in hairy cell leukemia: Diagnosis and treatment. European Journal of Haematology, 105(6), 682–691. DOI 10.1111/ejh.13505. [Google Scholar] [CrossRef]
26. Robak, E., Jesionek-Kupnicka, D., Robak, T. (2020). Skin changes in hairy cell leukemia. Annals of Hematology, 100(3), 615–625. DOI 10.1007/s00277-020-04349-z. [Google Scholar] [CrossRef]
27. AlEnazi, A., Alhedaithy, R., Alfayez, A., Alghonaim, Y. (2019). Acute profound sensorineural hearing loss as the initial manifestation of hairy cell leukemia, case report and literature review. International Journal of Surgery Case Reports, 60, 200–203. DOI 10.1016/j.ijscr.2019.05.056. [Google Scholar] [CrossRef]
28. Yam, L., Janckila, A., Li, C., Lam, W. (1987). Cytochemistry of tartrate-resistant acid phosphatase: 15 years’ experience. Leukemia, 1(4), 285–288. [Google Scholar]
29. Robbins, B. A., Ellison, D. J., Spinosa, J. C., Carey, C. A., Lukes, R. J. et al. (1993). Diagnostic application of two-color flow cytometry in 161 cases of hairy cell leukemia. Blood, 82(4), 1277–1287. DOI 10.1182/blood.V82.4.1277.1277. [Google Scholar] [CrossRef]
30. Del Giudice, I., Matutes, E., Morilla, R., Morilla, A., Owusu-Ankomah, K. et al. (2004). The diagnostic value of CD123 in B-cell disorders with hairy or villous lymphocytes. Haematologica, 89(3), 303–308. [Google Scholar]
31. El Achi, H., Dupont, E., Paul, S., Khoury, J. D. (2020). CD123 as a biomarker in hematolymphoid malignancies: Principles of detection and targeted therapies. Cancers, 12(11), 3087. DOI 10.3390/cancers12113087. [Google Scholar] [CrossRef]
32. Hounieu, H., Chittal, S. M., Al Saati, T., de Mascarel, A., Sabattini, E. et al. (1992). Hairy cell leukemia: Diagnosis of bone marrow involvement in paraffin-embedded sections with monoclonal antibody DBA.44. American Journal of Clinical Pathology, 98(1), 26–33. DOI 10.1093/ajcp/98.1.26. [Google Scholar] [CrossRef]
33. Salomon-Nguyen, F., Valensi, F., Troussard, X., Flandrin, G. (1996). The value of the monoclonal antibody, DBA44, in the diagnosis of B-lymphoid disorders. Leukemia Research, 20(11), 909–913. DOI 10.1016/0145-2126(95)00165-4. [Google Scholar] [CrossRef]
34. Falini, B., Tiacci, E., Liso, A., Basso, K., Sabattini, E. et al. (2004). Simple diagnostic assay for hairy cell leukaemia by immunocytochemical detection of annexin A1 (ANXA1). The Lancet, 363(9424), 1869–1871. DOI 10.1016/S0140-6736(04)16356-3. [Google Scholar] [CrossRef]
35. Matutes, E., Owusu-Ankomah, K., Morilla, R., Garcia Marco, J., Houlihan, A. et al. (1994). The immunological profile of B-cell disorders and proposal of a scoring system for the diagnosis of CLL. Leukemia, 8(10), 1640–1645. [Google Scholar]
36. Kohla, S., Ibrahim, F. A., Aldapt, M. B., ELSabah, H., Mohamed, S. et al. (2020). A rare case of hairy cell leukemia with unusual loss of CD123 associated with COVID-19 at the time of presentation. Case Reports in Oncology, 13(3), 1430–1440. DOI 10.1159/000512830. [Google Scholar] [CrossRef]
37. Jain, D., Dorwal, P., Gajendra, S., Pande, A., Mehra, S. et al. (2016). CD5 positive hairy cell leukemia: A rare case report with brief review of literature. Cytometry Part B: Clinical Cytometry, 90(5), 467–472. DOI 10.1002/cyto.b.21365. [Google Scholar] [CrossRef]
38. Wang, L., Tadros, A. S., Hoh, C. K., Wang, H. Y. (2016). CD10-Positive hairy cell leukemia involving multiple deep lymph nodes. Clinical Lymphoma Myeloma and Leukemia, 16(5), e51–e53. DOI 10.1016/j.clml.2016.02.041. [Google Scholar] [CrossRef]
39. Vittoria, L., Bozzi, F., Capone, I., Carniti, C., Lorenzini, D. et al. (2021). A rare biclonal hairy cell leukemia disclosed by an integrated diagnostic approach: A case report. Cytometry Part B: Clinical Cytometry, 100(6), 692–694. DOI 10.1002/cyto.b.21980. [Google Scholar] [CrossRef]
40. Jain, P., Kanagal-Shamanna, R., Konoplev, S., Zuo, Z., Estrov, Z. (2018). Biclonal IGHV-4-34 hairy cell leukemia variant and CLL-successful treatment with ibrutinib and venetoclax. American Journal of Hematology, 93(12), 1568–1569. DOI 10.1002/ajh.25264. [Google Scholar] [CrossRef]
41. Salem, D. A., Scott, D., McCoy, C. S., Liewehr, D. J., Venzon, D. et al. (2019). Differential expression of CD43, CD81, and CD200 in classic versus variant hairy cell leukemia. Cytometry Part B: Clinical Cytometry, 96. DOI 10.1002/cyto.b.21785. [Google Scholar] [CrossRef]
42. Tiacci, E., Trifonov, V., Schiavoni, G., Holmes, A., Kern, W. et al. (2011). BRAF mutations in hairy-cell leukemia. New England Journal of Medicine, 364(24), 2305–2315. DOI 10.1056/NEJMoa1014209. [Google Scholar] [CrossRef]
43. Xi, L., Arons, E., Navarro, W., Calvo, K. R., Stetler-Stevenson, M. et al. (2012). Both variant and IGHV4-34–expressing hairy cell leukemia lack the BRAF V600E mutation. Blood, 119(14), 3330–3332. DOI 10.1182/blood-2011-09-379339. [Google Scholar] [CrossRef]
44. Jain, P., Ok, C. Y., Konoplev, S., Patel, K. P., Jorgensen, J. et al. (2016). Relapsed refractory BRAF negative, IGHV 4-34–Positive variant of hairy cell leukemia: A distinct entity? Journal of Clinical Oncology, 34(7), e57–e60. DOI 10.1200/JCO.2013.50.9661. [Google Scholar] [CrossRef]
45. Tiacci, E., Pettirossi, V., Schiavoni, G., Falini, B. (2017). Genomics of hairy cell leukemia. Journal of Clinical Oncology, 35(9), 1002–1010. DOI 10.1200/JCO.2016.71.1556. [Google Scholar] [CrossRef]
46. Tschernitz, S., Flossbach, L., Bonengel, M., Roth, S., Rosenwald et al. (2014). Alternative BRAF mutations in BRAF V600E-negative hairy cell leukaemias. British Journal of Haematology, 165(4), 529–533. DOI 10.1111/bjh.12735. [Google Scholar] [CrossRef]
47. Cohn, A. L., Day, B. M., Abhyankar, S., McKenna, E., Riehl, T. et al. (2017). BRAFV600 mutations in solid tumors, other than metastatic melanoma and papillary thyroid cancer, or multiple myeloma: A screening study. OncoTargets and Therapy, 10, 965. DOI 10.2147/OTT. [Google Scholar] [CrossRef]
48. Langabeer, S. E., Quinn, F., O’Brien, D., McElligott, A. M., Kelly, J. et al. (2012). Incidence of the BRAFV600E mutation in chronic lymphocytic leukaemia and prolymphocytic leukaemia. Leukemia Research, 36(4), 483–484. DOI 10.1016/j.leukres.2011.12.015. [Google Scholar] [CrossRef]
49. Bohn, J. P., Salcher, S., Pircher, A., Untergasser, G., Wolf, D. (2021). The biology of classic hairy cell leukemia. International Journal of Molecular Sciences, 22(15), 7780. DOI 10.3390/ijms22157780. [Google Scholar] [CrossRef]
50. Dietrich, S., Hüllein, J., Lee, S. C. W., Hutter, B., Gonzalez et al. (2015). Recurrent CDKN1B (p27) mutations in hairy cell leukemia. Blood, 126(8), 1005–1008. DOI 10.1182/blood-2015-04-643361. [Google Scholar] [CrossRef]
51. Maitre, E., Bertrand, P., Maingonnat, C., Viailly, P. J., Wiber, M. et al. (2018). New generation sequencing of targeted genes in the classical and the variant form of hairy cell leukemia highlights mutations in epigenetic regulation genes. Oncotarget, 9(48), 28866–28876. DOI 10.18632/oncotarget.25601. [Google Scholar] [CrossRef]
52. Waterfall, J. J., Arons, E., Walker, R. L., Pineda, M., Roth, L. et al. (2014). High prevalence of MAP2K1 mutations in variant and IGHV4-34–expressing hairy-cell leukemias. Nature Genetics, 46(1), 8–10. DOI 10.1038/ng.2828. [Google Scholar] [CrossRef]
53. Weston-Bell, N. J., Tapper, W., Gibson, J., Bryant, D., Moreno, Y. et al. (2016). Exome sequencing in classic hairy cell leukaemia reveals widespread variation in acquired somatic mutations between individual tumours apart from the signature BRAFV(600)E lesion. PLoS One, 11(2), e0149162. DOI 10.1371/journal.pone.0149162. [Google Scholar] [CrossRef]
54. Arons, E., Zhou, H., Sokolsky, M., Gorelik, D., Potocka, K. et al. (2020). Expression of the muscle-associated gene MYF6 in hairy cell leukemia. PLoS One, 15(2), e0227586. DOI 10.1371/journal.pone.0227586. [Google Scholar] [CrossRef]
55. Teklie, Y., Bell, S., Idogun, P., Venigalla, M. (2021). A rare case of a patient with hairy cell leukemia developing blastoid marginal zone B-cell lymphoma. Cureus, 13(7), e16239. DOI 10.7759/cureus.16239. [Google Scholar] [CrossRef]
56. Forconi, F. (2011). Hairy cell leukaemia: Biological and clinical overview from immunogenetic insights. Hematological Oncology, 29(2), 55–66. DOI 10.1002/hon.975. [Google Scholar] [CrossRef]
57. Maral, S., Albayrak, M., Dagdas, S., Yıldız, A., Yıldırım, R. et al. (2020). Prognostic value of baseline serum lactate dehydrogenase level in patients with hairy cell leukemia. Clinical Lymphoma Myeloma and Leukemia, 20(9), e616–e621. DOI 10.1016/j.clml.2020.04.005. [Google Scholar] [CrossRef]
58. Forconi, F., Sozzi, E., Cencini, E., Zaja, F., Intermesoli, T. et al. (2009). Hairy cell leukemias with unmutated IGHV genes define the minor subset refractory to single-agent cladribine and with more aggressive behavior. Blood, 114(21), 4696–4702. DOI 10.1182/blood-2009-03-212449. [Google Scholar] [CrossRef]
59. Arons, E., Suntum, T., Stetler-Stevenson, M., Kreitman, R. J. (2009). VH4-34+ hairy cell leukemia, a new variant with poor prognosis despite standard therapy. Blood, 114(21), 4687–4695. DOI 10.1182/blood-2009-01-201731. [Google Scholar] [CrossRef]
60. Koldej, R. M., Prabahran, A., Tan, C. W., Ng, A. P., Davis, M. J. et al. (2021). Dissection of the bone marrow microenvironment in hairy cell leukaemia identifies prognostic tumour and immune related biomarkers. Scientific Reports, 11(1), 19056. DOI 10.1038/s41598-021-98536-1. [Google Scholar] [CrossRef]
61. Cornet, E., Delmer, A., Feugier, P., Garnache-Ottou, F., Ghez, D. et al. (2014). French Society of Haematology. Recommendations of the SFH (French Society of Haematology) for the diagnosis, treatment and follow-up of hairy cell leukaemia. Annals of Hematology, 93(12), 1977–1983. DOI 10.1007/s00277-014-2140-y. [Google Scholar] [CrossRef]
62. Robak, T., Matutes, E., Catovsky, D., Zinzani, P. L., Buske, C. (2015). Hairy cell leukaemia: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Annals of Oncology, 26(suppl 5), v100–v107. DOI 10.1093/annonc/mdv200. [Google Scholar] [CrossRef]
63. Grever, M. R., Abdel-Wahab, O., Andritsos, L. A., Banerji, V., Barrientos, J. et al. (2017). Consensus guidelines for the diagnosis and management of patients with classic hairy cell leukemia. Blood, 129(5), 553–560. DOI 10.1182/blood-2016-01-689422. [Google Scholar] [CrossRef]
64. Parry-Jones, N., Joshi, A., Forconi, F., Dearden, C., BSH guidelines committee (2020). Guideline for diagnosis and management of hairy cell leukaemia (HCL) and hairy cell variant (HCL-V). British Journal of Haematology, 191(5), 730–737. DOI 10.1111/bjh.17055. [Google Scholar] [CrossRef]
65. Troussard, X., Maitre, E., Cornet, E. (2022). Hairy cell leukemia 2022: Update on diagnosis, risk-stratification, and treatment. American Journal of Hematology. 97(2), 226–236 DOI 10.1002/ajh.26390. [Google Scholar] [CrossRef]
66. Paillassa, J., Troussard, X. (2020). Biology and treatment of hairy cell leukemia. Current Treatment Options in Oncology, 21(6), 44. DOI 10.1007/s11864-020-00732-0. [Google Scholar] [CrossRef]
67. Grever, M., Andritsos, L., Banerji, V., Barrientos, J. C., Bhat, S. et al. (2021). Hairy cell leukemia and COVID-19 adaptation of treatment guidelines. Leukemia, 35(7), 1864–1872. DOI 10.1038/s41375-021-01257-7. [Google Scholar] [CrossRef]
68. Andrulis, M., Penzel, R., Weichert, W., von Deimling, A., Capper, D. (2012). Application of a BRAFV600E mutation-specific antibody for the diagnosis of hairy cell leukemia. The American Journal of Surgical Pathology, 36(12), 1796–1800. DOI 10.1097/PAS.0b013e3182549b50. [Google Scholar] [CrossRef]
69. Akarca, A., Shende, V., Ramsay, A., Diss, T., Pane-Foix, M. et al. (2013). BRAF V600E mutation-specific antibody, a sensitive diagnostic marker revealing minimal residual disease in hairy cell leukaemia. British Journal of Haematology, 162(6), 848–851. DOI 10.1111/bjh.12429. [Google Scholar] [CrossRef]
70. Arons, E., Margulies, I., Sorbara, L., Raffeld, M., Stetler-Stevenson, M. et al. (2006). Minimal residual disease in hairy cell leukemia patients assessed by clone-specific polymerase chain reaction. Clinical Cancer Research, 12(9), 2804–2811. DOI 10.1158/1078-0432.CCR-05-2315. [Google Scholar] [CrossRef]
71. Broccoli, A., Terragna, C., Nanni, L., Martello, M., Armuzzi, S. et al. (2021). Droplet digital polymerase chain reaction for the assessment of disease burden in hairy cell leukemia. Hematological Oncology, 40(1), 57–62. DOI: 10.1002/hon.2932. [Google Scholar] [CrossRef]
72. Guerrini, F., Paolicchi, M., Ghio, F., Ciabatti, E., Grassi, S. et al. (2016). The droplet digital PCR: A new valid molecular approach for the assessment of BRAF V600E mutation in hairy cell leukemia. Frontiers in Pharmacology, 13(7), 363. DOI 10.3389/fphar.2016.00363. [Google Scholar] [CrossRef]
73. Ottou, G., Chandesris, F., Lhermitte, M. O., Callens, L., Beldjord, C. et al. (2014). Peripheral blood 8 colour flow cytometry monitoring of hairy cell leukaemia allows detection of high-risk patients. British Journal of Haematology, 166(1), 50–59. DOI 10.1111/bjh.12839. [Google Scholar] [CrossRef]
74. Troussard, X., Grever, M. R. (2021). The revised guidelines for the diagnosis and management of hairy cell leukaemia and the hairy cell leukaemia variant. British Journal of Haematology, 193(1), 11–14. DOI 10.1111/bjh.17201. [Google Scholar] [CrossRef]
75. Grever, M., Kopecky, K., Foucar, M. K., Head, D., Bennett, J. M. et al. (1995). Randomized comparison of pentostatin versus interferon alfa-2a in previously untreated patients with hairy cell leukemia: An intergroup study. Journal of Clinical Oncology, 13(4), 974–982. DOI 10.1200/JCO.1995.13.4.974. [Google Scholar] [CrossRef]
76. Kantarjian, H. M., Redman, J. R., Keating, M. J. (1990). Fludarabine phosphate therapy in other lymphoid malignancies. Seminars in Oncology, 17(5 Suppl 8), 66–70. [Google Scholar]
77. Kraut, E. H., Chun, H. G. (1991). Fludarabine phosphate in refractory hairy cell leukemia. American Journal of Hematology, 37(1), 59. DOI 10.1002/(ISSN)1096-8652. [Google Scholar] [CrossRef]
78. Spiers, A. S., Moore, D., Cassileth, P. A., Harrington, D. P., Cummings, F. J. et al. (1987). Remissions in hairy-cell leukemia with pentostatin (2′-deoxycoformycin). New England Journal of Medicine, 316(14), 825–830. DOI 10.1056/NEJM198704023161401. [Google Scholar] [CrossRef]
79. Ribeiro, P., Bouaffia, F., Peaud, P. Y., Blanc, M., Salles, B. et al. (1999). Long term outcome of patients with hairy cell leukemia treated with pentostatin. Cancer, 85, 65–71. DOI 10.1002/(ISSN)1097-0142. [Google Scholar] [CrossRef]
80. Maloisel, F., Benboubker, L., Gardembas, M., Coiffier, B., Divine et al. (2003). Long-term outcome with pentostatin treatment in hairy cell leukemia patients. A French retrospective study of 238 patients. Leukemia, 17(1), 45–51. DOI 10.1038/sj.leu.2402784. [Google Scholar] [CrossRef]
81. Thaler, J., Grünewald, K., Gattringer, C., Ho, A. D., Weyrer, K. et al. (1993). Long-term follow-up of patients with hairy cell leukaemia treated with pentostatin: Lymphocyte subpopulations and residual bone marrow infiltration. British Journal of Haematology, 84(1), 75–82. DOI 10.1111/j.1365-2141.1993.tb03027.x. [Google Scholar] [CrossRef]
82. Piro, D., Carrera, C. J., Carson, D. A., Beutler, E. (1990). Lasting remissions in hairy-cell leukemia induced by a single infusion of 2-chlorodeoxyadenosine. New England Journal of Medicine, 322, 1117–1121. DOI 10.1056/NEJM199004193221605. [Google Scholar] [CrossRef]
83. Saven, A., Piro, L. (1996). 2-Chlorodeoxyadenosine: A potent antimetabolite with major activity in the treatment of indolent lymphoproliferative disorders. Hematology and Cell Therapy, 38(Suppl 2), S93–101. [Google Scholar]
84. Zinzani, P. L., Tani, M., Marchi, E., Stefoni, V., Alinari, L. et al. (2004). Long-term follow-up of front-line treatment of hairy cell leukemia with 2-chlorodeoxyadenosine. Haematologica, 89(3), 309–313. [Google Scholar]
85. Cornet, E., Tomowiak, C., Tanguy-Schmidt, A., Lepretre, S., Dupuis, J. et al. (2014). Long-term follow-up and second malignancies in 487 patients with hairy cell leukaemia. British Journal of Haematology, 166(3), 390–400. DOI 10.1111/bjh.12908. [Google Scholar] [CrossRef]
86. Paillassa, J., Cornet, E., Noel, S., Tomowiak, C., Lepretre, S. et al. (2020). Analysis of a cohort of 279 patients with hairy-cell leukemia (HCL10 years of follow-up. Blood Cancer Journal, 10(5), 62. DOI 10.1038/s41408-020-0328-z. [Google Scholar] [CrossRef]
87. Else, M., Dearden, C. E., Catovsky, D. (2015). Long-term follow-up after purine analogue therapy in hairy cell leukaemia. Best Practice & Research Clinical Haematology, 28(4), 217–229. DOI 10.1016/j.beha.2015.09.004. [Google Scholar] [CrossRef]
88. Benz, R., Arn, K., Andres, M., Pabst, T., Baumann, M. et al. (2020). Prospective long-term follow-up after first-line subcutaneous cladribine in hairy cell leukemia: A SAKK trial. Blood Advances, 4(15), 3699–3707. DOI 10.1182/bloodadvances.2020002160. [Google Scholar] [CrossRef]
89. Van Hoef, M., Troussard, X. (2019). Cladribine in first line and relapsed hairy cell leukemia and the hairy cell leukemia variant. International Journal of Blood Disorders & Diseases, 3(1), 001–007. [Google Scholar]
90. Broccoli, A., Argnani, L., Nanni, L., Terragna, C., Sabattini, E. et al. (2021). The treatment of hairy cell leukemia with a focus on long lasting responses to cladribine: A 30-year experience. American Journal of Hematology, 96(10), 1204–1210. DOI 10.1002/ajh.26287. [Google Scholar] [CrossRef]
91. Paillassa, J., Troussard, X. (2021). Patients with relapsed/refractory hairy-cell leukemia. Cancer Reports, 12, e1495. DOI 10.1002/cnr2.1495. [Google Scholar] [CrossRef]
92. Chacko, J., Murphy, C., Duggan, C., O’Briain, D. S., Browne, P. V. et al. (1999). Weekly intermittent 2-CdA is less toxic and equally efficacious when compared to continuous infusion in hairy cell leukaemia. British Journal of Haematology, 105(4), 1145–1146. DOI 10.1111/j.1365-2141.1999.01523.x. [Google Scholar] [CrossRef]
93. Seymour, J., Talpaz, M., Kurzrock, R. (1997). Response duration and recovery of CD4+ lymphocytes following deoxycoformycin in interferon-resistant hairy cell leukemia:7-year follow-up. Leukemia, 11, 42–47. DOI 10.1038/sj.leu.2400513. [Google Scholar] [CrossRef]
94. Saven, A., Burian, C., Adusumalli, J., Koziol, J. A. (1999). Filgrastim for cladribine-induced neutropenic fever in patients with hairy cell leukemia. Blood, 93, 2471–2477. DOI 10.1182/blood.V93.8.2471. [Google Scholar] [CrossRef]
95. Seymour, J., Kurzrock, R., Freireich, E., Estey, E. (1994). 2-chlorodeoxyadenosine induces durable remissions and prolonged suppression of CD4+ lymphocyte counts in patients with hairy cell leukemia. Blood, 83(10), 2906–2911. DOI 10.1182/blood.V83.10.2906.2906. [Google Scholar] [CrossRef]
96. Juliusson, G., Lenkei, R., Liliemark, J. (1994). Flow cytometry of blood and bone marrow cells from patients with hairy cell leukemia: Phenotype of hairy cells and lymphocytes subset after treatment with 2-chlorodeoxyadenosine. Blood, 83(12), 3672–3681. DOI 10.1182/blood.V83.12.3672.3672. [Google Scholar] [CrossRef]
97. Raspadori, D., Rondelli, D., Birtolo, S., Lenoci, M., Nardi, G. et al. (1999). Long-lasting decrease of CD4+/CD45RA+T cells in HCL patients after 2-chlorodeoxyadenosine (2-CdA) treatment. Leukemia, 13(8), 1254–1257. DOI 10.1038/sj.leu.2401476. [Google Scholar] [CrossRef]
98. Juliusson, G., Liliemark, J. (1992). Rapid recovery from cytopenia in hairy cell leukemia after treatment with 2-chloro-2’-deoxyadenosine(CdARelation to opportunistic infections. Blood, 79, 888–894. DOI 10.1182/blood.V79.4.888.bloodjournal794888. [Google Scholar] [CrossRef]
99. Rutella, S., Sica, S., Rumi, C., Martucci, R., Etuk, B. et al. (1996). Hypereosinophilia during 2-chlorodeoxyadenosine treatment for hairy cell leukaemia. British Journal of Haematology, 92(2), 426–428. DOI 10.1046/j.1365-2141.1996.00869.x. [Google Scholar] [CrossRef]
100. Orchard, J. A., Bolam, S., Oscier, D. G. (1998). Association of myelodysplastic changes with purine analogues: Short report. British Journal of Haematology, 100(4), 677–679. DOI 10.1046/j.1365-2141.1998.00612.x. [Google Scholar] [CrossRef]
101. Ravandi, F., Jorgensen, J. L., O’Brien, S. M., Verstovsek, S., Koller, C. A. et al. (2006). Eradication of minimal residual disease in hairy cell leukemia. Blood, 107(12), 4658–4662. DOI 10.1182/blood-2005-11-4590. [Google Scholar] [CrossRef]
102. Ravandi, F., O’Brien, S., Jorgensen, J., Pierce, S., Faderl, S. et al. (2011). Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood, 118(14), 3818–3823. DOI 10.1182/blood-2011-04-351502. [Google Scholar] [CrossRef]
103. Chihara, D., Kantarjian, H., O’Brien, S., Jorgensen, J., Pierce, S. et al. (2016). Long-term durable remission by cladribine followed by rituximab in patients with hairy cell leukaemia. British Journal of Haematology, 174(5), 760–766. DOI 10.1111/bjh.14129. [Google Scholar] [CrossRef]
104. Chihara, D., Arons, E., Stetler-Stevenson, M., Yuan, C. M., Wang, H. W. et al. (2020). Randomized phase II study of first-line cladribine with concurrent or delayed rituximab in patients with hairy cell leukemia. Journal of Clinical Oncology, 38(14), 1527–1538. DOI 10.1200/JCO.19.02250. [Google Scholar] [CrossRef]
105. Quesada, J. R., Reuben, J., Manning, J. T., Hersh, E. M., Gutterman, J. U. (1984). Alpha interferon for induction of remission in hairy-cell leukemia. New England Journal of Medicine, 310(1), 15–18. DOI 10.1056/NEJM198401053100104. [Google Scholar] [CrossRef]
106. Smirnova, S. Yu., Al-Radi, L. S., Moiseeva, T. N., Gemdzhian, E. G., Yakutik, I. A. et al. (2021). Inhibitor of BRAFV600E mutation as a treatment option for hairy cell leukemia with deep neutropenia and infectious complications. Clinical Lymphoma Myeloma and Leukemia, 21(7), 427–430. DOI 10.1016/j.clml.2021.02.005. [Google Scholar] [CrossRef]
107. Tiacci, E., Park, J. H., de Carolis, L., Chung, S. S., Broccoli, A. et al. (2015). Targeting mutant BRAF in relapsed or refractory hairy-cell leukemia. New England Journal of Medicine, 373(18), 1733–1747. DOI 10.1056/NEJMoa1506583. [Google Scholar] [CrossRef]
108. Dietrich, S., Pircher, A., Endris, V., Peyrade, F., Wendtner, C. M. et al. (2016). BRAF inhibition in hairy cell leukemia with low-dose vemurafenib. Blood, 127(23), 2847–2855. DOI 10.1182/blood-2015-11-680074. [Google Scholar] [CrossRef]
109. Liebers, N., Roider, T., Bohn, J. P., Haberbosch, I., Pircher, A., Ferstl, B. et al. (2020). BRAF inhibitor treatment in classic hairy cell leukemia: A long-term follow-up study of patients treated outside clinical trials. Leukemia, 34(5), 1454–1457. DOI 10.1038/s41375-019-0646-y. [Google Scholar] [CrossRef]
110. Tiacci, E., De Carolis, L., Simonetti, E., Merluzzi, M., Bennati, A. et al. (2021). Safety and efficacy of the BRAF inhibitor dabrafenib in relapsed or refractory hairy cell leukemia: A pilot phase-2 clinical trial. Leukemia, 1–5. DOI 10.1038/s41375-021-01210-8. [Google Scholar] [CrossRef]
111. Shenoi, D. P., Andritsos, L. A., Blachly, J. S., Rogers, K. A., Moran, M. E. et al. (2019). Classic hairy cell leukemia complicated by pancytopenia and severe infection: A report of 3 cases treated with vemurafenib. Blood Advances, 3(2), 116–118. DOI 10.1182/bloodadvances.2018027466. [Google Scholar] [CrossRef]
112. Bohn, J., Pircher, A., Wanner, D., Vill, D., Foeger, B. et al. (2019). Low-dose vemurafenib in hairy cell leukemia patients with active infection. American Journal of Hematology, 94(6), E180–E182. DOI 10.1002/ajh.25474. [Google Scholar] [CrossRef]
113. Callahan, M. K., Rampal, R., Harding, J. J., Klimek, V. M., Chung, Y. R. et al. (2012). Progression of RAS-mutant leukemia during RAF inhibitor treatment. New England Journal of Medicine, 367(24), 2316–2321. DOI 10.1056/NEJMoa1208958. [Google Scholar] [CrossRef]
114. Simnica, D., Ittrich, H., Bockemeyer, C., Stein, A., Binder, M. (2020). Targeting the mutational landscape of bystander cells: Drug-promoted blood cancer from high-prevalence pre-neoplasias in patients on BRAF inhibitors. Frontiers in Oncology, 10, 540030. DOI 10.3389/fonc.2020.540030. [Google Scholar] [CrossRef]
115. Yaktapour, N., Meiss, F., Mastroianni, J., Zenz, T., Andrlova, H. et al. (2014). BRAF inhibitor–associated ERK activation drives development of chronic lymphocytic leukemia. Journal of Clinical Investigation, 124(11), 5074–5084. DOI 10.1172/JCI76539. [Google Scholar] [CrossRef]
116. Tiacci, E., De Carolis, L., Simonetti, E., Capponi, M., Ambrosetti, A. et al. (2021). Vemurafenib plus rituximab in refractory or relapsed hairy-cell leukemia. New England Journal of Medicine, 384(19), 1810–1823. DOI 10.1056/NEJMoa2031298. [Google Scholar] [CrossRef]
117. Robak, T., Janus, A., Jamroziak, K., Tiacci, E., Kreitman, R. J. (2021). Vemurafenib and rituximab in patients with hairy cell leukemia previously treated with moxetumomab pasudotox. Journal of Clinical Medicine, 10(13), 2800. DOI 10.3390/jcm10132800. [Google Scholar] [CrossRef]
118. Kreitman, R. J., Moreau, P., Hutchings, M., Gazzah, A., Blay, J. Y. et al. (2018). Treatment with combination of dabrafenib and trametinib in patients with recurrent/refractory BRAF V600E-mutated hairy cell leukemia (HCL). Blood, 132(Supplement 1), 391. DOI 10.1182/blood-2018-99-113135. [Google Scholar] [CrossRef]
119. Kreitman, R. J., Tallman, M. S., Robak, T., Coutre, S., Wilson, W. H. et al. (2018). Minimal residual hairy cell leukemia eradication with moxetumomab pasudotox: Phase I results and long-term follow-up. Blood, 131(21), 2331–2334. DOI 10.1182/blood-2017-09-803072. [Google Scholar] [CrossRef]
120. Kreitman, R. J., Dearden, C., Zinzani, P. L., Delgado, J., Karlin, L. et al. (2018). Moxetumomab pasudotox in relapsed/refractory hairy cell leukemia. Leukemia, 32(8), 1768–1777. DOI 10.1038/s41375-018-0210-1. [Google Scholar] [CrossRef]
121. Kreitman, R. J., Dearden, C., Zinzani, P. L., Delgado, J., Robak, T. et al. (2021). Moxetumomab pasudotox in heavily pre-treated patients with relapsed/refractory hairy cell leukemia (HCLLong-term follow-up from the pivotal trial. Journal of Hematology & Oncology, 14(1), 35. DOI 10.1186/s13045-020-01004-y. [Google Scholar] [CrossRef]
122. Yurkiewicz, I. R., Coutre, S., Ghesquieres, H., Pastan, I., Kreitman, R. J. (2021). Moxetumomab pasudotox as re-treatment for heavily-pretreated relapsed hairy cell leukemia. Leukemia & Lymphoma, 62(11), 2812–2814. DOI 10.1080/10428194.2021.1929959. [Google Scholar] [CrossRef]
123. Rogers, K. A., Andritsos, L. A., Wei, L., McLaughlin, E. M., Ruppert, A. S. et al. (2021). Phase 2 study of ibrutinib in classic and variant hairy cell leukemia. Blood, 137(25). DOI 10.1182/blood.2020009688. [Google Scholar] [CrossRef]
124. Vereertbrugghen, A., Colado, A., Gargiulo, E., Bezares, R. F., Fernández Grecco, H. et al. (2021). In vitro sensitivity to venetoclax and microenvironment protection in hairy cell leukemia. Frontiers in Oncology, 11, 598319. DOI 10.3389/fonc.2021.598319. [Google Scholar] [CrossRef]
125. Park, J. H., Winer, E. S., Huntington, S. F., von Keudell, G., Vemuri, S. et al. (2021). First line chemo-free therapy with the BRAF inhibitor vemurafenib combined with obinutuzumab is effective in patients with Hcl. Blood, 138, 43. DOI 10.1182/blood-2021-151074. [Google Scholar] [CrossRef]
126. Poret, N., Fu, Q., Guihard, S., Cheok, M., Miller, K. et al. (2015). CD38 in hairy cell leukemia is a marker of poor prognosis and a New target for therapy. Cancer Research, 75(18), 3902–3911. DOI 10.1158/0008-5472.CAN-15-0893. [Google Scholar] [CrossRef]
127. Troussard, X., Henry-Amar, M., Flandrin, G. (1994). Second cancer risk after interferon therapy? [letter; comment]. Blood, 84(9), 3242–3244. DOI 10.1182/blood.V84.9.3242.3242. [Google Scholar] [CrossRef]
128. Zheng, G., Chattopadhyay, S., Sud, A., Sundquist, K., Sundquist, J. et al. (2019). Second primary cancers in patients with acute lymphoblastic, chronic lymphocytic and hairy cell leukaemia. British Journal of Haematology, 185(2), 232–239. DOI 10.1111/bjh.15777. [Google Scholar] [CrossRef]
129. Kampmeier, P., Spielberger, R., Dickstein, J., Mick, R., Golomb, H. et al. (1994). Increased incidence of second neoplasms in patients treated with interferon alpha 2b for hairy cell leukemia: A clinicopathologic assessment [see comments]. Blood, 83(10), 2931–2938. DOI 10.1182/blood.V83.10.2931.2931. [Google Scholar] [CrossRef]
130. Traverse-Glehen, A., Baseggio, L., Callet-Bauchu, E., Morel, D., Gazzo, S. et al. (2008). Splenic red pulp lymphoma with numerous basophilic villous lymphocytes: A distinct clinicopathologic and molecular entity? Blood, 111(4), 2253–2260. DOI 10.1182/blood-2007-07-098848. [Google Scholar] [CrossRef]
131. Traverse-Glehen, A., Verney, A., Gazzo, S., Jallades, L., Chabane, K. et al. (2017). Splenic diffuse red pulp lymphoma has a distinct pattern of somatic mutations amongst B-cell malignancies. Leukemia & Lymphoma, 58(3) 666–675. DOI 10.1080/10428194.2016.1196813. [Google Scholar] [CrossRef]
132. Traverse-Glehen, A., Baseggio, L., Callet-Bauchu, E., Ffrench, M., Coiffier, B. et al. (2010). Hairy cell leukaemia-variant and splenic red pulp lymphoma: A single entity? British Journal of Haematology, 150(1), 113–116. DOI 10.1111/j.1365-2141.2010.08153.x. [Google Scholar] [CrossRef]
133. Yilmaz, E., Chhina, A., Nava, V. E., Aggarwal, A. (2021). A review on splenic diffuse Red pulp small B-cell lymphoma. Current Oncology, 28(6), 5148–5154. DOI 10.3390/curroncol28060431. [Google Scholar] [CrossRef]
134. Martinez, D., Navarro, A., Martinez-Trillos, A., Molina-Urra, R., Gonzalez-Farre, B. et al. (2016). NOTCH1, TP53, and MAP2K1 mutations in splenic diffuse Red pulp small B-cell lymphoma Are associated with progressive disease. The American Journal of Surgical Pathology, 40, 192–201. DOI 10.1097/PAS.0000000000000523. [Google Scholar] [CrossRef]
135. Suzuki, T., Miyoshi, H., Shimono, J., Kawamoto, K., Arakawa, F. et al. (2020). Clinicopathological analysis of splenic red pulp low-grade B-cell lymphoma. Pathology International, 70(5), 280–286. DOI 10.1111/pin.12909. [Google Scholar] [CrossRef]
136. Jallades, L., Baseggio, L., Sujobert, P., Huet, S., Chabane, K. et al. (2017). Exome sequencing identifies recurrent BCOR gene alterations and the absence of KLF2, TNFAIP3 and MYD88 mutations in splenic diffuse red pulp small B-cell lymphoma. Haematologica, 1758–1766. DOI 10.3324/haematol.2016.160192. [Google Scholar] [CrossRef]
137. Curiel-Olmo, S., Mondéjar, R., Almaraz, C., Mollejo, M., Cereceda, L. et al. (2017). Splenic diffuse red pulp small B-cell lymphoma displays increased expression of cyclin D3 and recurrent CCND3 mutations. Blood, 129(8), 1042–1045. DOI 10.1182/blood-2016-11-751024. [Google Scholar] [CrossRef]
138. Cheng, W. Y., Zhu, Y. M., Cheng, S., Chen, Y. S., Shen, Y. (2018). Development of B-cell prolymphocytic leukemia in a patient with splenic diffuse red pulp small B-cell lymphoma. Leukemia & Lymphoma, 59(8), 1990–1993. DOI 10.1080/10428194.2017.1405397. [Google Scholar] [CrossRef]
139. Cawley, J., Burns, G., Hayhoe, F. (1980). A chronic lymphoproliferative disorder with distinctive features: A distinct variant of hairy-cell leukaemia. Leukemia Research, 4(6), 547–559. DOI 10.1016/0145-2126(80)90066-1. [Google Scholar] [CrossRef]
140. Matutes, E., Wotherspoon, A., Catovsky, D. (2003). The variant form of hairy-cell leukaemia. Best Practice & Research Clinical Haematology, 16(1), 41–56. DOI 10.1016/S1521-6926(02)00086-5. [Google Scholar] [CrossRef]
141. Matutes, E., Martínez-Trillos, A., Campo, E. (2015). Hairy cell leukaemia-variant: Disease features and treatment. Best Practice & Research Clinical Haematology, 28(4), 253–263. DOI 10.1016/j.beha.2015.09.002. [Google Scholar] [CrossRef]
142. Angelova, E. A., Medeiros, L. J., Wang, W., Muzzafar, T., Lu, X., Khoury, J. D. et al. (2018). Clinicopathologic and molecular features in hairy cell leukemia-variant: Single institutional experience. Modern Pathology, 31(11), 1717–1732. DOI 10.1038/s41379-018-0093-8. [Google Scholar] [CrossRef]
143. Robak, T. (2006). Current treatment options in hairy cell leukemia and hairy cell leukemia variant. Cancer Treatment Reviews, 32(5), 365–376. DOI 10.1016/j.ctrv.2006.04.010. [Google Scholar] [CrossRef]
144. Hockley, S. L., Giannouli, S., Morilla, A., Wotherspoon, A., Morgan Gareth. J. (2010). Insight into the molecular pathogenesis of hairy cell leukaemia, hairy cell leukaemia variant and splenic marginal zone lymphoma, provided by the analysis of their IGH rearrangements and somatic hypermutation patterns. British Journal of Haematology, 148(4), 665–666. DOI 10.1111/j.1365-2141.2009.07962.x. [Google Scholar] [CrossRef]
145. Hockley, S. L., Morgan, G. J., Leone, P. E., Walker, B. A., Morilla, A. (2011). High-resolution genomic profiling in hairy cell leukemia-variant compared with typical hairy cell leukemia. Leukemia, 25(7), 1189–1192. DOI 10.1038/leu.2011.47. [Google Scholar] [CrossRef]
146. Durham, B. H., Getta, B., Dietrich, S., Taylor, J., Won, H. et al. (2017). Genomic analysis of hairy cell leukemia identifies novel recurrent genetic alterations. Blood, 130(14), 1644–1648. DOI 10.1182/blood-2017-01-765107. [Google Scholar] [CrossRef]
147. Locher, M., Jukic, E., Bohn, J., Untergasser, G., Steurer, M. et al. (2021). Clonal dynamics in a composite chronic lymphocytic leukemia and hairy cell leukemia-variant. Genes, Chromosomes and Cancer, 60(4), 287–292. DOI 10.1002/gcc.22925. [Google Scholar] [CrossRef]
148. Liu, Q., Harris, N., Epperla, N., Andritsos, L. A. (2021). Current and emerging therapeutic options for hairy cell leukemia variant. OncoTargets and Therapy, 14, 1797. DOI 10.2147/OTT.S242247. [Google Scholar] [CrossRef]
149. Al-Sarayfi, D., Meeuwes, F. O., Munnink, T. O., Plattel, W., Rosati, S. et al. (2022). Successful treatment of hairy cell leukemia variant with obinutuzumab. Annals of Hematology, 101(3), 703–704. DOI 10.1007/s00277-021-04559-z. [Google Scholar] [CrossRef]
150. Visentin, A., Imbergamo, S., Frezzato, F., Pizzi, M., Bertorelle, R. et al. (2017). Bendamustine plus rituximab is an effective first-line treatment in hairy cell leukemia variant: A report of three cases. Oncotarget, 8(66), 110727–110731. DOI 10.18632/oncotarget.21304. [Google Scholar] [CrossRef]
151. Chihara, D., Arons, E., Stetler-Stevenson, M., Yuan, C., Wang et al. (2021). Long term follow-up of a phase II study of cladribine with concurrent rituximab with hairy cell leukemia variant. Blood Advances, 5(23), 4807–4816. DOI 10.1182/bloodadvances.2021005039. [Google Scholar] [CrossRef]
152. Wiber, M., Maitre, E., Cornet, E., Salaün, V., Naguib, D., Troussard, X. (2019). Variant form of hairy cell leukemia. Clinical Case Reports, 7(6), 1161–1166. DOI 10.1002/ccr3.2176. [Google Scholar] [CrossRef]
153. Visentin, A., Imbergamo, S., Trimarco, V., Pravato, S., Romano Gargarella, L. et al. (2020). Ibrutinib in relapsed hairy cell leukemia variant: A case report and review of the literature. Hematological Oncology, 38(5), 823–826. DOI 10.1002/hon.2810. [Google Scholar] [CrossRef]
154. Bohn, J. P., Wanner, D., Steurer, M. (2017). Ibrutinib for relapsed refractory hairy cell leukemia variant. Leukemia & Lymphoma, 58(5), 1224–1226. DOI 10.1080/10428194.2016.1239262. [Google Scholar] [CrossRef]
155. Donzel, M., Baseggio, L., Fontaine, J., Pesce, F., Ghesquières, H. et al. (2021). New insights into the biology and diagnosis of splenic marginal zone lymphomas. Current Oncology, 28(5), 3430–3447. DOI 10.3390/curroncol28050297. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |