| Oncologie |
DOI: 10.32604/oncologie.2022.019415
REVIEW
Applications of CRISPR-Cas System in Tumor Biology
1Shenzhen Institute of Translational Medicine, Health Science Center, The First Affiliated Hospital of Shenzhen University, Shenzhen Second People’s Hospital, Shenzhen, 518035, China
2Shantou University Medical College, Shantou, 515041, China
*Corresponding Authors: Yuchen Liu. Email: liuyuchenmdcg@163.com; Weiren Huang. Email: pony8980@163.com
Received: 23 September 2021; Accepted: 16 November 2021
Abstract: The clustered regularly interspaced short palindromic repeats (CRISPR)-Cas system, which is an RNA-guided nuclease system, plays an important role in the adaptive immune response of bacteria, and it is a rapidly developing gene editing technology. It has been widely used in a variety of cells, microorganisms, plants, and animals. This technique has helped to overcome the limitations of previous gene editing methods, and it has promoted the development of synthetic biology, genetics, and proteomics. The ability of the CRISPR-Cas system to modify the genetic components of a system has led to various practical applications, such as base editing, transcription regulation, and epigenetic modification. Moreover, the CRISPR-Cas system can accelerate the development of animal tumor models, thereby promoting in vivo tumor research. In this review, first, we briefly introduce the CRISPR-Cas gene editing technology, followed by a discussion on how this technology plays a vital role in tumor modeling, researching the mechanism of cancer development, drug delivery system, therapy, and genome-wide screening for novel drug targets. Finally, we analyze the current technical problems of this system and highlight its future prospects in clinical applications. Our aim is to explore the applications of CRISPR in cancer research and treatment by discussing these research advances.
Keywords: CRISPR-Cas; tumor modeling construction; mechanism study; delivery system; immunotherapy; screening drug targets
Nomenclature
| CRISPR-Cas | Clustered regularly interspaced short palindromic repeats-associated |
| ZFN | Zinc-finger nucleases |
| TALEN | Transcription activator-like effector nucleases |
| gRNA | Guide RNA |
| crRNA | CRISPR RNA |
| RNP | Ribonucleoprotein |
| tracrRNA | Trans-activating CRISPR RNA |
| sgRNA | Single guide RNA |
| PAM | Protospacer adjacent motif |
| CRISPRa | CRISPR activation |
| CRISPRi | CRISPR interference |
| neoCOR | Neoplastic cerebral organoid |
| P53 | Tumor protein 53 gene |
| PTEN | Phosphatase and tensin homologue gene |
| RB1 | Retinoblastoma 1 |
| NF1 | Neurofibromin-1 gene |
| SCLC | Small cell lung cancer |
| PC | Prostate cancer |
| FRMD6 | FERM domain-containing protein 6 |
| ATT | Antiangiogenic therapy |
| GeCKO | Genome-wide CRISPR knockout |
| dCas9 | Dead Cas9 |
| CBE | Cytosine base editor |
| SHERLOCK | Specific High-Sensitivity Enzymatic Reporter Unlocking |
| RPA | Recombinant polymerase amplification |
| TME | Tumor microenvironment |
| CAR | Chimeric antigen receptor |
| TCR | T cell receptor |
| CRS | Cytokine release syndrome |
| CRES | CAR-T-cell-related encephalopathy syndrome |
| TanCARs | Tandem CARs |
| CSC | Cancer stem cell |
| GBM | Glioblastoma |
| TCRα | T cell receptor α |
| TCRβ | T cell receptor β |
| PD-1 | Death protein 1 |
| NYCE | NY-ESO-1 transduced CRISPR 3X edited cells |
| CTLA-4 | Cytotoxic T-lymphocyte-associated protein 4 |
| HPK1 | Hematopoietic progenitor kinase1 |
| hPSC | Human pluripotent stem cell |
| ESC | Embryonic stem cell |
| iPSC | Induced pluripotent stem cell |
| ITRs/IRs | Inverted end repeat sequences |
| AAV | Adeno-associated virus |
| DSB | DNA double-strand break |
| ABE | Adenine base editor |
| m6A | RNA N6-methyladenosine |
| miRNA | MicroRNA |
| METTL3 | Methyltransferase 3 |
| CRC | Colorectal cancer |
| lncRNA | Long non-coding RNA |
| GACAT3 | Gastric cancer associated transcript 3 |
| CTLs | Cytotoxic T lymphocytes |
| ATM | Ataxia telangiectasia mutated |
| PARP | Polyadenosine diphosphate-ribose polymerase |
| ATR | ATM and Rad3-related |
| pDNA | Plasmid DNA |
| LNP | Lipid nanoparticles |
| HTVI | Hydrodynamic tail-vein injection |
| NC | Nanocapsule |
| GSH | Glutathione |
| RPE | Retinal pigment epithelium |
| EV | Exosomes |
| ABP | Aptamer-binding protein |
| LBP | Lactose derived branch cationic biopolymer |
| HCC | Hepatocellular carcinoma |
| iPhos | Ionizable phospholipids |
| iPLNP | Multi-component lipid nanoparticles |
| SEND | Selective endogenous encapsidation for cellular delivery |
| PEG10 | Paternally expressed 10 |
| nCas9 | Cas9 nickase |
| NHEJ | Non-homologous end joining |
The clustered regularly interspaced short palindromic repeats (CRISPR)-Cas system is a precise, RNA-guided gene-editing technique. It has a great potential in the field of basic tumor study and a wide range of clinical applications. In an attempt to solve the existing clinical problems, many studies have used the CRISPR technology to establish cell, organoid, and animal cancer models [1,2] to study the underlying molecular mechanisms of occurrence and development of cancer, to screen drug-resistant strains [3,4], and to conduct genome-wide screening of drug-resistant genes in patients. This technology can screen targeted drugs with a high specificity, thereby improving the effects of gene therapy and immune cell therapy on cancer patients [5–11]. Therefore, the CRISPR technology has undoubtedly revolutionized biotechnology as well as medicine.
Previously, the gene-editing technologies that were widely used included zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs). The ZFNs are formed by the fusion of the zinc finger DNA-binding domain to the DNA-cutting domain of a nuclease [12]. Similarly, TALENs are obtained by the fusion of the DNA-binding domain of a TAL effector with the DNA cleavage domain of a nuclease. The DNA-binding element of the TALENs consists of a series of TALE subunits [13], and altering this DNA-binding domain alters the specificity of the DNA cutting site. The ZFNs and the TALENs have played an important role in the field of gene editing, but they are associated with problems, such as cytotoxicity, time-consuming design, or low editing efficiency [14]. In contrast to these programmable nuclease enzymes, the CRISPR-Cas system uses an RNA molecule (guide RNA, gRNA) to locate the target DNA sequence, and hence, it functions as an RNA-guided nuclease. The CRISPR-Cas gene array is mainly composed of a leader sequence, followed by short repeating sequences of similar length that are separated by unique interval sequences, and small clusters of cas genes [15]. This CRISPR-Cas system, which is a part of the natural immune system of bacteria, functions in three stages. During the adaptation stage, when exogenous plasmids or phages invade bacteria/archaea, some Cas proteins insert specific gene fragments from exogenous DNA into the original CRISPR sequence, known as the proto-spacer. Next, at the expression stage, CRISPR RNA (crRNA) expression and maturation occur, including transcription of repeat and spacer sequences, and pre-crRNA is formed. The sequence is stored with exogenous genetic information and further cleaved by the CRISPR-Cas system. Each repeating sequence-spacer sequence unit is separately cleaved and processed into crRNA. The crRNA interacts with Cas protein to form an RNA-protein complex. At the interference stage, each crRNA only contains a spacer sequence. When the foreign substance invades again, ribonucleoproteins (RNPs) scan the genetic material. Once the spacer sequence on the crRNA is matched with the original spacer sequence on the foreign invading DNA, Cas is activated and interferes with the foreign aid DNA, causing it to cleave [15]. The difference in the expression stage is that at the Cas9 loci, there is another sequence preceding Cas that can be transcribed into trans-activating CRISPR RNA (tracrRNA). crRNA is complementary to the tracrRNA sequences, and the two form a crRNA:tracrRNA double-stranded complex, which mediates site-specific DNA recognition and cleavage by Cas9. The crRNA:tracrRNA double-stranded complex was artificially redesigned into a single transcript, such as a single-guide RNA (sgRNA) or guide RNA chimera, and the important characteristics of the crRNA:tracrRNA double-stranded complex were incorporated into it. This could bind to Cas9 and recognize DNA targets [16].
The earliest discovered and the most widely used among the Cas proteins is the Cas9; thereafter, several Cas proteins of different sizes and with different targets and protospacer adjacent motif (PAM) preference sequences have been discovered [17]. The CRISPR-Cas system can be divided into two broad categories depending on whether it is a single- or multi-effector protein complex. The Class 1 system consists of multi-subunit protein-crRNA effector complexes, and it includes three Cas types, namely I, III, and IV [18,19]. On the contrary, the Class 2 system consists of single effector protein complexes, and it includes three Cas types, namely II, V, and VI [20,21]. The Class 2 systems are widely used in gene editing technology, especially type II Cas9, type V Cas12, and type VI Cas13. In addition to targeting DNA, Classes III, VI systems, as well as the modified RCas9 can specifically bind RNA, and Class V systems that can target both DNA and RNA [22].
The targeting specificity of the CRISPR-Cas gene editing system is determined by two conditions, namely the specific binding between the sgRNA and the genomic DNA sequences and the ability of the Cas proteins to specifically recognize specific short DNA sequences on the genomic DNA as PAM sequences (Fig. 1). Hence, by changing the sequence of the gRNA, we can target and edit almost all gene sequences, including DNA and RNA [23]. Concurrently, the development of various Cas derivatives has helped diversify the functions of the CRISPR/Cas system. Some examples are mentioned as follows. Gene editing: gene editing using the base editor, or prime editing for the insertion or deletion of short or long fragments, has been implemented [24–26]. Epigenetic modification: abnormal DNA methylation and its effect on gene expression can cause many complex human diseases, such as cancer [27]. The Cas protein is able to change its genetic expression without altering the DNA sequence by methylation with DNA [28]. At the transcriptional level, CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi) can be used to activate or inhibit RNA expression, respectively, from genes [29]. Therefore, CRISPR, with its high editing efficiency, high specificity, and simple design, is of groundbreaking and innovative significance in the field of gene editing.
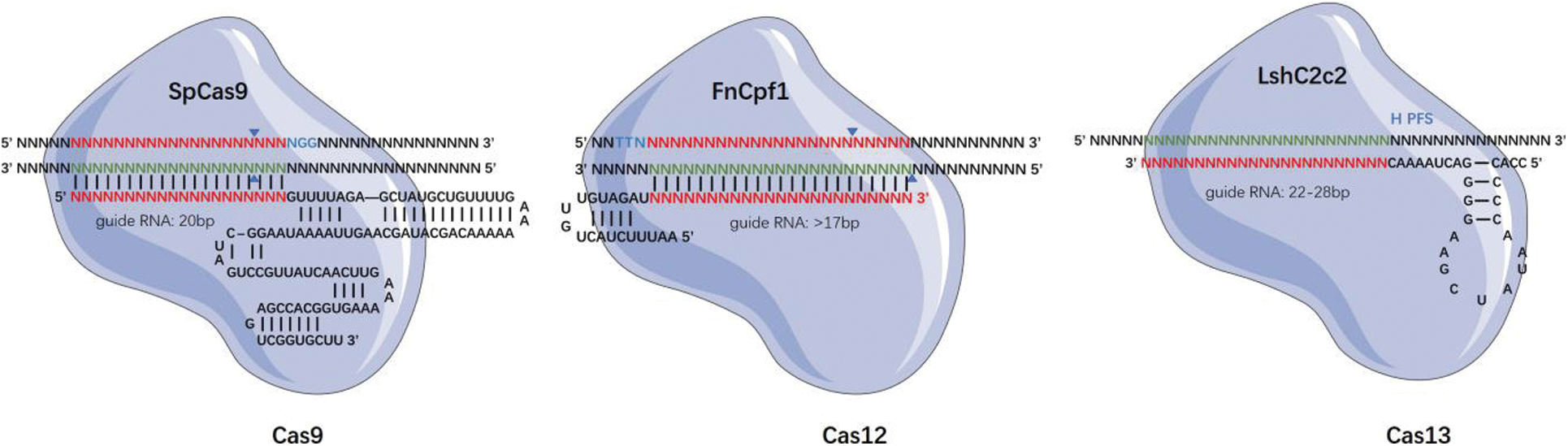
Figure 1: Structure of CRISPR Cas system
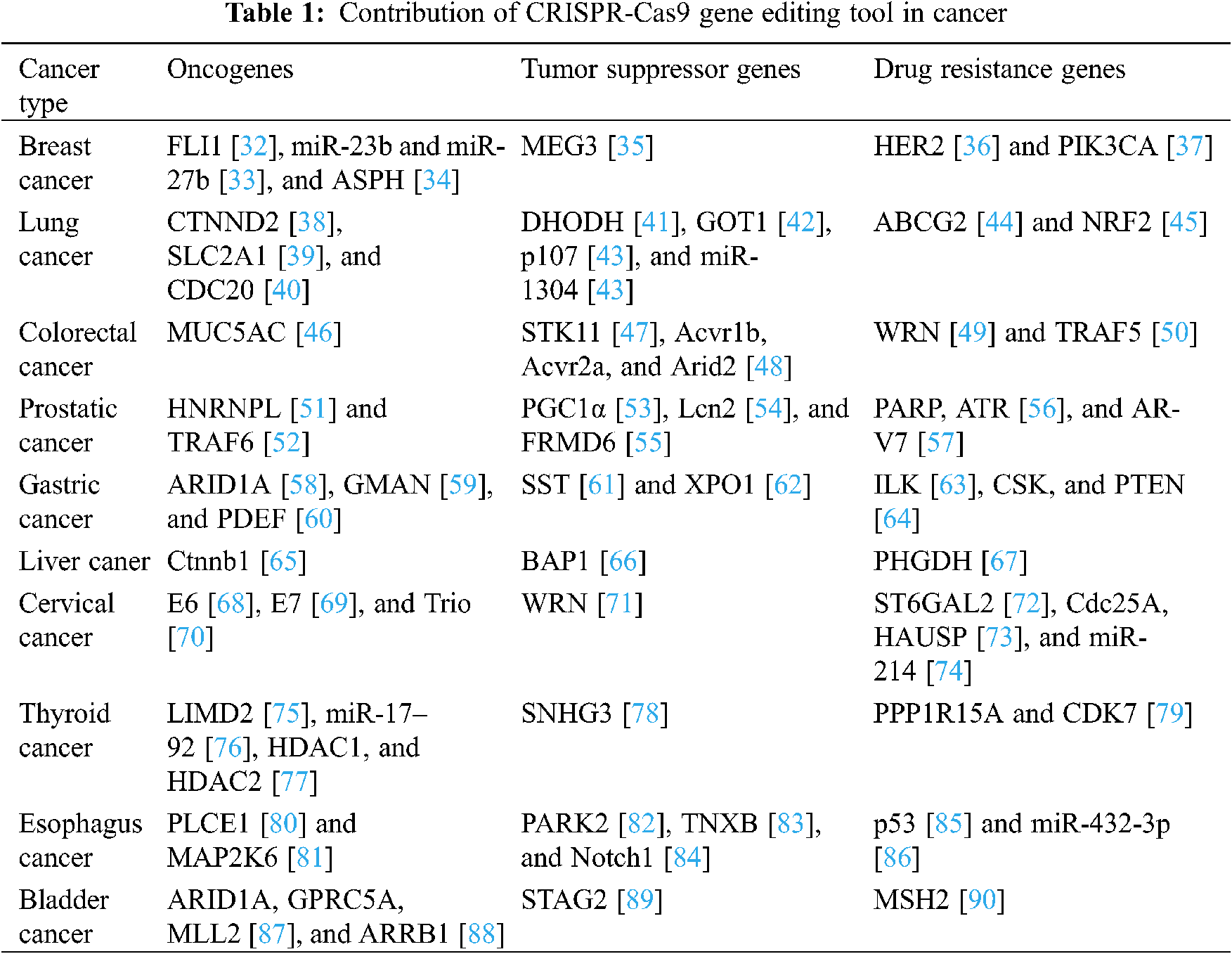
Cancer remains the leading cause of disease-related deaths despite rapid advances in diagnosis and treatment. In 2020, there were approximately 19.3 million new cancer cases worldwide, which is more than the 18.1 million cases detected in 2018 [30]. Therefore, it is particularly important to explore the underlying mechanisms of the occurrence, development, and spread of cancer and consequently develop appropriate treatments. The CRISPR-Cas system may be termed as a revolutionary “gene scissors”-based technology, which has brought a new dawn for cancer research and treatment [31]. In this review, we discuss the role of the CRISPR system in basic tumor research and its subsequent applications. The CRISPR-Cas9 system is highly effective in treating invasive cancers, and it is an important step in the search for a cure for cancer. The CRISPR-Cas9 system has been utilized by many researchers to study the characteristics of different cancers, to identify the mechanisms of drug resistance, to carry out research work related to cell death, functional genomics, signaling pathways, drug discovery, drug responses, cell therapy, and even in cancer treatment. The applications of the CRISPR-Cas9 gene editing technology in tumor research are summarized in Table 1.
With gene sequencing and whole-genome screening, we can identify the commonly mutated genes in cancer patients. Thereafter, based upon the knowledge of the mutated genes, the CRISPR-Cas technology can be used to simulate the tumor gene mutations via gene knockout, knockin, activation, or inhibition of the identified genes. This will help us to build relevant cellular or animal models rapidly and accurately for studying the genetic determinants as well as the underlying mechanisms of tumor development.
2.1 In Vitro Cellular Model Construction
Cellular models have a short cycle and a clean background, and they provide results easily. Neuroblastoma is a neuroextracranial tumor that most commonly affects children, but it has a poor prognosis. Certain studies indicate that the poor prognosis of neuroblastoma may be associated with mutations in ATRX, but it is difficult to confirm due to the lack of available models and preclinical studies. To assess the effect of the ATRX loss of function in neuroblastoma, George et al. [91] used the CRISPR-Cas9 technology to generate neuroblastoma cell lines with an ATRX mutation via homologous gene-targeting, and they used these cellular models to design innovative approaches that may be used for neuroblastoma treatment.
Organoids are essentially in vitro tissue constructs that mimic the patient’s tumor; therefore, organoid culture is a powerful tool for studying the characteristics of a cancerous tumor in an in vitro culture medium. Organoids reproduce many basic properties of primitive human tissues, including three-dimensional structure, multilineage differentiation, signal nodes, histology, and high-fidelity pathology. Hence, they can be used to construct disease models, personalize tumor therapy, explore cell growth and development mechanisms, and detect efficacy and toxicity [92]. They can grow to about 1 mm in diameter, resemble the patient’s tumor in shape, and share several molecular and genetic characteristics. Additionally, they can undergo genetic changes over time; this phenomenon is known as clonal evolution, and it is a major contributor to tumor progression and drug resistance [93]. Therefore, it represents a new method for investigating cancer biology [94]. Brain tumors are one of the deadliest and most devastating cancers, but their studies have been hampered due to genetic heterogeneity and a lack of models. A new model of neoplastic cerebral organoid (neoCOR) was established in vitro. In this model, oncogenic mutations were introduced in the organoids by transposon and CRISPR-Cas9 mediated mutations, thus, reproducing the development of brain tumors. The establishment of this model provided a valuable supplement to the basic and preclinical models that are currently used in brain tumor biology research [95]. Furthermore, to explore the causes of breast cancer, Cas9 was used to knockout four breast cancer-related genes, namely the tumor protein 53 gene (P53), phosphatase and tensin homologue gene (PTEN), Retinoblastoma 1 (RB1), and neurofibromin-1 gene (NF1), in breast progenitor cells. These mutant breast organoids gained the ability of long-term culture, formed estrogen-receptor positive luminal tumors, and were sensitive to chemotherapeutic drugs [96]. Therefore, the development of these organoid models with tumor-like characteristics provides a novel approach for further research in tumor biology.
2.2 In Vivo Model Construction
Animal models can show tumor growth, thereby facilitating the convenient and rapid detection of potential tumor- and metastasis-related genes and providing assistance in the investigation of tumor immune escape, drug resistance, and other mechanisms. Based on these functions, animal models are now considered an important tool for preclinical drug testing. Since mutations and deletions in the tumor suppressor genes Pten and p53 have been found in several human cancers, a Cas9 sgRNA system specifically targeting PTEN and P53 was administered into the liver of mice, thereby leading to the development of mice with tumor-like characteristics [97].
Small cell lung cancer (SCLC) is characterized by rapid disease progression, high recurrence after treatment, poor prognosis, poor treatment effects, and many other problems. Several SCLC-related gene mutations have been identified by sequencing, but their functions remain unclear. Some researchers have used the CRISPR-Cas9 technology to selectively mutate the identified genes in mice and construct SCLC mouse models; these models helped to explore the gene functions, thereby leading to the discovery of the tumor suppressor gene P107 [43].
Through RNA sequencing of prostate tissues in prostate cancer (PC) patients and normal individuals, Haldrup et al. [55] found that FRMD6 (FERM domain-containing protein 6) was abnormally hypermethylated and significantly downregulated in PC; moreover, it was associated with postoperative recurrence of PC. By knocking out or overexpressing FRMD6 in cells, it was observed that FRMD6 could inhibit cell proliferation, and the same phenomenon was seen in 3D spheres. Upon subcutaneous injection of the constructed FRMD6 knockout cell line into mice, they exhibited a faster rate of tumor growth than that seen in the control groups. Therefore, by using these animal models, the signaling pathway of FRMD6 was determined. Moreover, to study the role of FRMD6 in a non-tumorigenic environment, an FRMD6/PTEN double knockout in situ tumor mouse model was constructed. The results showed that the deletion of FRMD6 might accelerate the formation of tumor and play a driving role in the early stage of PC development. Therefore, FRMD6 has been identified as a novel tumor suppressor gene and a biomarker candidate in PC prognosis.
The establishment of animal models that can stably maintain the characteristics of tumor cells is conducive to further investigations on the tumor microenvironment and the mechanism underlying gene mutation as well as the development or screening of targeted drugs on this basis.
3 Studying the Mechanism of Tumor Development
The CRISPR-Cas9 technology can be used to study tumor genesis mechanisms, particularly the role of single nucleotide mutations, chromosomal ectopic events, and other factors involved in tumor development; additionally, it can be used to screen the functional genes present in tumor cells. These genetic associations correspond to the changes in gene function that occur in tumorigenesis, thereby helping to study the disease mechanism [98]. Numerous studies have shown that in the case of benign tumors, angiogenesis is rare and vascular growth is slow; on the contrary, most malignant tumors exhibit dense angiogenesis and rapid vascular growth [99]. Therefore, angiogenesis plays an important role in tumor development and metastasis, and inhibiting this process will significantly prevent the development, diffusion, and metastasis of tumor tissues. Incidentally, antiangiogenic therapy (ATT) has been successfully used to inhibit abnormal angiogenesis in the clinical treatment of tumors. However, the cancerous tumors often portray resistance to ATT. The CRISPR-Cas9 technology can be used to study the mechanisms of tumor angiogenesis as well as the probable causes of drug resistance [100,101]. The ARID1A mutation is one of the most common molecular aberrations in human cancers, but its carcinogenic mechanism is not clear. The ARID1A knockout model was established in TP53−/− human gastric organs. These engineered ARID1A deficient organs reflected several clinicopathological features of ARID1A mutant gastric cancer. Together with regulatory network-based analysis and high-throughput drug screening, researchers used this human organoid model to discover the potential mechanism of ARID1A in gastric epithelial carcinogenesis [58].
Another application of the CRISPR-Cas9 technology is the development of genome-scale CRISPR knockout (GeCKO) libraries, which are capable of rapidly producing loss-of-function mutants in human or mouse cells and subsequently screening for the desired phenotypes. These libraries can be used to identify the genes necessary for cell viability in cancer and pluripotent stem cells as well as to screen for missing genes related to drug resistance of the cancer cells [102].
Many CRISPR derivatives have also played an important role in cancer research. Upon fusion of the dead Cas9 (dCas9) protein with a transcriptional activator or suppressor protein domain and its subsequent association with gRNA sequences, it is possible to target the upstream regulatory regions of genes. This allows the transcription regulatory complex to effectively control the expression level of genes, and then explore their functions. The construction of a genome-wide targeted gRNA library and the upregulated expression of the whole genome by the cooperative activity of dCas9 and transcriptional activator proteins can be applied to high-throughput and rapid functional acquisition screening. The single-base editor, namely the cytosine base editor (CBE) is used to generate nucleotide mutations of multiple DNA damage response genes, which leads to adaptive changes in the cells when DNA damage occurs to screen and identify the DNA damage responses [103].
4 CRISPR in Cancer Diagnostics
Early diagnosis of cancer can facilitate early detection and treatment of patients, thereby reducing the physical and economic burden of patients. The existing methods of cancer diagnosis have certain limitations with respect to sensitivity, specificity, cost, and time. The ability to rapidly detect nucleic acids in a highly sensitive and single-base-specific manner using a portable platform may help improve disease diagnosis and surveillance, epidemiology, and general laboratory tasks. The Cas13a protein can remain active after cleaving the target RNA and continue to cleave other non-target RNAs. Gootenberg et al. developed a CRISPR-based diagnostic system named SHERLOCK (Specific High-Sensitivity Enzymatic Reporter Unlocking). The system combines Cas13 with recombinant polymerase amplification (RPA), which transcribes amplified DNA into RNA using T7 polymerase, which is then detected by Cas13a. Two human tumor gene hotspot mutations (EGFR L858R and BRAF V600E) were selected. SHERLOCK could distinguish between the normal genotype and the hotspot mutation, and the two human tumor gene mutations could be detected with 0.1% sensitivity [104]. The assay involved the addition of all reagents, including CRISPR-Cas12a and its guide RNA, fluorescent reporter molecules, and the RPA system, in a single reaction. When heated to body temperature, the copy number of the target DNA was rapidly increased via RPA, which made it easier for Cas12a to detect and cleave the target DNA, followed by the arbitrary cleavage of a nearby single-stranded DNA, which induced fluorescence in the reporter molecule. The technique can be used for the instantaneous and easy detection of small quantities of DNA in clinical samples. DETECTR is a technology based on DNA testing that can potentially be used for any type of rapid diagnosis, including the diagnosis of cancer and infectious diseases [105].
Cancer is a genomic disease characterized by an overall genomic instability, a large accumulation of point mutations, and structural changes during tumor progression. These genomic mutations may produce tumor antigens that may be recognized by the immune system as non-autoantigens, thereby triggering cellular immune responses. Therefore, the immune system plays a crucial role in immune surveillance, as immune cells from the adaptive and innate immune systems infiltrate the tumor microenvironment (TME) and help to regulate the tumor progression [106]. Immune cell therapy has shown excellent efficacy in the treatment of cancer, and a variety of immune cell therapies have been successfully tested in clinical trials.
As an emerging gene editing technology, the CRISPR-Cas9 system has been widely used in the field of tumor therapy for targeted knockout of tumor immune checkpoint molecules or for rapid and simple gene editing, which significantly reduce the operational difficulty of tumor immunotherapy and greatly promote the development of research in this field.
5.1 Adoptive Cell Transfer Therapy (ACT)
The ACT is used to isolate immunoactive cells from tumor patients, amplify them, identify their functions in vitro, and deliver them back to the patients to directly kill the tumors or to stimulate immune responses that will ultimately kill the tumor cells [107]. The most widely used ACT techniques include the chimeric antigen receptor (CAR) T cell therapy and T cell receptor (TCR) T cell therapy that have been engineered using the CRISPR-Cas9 technology [108,109]. This technique is the most mature and widely used immune cell therapy that has shown good targeting, tumor destruction, and persistence in both in vivo and in vitro situations [110].
The CAR-T cell therapy works by recognizing membrane surface antigens, and it is highly effective against hematological tumors [111]. The combination of radiotherapy and CAR-T cell therapy may enhance the therapeutic effects [108]. A number of clinical trials have been conducted or are underway to address the safety, feasibility, and efficacy of CAR-T cell therapy in patients with hematological malignancies or solid tumors. CAR-T cell therapy has been shown to be effective in hematoma owing to the fact that hematoma tumor cells have an ancestral target, CD19 (found only in tumor cells but not in normal cells), which can be used to guide CAR-T cells to detect and destroy cancer cells in the treatment of hematological malignancies. CAR-T cell therapy is an important breakthrough in the treatment of leukemia and lymphoma, but unlike other cancer therapies, its success has been accompanied by drastic side effects, including cytokine release syndrome (CRS) and CAR-T-cell-related encephalopathy syndrome (CRES) [112]. A Phase II trial performed with 74 participants with recurrent or refractory Mantle cell Lymphoma showed that KTE-X19 CAR-T cell therapy induced lasting remission, but the intervention also induced serious and life-threatening toxic effects, which was consistent with findings from previous reports [113]. Anti-CD19 CAR-T cell therapy can lead to severe CRS and neurotoxicity. Ying et al. generated a new type of CD19 BBz (86) CAR-T cells with costimulation, which showed lower cytokine levels, higher anti-apoptotic molecule production, and slower proliferation than other cell types previously studied. In clinical trials, CD19 BBz (86) CAR-T cell therapy produced an effective and lasting antilymphoma response without causing neurotoxicity or severe CRS [114]. To reduce the failure rate and recurrence rate after CAR-T cell treatment, Tong et al. [115] designed a series of tandem CARs (TanCARs). It was found that TanCAR-T cells possess double antigens targeting CD19 and CD20. In clinical trials, the safety and tolerance of TanCAR-T cells were evaluated. The results showed that TanCAR-T cells mediated a long-lasting and effective antitumor response without causing CRES of Grade 3 or a higher grade.
A clinical study evaluated the safety and efficacy of CD30 CAR-T cell therapy in patients with recurrent/refractory Hodgkin’s lymphoma. The results showed that the effects of autologous CD30 CAR-T cell therapy were long-lasting, and the therapy achieved complete remission and showed good safety. They suggested that CD30 CAR-T cells may be a safe and effective choice for patients with Hodgkin’s lymphoma and other lymphomas expressing CD30. At present, autologous CAR-T therapy has achieved clinical and commercial success. The development of more targets is an important strategy to benefit more patients and expand the applicability of the therapy. In addition, general CAR-T therapy is an important direction of CAR-T in the future. We expect these leading technologies to be transformed into products of significant utility and provide patients with a wider range of treatment options [116].
Certain results have also been reported in the treatment of solid tumors, such as gastrointestinal tumors [117]. CD133 is a marker of cancer stem cells (CSCs). CSCs are cancer cells with stem cell-like properties, such as self-replication and multicellular differentiation. These cells are considered to have the potential to form tumors and develop cancer. They are important cellular sources in the process of cancer metastasis. Singh et al. designed three immunotherapy methods based on human anti-CD133 antibody fragments and studied their targeting effect on CD133+ cells in a glioblastoma (GBM) humanized mouse model. CD133 CAR-T cells showed highest effectiveness in the GBM mouse model, without exerting any adverse effects on normal CD133+ hematopoietic stem cells, and did not induce acute systemic toxicity in a GBM humanized mouse model in situ [118]. CD133 CAR-T cell therapy also showed antitumor activity and safety in the clinical treatment of liver cancer [119].
Likewise, TCR-T therapy has made significant breakthroughs in clinical trials of melanoma and synovial sarcoma, and it has achieved preliminary efficacy in treating other advanced solid tumors [120]. Compared to the 2D-cultured cells, the 3D-cultured cells are more similar to the in vivo physiological conditions, especially the cell-cell interactions, cell proliferation, anaerobic conditions, and gene expression. Studies have shown that the 3D cell culture technology is a potential model for the pre-drug testing or drug screening of TCR-T and CAR-T based immunotherapy [121]. The researchers used CRISPR to knock out three genes in T cells, namely the T cell receptor α (TCRα) and T cell receptor β (TCRβ), two genes that encode the endogenous TCR chain, and PDCD1, a gene that encodes the cell death protein 1 (PD-1). Moreover, one cancer-specific TCR transgene, NY-ESO-1, was inserted. Therefore, the modified T cells were named NYCE (NY-ESO-1 transduced CRISPR 3X edited cells). Eventually, after four rounds of editing, the T cells were injected back into the patient. The results showed that the engineered T cells had the ability to kill tumor cells in vivo, and there were no obvious immune rejections or related toxicity [122].
5.2 Antibody Targeting Therapy
Immune checkpoint molecules are the key factors that regulate the balance between the activation and suppression of immune responses. Cancer cells typically evade immune surveillance by upregulating the immune checkpoint molecules; hence, blocking this process can significantly enhance anti-tumor immune responses. The PD-1 and the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) are well-known immune checkpoints that can significantly inhibit the activity of T cells, thus making it easy for the tumor cells to escape from the body’s immune mechanism; this leads to the poor results of T cell therapy [123]. At present, to improve the therapeutic effects of T cells, it is common to use immune checkpoint inhibitors, which are antibodies generated against the common immune check-points, and which form an effective method to overcome the inhibitions of the T cell immune response. Anti-tumor immune responses can also be achieved by using the CRISPR-Cas9 gene editing technology to knock out the genes that are involved in negative immune regulation. For instance, knockout of immune checkpoints using targeted gene editing techniques has been a promising strategy for improving the outcome of the CAR-T therapy in solid tumors [123]. Moreover, in the first human phase I clinical trial of CRISPR-Cas9 gene-edited T cell therapy in patients with advanced non-small cell lung cancer, Lu et al. [124] demonstrated that such a targeted immunotherapy is clinically feasible. Likewise, Si et al. [125] conducted a comprehensive study on the role of hematopoietic progenitor kinase1 (HPK1) and its related mechanisms in the regulation of T cell dysfunction. They found that gene depletion, pharmacological inhibition, or proteolysis targeting chimeric mediated degradation of HPK1 enhanced the efficacy of CAR-T cell-based immunotherapy in a variety of hematological and solid tumor mouse models. HPK1 is an attractive drug target for improving immunotherapy. The CD-19 HPK1KO CAR-T cells prepared by the CRISPR-Cas9 technology have been used in clinical trials, which are a part of the second clinical exploration in the world to improve T cell function through gene editing [125].
Unlike tumor immunotherapy, which targets immune checkpoints, tumor vaccines utilize antigen-antibody responses [126]. Cancer vaccines are of two main types: prophylactic and therapeutic. Prophylactic vaccines prevent cancer by preventing certain viral or bacterial infections or killing the corresponding pathogens, whereas therapeutic vaccines restrict tumor cell proliferation or kill tumor cells by activating specific immune functions in the body through cancer cell antigens. Tumor antigens that are generated after cell canceration can be used as the target for the adaptive immune response; this is the basis for preparing tumor vaccines. Additionally, sequencing mutated genes and obtaining new antigens help to prepare personalized tumor vaccines. These personalized cancer vaccines are an emerging treatment option that uses a patient’s own cancer cells to develop vaccines that are designed to train the immune system to recognize and destroy the cancer cells [127,128]. NeoVax, a personalized cancer vaccine, has shown excellent results in clinical trials with patients with melanoma and produced long-lasting immune responses [129]. Along with immune checkpoint inhibitors or other cancer therapies, personalized cancer vaccines may show a high clinical efficacy [130]. Ravindranathan et al. [131] used the CRISPR-Cas9 technology to screen cytokines affecting the immunogenicity of breast cancer cells and enhance the effectiveness of autologous tumor vaccines. Guo et al. [132] found that STEAP1 was expressed at high levels in both human and mouse prostate cancer cells, and constructed a fusion protein vaccine that could inhibit tumor growth and improve the tumor microenvironment in vivo. When the antigenic target was knocked out using the CRISPR-Cas9 system, the immune response against prostate cancer was considerably suppressed. There is a broad scope for further explorations in tumor vaccine development.
5.4 Cell Therapy Based on Pluripotent Stem Cells
Human pluripotent stem cells (hPSCs), such as embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), have the potential for self-renewal and differentiation, thereby making them the most advantageous sources of cells for cell therapy [133]. One of the most significant obstacles in the clinical application of gene-edited hPSCs is the risk of cancer development [134,135]. Two drugs have been developed to inhibit the normal functions of hPSCs. The first drug depletes undifferentiated hPSCs, and the second drug kills cells derived from hPSCs [136]. The two main categories of hPSCs that are currently being used in clinical research are ESC and iPSC, and they have also been used in several clinical trials. It is expected that these hPSCs will provide scientists with an unprecedented opportunity to develop cell therapies for refractory diseases and injuries [137]. Methods for iPSC-based immunotherapy, which involves iPSC-derived MSCs and iPSC-derived NK cells, have been developed by organizations in the private sector for the treatment of hematological malignancies [138]. In patients with acute myeloid leukemia, the CD33 protein is expressed at high levels on the surface of tumor cells; however, treatments targeting CD33 may damage healthy cells. CD33 knockout in hPSCs, which induces hematopoietic resistance to CD33-targeted therapy and specific targeting of AML using CAR-T cells, can effectively eliminate leukemia without causing myelotoxicity [139].
5.5 Identification of Potential Specific Targets
The CRISPR technology can screen for targets that can improve the immune cell functioning, thereby improving immunotherapy [9,140]. Ye et al. [141] embedded an sgRNA and the “Sleeping Beauty” transposon in between the inverted end repeat sequences (ITRs/IRs) in the genome of adeno-associated virus (AAV). Transfection into mouse T cells and an effective gene editing screening can improve immunotherapy targets in glioblastoma mouse models. Therefore, by using the CRISPR screening in vivo, an epigenetic transcription network was established to screen new targets for enhanced immunotherapy [142].
At present, most of the anticancer drugs require repeated administration; this not only increases the treatment cost and the drug toxicity, but also reduces the patient’s quality of life to a great extent. However, with the progress of technology, it has been demonstrated that the CRISPR-Cas9 gene editing system has the potential to destroy the tumor survival genes permanently; this can overcome the difficulties of traditional cancer treatment procedures, improve the efficacy of the treatment, and unlock new approaches for cancer treatment [143].
Fusion genes are present in all major types of cancer cells. It can inhibit tumor formation by inducing death of cancer cells containing fusion genes. In fact, the CRISPR-Cas9 gene editing system has successfully eliminated tumor cells in cell lines and mouse models of Ewing sarcoma and chronic myeloid leukemia by inducing cell death in the cancer cells containing fusion genes [144].
The use of the CRISPR technology to edit genes for the treatment of diseases caused by gene dysfunction is quite efficient. Theoretically, the CRISPR-Cas9 gene editing technology can permanently disrupt the tumor survival genes, thereby overcoming the repeated dose limits of traditional cancer therapies and improving the effectiveness of treatment. This is a promising option for treating the root cause of several human diseases. Undoubtedly, the CRISPR technology plays an important role in the treatment of numerous diseases, including genetic diseases [145].
At present, the CRISPR technology for gene therapy is mostly based on generating a DNA double-strand break (DSB) and inducing the cell DNA repair pathway, thereby resulting in a change in the target site sequence. Most diseases, including cancer, are caused by point mutations. Therefore, it is necessary to develop tools that can target specific point mutation sites. At present, the existing single base editing technologies include the CBE, which can convert C:G to T:A and the adenine base editor (ABE) that can convert A:T to G:C [146–149]. The newly discovered prime editor can carry out accurate gene editing, including long fragment replacement, insertion, and deletion, by editing the sgRNA and introducing a template at a DSB [150,151]. It is very important to explore the potential of guided editing to realize this application, including the research and treatment of genetic diseases [152].
In general, gene therapy is still at the pre-clinical exploration stage, and the criteria for estimating its safety, efficacy, and ability to exert long-lasting effects should be further refined. Cell and gene therapy may be truly life-changing and/or yield life-saving outcomes. Current regulatory models that require tests to be conducted on a large number of patients for determining the safety and efficacy of treatment methods are inappropriate for therapeutic techniques that target mutations found in a single patient or in a very small patient population. The development of a single composition that can be used to treat a larger patient population is a promising strategy. In the near future, the field of innovations in cell and gene therapy will face unique challenges, with the shift of traditional therapies to personalized therapies. This will be accompanied by an unprecedented level of control over nucleic acid delivery, immune system regulation, and precise manipulation of the human genome. Concurrently, insights on a new world of technological potential have inspired entirely new areas of research, such as synthetic biology, cell reprogramming, and high-throughput functional genomics, that will undoubtedly continue to reshape the face of biomedical research.
7 CRISPR Screening for Novel Drug Targets
The CRISPR technology can also accelerate the discovery of genes that may be targeted for new cancer treatments. Genome-wide screening in cancer models allows a large-scale screening of cancer-specific genes as well as the genes necessary for growth and development. By using the CRISPR-Cas9 screening in the cancer models, we can systematically study the carcinogenesis, development, and metabolic dependence of cancer cells to identify the potential therapeutic pathways [153,154]. It is possible to integrate the drug sensitivity of specific anticancer drugs with genome-wide CRISPR loss-of-function screening in cell lines to investigate the cellular pathways that support drug sensitivity; identification of these protein-protein networks facilitate drug development [155,156]. Multiple drug combinations have been discovered based upon the mechanism of drug resistance in tumor cells [3,56,157].
Over the years, there has been an increase in the number of mutated genes observed in cancer, but the functional genes, i.e., the genes necessary for the survival and proliferation of cancer cells, are largely unknown. To identify these cancer driver genes, lung cancer cells were cultured in both 2D monolayers and 3D lung cancer spheroids, followed by a genome-wide screening that revealed that some genes were necessary for cell growth in the 3D spheroids but not in the 2D levels [158]. Behan et al. [159] used the CRISPR-Cas9 technology to disrupt every gene in more than 300 cancer models developed from 30 types of cancer to identify the genes necessary for cancer cell survival. Additionally, a new system was developed to prioritize the 600 most promising drug targets for developing novel cancer treatments. These results can accelerate the development of targeted therapies and help scientists to complete the development of a Cancer Dependency Map, which is a detailed rulebook for enlisting the hundreds of cancer gene dependencies, and which can help to develop targeted cancer treatment for patients [159].
The RNA N6-methyladenosine (m6A) modification can regulate gene expression and play an important role in many life processes. Previously, a study has shown that m6A modification plays a vital role in the occurrence and development of human tumors by regulating RNA stability, microRNA (miRNA) processing, mRNA cutting, and translation [160]. Using CRISPR-Cas9 knockout screening, methyltransferase 3 (METTL3) was identified as an important m6A regulatory enzyme in colorectal cancer (CRC). Silencing METTL3 inhibited the occurrence of CRC tumors at the cellular, organoid and mouse levels. The signal pathway of mettl3 promoting CRC was determined by m6A sequencing, RNA sequencing, ribosome sequencing and functional verification. The efficacy of targeting the METTL3 in the treatment of CRC was established in CRC cell lines, patient-derived CRC-like organs, and METTL3 knockout mouse models [161]. Ever since it was discovered that RNA-guided RNA targeting the CRISPR/Cas effector Cas13 (previously known as C2c2) can be used for RNA knockout in mammalian cells, the use of this technique in cancer research has increased considerably [162]. Long non-coding RNAs (lncRNAs) are closely associated with the occurrence of cancer. They can regulate DNA methylation, histone modification, chromatin remodeling, and act as precursors of miRNAs through a variety of pathways [163]. Incidentally, the lncRNA gastric cancer associated transcript 3 (GACAT3) was significantly expressed in bladder cancer tissues. After knockout of GACAT3 by CRISPR-Cas13 technology, it was found that cell proliferation ability was inhibited and enhanced the apoptosis of the cancer cells. Therefore, GACAT3 is a potential target for bladder cancer diagnosis and therapy [164].
Metastasis is one of the major factors that makes cancer treatment difficult, and it is associated with the poor prognosis of cancer. In a recent study, the genes encoding cell surface proteins were screened by Cas9-based CRISPRa to identify the cell surface molecules that enhance metastatic abilities of cancer cells. The scientists found that overexpression of the gene LRRN4CL increased the ability of mouse as well as human melanoma cells to metastasize to the lungs. They also tested the cellular models of colon cancer, breast cancer, and bladder cancer, and they confirmed that LRRN4CL was transferred to all lungs in all these cells. This is the first time that LRRN4CL has been associated with cancer. Moreover, the results show that reducing the LRRN4CL expression can help to prevent metastasis to the lungs, thereby making it a potential drug target [165].
To screen the genes and signaling pathways that enable cancer cells to escape destruction by the host’s immune system, a group of genetically diverse mouse cancer cell lines that were cultured in the presence of cytotoxic T lymphocytes (CTLs) were screened using genome-wide CRISPR technology. A total of 182 genes were identified in this experiment, the individual interference of which increased either the sensitivity or the resistance of the cancer cells to CTL-mediated toxicity. Further exploration revealed the coordinated manner in which the identified genes and signaling pathways function in cancer cells to evade the action of CTLs [166].
To study the characteristics of ataxia telangiectasia mutated (ATM)-deficient fatal PC and discuss its comprehensive lethal treatment strategy, the ATM function was impaired in modified in vitro cell line models by using CRISPR-Cas9 technology. These models were used to test drug sensitivity and function analysis, followed by validation in patient-derived models. The results showed that the combined inhibition of polyadenosine diphosphate-ribose polymerase (PARP) and ATR (ATM and Rad3-related) had good antitumor activity in these models [56].
8 Delivery of CRISPR-Cas in vivo
Although the CRISPR gene-editing technology is already being used in clinical trials, the efficiency of its safe delivery remains a major challenge. Commonly used delivery methods include physical methods, viral vectors, and non-viral vectors. Currently, therapeutic technologies based on the CRISPR-Cas9 system are mainly realized through targeted delivery [7]. The various ways in which the CRISPR system can be delivered is discussed in the following section (Fig. 2).
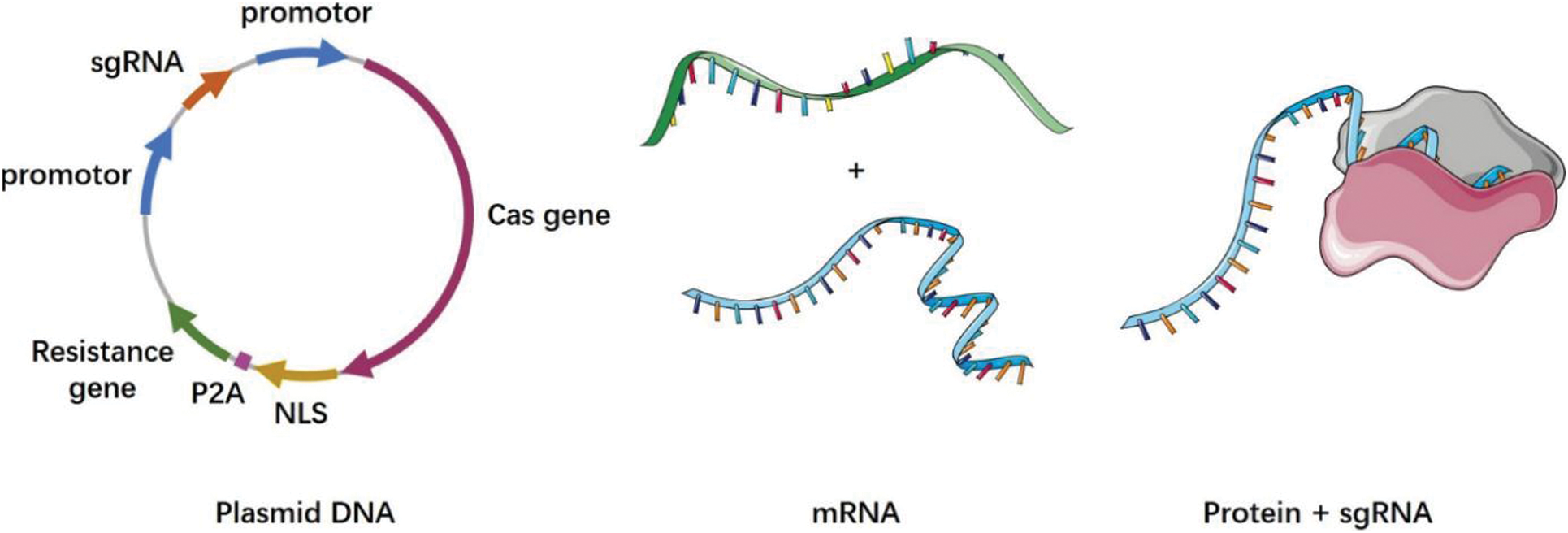
Figure 2: CRISPR cas system delivery forms
Plasmid DNA (pDNA): a plasmid containing the Cas9 and sgRNA sequences; mRNA: Cas9 is transcribed into mRNA in vitro, and then delivered into cells via a vector with the sgRNA; protein+sgRNA: Cas9 is translated into the protein in vitro, and then delivered into cells via a vector with the sgRNA
The CRISPR-Cas9 system encoding the Cas9 protein and the specific sgRNA that is required by the CRISPR-Cas9 system are integrated into the same plasmid DNA vector. This technique has the advantages of a simple operation and low cost, and is widely used [167]. In one experiment, a β-catenin-targeting CRISPR-Cas9 plasmid was constructed, which was modified by aptamer and enriched significantly in tumor nuclei. This system can effectively eliminate β-catenin and inhibit the Wnt/β-catenin pathway, reversing the immunosuppressive effect of tumors [168]. However, the large amount of encoded pDNA increases the delivery difficulty, and the long-term expression of Cas9 in the target cells may lead to an increase in the off-target effects and immune responses [169,170].
The off-target effect of the CRISPR-Cas9 system can be significantly reduced by transcribing the Cas9 mRNA in vitro and directly delivering the Cas9 mRNA to cells. This ensures a controlled dosage of the mRNA introduction as well as a rapid initiation of the gene editing process [171]. Owing to the poor stability of mRNA, a special mechanism is required to protect it from extracellular nuclease degradation during delivery [172–174]. In a clinical trial, Gillmore et al. [175] delivered sgRNA targeting the TTR gene and optimized the mRNA sequences of the spCas9 protein to the liver via a lipid nanoparticle (LNP) delivery vector for the treatment of transthyroxine protein amyloidosis. Phase I clinical trials showed good efficacy and safety of the treatment method.
8.1.3 Cas Protein and Guide RNA
To minimize the off-target effects of the CRISPR-Cas9 system, the most efficient approach for its delivery is to directly introduce the Cas9 protein and its specific sgRNA instead of the intracellular expression of pDNA or mRNA for Cas9 protein and sgRNA [176,177]. RNP-based delivery methods can strike a good balance between reducing cytotoxicity and maintaining genome rearrangement efficiency [178]. Two essential conditions for the direct delivery of the Cas9 protein and efficient genome editing are vector assistance for successful intracellular delivery of Cas9 protein and efficient lysosome escape. A nanodelivery system loaded with Cas9 can assist the Cas9 protein to enter the target cell, where it can be rapidly degraded by endogenous proteases immediately after cutting the target to achieve higher gene editing efficiency and lower off-target effects. There are three main methods of protein delivery, namely direct conjugation of proteins and carriers, physical adsorption of proteins and carriers based on charge or hydrophilic interactions, and protein nanocapsules based on emulsion dispersion [179]. Currently, majority of the Cas9 protein delivery systems are in the form of RNP [180].
Direct in vivo application of CRISPR-Cas9 system requires delivery systems that can target the CRISPR-Cas components to specific tissues or cells in the human body without triggering immune activation or causing high frequencies of off-target effects. Currently, physical methods, lentivirus, adenovirus, AAV, and LNP are being used as delivery vectors for human gene therapy; however, some of these delivery vectors are associated with problems like random integration into the genome, poor efficiency, and triggering unnecessary immune responses. At present, the most commonly used physical methods for the CRISPR delivery include microinjection [181], electroporation [182], and hydrodynamic tail-vein injection (HTVI) [183]. These methods are not applicable in case of the in vivo clinical trials because of their high experimental conditions, operation techniques, and subjects. Retroviruses, adenoviruses, and AAV are three major groups of viruses that have been used extensively to provide genetic material for treatment purposes. However, building viral vectors is a laborious and costly process with certain problems, such as insertion size limitations as well as the risk of developing cancer or other immune responses in humans. For example, retroviruses can cause insertional mutations that lead to cancer [184], and high intravenous doses of AAV for gene therapy can also cause toxicity [185]. Compared to these viral vectors, the non-viral vectors have advantages of low immunogenicity and no endogenous viral recombination, thereby minimizing the chances of short- and long-term adverse reactions; moreover, they have lower payload constraints and are generally easier to synthesize and produce on a large scale [186]. Therefore, many studies have focused on non-viral vectors, including polymer-liposome nanomaterials and emerging exosomes. Nevertheless, the safe and effective delivery of the CRISPR-Cas9 system into organisms, followed by a successful gene editing function still faces several challenges.
The CRISPR-lipid nanoparticle, which is a new delivery system based on lipid nanoparticles, has been developed to specifically target cancer cells and destroy them through genetic manipulation [187]. The system carries a genetic messenger (mRNA) that encodes the Cas9 protein, which acts as the “molecular scissors” that cuts into the DNA of cancer cells, thereby disabling it and permanently preventing replication. This method does not involve any chemotherapy drugs, and hence, does not have any side effects; moreover, the cancer cells treated in this way will never be active again. To test the feasibility of using this technology to treat cancer, two of the deadliest cancers, glioblastoma and metastatic ovarian cancer, were studied. The overall survival was improved in mice treated with CRISPR-LNPs in both the cancer models. Therefore, this technology reveals many new possibilities for treating various types of cancer as well as rare genetic diseases and chronic viral diseases such as AIDS [187].
Chen et al. developed a nanocapsule (NC) that was synthesized from a shearable, covalently cross-linked polymer coating of glutathione (GSH) that can encapsulate a preassembled RNP complex of Cas9 and sgRNA. These NCs effectively performed a targeted gene editing in vitro, without any significant cytotoxicity. Additionally, after local administration, the NCs exhibited powerful gene-editing functions in the mouse retinal pigment epithelium (RPE) tissue and skeletal muscles. Hence, these customizable NCs provide a nanoplatform that effectively delivers CRISPR-RNP complexes for somatic cell gene editing in vitro as well as in vivo [188].
Recently, exosomes/extracellular vesicles (EVs) are being increasingly used as delivery vectors for small-molecule therapies, proteins, RNA, DNA, and potentially new CRISPR technologies because they form a natural means of intercellular communication that is biocompatible, immunoinert, and effective in reaching their targets [189]. The RNPs were integrated into the EVs by the specific interaction between an RNA adaptor and an aptamer-binding protein (ABP). The RNA adaptor was inserted into an sgRNA, and the ABP COM was fused at both ends of the exosome-rich tetra protein CD63. A three-component complex consisting of CD63-COM fusion protein, modified sgRNA, and Cas9 or ABE was formed as a result of the abovementioned interaction, and the Cas9 and ABE RNPs were enriched into the EVs. This system can deliver gene modifications to multiple sites with high efficiency and safety [190].
Qi et al. [191] developed a lactose-derived branched cationic biopolymer (LBP) with a large number of reducible disulfide bonds and hydroxyl groups as a potential delivery vector of the CRISPR-Cas9 system for efficient in vivo genome editing in the treatment of hepatocellular carcinoma (HCC) in situ. The LBP-mediated delivery of pCas9-survivin demonstrated effective gene editing in the in vitro conditions by inducing apoptosis and inhibiting the proliferation of HCC cells. Furthermore, the LBP/pCas9 complex significantly enhanced the antitumor effect of the drug by inducing survivin knockout. Hence, a safe method was developed to efficiently edit the genome of the CRISPR-Cas9 system in vitro and in vivo.
In a recent study, the researchers have explored the combinatorial synthesis of multi-tailed ionizable phospholipids (iPhos) that can deliver messenger RNA or mRNA/sgRNA in vivo for gene editing. The structure-activity relationship shows that the chemical structure of iPhos can control the efficacy and organ selectivity in the in vivo conditions. Furthermore, the iPhos lipids cooperate with various auxiliary lipids to form multi-component lipid nanoparticles (iPLNPs) for selective organ targeting. Zwitterions, ionizable cations, and permanent cationic auxiliary lipids can carry out tissue-specific mRNA transmission and CRISPR-Cas9 gene editing in the spleen, liver, and lungs after intravenous administration [192]. This discovery proves that the regulation of the chemical structure of the lipids as well as the nanoparticle components can achieve organ selectivity. This, in turn, provides an idea for the development of next-generation gene vectors, and it can promote the research for organ-specific disease treatment based on mRNA delivery and the CRISPR-Cas gene editing.
Recently, Feng Zhang’s team developed a new RNA delivery platform named selective endogenous encapsidation for cellular delivery (SEND). The paternally expressed 10 (PEG10) protein, which is a retrovirus-like protein engineered to bind to its mRNA and form a protective globule around it, is used to package and deliver RNA. Using SEND, the CRISPR-Cas9 system was successfully delivered to human and mouse cells in mRNA form, and specific genes were edited. Compared with other delivery vectors, the immune response was lower and the safety was greater [145].
Since the birth of the CRISPR technology, there have been two main obstacles hindering its clinical application, namely the off-target effects and the low delivery efficiency. Several variants of Cas proteins have been developed until now, among which the most widely used ones are dCas9 [193] and Cas9 nickase (nCas9) [194], which are inactivated at the restriction site, and xCas9 [195], spCas9-NG [196], SpG, and SpRY [197], which have a wide PAM sequence and high specificity. The initial concerns about the off-target activities have been overcome with the improvements in the CRISPR-Cas system and its delivery strategies [195,198–200]. At present, the most serious problem is the genome damage caused by DSBs and the toxicity to organisms.
The CRISPR system can activate the p53, thereby leading to DNA damage [134,135]. In a comprehensive systematic study of mouse and human cells, the researchers found that the CRISPR-Cas9 system caused a wide range of mutations and quite a few genetic rearrangements, such as DNA deletions and insertions in a number of cells; these changes can cause important genes to be turned on or off, thereby leading to pathogenic consequences [201]. Xu et al. found that the Cas9 protein as well as its various mutations could directly bind to KU86, which is a key factor in the DNA repair pathway, and interfere with the formation of the DNA-PK complex; this can inhibit DNA damage repair mediated by the non-homologous end joining (NHEJ) repair pathway and lead to DNA damage and genomic instability [202]. Hence, further research and specificity tests are needed before the CRISPR-Cas9 system can be used clinically.
9.2 Toxicity of CRISPR-Cas System
Previous studies have shown that the CRISPR-Cas9 gene editing can lead to the activation of p53, which, in turn, causes DNA damage; therefore, it is possible that the p53 in the successfully gene-edited cells is defective. This increases the risk of cancer, and it can potentially cause cancer in individuals who have been treated by these gene-edited cells [134,135]. It was subsequently discovered that CRISPR-Cas9 edited mice exhibited frequent and large base deletions in their fertilized eggs [203,204]. Wagner et al. found that almost all healthy human subjects possessed T cells that reacted against Cas proteins; this is possibly because of the pre-existing immune memory against Cas9 protein since Streptococcus infection is quite common in humans. Incidentally, the Cas molecules from other bacteria also produce a similar immune response, which may be due to the high degree of similarity between these enzymes. Hence, these immune cells may produce adverse immune responses during gene therapy, thus compromising the safety and effectiveness of the CRISPR technology [205]. These studies suggest that the body’s immune response can disrupt gene therapy and pose a health threat to the patients who are receiving it. If antibodies against Cas9 bind to the Cas9 in blood, then the Cas9 can be inactivated even before it performs the gene editing. Additionally, the Cas9-specific T cells may actively eliminate the Cas9-modified cells, resulting in treatment failure [206]. Recently, Leibowitz reported that DSBs introduced during CRISPR-Cas9 gene editing may lead to chromosome fragmentation, which is a highly destructive form of genome rearrangement that may lead to the emergence of oncogenic fusion proteins or dysregulation of specific gene expression [207]. However, we can avoid immune rejection by searching for a new Cas9 enzyme from a bacterium that has never infected humans or by artificially redesigning the Cas9 enzyme.
The discovery of a series of problems caused by the CRISPR-Cas system indicates that our understanding of the mechanisms of CRISPR-Cas9 gene editing technology is still insufficient, and we need to be cautious regarding the safety evaluation of this technology.
In the past ten years, the CRISPR technology has developed by leaps and bounds from the initial gene knockout to modular tools, and today, it plays a significant role in disease treatment and research. For example, it has applications in transcriptional regulation [29,208], epigenetic modification [28,209], chromatin imaging [210,211] and base editing tools [212]. The key challenge is to apply new technologies for clinical treatment. It is believed that the CRISPR technology will be more widely used in disease treatment in the future with the development of safe and effective gene editing tools as well as improved delivery systems. The development of DNA sequencing and its large-scale applications have led to the recognition of genetic variation between the patient population and the general population; moreover, it has expanded our understanding of how genetic variation relates to disease predisposition, disease development, and treatment response [156]. These advances have spurred the development of personalized or precision medicine. The CRISPR-Cas system is also suitable for large-scale screening, and it has been used in a variety of settings. It has shown good results in the construction of disease models, drug development, screening of drug targets, and preclinical research. The CRISPR-Cas9 gene editing system also functions as a self-regulating switch for the CRISPR-Cas9 gene array. Its role in turning on or turning down CRISPR-Cas9 activity could help scientists to develop new ways of genetically engineering cells for research purposes [213]. The development and application of smaller Cas proteins, which ease the delivery process, can be developed as a varied genetic engineering tool for gene regulation and therapy [214]. With the progress in research, the CRISPR-Cas system has been used in clinical applications, and it is believed that with the improvement of the safety measures and the development of personalized therapy, the large-scale use of this technology in disease treatment will soon be possible.
Author Contributions: MD-M is the main author of the review, completing the collection and analysis of relevant literature and writing the draft of the paper. YC-L and WR-H are the project leaders, supervising the writing and revision of the paper. The manuscript was reviewed and approved by all coauthors.
Ethics Approval and Informed Consent Statement: Ethics approval and informed consent statements are not covered in this article.
Availability of Data and Materials: All data are included in this published article.
Funding Statement: This work was supported by the National Key R&D Program of China (2021YFA0911600 and 2019YFA0906000), the National Natural Science Foundation of China (81772737, 81772736, 81972867), the National Science Foundation Projects of Guangdong Province (2018B030306023), the Shenzhen Municipal Government of China (RCYX20200714114701035, JCYJ20180507184642475, JCYJ20200109120016553), and the Sanming Project of Shenzhen Health and Family Planning Commission, China (SZSM202011017).
Conflicts of Interest: No conflicts of interest exist for any co-author on this manuscript. Professor YC-L is a member of the editorial board of Oncologie. He did not participate in the peer review process.
1. Yin, H., Xue, W., Anderson, D. G. (2019). CRISPR-Cas: A tool for cancer research and therapeutics. Nature Reviews: Clinical Oncology, 16(5), 281–295. DOI 10.1038/s41571-019-0166-8. [Google Scholar] [CrossRef]
2. Ray, S. K., Mukherjee, S. (2020). Genome editing with CRISPR-Cas9: A budding biological contrivance for colorectal carcinoma research and its perspective in molecular medicine. Current Molecular Medicine, 21(6), 462–475. DOI 10.2174/1566524020666201119143943. [Google Scholar] [CrossRef]
3. Han, K., Jeng, E. E., Hess, G. T., Morgens, D. W., Li, A. et al. (2017). Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nature Biotechnology, 35(5), 463–474. DOI 10.1038/nbt.3834. [Google Scholar] [CrossRef]
4. Kong, X., Kuilman, T., Shahrabi, A., Boshuizen, J., Kemper, K. et al. (2017). Cancer drug addiction is relayed by an ERK2-dependent phenotype switch. Nature, 550(7675), 270–274. DOI 10.1038/nature24037. [Google Scholar] [CrossRef]
5. Kurata, M., Yamamoto, K., Moriarity, B. S., Kitagawa, M., Largaespada, D. A. (2018). CRISPR/Cas9 library screening for drug target discovery. Journal of Human Genetics, 63(2), 179–186. DOI 10.1038/s10038-017-0376-9. [Google Scholar] [CrossRef]
6. Liu, D., Zhao, X., Tang, A., Xu, X., Liu, S. et al. (2020). CRISPR screen in mechanism and target discovery for cancer immunotherapy. Biochimica et Biophysica Acta-Reviews on Cancer, 1874(1), 188378. DOI 10.1016/j.bbcan.2020.188378. [Google Scholar] [CrossRef]
7. Song, X., Liu, C., Wang, N., Huang, H., He, S. et al. (2021). Delivery of CRISPR/Cas systems for cancer gene therapy and immunotherapy. Advanced Drug Delivery Reviews, 168, 158–180. DOI 10.1016/j.addr.2020.04.010. [Google Scholar] [CrossRef]
8. Moreno, A. M., Fu, X., Zhu, J., Katrekar, D., Shih, Y. V. et al. (2018). In situ gene therapy via AAV-CRISPR-Cas9-mediated targeted gene regulation. Molecular Therapy, 26(7), 1818–1827. DOI 10.1016/j.ymthe.2018.04.017. [Google Scholar] [CrossRef]
9. Manguso, R. T., Pope, H. W., Zimmer, M. D., Brown, F. D., Yates, K. B. et al. (2017). In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature, 547(7664), 413–418. DOI 10.1038/nature23270. [Google Scholar] [CrossRef]
10. Dong, M. B., Wang, G., Chow, R. D., Ye, L., Zhu, L. et al. (2019). Systematic immunotherapy target discovery using genome-scale in vivo CRISPR screens in CD8 T cells. Cell, 178(5), 1189–1204.e23. DOI 10.1016/j.cell.2019.07.044. [Google Scholar] [CrossRef]
11. Dufva, O., Koski, J., Maliniemi, P., Ianevski, A., Klievink, J. et al. (2020). Integrated drug profiling and CRISPR screening identify essential pathways for CAR T-cell cytotoxicity. Blood, 135(9), 597–609. DOI 10.1182/blood.2019002121. [Google Scholar] [CrossRef]
12. Carroll, D. (2011). Genome engineering with zinc-finger nucleases. Genetics, 188(4), 773–782. DOI 10.1534/genetics.111.131433. [Google Scholar] [CrossRef]
13. Kim, H., Kim, J. S. (2014). A guide to genome engineering with programmable nucleases. Nature Reviews: Genetics, 15(5), 321–334. DOI 10.1038/nrg3686. [Google Scholar] [CrossRef]
14. Gasiunas, G., Siksnys, V. (2013). RNA-Dependent DNA endonuclease Cas9 of the CRISPR system: Holy grail of genome editing? Trends in Microbiology, 21(11), 562–567. DOI 10.1016/j.tim.2013.09.001. [Google Scholar] [CrossRef]
15. Amitai, G., Sorek, R. (2016). CRISPR-Cas adaptation: Insights into the mechanism of action. Nature Reviews: Microbiology, 14(2), 67–76. DOI 10.1038/nrmicro.2015.14. [Google Scholar] [CrossRef]
16. Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A. et al. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science, 337(6096), 816–821. DOI 10.1126/science.1225829. [Google Scholar] [CrossRef]
17. Porteus, M. H., Baltimore, D. (2003). Chimeric nucleases stimulate gene targeting in human cells. Science, 300(5620), 763. DOI 10.1126/science.1078395. [Google Scholar] [CrossRef]
18. Makarova, K. S., Zhang, F., Koonin, E. V. (2017). SnapShot: Class 1 CRISPR-Cas systems. Cell, 168(5), 946–946.e1. DOI 10.1016/j.cell.2017.02.018. [Google Scholar] [CrossRef]
19. Makarova, K. S., Wolf, Y. I., Alkhnbashi, O. S., Costa, F., Shah, S. A. et al. (2015). An updated evolutionary classification of CRISPR-Cas systems. Nature Reviews: Microbiology, 13(11), 722–736. DOI 10.1038/nrmicro3569. [Google Scholar] [CrossRef]
20. Shmakov, S., Smargon, A., Scott, D., Cox, D., Pyzocha, N. et al. (2017). Diversity and evolution of Class 2 CRISPR-Cas systems. Nature Reviews: Microbiology, 15(3), 169–182. DOI 10.1038/nrmicro.2016.184. [Google Scholar] [CrossRef]
21. Makarova, K. S., Zhang, F., Koonin, E. V. (2017). SnapShot: Class 2 CRISPR-Cas systems. Cell, 168(1–2), 328–328.e1. DOI 10.1016/j.cell.2016.12.038. [Google Scholar] [CrossRef]
22. Abudayyeh, O. O., Gootenberg, J. S., Konermann, S., Joung, J., Slaymaker, I. M. et al. (2016). C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science, 353(6299), aaf5573. DOI 10.1126/science.aaf5573. [Google Scholar] [CrossRef]
23. Yin, H., Kauffman, K. J., Anderson, D. G. (2017). Delivery technologies for genome editing. Nature Reviews: Drug Discovery, 16(6), 387–399. DOI 10.1038/nrd.2016.280. [Google Scholar] [CrossRef]
24. Nelson, J. W., Randolph, P. B., Shen, S. P., Everette, K. A., Chen, P. J. et al. (2021). Engineered pegRNAs improve prime editing efficiency. Nature Biotechnology. DOI 10.1038/s41587-021-01039-7. [Google Scholar] [CrossRef]
25. Anzalone, A. V., Koblan, L. W., Liu, D. R. (2020). Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nature Biotechnology, 38(7), 824–844. DOI 10.1038/s41587-020-0561-9. [Google Scholar] [CrossRef]
26. Chen, P. J., Hussmann, J. A., Yan, J., Knipping, F., Ravisankar, P. et al. (2021). Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell, 184(22), 5635–5652.e29. DOI 10.1016/j.cell.2021.09.018. [Google Scholar] [CrossRef]
27. Saghafinia, S., Mina, M., Riggi, N., Hanahan, D., Ciriello, G. (2018). Pan-cancer landscape of aberrant DNA methylation across human tumors. Cell Reports, 25(4), 1066–1080.e8. DOI 10.1016/j.celrep.2018.09.082. [Google Scholar] [CrossRef]
28. Liu, X. S., Wu, H., Ji, X., Stelzer, Y., Wu, X. et al. (2016). Editing DNA methylation in the mammalian genome. Cell, 167(1), 233–247.e17. DOI 10.1016/j.cell.2016.08.056. [Google Scholar] [CrossRef]
29. Gilbert, L. A., Horlbeck, M. A., Adamson, B., Villalta, J. E., Chen, Y. et al. (2014). Genome-scale CRISPR-mediated control of gene repression and activation. Cell, 159(3), 647–661. DOI 10.1016/j.cell.2014.09.029. [Google Scholar] [CrossRef]
30. Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I. et al. (2021). Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians, 71(3), 209–249. DOI 10.3322/caac.21660. [Google Scholar] [CrossRef]
31. Knott, G. J., Doudna, J. A. (2018). CRISPR-Cas guides the future of genetic engineering. Science, 361(6405), 866–869. DOI 10.1126/science.aat5011. [Google Scholar] [CrossRef]
32. Chen, N., Zhao, G., Yan, X., Lv, Z., Yin, H. et al. (2018). A novel FLI1 exonic circular RNA promotes metastasis in breast cancer by coordinately regulating TET1 and DNMT1. Genome Biology, 19(1), 218. DOI 10.1186/s13059-018-1594-y. [Google Scholar] [CrossRef]
33. Hannafon, B. N., Cai, A., Calloway, C. L., Xu, Y. F., Zhang, R. et al. (2019). miR-23b and miR-27b are oncogenic microRNAs in breast cancer: Evidence from a CRISPR/Cas9 deletion study. BMC Cancer, 19(1), 642. DOI 10.1186/s12885-019-5839-2. [Google Scholar] [CrossRef]
34. Lin, Q., Chen, X., Meng, F., Ogawa, K., Li, M. et al. (2019). ASPH-Notch axis guided exosomal delivery of prometastatic secretome renders breast cancer multi-organ metastasis. Molecular Cancer, 18(1), 156. DOI 10.1186/s12943-019-1077-0. [Google Scholar] [CrossRef]
35. Budkova, Z., Sigurdardottir, A. K., Briem, E., Bergthorsson, J. T., Sigurdsson, S. et al. (2020). Expression of ncRNAs on the DLK1-DIO3 locus is associated with basal and mesenchymal phenotype in breast epithelial progenitor cells. Frontiers in Cell and Developmental Biology, 8, 461. DOI 10.3389/fcell.2020.00461. [Google Scholar] [CrossRef]
36. Li, X., Xu, Y., Ding, Y., Li, C., Zhao, H. et al. (2018). Posttranscriptional upregulation of HER3 by HER2 mRNA induces trastuzumab resistance in breast cancer. Molecular Cancer, 17(1), 113. DOI 10.1186/s12943-018-0862-5. [Google Scholar] [CrossRef]
37. Cai, Y., Xu, G., Wu, F., Michelini, F., Chan, C. et al. (2021). Genomic alterations in PIK3CA-mutated breast cancer result in mTORC1 activation and limit the sensitivity to PI3Kα inhibitors. Cancer Research, 81(9), 2470–2480. DOI 10.1158/0008-5472.CAN-20-3232. [Google Scholar] [CrossRef]
38. Huang, F., Chen, J., Wang, Z., Lan, R., Fu, L. et al. (2018). δ-Catenin promotes tumorigenesis and metastasis of lung adenocarcinoma. Oncology Reports, 39(2), 809–817. DOI 10.3892/or.2017.6140. [Google Scholar] [CrossRef]
39. Zhou, J., Wang, D., Tang, D., Huang, W. (2020). Abnormal activations of super-enhancers enhance the carcinogenicity in lung adenocarcinoma. Cancer Management and Research, 12, 8509–8518. DOI 10.2147/CMAR.S258497. [Google Scholar] [CrossRef]
40. Yi, J., Wei, X., Li, X., Wan, L., Dong, J. et al. (2018). A Genome-wide comprehensive analysis of alterations in driver genes in non-small-cell lung cancer. Anti-Cancer Drugs, 29(1), 10–18. DOI 10.1097/CAD.0000000000000571. [Google Scholar] [CrossRef]
41. Li, L., Ng, S. R., Colón, C. I., Drapkin, B. J., Hsu, P. P. et al. (2019). Identification of DHODH as a therapeutic target in small cell lung cancer. Science Translational Medicine, 11(517), eaaw7852. DOI 10.1126/scitranslmed.aaw7852. [Google Scholar] [CrossRef]
42. Zhou, X., Curbo, S., Li, F., Krishnan, S., Karlsson, A. (2018). Inhibition of glutamate oxaloacetate transaminase 1 in cancer cell lines results in altered metabolism with increased dependency of glucose. BMC Cancer, 18(1), 559. DOI 10.1186/s12885-018-4443-1. [Google Scholar] [CrossRef]
43. Ng, S. R., Rideout, W. M., Akama-Garren, E. H., Bhutkar, A., Mercer, K. L. et al. (2020). CRISPR-Mediated modeling and functional validation of candidate tumor suppressor genes in small cell lung cancer. Proceedings of the National Academy of Sciences of the United States of America, 117(1), 513–521. DOI 10.1073/pnas.1821893117. [Google Scholar] [CrossRef]
44. Kovacsics, D., Brózik, A., Tihanyi, B., Matula, Z., Borsy, A. et al. (2020). Precision-engineered reporter cell lines reveal ABCG2 regulation in live lung cancer cells. Biochemical Pharmacology, 175, 113865. DOI 10.1016/j.bcp.2020.113865. [Google Scholar] [CrossRef]
45. Bialk, P., Wang, Y., Banas, K., Kmiec, E. B. (2018). Functional gene knockout of NRF2 increases chemosensitivity of human lung cancer a549 cells in vitro and in a xenograft mouse model. Molecular Therapy-Oncolytics, 11, 75–89. DOI 10.1016/j.omto.2018.10.002. [Google Scholar] [CrossRef]
46. Pothuraju, R., Rachagani, S., Krishn, S. R., Chaudhary, S., Nimmakayala, R. K. et al. (2020). Molecular implications of MUC5AC-CD44 axis in colorectal cancer progression and chemoresistance. Molecular Cancer, 19(1), 37. DOI 10.1186/s12943-020-01156-y. [Google Scholar] [CrossRef]
47. Michels, B. E., Mosa, M. H., Streibl, B. I., Zhan, T., Menche, C. et al. (2020). Pooled in vitro and in vivo CRISPR-Cas9 screening identifies tumor suppressors in human colon organoids. Cell Stem Cell, 26(5), 782–792.e787. DOI 10.1016/j.stem.2020.04.003. [Google Scholar] [CrossRef]
48. Takeda, H., Kataoka, S., Nakayama, M., Ali, M. A. E., Oshima, H. et al. (2019). CRISPR-Cas9-mediated gene knockout in intestinal tumor organoids provides functional validation for colorectal cancer driver genes. Proceedings of the National Academy of Sciences of the United States of America, 116(31), 15635–15644. DOI 10.1073/pnas.1904714116. [Google Scholar] [CrossRef]
49. Picco, G., Cattaneo, C. M., van Vliet, E. J., Crisafulli, G., Rospo, G. et al. (2021). Werner helicase is a synthetic-lethal vulnerability in mismatch repair-deficient colorectal cancer refractory to targeted therapies, chemotherapy, and immunotherapy. Cancer Discovery, 11(8), 1923–1937. DOI 10.1158/2159-8290.CD-20-1508. [Google Scholar] [CrossRef]
50. Lan, H., Liu, Y., Liu, J., Wang, X., Guan, Z. et al. (2021). Tumor-associated macrophages promote oxaliplatin resistance via METTL3-mediated m(6)A of TRAF5 and necroptosis in colorectal cancer. Molecular Pharmaceutics, 18(3), 1026–1037. DOI 10.1021/acs.molpharmaceut.0c00961. [Google Scholar] [CrossRef]
51. Fei, T., Chen, Y., Xiao, T., Li, W., Cato, L. et al. (2017). Genome-wide CRISPR screen identifies HNRNPL as a prostate cancer dependency regulating RNA splicing. Proceedings of the National Academy of Sciences of the United States of America, 114(26), E5207–E5215. DOI 10.1073/pnas.1617467114. [Google Scholar] [CrossRef]
52. Aripaka, K., Gudey, S. K., Zang, G., Schmidt, A., Åhrling, S. S. et al. (2019). TRAF6 function as a novel co-regulator of Wnt3a target genes in prostate cancer. EBioMedicine, 45, 192–207. DOI 10.1016/j.ebiom.2019.06.046. [Google Scholar] [CrossRef]
53. Valcarcel-Jimenez, L., Macchia, A., Crosas-Molist, E., Schaub-Clerigué, A., Camacho, L. et al. (2019). PGC1α suppresses prostate cancer cell invasion through ERRα transcriptional control. Cancer Research, 79(24), 6153–6165. DOI 10.1158/0008-5472.CAN-19-1231. [Google Scholar] [CrossRef]
54. Rahimi, S., Roushandeh, A. M., Ebrahimi, A., Samadani, A. A., Kuwahara, Y. et al. (2019). CRISPR/Cas9-mediated knockout of Lcn2 effectively enhanced CDDP-induced apoptosis and reduced cell migration capacity of PC3 cells. Life Sciences, 231, 116586. DOI 10.1016/j.lfs.2019.116586. [Google Scholar] [CrossRef]
55. Haldrup, J., Strand, S. H., Cieza-Borrella, C., Jakobsson, M. E., Riedel, M. et al. (2021). FRMD6 has tumor suppressor functions in prostate cancer. Oncogene, 40(4), 763–776. DOI 10.1038/s41388-020-01548-w. [Google Scholar] [CrossRef]
56. Neeb, A., Herranz, N., Arce-Gallego, S., Miranda, S., Buroni, L. et al. (2021). Advanced prostate cancer with ATM loss: PARP and ATR inhibitors. European Urology, 79(2), 200–211. DOI 10.1016/j.eururo.2020.10.029. [Google Scholar] [CrossRef]
57. Kawamura, N., Nimura, K., Saga, K., Ishibashi, A., Kitamura, K. et al. (2019). SF3B2-Mediated RNA splicing drives human prostate cancer progression. Cancer Research, 79(20), 5204–5217. DOI 10.1158/0008-5472.CAN-18-3965. [Google Scholar] [CrossRef]
58. Lo, Y. H., Kolahi, K. S., Du, Y., Chang, C. Y., Krokhotin, A. et al. (2021). A CRISPR/Cas9-engineered ARID1A-deficient human gastric cancer organoid model reveals essential and nonessential modes of oncogenic transformation. Cancer Discovery, 11(6), 1562–1581. DOI 10.1158/2159-8290.CD-20-1109. [Google Scholar] [CrossRef]
59. Zhuo, W., Liu, Y., Li, S., Guo, D., Sun, Q. et al. (2019). Long noncoding RNA GMAN, up-regulated in gastric cancer tissues, is associated with metastasis in patients and promotes translation of ephrin A1 by competitively binding GMAN-AS. Gastroenterology, 156(3), 676–691.e11. DOI 10.1053/j.gastro.2018.10.054. [Google Scholar] [CrossRef]
60. Zhang, Y. Q., Pei, J. H., Shi, S. S., Guo, X. S., Cui, G. Y. et al. (2019). CRISPR/Cas9-mediated knockout of the PDEF gene inhibits migration and invasion of human gastric cancer AGS cells. Biomedicine and Pharmacotherapy, 111, 76–85. DOI 10.1016/j.biopha.2018.12.048. [Google Scholar] [CrossRef]
61. Chen, W., Ding, R., Tang, J., Li, H., Chen, C. et al. (2020). Knocking out SST gene of BGC823 gastric cancer cell by CRISPR/Cas9 enhances migration, invasion and expression of SEMA5A and KLF2. Cancer Management and Research, 12, 1313–1321. DOI 10.2147/CMAR. [Google Scholar] [CrossRef]
62. Gruffaz, M., Yuan, H., Meng, W., Liu, H., Bae, S. et al. (2019). CRISPR-Cas9 screening of kaposi’s sarcoma-associated herpesvirus-transformed cells identifies XPO1 as a vulnerable target of cancer cells. mBio, 10(3), e00866–19. DOI 10.1128/mBio.00866-19. [Google Scholar] [CrossRef]
63. Chen, J., Bell, J., Lau, B. T., Whittaker, T., Stapleton, D. et al. (2019). A functional CRISPR/Cas9 screen identifies kinases that modulate FGFR inhibitor response in gastric cancer. Oncogenesis, 8(5), 33. DOI 10.1038/s41389-019-0145-z. [Google Scholar] [CrossRef]
64. Ning, G., Zhu, Q., Kang, W., Lee, H., Maher, L. et al. (2021). A novel treatment strategy for lapatinib resistance in a subset of HER2-amplified gastric cancer. BMC Cancer, 21(1), 923. DOI 10.1186/s12885-021-08283-9. [Google Scholar] [CrossRef]
65. To, J. C., Chiu, A. P., Tschida, B. R., Lo, L. H., Chiu, C. H. et al. (2021). ZBTB20 regulates WNT/CTNNB1 signalling pathway by suppressing PPARG during hepatocellular carcinoma tumourigenesis. JHEP Reports, 3(2), 100223. DOI 10.1016/j.jhepr.2020.100223. [Google Scholar] [CrossRef]
66. Artegiani, B., van Voorthuijsen, L., Lindeboom, R. G. H., Seinstra, D., Heo, I. et al. (2019). Probing the tumor suppressor function of BAP1 in CRISPR-engineered human liver organoids. Cell Stem Cell, 24(6), 927–943.e926. DOI 10.1016/j.stem.2019.04.017. [Google Scholar] [CrossRef]
67. Wei, L., Lee, D., Law, C. T., Zhang, M. S., Shen, J. et al. (2019). Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for sorafenib resistance in HCC. Nat Commun, 10(1), 4681. DOI 10.1038/s41467-019-12606-7. [Google Scholar] [CrossRef]
68. Yoshiba, T., Saga, Y., Urabe, M., Uchibori, R., Matsubara, S. et al. (2019). CRISPR/Cas9-mediated cervical cancer treatment targeting human papillomavirus E6. Oncology Letters, 17(2), 2197–2206. DOI 10.3892/ol.2018.9815. [Google Scholar] [CrossRef]
69. Gao, X., Jin, Z., Tan, X., Zhang, C., Zou, C. et al. (2020). Hyperbranched poly (β-amino ester) based polyplex nanopaticles for delivery of CRISPR/Cas9 system and treatment of HPV infection associated cervical cancer. Journal of Controlled Release, 321, 654–668. DOI 10.1016/j.jconrel.2020.02.045. [Google Scholar] [CrossRef]
70. Hou, C., Zhuang, Z., Deng, X., Xu, Y., Zhang, P. et al. (2018). Knockdown of trio by CRISPR/Cas9 suppresses migration and invasion of cervical cancer cells. Oncology Reports, 39(2), 795–801. DOI 10.3892/or.2017.6117. [Google Scholar] [CrossRef]
71. James, C. D., Das, D., Morgan, E. L., Otoa, R., Macdonald, A. et al. (2020). Werner syndrome protein (WRN) regulates cell proliferation and the human papillomavirus 16 life cycle during epithelial differentiation. mSphere, 5(5), e00858-20. DOI 10.1128/mSphere.00858-20. [Google Scholar] [CrossRef]
72. Tian, X., Wang, X., Cui, Z., Liu, J., Huang, X. et al. (2021). A Fifteen-gene classifier to predict neoadjuvant chemotherapy responses in patients with stage IB to IIB squamous cervical cancer. Advanced Science, 8(10), 2001978. DOI 10.1002/advs.202001978. [Google Scholar] [CrossRef]
73. Das, S., Chandrasekaran, A. P., Jo, K. S., Ko, N. R., Oh, S. J. et al. (2020). HAUSP stabilizes Cdc25A and protects cervical cancer cells from DNA damage response. Biochimica et Biophysica Acta–Molecular Cell Research, 1867(12), 118835. DOI 10.1016/j.bbamcr.2020.118835. [Google Scholar] [CrossRef]
74. Sen, P., Ghosal, S., Hazra, R., Mohanty, R., Arega, S. et al. (2020). CRISPR-Mediated knockdown of miR-214 modulates cell fate in response to anti-cancer drugs in HPV-negative and HPV-positive cervical cancer cells. Journal of Biosciences, 45, 80. DOI 10.1007/s12038-020-00054-1. [Google Scholar] [CrossRef]
75. Araldi, R. P., De Melo, T. C., Levy, D., De Souza, D. M., Maurício, B. et al. (2020). LIMD2 regulates Key steps of metastasis cascade in papillary thyroid cancer cells via MAPK crosstalk. Cells, 9(11), 2522. DOI 10.3390/cells9112522. [Google Scholar] [CrossRef]
76. Fuziwara, C. S., Saito, K. C., Kimura, E. T. (2020). Thyroid follicular cell loss of differentiation induced by MicroRNA miR-17–92 cluster is attenuated by CRISPR/Cas9n gene silencing in anaplastic thyroid cancer. Thyroid, 30(1), 81–94. DOI 10.1089/thy.2018.0601. [Google Scholar] [CrossRef]
77. Lin, C. L., Tsai, M. L., Lin, C. Y., Hsu, K. W., Hsieh, W. S. et al. (2019). HDAC1 and HDAC2 double knockout triggers cell apoptosis in advanced thyroid cancer. International Journal of Molecular Sciences, 20(2), 454. DOI 10.3390/ijms20020454. [Google Scholar] [CrossRef]
78. Duan, Y., Wang, Z., Xu, L., Sun, L., Song, H. et al. (2020). lncRNA SNHG3 acts as a novel tumor suppressor and regulates tumor proliferation and metastasis via AKT/mTOR/ERK pathway in papillary thyroid carcinoma. Journal of Cancer, 11(12), 3492–3501. DOI 10.7150/jca.42070. [Google Scholar] [CrossRef]
79. Cao, X., Dang, L., Zheng, X., Lu, Y., Lu, Y. et al. (2019). Targeting super-enhancer-driven oncogenic transcription by CDK7 inhibition in anaplastic thyroid carcinoma. Thyroid, 29(6), 809–823. DOI 10.1089/thy.2018.0550. [Google Scholar] [CrossRef]
80. Zhai, S., Liu, C., Zhang, L., Zhu, J., Guo, J. et al. (2017). PLCE1 promotes esophageal cancer cell progression by maintaining the transcriptional activity of snail. Neoplasia, 19(3), 154–164. DOI 10.1016/j.neo.2016.12.007. [Google Scholar] [CrossRef]
81. Lin, S., Liu, K., Zhang, Y., Jiang, M., Lu, R. et al. (2018). Pharmacological targeting of p38 MAP-kinase 6 (MAP2K6) inhibits the growth of esophageal adenocarcinoma. Cellular Signalling, 51, 222–232. DOI 10.1016/j.cellsig.2018.08.008. [Google Scholar] [CrossRef]
82. Zhou, X., Li, Y., Wang, W., Wang, S., Hou, J. et al. (2020). Regulation of Hippo/YAP signaling and esophageal squamous carcinoma progression by an E3 ubiquitin ligase PARK2. Theranostics, 10(21), 9443–9457. DOI 10.7150/thno.46078. [Google Scholar] [CrossRef]
83. Yang, N., Tian, J., Wang, X., Mei, S., Zou, D. et al. (2020). A functional variant in TNXB promoter associates with the risk of esophageal squamous-cell carcinoma. Molecular Carcinogenesis, 59(4), 439–446. DOI 10.1002/mc.23166. [Google Scholar] [CrossRef]
84. Sawangarun, W., Mandasari, M., Aida, J., Morita, K. I., Kayamori, K. et al. (2018). Loss of Notch1 predisposes oro-esophageal epithelium to tumorigenesis. Experimental Cell Research, 372(2), 129–140. DOI 10.1016/j.yexcr.2018.09.019. [Google Scholar] [CrossRef]
85. Liu, D. S., Read, M., Cullinane, C., Azar, W. J., Fennell, C. M. et al. (2015). APR-246 potently inhibits tumour growth and overcomes chemoresistance in preclinical models of oesophageal adenocarcinoma. Gut, 64(10), 1506–1516. DOI 10.1136/gutjnl-2015-309770. [Google Scholar] [CrossRef]
86. Akdemir, B., Nakajima, Y., Inazawa, J., Inoue, J. (2017). miR-432 induces NRF2 stabilization by directly targeting KEAP1. Molecular Cancer Research, 15(11), 1570–1578. DOI 10.1158/1541-7786.MCR-17-0232. [Google Scholar] [CrossRef]
87. Yang, Z., Li, C., Fan, Z., Liu, H., Zhang, X. et al. (2017). Single-cell sequencing reveals variants in ARID1A, GPRC5A and MLL2 driving self-renewal of human bladder cancer stem cells. European Urology, 71(1), 8–12. DOI 10.1016/j.eururo.2016.06.025. [Google Scholar] [CrossRef]
88. Kallifatidis, G., Smith, D. K., Morera, D. S., Gao, J., Hennig, M. J. et al. (2019). Β-Arrestins regulate stem cell-like phenotype and response to chemotherapy in bladder cancer. Molecular Cancer Therapeutics, 18(4), 801–811. DOI 10.1158/1535-7163.MCT-18-1167. [Google Scholar] [CrossRef]
89. Nie, Z., Gao, W., Zhang, Y., Hou, Y., Liu, J. et al. (2019). STAG2 Loss-of-function mutation induces PD-l1 expression in U2OS cells. Annals of Translational Medicine, 7(7), 127. DOI 10.21037/atm.2019.02.23. [Google Scholar] [CrossRef]
90. Goodspeed, A., Jean, A., Costello, J. C. (2019). A Whole-genome CRISPR screen identifies a role of MSH2 in cisplatin-mediated cell death in muscle-invasive bladder cancer. European Urology, 75(2), 242–250. DOI 10.1016/j.eururo.2018.10.040. [Google Scholar] [CrossRef]
91. George, S. L., Lorenzi, F., King, D., Hartlieb, S., Campbell, J. et al. (2020). Therapeutic vulnerabilities in the DNA damage response for the treatment of ATRX mutant neuroblastoma. EBioMedicine, 59, 102971. DOI 10.1016/j.ebiom.2020.102971. [Google Scholar] [CrossRef]
92. Foley, K. E. (2017). Organoids: A better in vitro model. Nature Methods, 14(6), 559–562. DOI 10.1038/nmeth.4307. [Google Scholar] [CrossRef]
93. Lee, S. H., Hu, W., Matulay, J. T., Silva, M. V., Owczarek, T. B. et al. (2018). Tumor evolution and drug response in patient-derived organoid models of bladder cancer. Cell, 173(2), 515–528.e17. DOI 10.1016/j.cell.2018.03.017. [Google Scholar] [CrossRef]
94. Lo, Y. H., Karlsson, K., Kuo, C. J. (2020). Applications of organoids for cancer biology and precision medicine. Nature Cancer, 1(8), 761–773. DOI 10.1038/s43018-020-0102-y. [Google Scholar] [CrossRef]
95. Bian, S., Repic, M., Guo, Z., Kavirayani, A., Burkard, T. et al. (2018). Genetically engineered cerebral organoids model brain tumor formation. Nature Methods, 15(8), 631–639. DOI 10.1038/s41592-018-0070-7. [Google Scholar] [CrossRef]
96. Dekkers, J. F., Whittle, J. R., Vaillant, F., Chen, H. R., Dawson, C. et al. (2020). Modeling breast cancer using CRISPR-Cas9-mediated engineering of human breast organoids. Journal of the National Cancer Institute, 112(5), 540–544. DOI 10.1093/jnci/djz196. [Google Scholar] [CrossRef]
97. Xue, W., Chen, S., Yin, H., Tammela, T., Papagiannakopoulos, T. et al. (2014). CRISPR-Mediated direct mutation of cancer genes in the mouse liver. Nature, 514(7522), 380–384. DOI 10.1038/nature13589. [Google Scholar] [CrossRef]
98. Wang, X., Hayes, J. E., Xu, X., Gao, X., Mehta, D. et al. (2021). Validation of prostate cancer risk variants rs10993994 and rs7098889 by CRISPR/Cas9 mediated genome editing. Gene, 768, 145265. DOI 10.1016/j.gene.2020.145265. [Google Scholar] [CrossRef]
99. Zhu, C., Kros, J. M., Cheng, C., Mustafa, D. (2017). The contribution of tumor-associated macrophages in glioma neo-angiogenesis and implications for anti-angiogenic strategies. Neuro-Oncology, 19(11), 1435–1446. DOI 10.1093/neuonc/nox081. [Google Scholar] [CrossRef]
100. Muoio, M. G., Talia, M., Lappano, R., Sims, A. H., Vella, V. et al. (2021). Activation of the S100a7/RAGE pathway by IGF-1 contributes to angiogenesis in breast cancer. Cancers, 13(4), 621. DOI 10.3390/cancers13040621.
101. He, M. Y., Halford, M. M., Liu, R., Roy, J. P., Grant, Z. L. et al. (2021). Three-dimensional CRISPR screening reveals epigenetic interaction with anti-angiogenic therapy. Communications Biology, 4(1), 878. DOI 10.1038/s42003-021-02397-3.
102. Shalem, O., Sanjana, N. E., Hartenian, E., Shi, X., Scott, D. A. et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science, 343(6166), 84–87. DOI 10.1126/science.1247005.
103. Cuella-Martin, R., Hayward, S. B., Fan, X., Chen, X., Huang, J. W. et al. (2021). Functional interrogation of DNA damage response variants with base editing screens. Cell, 184(4), 1081–1097.e19. DOI 10.1016/j.cell.2021.01.041.
104. Gootenberg, J. S., Abudayyeh, O. O., Lee, J. W., Essletzbichler, P., Dy, A. J. et al. (2017). Nucleic acid detection with CRISPR-Cas13a/C2c2. Science, 356(6336), 438–442. DOI 10.1126/science.aam9321.
105. Chen, J. S., Ma, E., Harrington, L. B., da Costa, M., Tian, X. et al. (2018). CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science, 360(6387), 436–439. DOI 10.1126/science.aar6245.
106. Zhang, Y., Zhang, Z. (2020). The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cellular & Molecular Immunology, 17(8), 807–821. DOI 10.1038/s41423-020-0488-6.
107. June, C. H., O’connor, R. S., Kawalekar, O. U., Ghassemi, S., Milone, M. C. (2018). CAR T cell immunotherapy for human cancer. Science, 359(6382), 1361–1365. DOI 10.1126/science.aar6711.
108. Murty, S., Haile, S. T., Beinat, C., Aalipour, A., Alam, I. S. et al. (2020). Intravital imaging reveals synergistic effect of CAR T-cells and radiation therapy in a preclinical immunocompetent glioblastoma model. Oncoimmunology, 9(1), 1757360. DOI 10.1080/2162402X.2020.1757360.
109. Lynn, R. C., Weber, E. W., Sotillo, E., Gennert, D., Xu, P. et al. (2019). C-Jun overexpression in CAR T cells induces exhaustion resistance. Nature, 576(7786), 293–300. DOI 10.1038/s41586-019-1805-z.
110. Hong, M., Clubb, J. D., Chen, Y. Y. (2020). Engineering CAR-T cells for next-generation cancer therapy. Cancer Cell, 38(4), 473–488. DOI 10.1016/j.ccell.2020.07.005.
111. Qin, H., Yang, L., Chukinas, J. A., Shah, N., Tarun, S. et al. (2021). Systematic preclinical evaluation of CD33-directed chimeric antigen receptor T cell immunotherapy for acute myeloid leukemia defines optimized construct design. J Immunother Cancer, 9(9), e003149. DOI 10.1136/jitc-2021-003149.
112. Neelapu, S. S., Tummala, S., Kebriaei, P., Wierda, W., Gutierrez, C. et al. (2018). Chimeric antigen receptor T-cell therapy-assessment and management of toxicities. Nature Reviews: Clinical Oncology, 15(1), 47–62. DOI 10.1038/nrclinonc.2017.148.
113. Wang, M., Munoz, J., Goy, A., Locke, F. L., Jacobson, C. A. et al. (2020). KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. New England Journal of Medicine, 382(14), 1331–1342. DOI 10.1056/NEJMoa1914347.
114. Ying, Z., Huang, X. F., Xiang, X., Liu, Y., Kang, X. et al. (2019). A safe and potent anti-CD19 CAR T cell therapy. Nature Medicine, 25(6), 947–953. DOI 10.1038/s41591-019-0421-7.
115. Tong, C., Zhang, Y., Liu, Y., Ji, X., Zhang, W. et al. (2020). Optimized tandem CD19/CD20 CAR-engineered T cells in refractory/relapsed B-cell lymphoma. Blood, 136(14), 1632–1644. DOI 10.1182/blood.2020005278.
116. Ramos, C. A., Grover, N. S., Beaven, A. W., Lulla, P. D., Wu, M. F. et al. (2020). Anti-CD30 CAR-T cell therapy in relapsed and refractory hodgkin lymphoma. Journal of Clinical Oncology, 38(32), 3794–3804. DOI 10.1200/JCO.20.01342.
117. Anwar, M. Y., Williams, G. R., Paluri, R. K. (2021). CAR T cell therapy in pancreaticobiliary cancers: A focused review of clinical data. Journal of Gastrointestinal Cancer, 52(1), 1–10. DOI 10.1007/s12029-020-00457-1.
118. Vora, P., Venugopal, C., Salim, S. K., Tatari, N., Bakhshinyan, D. et al. (2020). The rational development of CD133-targeting immunotherapies for glioblastoma. Cell Stem Cell, 26(6), 832–844.e6. DOI 10.1016/j.stem.2020.04.008.
119. Dai, H., Tong, C., Shi, D., Chen, M., Guo, Y. et al. (2020). Efficacy and biomarker analysis of CD133-directed CAR T cells in advanced hepatocellular carcinoma: A single-arm, open-label, phase II trial. Oncoimmunology, 9(1), 1846926. DOI 10.1080/2162402X.2020.1846926.
120. Zhao, L., Cao, Y. J. (2019). Engineered T cell therapy for cancer in the clinic. Frontiers in Immunology, 10, 2250. DOI 10.3389/fimmu.2019.02250.
121. Brancato, V., Oliveira, J. M., Correlo, V. M., Reis, R. L., Kundu, S. C. (2020). Could 3D models of cancer enhance drug screening? Biomaterials, 232, 119744. DOI 10.1016/j.biomaterials.2019.119744.
122. Stadtmauer, E. A., Fraietta, J. A., Davis, M. M., Cohen, A. D., Weber, K. L. et al. (2020). CRISPR-Engineered T cells in patients with refractory cancer. Science, 367(6481), eaba7365. DOI 10.1126/science.aba7365.
123. Eyquem, J., Mansilla-Soto, J., Giavridis, T., van der Stegen, S. J., Hamieh, M. et al. (2017). Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature, 543(7643), 113–117. DOI 10.1038/nature21405.
124. Lu, Y., Xue, J., Deng, T., Zhou, X., Yu, K. et al. (2020). Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nature Medicine, 26(5), 732–740. DOI 10.1038/s41591-020-0840-5.
125. Si, J., Shi, X., Sun, S., Zou, B., Li, Y. et al. (2020). Hematopoietic progenitor kinase1 (HPK1) mediates T cell dysfunction and is a druggable target for T cell-based immunotherapies. Cancer Cell, 38(4), 551–566.e11. DOI 10.1016/j.ccell.2020.08.001.
126. Hu, Z., Ott, P. A., Wu, C. J. (2018). Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nature Reviews: Immunology, 18(3), 168–182. DOI 10.1038/nri.2017.131.
127. Sahin, U., Türeci, Ö. (2018). Personalized vaccines for cancer immunotherapy. Science, 359(6382), 1355–1360. DOI 10.1126/science.aar7112.
128. Blass, E., Ott, P. A. (2021). Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nature Reviews: Clinical Oncology, 18(4), 215–229. DOI 10.1038/s41571-020-00460-2.
129. Hu, Z., Leet, D. E., Allesøe, R. L., Oliveira, G., Li, S. et al. (2021). Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nature Medicine, 27(3), 515–525. DOI 10.1038/s41591-020-01206-4.
130. Ott, P. A., Hu, Z., Keskin, D. B., Shukla, S. A., Sun, J. et al. (2017). An immunogenic personal neoantigen vaccine for patients with melanoma. Nature, 547(7662), 217–221. DOI 10.1038/nature22991.
131. Ravindranathan, S., Nguyen, K. G., Kurtz, S. L., Frazier, H. N., Smith, S. G. et al. (2018). Tumor-derived granulocyte colony-stimulating factor diminishes efficacy of breast tumor cell vaccines. Breast Cancer Research, 20(1), 126. DOI 10.1186/s13058-018-1054-3.
132. Guo, L., Xie, H., Zhang, Z., Wang, Z., Peng, S. et al. (2021). Fusion protein vaccine based on Ag85B and STEAP1 induces a protective immune response against prostate cancer. Vaccines (Basel), 9(7), 786. DOI 10.3390/vaccines9070786.
133. Buzhor, E., Leshansky, L., Blumenthal, J., Barash, H., Warshawsky, D. et al. (2014). Cell-based therapy approaches: The hope for incurable diseases. Regenerative Medicine, 9(5), 649–672. DOI 10.2217/rme.14.35.
134. Ihry, R. J., Worringer, K. A., Salick, M. R., Frias, E., Ho, D. et al. (2018). P53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nature Medicine, 24(7), 939–946. DOI 10.1038/s41591-018-0050-6.
135. Haapaniemi, E., Botla, S., Persson, J., Schmierer, B., Taipale, J. (2018). CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nature Medicine, 24(7), 927–930. DOI 10.1038/s41591-018-0049-z.
136. Martin, R. M., Fowler, J. L., Cromer, M. K., Lesch, B. J., Ponce, E. et al. (2020). Improving the safety of human pluripotent stem cell therapies using genome-edited orthogonal safeguards. Nature Communications, 11(1), 2713. DOI 10.1038/s41467-020-16455-7.
137. Yamanaka, S. (2020). Pluripotent stem cell-based cell therapy-promise and challenges. Cell Stem Cell, 27(4), 523–531. DOI 10.1016/j.stem.2020.09.014.
138. Flahou, C., Morishima, T., Takizawa, H., Sugimoto, N. (2021). Fit-for-all iPSC-derived cell therapies and their evaluation in humanized mice with NK cell immunity. Frontiers in Immunology, 12, 662360. DOI 10.3389/fimmu.2021.662360.
139. Kim, M. Y., Yu, K. R., Kenderian, S. S., Ruella, M., Chen, S. et al. (2018). Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell, 173(6), 1439–1453.e19. DOI 10.1016/j.cell.2018.05.013.
140. Wang, D., Prager, B. C., Gimple, R. C., Aguilar, B., Alizadeh, D. et al. (2021). CRISPR screening of CAR T cells and cancer stem cells reveals critical dependencies for cell-based therapies. Cancer Discovery, 11(5), 1192–1211. DOI 10.1158/2159-8290.CD-20-1243.
141. Ye, L., Park, J. J., Dong, M. B., Yang, Q., Chow, R. D. et al. (2019). In vivo CRISPR screening in CD8 T cells with AAV-sleeping beauty hybrid vectors identifies membrane targets for improving immunotherapy for glioblastoma. Nature Biotechnology, 37(11), 1302–1313. DOI 10.1038/s41587-019-0246-4.
142. Li, J., Yuan, S., Norgard, R. J., Yan, F., Sun, Y. H. et al. (2021). Epigenetic and transcriptional control of the epidermal growth factor receptor regulates the tumor immune microenvironment in pancreatic cancer. Cancer Discovery, 11(3), 736–753. DOI 10.1158/2159-8290.CD-20-0519.
143. Jiang, F., Doudna, J. A. (2017). CRISPR-Cas9 structures and mechanisms. Annu Rev Biophys, 46, 505–529. DOI 10.1146/annurev-biophys-062215-010822.
144. Martinez-Lage, M., Torres-Ruiz, R., Puig-Serra, P., Moreno-Gaona, P., Martin, M. C. et al. (2020). In vivo CRISPR/Cas9 targeting of fusion oncogenes for selective elimination of cancer cells. Nature Communications, 11(1), 5060. DOI 10.1038/s41467-020-18875-x.
145. Frangoul, H., Altshuler, D., Cappellini, M. D., Chen, Y. S., Domm, J. et al. (2021). CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. New England Journal of Medicine, 384(3), 252–260. DOI 10.1056/NEJMoa2031054.
146. Kurt, I. C., Zhou, R., Iyer, S., Garcia, S. P., Miller, B. R. et al. (2021). CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nature Biotechnology, 39(1), 41–46. DOI 10.1038/s41587-020-0609-x.
147. Zhao, D., Li, J., Li, S., Xin, X., Hu, M. et al. (2021). Glycosylase base editors enable C-to-A and C-to-G base changes. Nature Biotechnology, 39(1), 35–40. DOI 10.1038/s41587-020-0592-2.
148. Jeong, Y. K., Lee, S., Hwang, G. H., Hong, S. A., Park, S. E. et al. (2021). Adenine base editor engineering reduces editing of bystander cytosines. Nature Biotechnology, 39(11), 1426–1433. DOI 10.1038/s41587-021-00943-2.
149. Jin, S., Fei, H., Zhu, Z., Luo, Y., Liu, J. et al. (2020). Rationally designed APOBEC3B cytosine base editors with improved specificity. Molecular Cell, 79(5), 728–740.e6. DOI 10.1016/j.molcel.2020.07.005.
150. Anzalone, A. V., Randolph, P. B., Davis, J. R., Sousa, A. A., Koblan, L. W. et al. (2019). Search-and-replace genome editing without double-strand breaks or donor DNA. Nature, 576(7785), 149–157. DOI 10.1038/s41586-019-1711-4.
151. Kim, D. Y., Moon, S. B., Ko, J. H., Kim, Y. S., Kim, D. (2020). Unbiased investigation of specificities of prime editing systems in human cells. Nucleic Acids Research, 48(18), 10576–10589. DOI 10.1093/nar/gkaa764.
152. Kantor, A., Mcclements, M. E., Maclaren, R. E. (2020). CRISPR-Cas9 DNA base-editing and prime-editing. International Journal of Molecular Sciences, 21(17), 6240. DOI 10.3390/ijms21176240.
153. Biancur, D. E., Kapner, K. S., Yamamoto, K., Banh, R. S., Neggers, J. E. et al. (2021). Functional genomics identifies metabolic vulnerabilities in pancreatic cancer. Cell Metabolism, 33(1), 199–210.e8. DOI 10.1016/j.cmet.2020.10.018.
154. Kawasaki, K., Toshimitsu, K., Matano, M., Fujita, M., Fujii, M. et al. (2020). An organoid biobank of neuroendocrine neoplasms enables genotype-phenotype mapping. Cell, 183(5), 1420–1435.e21. DOI 10.1016/j.cell.2020.10.023.
155. Gonçalves, E., Segura-Cabrera, A., Pacini, C., Picco, G., Behan, F. M. et al. (2020). Drug mechanism-of-action discovery through the integration of pharmacological and CRISPR screens. Molecular Systems Biology, 16(7), e9405. DOI 10.15252/msb.20199405.
156. Fellmann, C., Gowen, B. G., Lin, P. C., Doudna, J. A., Corn, J. E. (2017). Cornerstones of CRISPR-Cas in drug discovery and therapy. Nature Reviews: Drug Discovery, 16(2), 89–100. DOI 10.1038/nrd.2016.238.
157. Nguyen, T. T., Ramsay, L., Ahanfeshar-Adams, M., Lajoie, M., Schadendorf, D. et al. (2021). Mutations in the IFNγ-JAK-STAT pathway causing resistance to immune checkpoint inhibitors in melanoma increase sensitivity to oncolytic virus treatment. Clinical Cancer Research, 27(12), 3432–3442. DOI 10.1158/1078-0432.CCR-20-3365.
158. Han, K., Pierce, S. E., Li, A., Spees, K., Anderson, G. R. et al. (2020). CRISPR screens in cancer spheroids identify 3D growth-specific vulnerabilities. Nature, 580(7801), 136–141. DOI 10.1038/s41586-020-2099-x.
159. Behan, F. M., Iorio, F., Picco, G., Gonçalves, E., Beaver, C. M. et al. (2019). Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature, 568(7753), 511–516. DOI 10.1038/s41586-019-1103-9.
160. Zeng, C., Huang, W., Li, Y., Weng, H. (2020). Roles of METTL3 in cancer: Mechanisms and therapeutic targeting. Journal of Hematology & Oncology, 13(1), 117. DOI 10.1186/s13045-020-00951-w.
161. Chen, H., Gao, S., Liu, W., Wong, C. C., Wu, J. et al. (2021). RNA N(6)-Methyladenosine methyltransferase METTL3 facilitates colorectal cancer by activating the m(6)A-GLUT1-mTORC1 axis and is a therapeutic target. Gastroenterology, 160(4), 1284–1300.e16. DOI 10.1053/j.gastro.2020.11.013.
162. Abudayyeh, O. O., Gootenberg, J. S., Essletzbichler, P., Han, S., Joung, J. et al. (2017). RNA targeting with CRISPR-Cas13. Nature, 550(7675), 280–284. DOI 10.1038/nature24049.
163. Bhan, A., Soleimani, M., Mandal, S. S. (2017). Long noncoding RNA and cancer: A New paradigm. Cancer Research, 77(15), 3965–3981. DOI 10.1158/0008-5472.CAN-16-2634.
164. Zhang, Z., Chen, J., Zhu, Z., Zhu, Z., Liao, X. et al. (2020). CRISPR-Cas13-mediated knockdown of lncRNA-GACAT3 inhibited cell proliferation and motility, and induced apoptosis by increasing p21, Bax, and E-cadherin expression in bladder cancer. Frontiers in Molecular Biosciences, 7, 627774. DOI 10.3389/fmolb.2020.627774.
165. Van Der Weyden, L., Harle, V., Turner, G., Offord, V., Iyer, V. et al. (2021). CRISPR activation screen in mice identifies novel membrane proteins enhancing pulmonary metastatic colonisation. Communications Biology, 4(1), 395. DOI 10.1038/s42003-021-01912-w.
166. Lawson, K. A., Sousa, C. M., Zhang, X., Kim, E., Akthar, R. et al. (2020). Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature, 586(7827), 120–126. DOI 10.1038/s41586-020-2746-2.
167. Yin, H., Kanasty, R. L., Eltoukhy, A. A., Vegas, A. J., Dorkin, J. R. et al. (2014). Non-viral vectors for gene-based therapy. Nature Reviews: Genetics, 15(8), 541–555. DOI 10.1038/nrg3763.
168. He, X. Y., Ren, X. H., Peng, Y., Zhang, J. P., Ai, S. L. et al. (2020). Aptamer/Peptide-functionalized genome-editing system for effective immune restoration through reversal of PD-L1-mediated cancer immunosuppression. Advanced Materials, 32(17), e2000208. DOI 10.1002/adma.202000208.
169. Glass, Z., Lee, M., Li, Y., Xu, Q. (2018). Engineering the delivery system for CRISPR-based genome editing. Trends in Biotechnology, 36(2), 173–185. DOI 10.1016/j.tibtech.2017.11.006.
170. Wang, P., Zhang, L., Zheng, W., Cong, L., Guo, Z. et al. (2018). Thermo-triggered release of CRISPR-Cas9 system by lipid-encapsulated gold nanoparticles for tumor therapy. Angewandte Chemie International Edtion, 57(6), 1491–1496 (in English). DOI 10.1002/anie.201708689.
171. Zhang, S., Shen, J., Li, D., Cheng, Y. (2021). Strategies in the delivery of Cas9 ribonucleoprotein for CRISPR/Cas9 genome editing. Theranostics, 11(2), 614–648. DOI 10.7150/thno.47007.
172. Miller, J. B., Zhang, S., Kos, P., Xiong, H., Zhou, K. et al. (2017). Non-viral CRISPR/Cas gene editing in vitro and in vivo enabled by synthetic nanoparticle Co-delivery of Cas9 mRNA and sgRNA. Angewandte Chemie International Edtion. in English, 56(4), 1059–1063. DOI 10.1002/anie.201610209.
173. Finn, J. D., Smith, A. R., Patel, M. C., Shaw, L., Youniss, M. R. et al. (2018). A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Reports, 22(9), 2227–2235.
174. Liu, J., Chang, J., Jiang, Y., Meng, X., Sun, T. et al. (2019). Fast and efficient CRISPR/Cas9 genome editing in vivo enabled by bioreducible lipid and messenger RNA nanoparticles. Advanced Materials, 31(33), e1902575. DOI 10.1002/adma.201902575.
175. Gillmore, J. D., Gane, E., Taubel, J., Kao, J., Fontana, M. et al. (2021). CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. New England Journal of Medicine, 385(6), 493–502. DOI 10.1056/NEJMoa2107454.
176. Xu, X., Wan, T., Xin, H., Li, D., Pan, H. et al. (2019). Delivery of CRISPR/Cas9 for therapeutic genome editing. Journal of Gene Medicine, 21(7), e3107. DOI 10.1002/jgm.3107.
177. Wilbie, D., Walther, J., Mastrobattista, E. (2019). Delivery aspects of CRISPR/Cas for in vivo genome editing. Accounts of Chemical Research, 52(6), 1555–1564. DOI 10.1021/acs.accounts.9b00106.
178. Lattanzi, A., Meneghini, V., Pavani, G., Amor, F., Ramadier, S. et al. (2019). Optimization of CRISPR/Cas9 delivery to human hematopoietic stem and progenitor cells for therapeutic genomic rearrangements. Molecular Therapy, 27(1), 137–150. DOI 10.1016/j.ymthe.2018.10.008.
179. Lv, J., Fan, Q., Wang, H., Cheng, Y. (2019). Polymers for cytosolic protein delivery. Biomaterials, 218, 119358. DOI 10.1016/j.biomaterials.2019.119358.
180. Lyu, P., Javidi-Parsijani, P., Atala, A., Lu, B. (2019). Delivering Cas9/sgRNA ribonucleoprotein (RNP) by lentiviral capsid-based bionanoparticles for efficient ‘hit-and-run’ genome editing. Nucleic Acids Research, 47(17), e99. DOI 10.1093/nar/gkz605.
181. Shen, B., Zhang, W., Zhang, J., Zhou, J., Wang, J. et al. (2014). Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nature Methods, 11(4), 399–402. DOI 10.1038/nmeth.2857.
182. Chen, S., Lee, B., Lee, A. Y., Modzelewski, A. J., He, L. (2016). Highly efficient mouse genome editing by CRISPR ribonucleoprotein electroporation of zygotes. Journal of Biological Chemistry, 291(28), 14457–14467. DOI 10.1074/jbc.M116.733154.
183. Niola, F., Dagnæs-Hansen, F., Frödin, M. (2019). In vivo editing of the adult mouse liver using CRISPR/Cas9 and hydrodynamic tail vein injection. Methods in Molecular Biology, 1961, 329–341. DOI 10.1007/978-1-4939-9170-9.
184. Nguyen, G. N., Everett, J. K., Kafle, S., Roche, A. M., Raymond, H. E. et al. (2021). A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nature Biotechnology, 39(1), 47–55. DOI 10.1038/s41587-020-0741-7.
185. Hinderer, C., Katz, N., Buza, E. L., Dyer, C., Goode, T. et al. (2018). Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Human Gene Therapy, 29(3), 285–298. DOI 10.1089/hum.2018.015.
186. Li, L., He, Z. Y., Wei, X. W., Gao, G. P., Wei, Y. Q. (2015). Challenges in CRISPR/CAS9 delivery: Potential roles of nonviral vectors. Human Gene Therapy, 26(7), 452–462. DOI 10.1089/hum.2015.069.
187. Rosenblum, D., Gutkin, A., Kedmi, R., Ramishetti, S., Veiga, N. et al. (2020). CRISPR-Cas9 genome editing using targeted lipid nanoparticles for cancer therapy. Science Advances, 6(47), eabc9450. DOI 10.1126/sciadv.abc9450.
188. Chen, G., Abdeen, A. A., Wang, Y., Shahi, P. K., Robertson, S. et al. (2019). A biodegradable nanocapsule delivers a Cas9 ribonucleoprotein complex for in vivo genome editing. Nature Nanotechnology, 14(10), 974–980. DOI 10.1038/s41565-019-0539-2.
189. De Voogt, W. S., Tanenbaum, M. E., Vader, P. (2021). Illuminating RNA trafficking and functional delivery by extracellular vesicles. Advanced Drug Delivery Reviews, 174, 250–264. DOI 10.1016/j.addr.2021.04.017.
190. Yao, X., Lyu, P., Yoo, K., Yadav, M. K., Singh, R. et al. (2021). Engineered extracellular vesicles as versatile ribonucleoprotein delivery vehicles for efficient and safe CRISPR genome editing. Journal of Extracellular Vesicles, 10(5), e12076. DOI 10.1002/jev2.12076.
191. Qi, Y., Liu, Y., Yu, B., Hu, Y., Zhang, N. et al. (2020). A Lactose-derived CRISPR/Cas9 delivery system for efficient genome editing in vivo to treat orthotopic hepatocellular carcinoma. Advanced Science (Weinh), 7(17), 2001424. DOI 10.1002/advs.202001424.
192. Liu, S., Cheng, Q., Wei, T., Yu, X., Johnson, L. T. et al. (2021). Membrane-destabilizing ionizable phospholipids for organ-selective mRNA delivery and CRISPR-Cas gene editing. Nat Mater, 20(5), 701–710. DOI 10.1038/s41563-020-00886-0.
193. Gilbert, L. A., Larson, M. H., Morsut, L., Liu, Z., Brar, G. A. et al. (2013). CRISPR-Mediated modular RNA-guided regulation of transcription in eukaryotes. Cell, 154(2), 442–451. DOI 10.1016/j.cell.2013.06.044.
194. Richardson, C. D., Ray, G. J., Dewitt, M. A., Curie, G. L., Corn, J. E. (2016). Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nature Biotechnology, 34(3), 339–344. DOI 10.1038/nbt.3481.
195. Hu, J. H., Miller, S. M., Geurts, M. H., Tang, W., Chen, L. et al. (2018). Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature, 556(7699), 57–63. DOI 10.1038/nature26155.
196. Nishimasu, H., Shi, X., Ishiguro, S., Gao, L., Hirano, S. et al. (2018). Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science, 361(6408), 1259–1262. DOI 10.1126/science.aas9129.
197. Walton, R. T., Christie, K. A., Whittaker, M. N., Kleinstiver, B. P. (2020). Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science, 368(6488), 290–296. DOI 10.1126/science.aba8853.
198. Akcakaya, P., Bobbin, M. L., Guo, J. A., Malagon-Lopez, J., Clement, K. et al. (2018). In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature, 561(7723), 416–419. DOI 10.1038/s41586-018-0500-9.
199. Kleinstiver, B. P., Pattanayak, V., Prew, M. S., Tsai, S. Q., Nguyen, N. T. et al. (2016). High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature, 529(7587), 490–495. DOI 10.1038/nature16526.
200. Kocak, D. D., Josephs, E. A., Bhandarkar, V., Adkar, S. S., Kwon, J. B. et al. (2019). Increasing the specificity of CRISPR systems with engineered RNA secondary structures. Nature Biotechnology, 37(6), 657–666. DOI 10.1038/s41587-019-0095-1.
201. Kosicki, M., Tomberg, K., Bradley, A. (2018). Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nature Biotechnology, 36(8), 765–771. DOI 10.1038/nbt.4192.
202. Xu, S., Kim, J., Tang, Q., Chen, Q., Liu, J. et al. (2020). CAS9 is a genome mutator by directly disrupting DNA-PK dependent DNA repair pathway. Protein Cell, 11(5), 352–365. DOI 10.1007/s13238-020-00699-6.
203. Adikusuma, F., Piltz, S., Corbett, M. A., Turvey, M., Mccoll, S. R. et al. (2018). Large deletions induced by Cas9 cleavage. Nature, 560(7717), E8–E9. DOI 10.1038/s41586-018-0380-z.
204. Song, Y., Liu, Z., Zhang, Y., Chen, M., Sui, T. et al. (2020). Large-fragment deletions induced by Cas9 cleavage while not in the BEs system. Molecular Therapy-Nucleic Acids, 21, 523–526. DOI 10.1016/j.omtn.2020.06.019.
205. Hakim, C. H., Kumar, S. R. P., Pérez-López, D. O., Wasala, N. B., Zhang, D. et al. (2021). Cas9-specific immune responses compromise local and systemic AAV CRISPR therapy in multiple dystrophic canine models. Nat Commun, 12(16769. DOI 10.1038/s41467-021-26830-7.
206. Charlesworth, C. T., Deshpande, P. S., Dever, D. P., Camarena, J., Lemgart, V. T. et al. (2019). Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nature Medicine, 25(2249–254. DOI 10.1038/s41591-018-0326-x.
207. Leibowitz, M. L., Papathanasiou, S., Doerfler, P. A., Blaine, L. J., Sun, L. et al. (2021). Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nature Genetics, 53(6), 895–905. DOI 10.1038/s41588-021-00838-7.
208. Mahas, A., Neal Stewart, C., Jr., Mahfouz, M. M. (2018). Harnessing CRISPR/Cas systems for programmable transcriptional and post-transcriptional regulation. Biotechnology Advances, 36(1), 295–310. DOI 10.1016/j.biotechadv.2017.11.008.
209. Nuñez, J. K., Chen, J., Pommier, G. C., Cogan, J. Z., Replogle, J. M. et al. (2021). Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell, 184(9), 2503–2519.e17. DOI 10.1016/j.cell.2021.03.025.
210. Pulecio, J., Verma, N., Mejía-Ramírez, E., Huangfu, D., Raya, A. (2017). CRISPR/Cas9-based engineering of the epigenome. Cell Stem Cell, 21(4), 431–447. DOI 10.1016/j.stem.2017.09.006.
211. Liu, X., Zhang, Y., Chen, Y., Li, M., Zhou, F. et al. (2017). In situ capture of chromatin interactions by biotinylated dCas9. Cell, 170(5), 1028–1043.e19. DOI 10.1016/j.cell.2017.08.003.
212. Zafra, M. P., Schatoff, E. M., Katti, A., Foronda, M., Breinig, M. et al. (2018). Optimized base editors enable efficient editing in cells, organoids and mice. Nature Biotechnology, 36(9), 888–893. DOI 10.1038/nbt.4194.
213. Workman, R. E., Pammi, T., Nguyen, B. T. K., Graeff, L. W., Smith, E. et al. (2021). A natural single-guide RNA repurposes Cas9 to autoregulate CRISPR-Cas expression. Cell, 184(3), 675–688.e19. DOI 10.1016/j.cell.2020.12.017.
214. Xu, X., Chemparathy, A., Zeng, L., Kempton, H. R., Shang, S. et al. (2021). Engineered miniature CRISPR-Cas system for mammalian genome regulation and editing. Molecular Cell, 81(20), 4333–4345.e4. DOI 10.1016/j.molcel.2021.08.008.
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |