| Oncologie |
DOI: 10.32604/oncologie.2021.018647
ARTICLE
A Retrospective Analysis of 94 Patients with Hemophagocytic Lymphohistiocytosis of Unknown Etiology from a Single Center
Department of Hematology, Beijing Friendship Hospital, Capital Medical University, Beijing, China
*Corresponding Author: Zhao Wang. Email: wangzhao@ccmu.edu.cn
Received: 08 August 2021; Accepted: 01 November 2021
Abstract: Despite extensive work-ups, some patients have been diagnosed with hemophagocytic lymphohistiocytosis (HLH) of unknown etiology. For HLH of unknown etiology, to investigate the clinical features and the factors that may affect the prognosis, we retrospectively reviewed the medical records of 94 patients hospitalized from January 2014 to December 2019. Survival times were evaluated until April 2020. For the 94 patients, the underlying causes of their diseases remained unclear at the end of the follow-up period, and the 1-, 3-, and 6-month survival rates, and the overall survival (OS) rates were 86.2%, 78.7%, 73.4%, and 70.2%, respectively. The multivariate analysis showed that the absence of an overall response or remission within 1 month after initial induction therapy (hazard ratio [HR], 11.914; 95% confidence interval [CI], 3.114–45.584; p < 0.001) and the refractory and relapsed HLH (HR, 16.003; 95% CI, 2.425–105.598; p = 0.004) were independently associated with adverse prognoses. The OR rate within 1 month occurred in 86.2% of the patients with doxorubicin-etoposide-methylprednisolone (DEP) regimen as initial induction treatments, while, that occurred in 58.46% of the patients with non-DEP regimens (p = 0.008). The OS rates did not differ between the patients with DEP regimens and non-DEP regimens (p = 0.252). These findings suggest that HLH of unknown etiology has relatively good OS rate and prognosis overall. Absence of an OR within 1 month after initial induction therapy and refractory and relapsed HLH were independently associated with adverse prognoses. The DEP regimen indeed could improve the OR rate, but it failed to show an OS benefit.
Keywords: Hemophagocytic lymphohistiocytosis; unknown etiology; doxorubicin-etoposide-methylprednisolone regimen; prognosis; remission; complete response; partial response; survival rate
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening syndrome involving excessive immune activation, and it is characterized by an inflammatory cytokine storm that causes multiorgan dysfunction. Excessive, but defective, immune activation leads to the atypical clinical manifestations and characteristic laboratory findings of HLH, including fever, hemocytopenia, splenomegaly, hypertriglyceridemia, hyperferritinemia, hypofibrinogenemia, and hemophagocytosis in bone marrow, spleen, or lymph nodes [1,2]. HLH is categorized as either primary or secondary HLH. Primary or familial HLH (FHL) has an autosomal recessive inheritance. Mutations of genes, including PRF1, UNC13D, STX11 and STXBP2, RAB27, CHS1/LYST, and AP13β1, are the major causes of the congenital immune deficiency syndrome associated with HLH. SH2D1A and BIRC4 gene mutations are associated with Epstein-Barr virus (EBV)-driven HLH. Secondary HLH is usually associated with viral, bacterial, fungal, and parasitic infections, autoimmune diseases, and malignant disorders. The related patients often have no family history of HLH or known genetic defects [3,4]. The clarification of the mechanisms underlying the development of HLH in some patients is required but hard, because HLH has a heterogeneous etiology, and it is associated with nonspecific clinical and laboratory findings. For unexplained HLH, the proportions of patients, the treatments, the related prognostic factors and the outcomes differ among research institutions [5–8]. Systematic studies investigating the clinical characteristics and prognoses of patients with HLH of unknown etiology are absent. To explore the clinical features and possible factors that may affect the prognosis of HLH of unknown etiology, we conducted a retrospective study that involved 105 patients with HLH of unknown etiology who were hospitalized from January 2014 to December 2019 at Beijing Friendship hospital.
From January 2014 to December 2019, 105 patients who met the HLH-2004 diagnostic criteria for HLH and whose underlying causes of HLH were not determined within 1 month, despite extensive examinations, were enrolled in the study. All the 94 patients had undergone HLH-associated Sanger sequencing, including PRF1, UNC13D, STX11, STXBP2, SH2D1A, BIRC4, RAB27A, LYST, ADTB3A, ITK, CD27 and XMEN, which corresponded to FHL2-FHL5, XLP-1, XLP-2, Griscelli syndeome 2(GS-2), Chediak-Higashi syndrome 1(CHS-1), Hermansky-Pudlak syndrome lI (HPS-II) and other EBV drived HLH, respectively [9]. They also undergone investigations into common pathogens that cause HLH, including bacteria, fungi, viruses, atypical pathogens, and parasites in the blood, sputum, urine, feces, and tissues, if needed, assessments of parameters related to rheumatic and immunologic diseases, positron emission tomography/computed tomography (PET/CT), lymph node and suspected lesion biopsies, if necessary, bone marrow smears, flow cytometric analyses, and bone marrow biopsies to exclude primary HLH, infection, rheumatic and immunologic diseases, and malignant tumors, especially lymphomas. Given that diagnosing lymphomas is difficult in some patients, we excluded patients whose PET/CT or bone marrow flow cytometry examinations indicated suspected lymphomas. Based on our research group’s experience, we defined HLH of unknown etiology as patients with HLH who underwent all the aforementioned examinations and whose underlying causes of HLH remained unclear within 1 month. The patients’ clinical characters and laboratory findings were retrospectively evaluated by reviewing their medical records.
Initial induction treatments included the DEP regimen and non-DEP regimens. The DEP regimen was administrated as: Liposomal doxorubicin: 25 mg/m2/d d1; VP-16: 100 mg/m2/d d1; methylprednisolone: 2 mg/kg/d d1-3, 0.75 mg/kg/d d4-7, 0.25 mg/kg/d d8-10, and 0.1 mg/kg/d until the next cycle. Generally, 2 weeks is a cycle, and 3 to 6 cycles of DEP regimen were administrated to HLH of unknown etiology based on our experience. The non-DEP regimens, which included the HLH-94 and HLH-2004 protocols, and glucocorticoid and intravenous immunoglobulin therapy.
In this study, 35 of the 94 patients suffered from refractory and relapsed HLH. For patients with refractory and relapsed HLH, they also adopted the DEP regimen or non-DEP regimens as initial induction treatment. However, they adopted other salvage therapies after HLH relapse: 19 patients adopted DEP regimen (Liposomal doxorubicin + etoposide + methylprednisolone), and 14 of 19 patients died; 1 patient adopted Ru-DEP regimen (ruxolitinib + DEP); 3 patients adopted high-dose glucocorticosteroid + etoposide +/- cyclosporine A; 1 patient adopted ECHOP-like regimens (etoposide + cyclophosphamide + epirubicin + vincristine + glucocorticosteroid); 1 patient adopted ECOP-like regimens (etoposide + cyclophosphamide + vincristine + glucocorticosteroid); 1 patient adopted splenectomy; 9 patients died after HLH relapse without receiving any salvage therapies. The patients were not able to undergo hematopoietic stem cell transplantation for various reasons.
The patients’ treatment was assessed using the criteria proposed by Marsh et al. [10], with modifications based on our research group’s experience [5]. A complete response (CR) was defined as the normalization of the clinical symptoms and laboratory findings associated with HLH. A partial response (PR) was defined as a ≥25% improvement in ≥2 symptoms and laboratory tests, including the soluble interleukin (IL)-2 receptor-α (sCD25), ferritin, and triglyceride levels. For patients with initial absolute neutrophil counts (ANCs) <0.5 × 109/L, a response was defined as values that were elevated to >0.5 × 109/L; for patients with initial ANCs of 0.5–2.0 × 109/L, a response was defined as values that were elevated to >5.0 × 109/L; and for patients with alanine aminotransferase (ALT) levels >400 U/L, a response was defined as a 50% decrease in the absence of hemophagocytosis. Additionally, for patients with either a CR or a PR, their body temperature had to revert to the normal range. No response (NR) was defined as a failure to achieve a PR. Overall response (OR) or remission was defined as a success to achieve a CR or PR.
There are no accepted diagnostic criteria for refractory and relapsed HLH at present. It is generally recognized that absence of response at 2 to 3 weeks is often a sign of refractory HLH [11]. Therefore, in this study, we defined refractory HLH as patients who did not achieve at least PRs 2 weeks after initial standard. Relapsed HLH was defined as patients who had experienced relapses of HLH after initial remissions achieved with standard HLH therapy and had failed to achieve CRs with salvage therapies.
The data were analyzed using IBM®SPSS® software, version 23.0 (IBM Corporation, Armonk, NY, USA). The continuous variables are presented as the medians and the 25th–75th percentiles. The categorical variables are presented as proportions. Overall survival (OS) was calculated from the date of an HLH diagnosis until death or April 2020. The comparison of ORR of DEP and non-DEP regimen was performed using Chi-square text. The survival curves were estimated by using the Kaplan–Meier method, and differences between the curves were tested for statistical significance using the log-rank test. Univariate analyses were performed using KM method. Multivariate analyses were performed using the Cox regression method. A value of p < 0.05 was considered statistically significant.
3.1 Clinical and Laboratory Characteristics
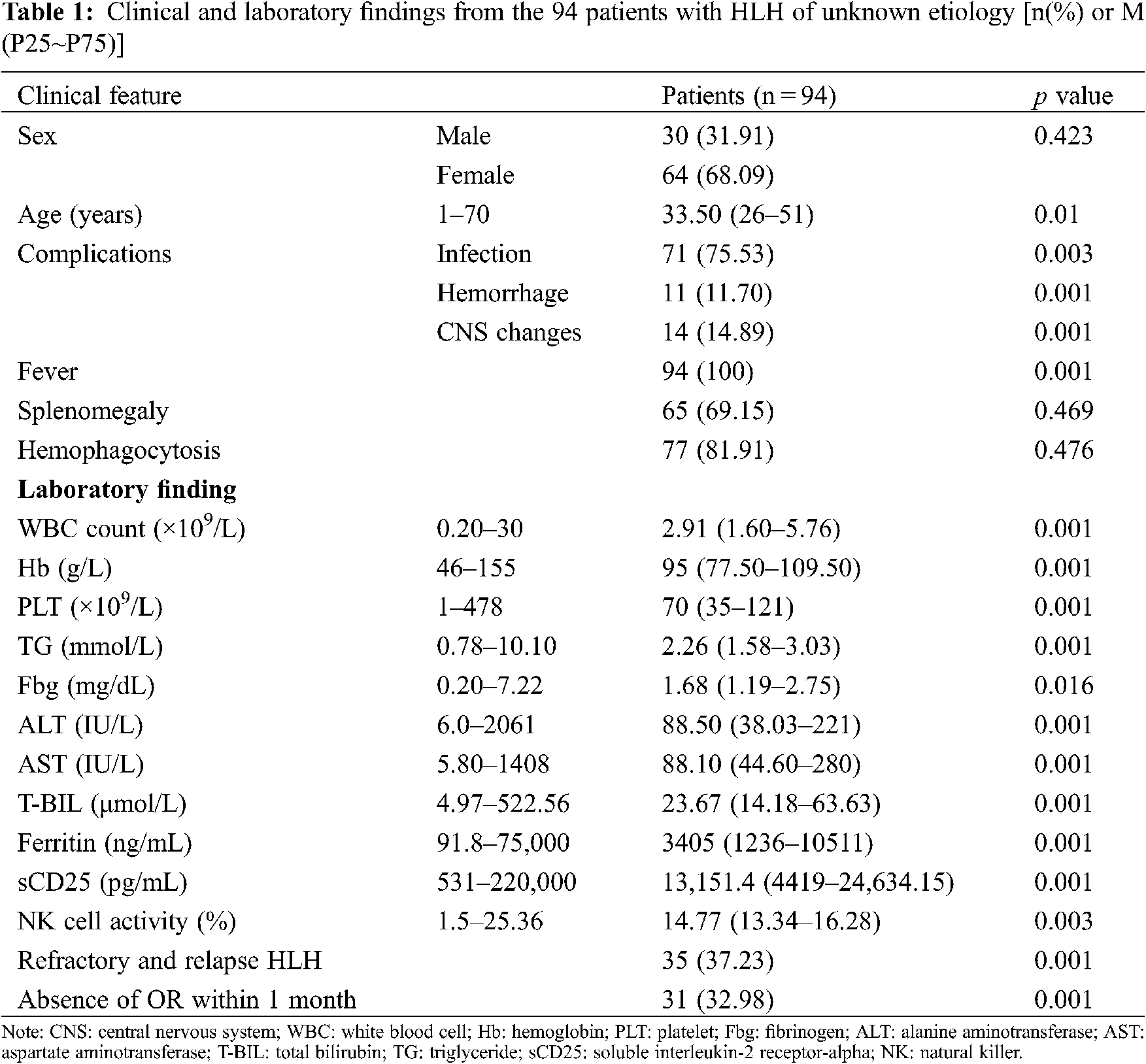
From January 2014 to December 2019, 930 patients with HLH were hospitalized at our center. Of these patients, 105 for whom the underlying causes of HLH were not identified during early diagnoses were enrolled in this study. 11 patients’ underlying causes of HLH were identified eventually, and they comprised 4 patients with lymphomas, 6 patients with rheumatic and immunologic diseases, and 1 patient with typhoid fever. The underlying causes of HLH remained unidentified for the remaining 94 patients at the end of the follow-up period. Of these patients with HLH of unknown etiology, 30 (31.91%) were male and 64 (68.09%) were female. The age of the patients ranged from 1 to 70 years, and their median age was 33.5 years (interquartile range, 26–51 years). Complications of the 94 patients, 71patients (75.53%) had infections, 11 patients (11.7%) had hemorrhage, and 14 patients (14.89%) had central nervous system changes during the clinical course. All of the patients with HLH of unknown etiology presented with fever, and 88.3% had increased serum ferritin levels, 86.2% had increased sCD25 levels, 77.7% had splenomegaly, and 77.8% had the hemophagocytic phenomenon. Lower blood cell levels, low fibrinogenemia levels, hypertriglyceridemia, and reduced natural killer (NK) cell activity were present in 36.2%, 33%, 20.2%, and 12.8% of the patients, respectively. The clinical features of and the laboratory findings from the 94 patients with HLH of unknown etiology are shown in Table 1. Multivariate analysis of risk factors for hemophagocytic lymphohistiocytosis of unknown etiology are shown in Table 2.
3.2 Initial Induction Treatment of Hemophagocytic Lymphohistiocytosis of Unknown Etiology
Of the 94 patients with HLH of unknown etiology, 29 were administered the doxorubicin-etoposide-methylprednisolone (DEP) regimen as initial induction treatment, and 65 were administered non-DEP regimens. Of the 29 patients who received the DEP regimen, 9 (31.03%) achieved CRs, 16 (55.71%) achieved PRs, and 4 (13.79%) had NRs at 1 month after initial induction treatment; the overall response (OR) rate was 86.2%. Of the 65 patients who received non-DEP regimens as initial induction therapy, 15 (23.08%) achieved CRs, 23 (35.38%) achieved PRs, and 27 (36.92%) had NRs at 1 month after treatment; the OR rate was 58.46%. The OR rate of using DEP regimen as initial induction treatment was higher than using non-DEP regimens (p = 0.008). The OS rates for the patients who received the DEP regimen and non-DEP regimens were 79.3% and 66.2%, respectively; this difference was not significant (p = 0.252) (Fig. 1).
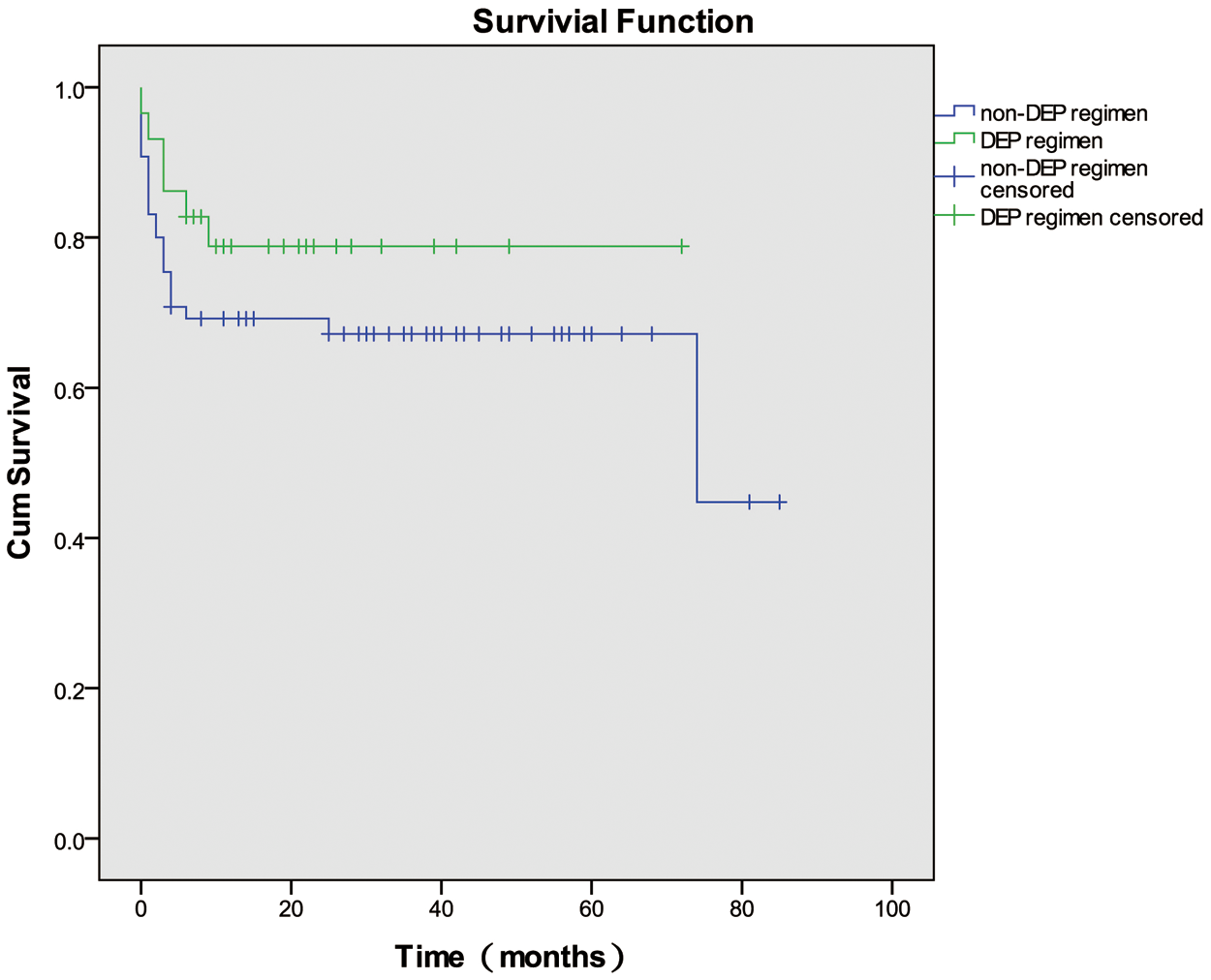
Figure 1: Overall survival curves for the patients who were administered the doxorubicin-etoposide-methylprednisolone (DEP) regimen and non-DEP regimens. DEP: doxorubicin-etoposide-methylprednisolone
3.3 Survival Analysis of Hemophagocytic Lymphohistiocytosis of Unknown Etiology
Survival was assessed until April 2020. Among the 94 patients evaluated, the 1-, 3-, and 6-month survival rates, and OS rate were 86.2%, 78.7%, 73.4%, and 70.2%, respectively. The OS curve is depicted in Fig. 2.
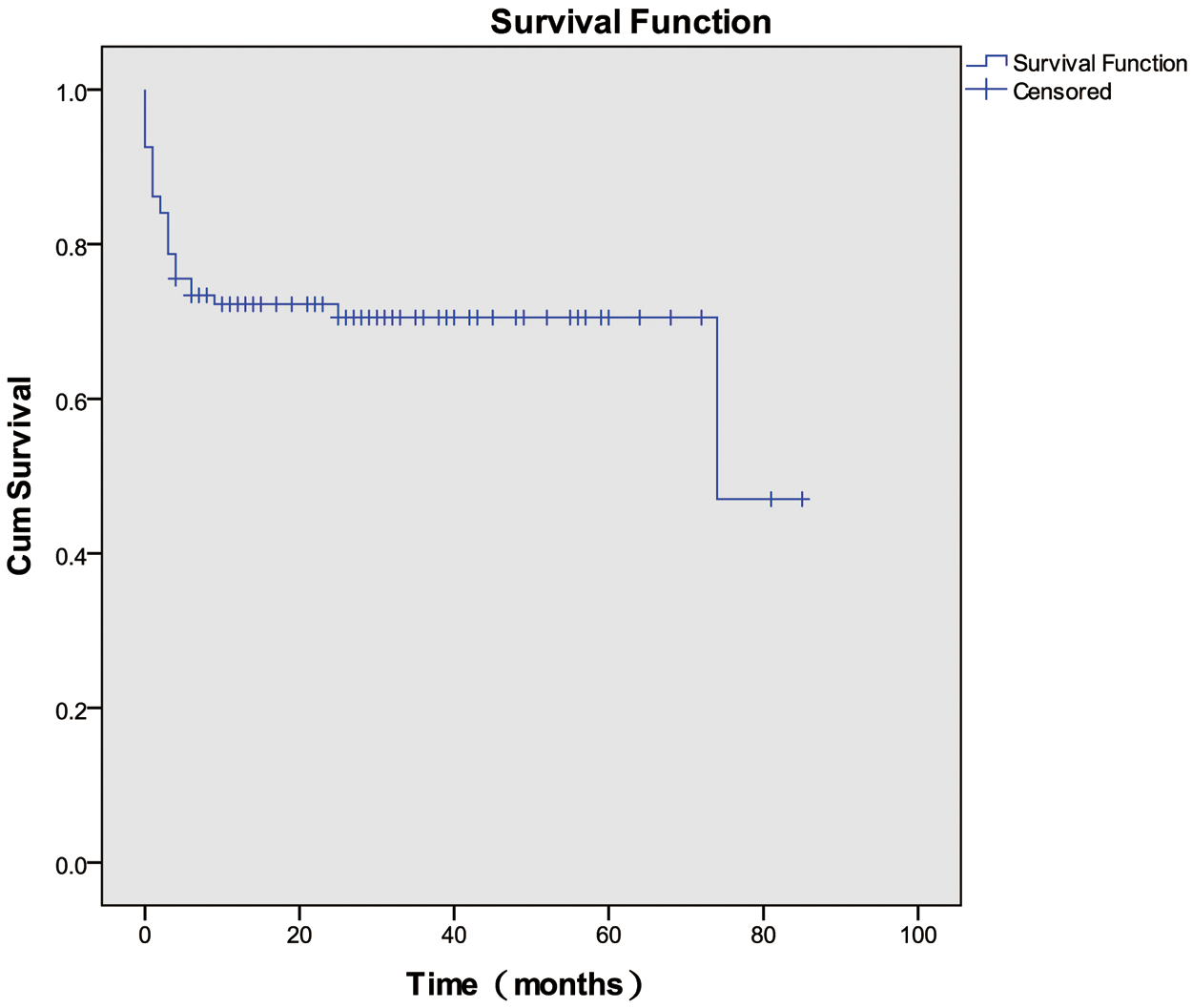
Figure 2: Overall survival among patients with HLH of unknown etiology
3.4 Factors Influencing the Prognosis of Hemophagocytic Lymphohistiocytosis of Unknown Etiology
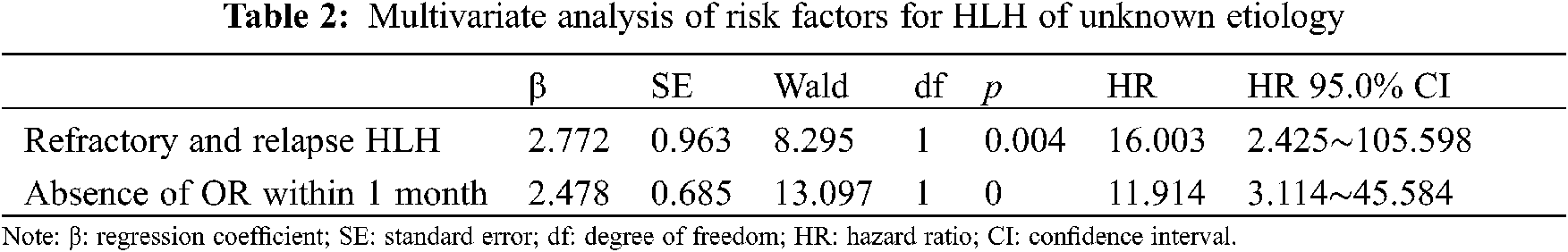
The univariate analysis identified several factors that may influence the survival of patients with HLH of unknown etiology, including age, sex, complications, the white blood cell count, and the hemoglobin, platelet, triglyceride, fibrinogen, ALT, aspartate aminotransferase, sCD25, ferritin, and NK cell activity levels, whether an OR or remission was achieved within 1 month after initial induction therapy, and whether patients experienced refractory and relapsed HLH. The multivariate analyses showed that failure to achieve an OR or remission within 1 month after initial induction therapy (hazard ratio [HR], 11.914; 95% confidence interval [CI], 3.114–45.584; p < 0.001) and refractory and relapsed HLH (HR, 16.003; 95% CI, 2.425–105.598; p = 0.004) were independently associated with adverse prognoses.
HLH is a heterogeneous disease. The difficulties of diagnosing HLH and identifying the underlying causes of HLH are associated with multiple confounding medical illnesses and imprecise diagnostic criteria. In addition, a small number of patients are diagnosed with HLH of unknown etiology, because of the limitations of current medical examination techniques, the rapid progression of the disease, and complications that include thrombocytopenia, coagulation disorders, and bleeding, which impede biopsies of suspicious lesions. Lymphoma is one of the most common diseases that cause adult HLH. A review of 162 patients with HLH showed that 56% had non-Hodgkin lymphomas [12]. Recommendations for the management of HLH in adults indicated that about 40%–70% of HLH in adults was associated with malignancy, particularly Hodgkin and non-Hodgkin lymphomas [13]. However, some patients do not have prominent masses or enlarged lymph nodes, and diagnosing lymphoma-associated HLH is difficult. Some lymphoma-associated HLH may be misdiagnosed as HLH of unknown etiology. Tabata et al. [14,15] reported a patient with HLH of unknown etiology whose autopsy revealed a disseminated, diffuse large B-cell lymphoma. However, no positive evidence of HLH was found before the autopsy, except for an extremely high serum sIL-2R level relative to the serum ferritin level. These authors subsequently reviewed 110 Japanese patients with HLH retrospectively, and they proposed that the serum sIL-2R level and the sIL-2R/ferritin ratio may be useful markers for predicting underlying malignant lymphomas in patients with hemophagocytic syndrome [14,15]. Wang et al. studied 19 patients with HLH of unknown etiology, and histopathologic examinations of the patients’ spleens identified lymphomas in 7 patients, which suggested that primary splenic lymphomas may underlie the development of HLH in some patients and that splenectomy could guide the diagnosis and treatment of HLH [16]. In our study, 105 patients fulfilled the study’s inclusion criteria during their early diagnoses, and of these, the underlying causes of HLH were eventually determined for 11 patients, of whom, 4 had lymphomas, 6 had autoimmune diseases, and 1 had typhoid fever. 2 of 4 patients who were diagnosed with hepatosplenic non-Hodgkin T-cell lymphomas following splenectomy, died as a consequence of HLH progression; these patients survived 2 months and 10 months. 1patient who was diagnosed with a non-Hodgkin B-cell lymphoma based on a pararenal lymph node biopsy, survived the follow-up period. An ascites smear led to a diagnosis of a non-Hodgkin T-cell lymphoma in 1 patient; this patient’s lymphoma progressed and he died after 36 months. The patients with the autoimmune diseases and typhoid fever survived the follow-up period. These findings indicate that some patients with HLH of unknown etiology may have underlying lymphomas, even if clinical and imaging manifestations and pathologic evidence are absent. Moreover, unidentified infections, autoimmune disease, and unknown genetic anomalies may exist that require more extensive work-ups and multidisciplinary collaborations.
Mortality among adult patients with HLH differs according to the different causes of the disease, and it ranges from 20% to 88% [13]. The prognoses, OS rate, and ORR to treatment vary among different institutions. Yanagisawa et al. [8] studied the outcomes of the HLH-2004 protocol administered to children with EBV-driven HLH, FHL, and HLH of unknown etiology. These investigators showed that the 3-year OS rate for children with HLH of unknown etiology was significantly lower (56.2%) than the 3-year OS rates for children with EBV-driven HLH (85.3%) and FHL (66.7%). Furthermore, the prognosis of children with HLH of unknown etiology after hematopoietic stem cell transplantation was much worse (25%) than the prognoses of children with EBV-HLH (66.7%) or FHL (85.7%) [8]. Li et al. found that patients with HLH of unknown origin had a poor survival rate (17%) [7,17]. The reasons for the poor prognoses of patients with HLH of unknown etiology at these institutions may relate to the studies involving children whose conditions were poor at diagnosis and their admission to intensive care units [6]. The findings from a multicenter study conducted by Wang et al. showed an OS rate of 87.5% for patients with HLH of unknown etiology, which is much higher than the OS rates determined for other types of secondary HLH [5]. In this study, our analysis showed that the 1-month, 3-month, and 6-month survival rates, and OS rate were 86.2%, 78.7%, 73.4%, and 70.2%, respectively, which indicated that the OS rate and the prognoses of patients with HLH of unknown etiology were relatively good overall.
HLH is characterized by a hyperinflammatory phenotype. Irrespective of the underlying causes, the early suppression of the life-threatening inflammatory processes with initial induction therapy is elementary, and it impacts on the prognosis of the disease [11,18]. In the present study, the multivariate analysis showed that the failure to achieve an OR within 1 month after initial induction therapy and the refractory and relapsed HLH were independently associated with an adverse prognosis. The use of the DEP regimen as a salvage therapy for adult refractory HLH was proposed by the personnel at our center, because it had significantly improved the response rate (76%) in the adult patients, who failed to respond to the HLH-94 or HLH-04 protocols [5]. In this study, the OS rates for the patients with the DEP and non-DEP regimens were 79.3% and 66.2%, respectively; these findings, in addition to the 1-month OR rate, highlight the potential benefit of using the DEP regimen as initial induction treatment in patients with HLH of unknown etiology. However, the DEP regimen could improve the OR rate, but it failed to show an OS benefit. Overall, this revealed that DEP could be a potential optimal choice for the related patients. Some researchers have also suggested that, for the patients with relapsed HLH of unknown etiology, splenectomy can improve the clinical symptoms and survival by changing the percentage of NK cells as well as the IL-21 and IL-1α levels [19]. Another study showed that, splenectomy could improve the survival of the patients with HLH and unknown underlying diseases, and, to achieve optimal results, splenectomy should be performed during the initial stages of the related diseases. However, splenectomy did not prolong the survival time for all the patients with HLH, particularly for those with definite diagnoses [20]. In our study, 6 of 105 patients underwent splenectomies for splenomegaly. In the 6 patients, two were diagnosed with lymphomas based on their histopathology findings, and they died as a consequence of primary disease progression. Moreover, 4 were not diagnosed with lymphomas, and their clinical manifestations and laboratory findings improved, and they survived the follow-up period; 2 achieved CRs, and they survived 35 and 11 months, respectively, and, 1 achieved a PR and survived 42 months, and, 1 had NR and survived 60 months. Hence, splenectomy may be a useful measure for diagnosing and treating patients with HLH of unknown etiology. The mechanism for splenectomy in the treatment of HLH of unknown etiology needs further studies.
HLH is a heterogeneous disease, and it may have many unknown etiologies, as a consequence of the limitations of medical technologies. For patients with HLH, the treatments and prognoses vary due to the different underlying etiologies. Therefore, further researches into the patient assessments and disease indicators is necessary to identify the causes of HLH. HLH of unknown etiology has relatively good OS rate and prognosis overall. Early suppression of the life-threatening inflammatory processes with initial induction therapy is important, and DEP regimen is a potential optimal choice for adult patients with HLH of unknown etiology.
Author Contributions: Xiaodan He designed and performed the research and wrote the paper; Zhao Wang designed the research and supervised the report; Jingshi Wang provided clinical advice and supervised the report.
Availability of Data and Materials: The datasets used during the current study are available from the corresponding author on request.
Ethical Approval and Informed Consent Statement: All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This study was approved by the independent Ethics Committee of Beijing Friendship hospital (Document No. 2021-P2-386-01). Written informed consent was obtained from each patient or his (or her) guardian.
Funding Statement: This work was supported by grants from the National Natural Science Foundation of China (Grant No. 81871633), the Beijing Natural Science Foundation (Grant No. 7181003) and Beijing Municipal Administration of Hospital Incubating Program (Code: PX2018003).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Henter, J. I., Horne, A., Aricó, M., Egeler, R. M., Filipovich, A. H. et al. (2007). Hlh-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatric Blood & Cancer, 48, 124–131. DOI 10.1002/(ISSN)1545-5017. [Google Scholar] [CrossRef]
2. Hayden, A., Park, S., Giustini, D., Lee, A. Y. Y., Chen, L. Y. C. (2016). Hemophagocytic syndromes (HPSs) including hemophagocytic lymphohistiocytosis (HLH) in adults: A systematic scoping review. Blood Reviews, 30, 411–420. DOI 10.1016/j.blre.2016.05.001. [Google Scholar] [CrossRef]
3. Sieni, E., Cetica, V., Hackmann, Y., Coniglio, M. L., da Ros, M. et al. (2014). Familial hemophagocytic lymphohistiocytosis: When rare diseases shed light on immune system functioning. Front Immunol, 5. DOI 10.3389/fimmu.2014.00167. [Google Scholar] [CrossRef]
4. Janka, G. E. (2017). Familial and acquired hemophagocytic lymphohistiocytosis. European Journal of Pediatrics, 166, 95–109. DOI 10.1007/s00431-006-0258-1. [Google Scholar] [CrossRef]
5. Wang, Y., Huang, W., Hu, L., Cen, X., Li, L. et al. (2015). Multicenter study of combination dep regimen as a salvage therapy for adult refractory hemophagocytic lymphohistiocytosis. Blood, 126, 2186–2192. DOI 10.1182/blood-2015-05-644914. [Google Scholar] [CrossRef]
6. Grom, A. A. (2003). Macrophage activation syndrome and reactive hemophagocytic lymphohistiocytosis: The same entities? Current Opinion in Rheumatology, 15, 587–590. DOI 10.1097/00002281-200309000-00011. [Google Scholar] [CrossRef]
7. Li, J., Wang, Q., Zheng, W., Ma, J., Zhang, W. et al. (2014). Hemophagocytic lymphohistiocytosis: Clinical analysis of 103 adult patients. Medicine (Baltimore), 93, 100–105. DOI 10.1097/MD.0000000000000022. [Google Scholar] [CrossRef]
8. Yanagisawa, R., Nakazawa, Y., Matsuda, K., Yasumi, T., Kanegane, H. et al. (2019). Outcomes in children with hemophagocytic lymphohistiocytosis treated using HLH-2004 protocol in Japan. International Journal of Hematology, 109, 206–213. DOI 10.1007/s12185-018-02572-z. [Google Scholar] [CrossRef]
9. Ganna, S. W., Marsh, R. A. (2020). Pediatric hemophagocytic lymphohistiocytosis. Blood, 135, 1332–1343. DOI 10.1182/blood.2019000936. [Google Scholar] [CrossRef]
10. Marsh, R. A., Allen, C. E., McClain, K. L., Weinstein, J. L., Kanter, J. et al. (2013). Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatric Blood & Cancer, 60, 101–109. DOI 10.1002/pbc.24188. [Google Scholar] [CrossRef]
11. Jordan, M. B., Allen, C. E., Weitzman, S., Filipovich, A. H., McClain, K. L. (2011). How I treat hemophagocytic lymphohistiocytosis. Blood, 118, 4041–4052. DOI 10.1182/blood-2011-03-278127. [Google Scholar] [CrossRef]
12. Rivière, S., Galicier, L., Coppo, P., Marzac, C., Aumont, C. et al. (2014). Reactive hemophagocytic syndrome in adults: A retrospective analysis of 162 patients. The American Journal of Medicine, 127, 1118–1125. DOI 10.1016/j.amjmed.2014.04.034. [Google Scholar] [CrossRef]
13. La Rosée, P., Horne, A., Hines, M., von Bahr Greenwood, T., Machowicz, R. et al. (2019). Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood, 133, 2465–2477. DOI 10.1182/blood.2018894618. [Google Scholar] [CrossRef]
14. Tabata, R., Tabata, C., Kimura, T., Nagai, T., Yasumizu, R. (2011). Prominent granulomas in bone marrow in disseminated lymphoma with hemophagocytic syndrome. Annals of Hematology, 90, 1365–1367. DOI 10.1007/s00277-011-1187-2. [Google Scholar] [CrossRef]
15. Tabata, C., Tabata, R. (2012). Possible prediction of underlying lymphoma by high sil-2r/ferritin ratio in hemophagocytic syndrome. Annals of Hematology, 91, 63–71. DOI 10.1007/s00277-011-1239-7. [Google Scholar] [CrossRef]
16. Wang, J. S., Wang, Y. N., Wu, L., Wang, Z. (2015). Splenectomy as a treatment for adults with relapsed hemophagocytic lymphohistiocytosis of unknown cause. Annals of Hematology, 94, 753–760. DOI 10.1007/s00277-014-2276-9. [Google Scholar] [CrossRef]
17. Li, F., Yang, Y., Jin, F., Dehoedt, C., Rao, J. et al. (2015). Clinical characteristics and prognostic factors of adult hemophagocytic syndrome patients: A retrospective study of increasing awareness of a disease from a single-center in China. Orphanet Journal of Rare Diseases, 10, 20. DOI 10.1186/s13023-015-0224-y. [Google Scholar] [CrossRef]
18. Schram, A. M., Berliner, N. (2015). How i treat hemophagocytic lymphohistiocytosis in the adult patient. Blood, 125, 2908–2914. DOI 10.1182/blood-2015-01-551622. [Google Scholar] [CrossRef]
19. Wang, J., Han, W., Gao, Z., Wang, Y., Wu, L. et al. (2017). Elevation of cd16 + cd56 + nk-cells and down-regulation of serum interleukin-21 (il-21) and il-1α after splenectomy in relapsed hemophagocytic lymphohistiocytosis of unknown cause. Hematology, 22, 477–483. DOI 10.1080/10245332.2017.1311443. [Google Scholar] [CrossRef]
20. Ma, J., Jiang, Z., Ding, T., Xu, H., Song, J. et al. (2017). Splenectomy as a diagnostic method in lymphoma-associated hemophagocytic lymphohistiocytosis of unknown origin. Blood Cancer Journal, 7, e534–e534. DOI 10.1038/bcj.2016.125. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |