 Open Access
Open Access
ARTICLE

Insilico Anticancer Peptide Prediction from Curcuma longa
1 Centre for Knowledge Management & e-Governance, Atal Bihari Vajpayee Institute of Good Governance & Policy Analysis, Bhopal, India
2 Department of Bioinformatics, Maulana Azad National Institute of Technology, Bhopal, India
* Corresponding Author: Jyoti Kant Choudhari. Email:
Molecular & Cellular Biomechanics 2022, 19(4), 191-208. https://doi.org/10.32604/mcb.2022.023911
Received 18 May 2022; Accepted 02 August 2022; Issue published 27 December 2022
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Cancer is the second biggest cause of death globally, and the use of therapeutic peptides to specifically target and destroy cancer cells has gotten much interest. Cancer peptides or vaccinations are utilized to treat cancer nowadays, apart from chemotherapy, which has significant discomfort, side effects and costly. It is time demanding to identify and predict potential anticancer peptides using computational biology approaches. Thus, 3-D molecular modeling is being used to find possible ACP candidates. In this research, Curcuma longa has predicted peptide sequences were docked on breast cancer receptors and used a molecular docking technique to assess the anticipated peptides’ binding affinities to MHC molecules. A similar approach was utilized to simulate the interactions of the chosen peptide with the TCR. Additionally, the Pep10 LIRQHVASNIGIAKSKIREPIV was examined, and our findings indicated interaction with MHC classes I and II. However, the maximum binding energy was obtained with TCR at 695.61, giving strength through eight hydrogen bonds. Similarly, the Pep20, GAIIGNRKIKLQPHIIIRID, the projected, has the most significant overall binding energy with MHC classes I and II but a lower global E total value with TCR, namely −600.97 kj/Mol, and also four hydrogen bonds. This research could lead to the development of novel anticancer drugs based on the anticancer activity of the Curcuma longa medicinal plant.Keywords
Cancer therapies have increasingly been associated with surgery, chemotherapy, radiation, and hormone therapy [1,2]. These therapies are critical in treating cancer initially, but the danger of recurrence may continue throughout the dosage used, resulting in drug resistance [3]. In this modern era, novel approaches such as cancer vaccines are promising areas to challenge this condition actively as a clinical therapeutic to recognize the antigens by inducing the immune system. Cancer vaccines are promising because they can help the immune system recognize the antigens [4]. Peptide engineering creates shorter peptide fragments that target specific immune responses, such as peptide vaccines or peptidomimetics [5,6].
The interactions between the compounds and selected targets have been analyzed by insilico modeling to verify potential binding for anticancer drug targets. Docking and binding affinity calculations were carried out and compared with known anticancer agents. Regarding other immunogens, selective T and B cell-mediated positive primary immunological response is evoked by the peptide vaccines with antigenic epitopes ranging from 6–15 amino acids [7]. Antigen presentation processing pathways in the case of T-cell mediated immune response reveal that the peptide antigens are presented on the outer surface of cancer cells specific to T-cell receptors (TCR) recognized by Major Histocompatibility Complex (MHC) of class I and class II molecules [8]. Proteasome machinery is active endogenously in the Antigen-presenting cells (APCs) resulting from converting large protein fragments into short peptides with 6–10 amino acids and presenting this peptide in association with class I MHC molecules recognized by CD8+ cytotoxic T lymphocytes (CTL) [9]. Simultaneously, long peptides ranging from 11–30 amino acids were produced from exogenous antigens processing pathways combined with Class II MHC molecules, accepted by helper (CD4+) T cells [10].
X-ray crystallographic studies revealed the presence of class I MHC/peptide complexes which depict the information about the conformation of a peptide in 3-dimensional space. It is network formation in terms of bonding between peptides, and complementary MHC residues with hydrogen bonds have an affinity to hold this contact tightly [11]. Electrostatic interactions and hydrogen bonding with peptide side chains are common. However, the specific binding is governed by the interaction between charged amino acids and carboxyl-termini of peptides with the binding groove of polar residues at both ends of MHC residues [12]. When the MHC-peptide complex has been formed, the maximum portion of central residues of the peptides disclosed in the MHC complexes is identified by TCR [13].
In the engineering of peptide vaccines, the aim should be to produce efficient induction of either B-cell or cytotoxic T-cell responses. The main aspect of this is capable helper T cell responses, which are necessary for the immune system to control humoral or adaptive immunity [14,15]. So, the challenging opportunities exit remain here to distinguish active epitopes from the peptides that can evoke specific immune responses that encounter T cells to provide defensive immunity [16]. MHC is a widely diversified protein in the human population, so it is vital to deal with the candidate peptide vaccine by the APCs. The tremendous importance of peptide vaccines deals with saving time and expense reduction to recognize reliable identification of peptide vaccines because, in nature, MHC alleles and candidate peptides are found to be diverse [17].
In bioinformatics, molecular docking is a widely utilized methodology for structure-based rational drug designing. It has all the features needed to become a complete tool and solve the binding prediction for MHC and peptide affinity. Most successful attempts have been made with small molecules as ligands with molecular docking. Peptide docking is also a central thirsty area with promising outcomes to produce and subsequently find the best match with MHC classes I and II complexes. This bioinformatics approach was a better, rapid, accurate, and reliable way to predict peptide binding with MHC molecules [18,19].
This study selected only 20 peptide sequences for the three-dimensional modeling and molecular docking analysis against breast cancer. We enforced a molecular docking approach to determine the binding affinities of the predicted peptides with MHC molecules. A similar approach is used to model the interactions between the selected peptide with TCR α and β.
In this study, 165 protein sequences were downloaded from NCBI using the keyword “Curcuma longa.” Protein sequences have their uniqueness and visual language, retaining a wide variety of means, and from where we find some unique peptide designs [20]. The enormous peptide will be generated using these protein sequences insilico digestion, further evaluating the performance of our methodology. The prediction tools available in the CAMP database were utilized. It is based on various machine learning algorithms such as Support Vector Machine (SVM), Random forest (RF), Artificial neural network (ANN) and Discriminant Analysis (DA). The results accuracy for different statistical models ranges from 87% to 93%, and their antimicrobial activity is independent of length. The antibacterial potential of Curcuma longa protein fragments generated following proteolysis was evaluated using four statistical models from the CAMP database. The results were used to manually select peptides where a positive result was reported throughout the four statistical models. Thus, 20 potentially antimicrobial peptides were identified with a prediction value greater than 0.45 for at least three different statistical models. Further, all these peptides have been selected for further processing. Fig. 1 depicts the Insilico analysis process step-by-step.

Figure 1: Workflow of insilico anticancer peptide design and analysis
The 3D structure of the peptide sequences is predicted using the PEP-FOLD server. PEP-FOLD is an online tool for predicting the 3D conformation of peptides [21]. It allows the linear and disulfide bonded cyclic peptide with a limitation of 9–36 amino acids for modeling. All models are visualized using the Discovery Studio 2.5 software [22,23].
2.3 Prediction of Anticancer and Toxicity Properties
The AntiCP is a server that predicts the anticancer effects of peptides. It is used to predict the anticancer properties of peptides [23], and The Toxinpred server predicts the peptides’ toxicity. It employs a machine learning technique based on the SVM classifier [24]. Algorithms for both the tool are based on machine learning multidimensional techniques. The quantitative matrix belongs to peptides’ properties as valuable features related to the anticancer and toxicity. This server is based on the training and testing of specific features related to cancer and toxic protein datasets.
The protein-peptide docking of 20 peptides with MHC-I (1i4f, 1a07), MHC-II (1bx2, 1fv1, 1h15, 1a6a, 1j8h, 1jk8, 31qz), and TCR (3mff) was performed to determine their overall binding affinities using Hex 8.0 software. Figs. 2A–2K depict the 3D structures of MHC-I, MHC-II, and TCR αβ. It is a well-known tool for calculating and presenting possible docking pairs of protein and peptide molecules in an interactive molecular graphics computer. In this docking procedure, the ligand molecules assume to be rigid and can superimpose pairs of molecules using only their 3D shapes. Hex is one of the few docking programs based on the spherical polar fourier (SPF) correlations to accelerate the calculations and has built-in graphics to view the results. Furthermore, it is the first protein docking program to use modern graphic processor units (GPUs) to accelerate the calculation.

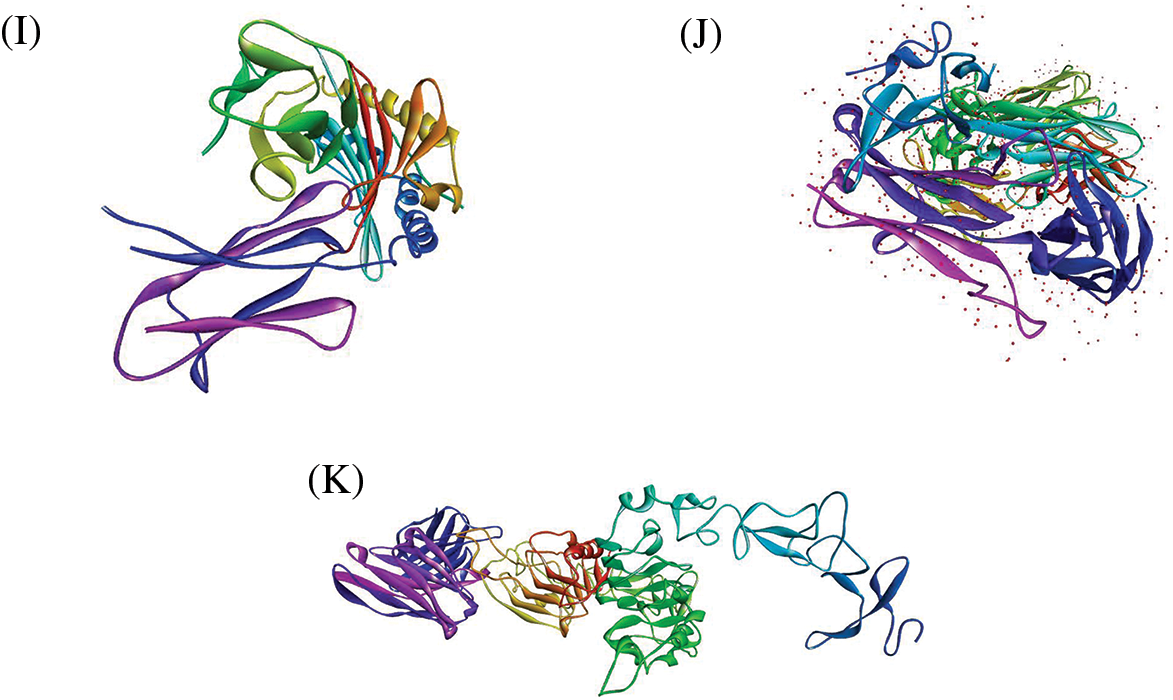
Figure 2: 3D structures of (A) MHC-I (PDB Id: 1a6a) (B) MHC-I (PDB Id: 1a07) (C) MHC-II (PDB Id: 1bx2) (D) MHC-II (PDB Id: 1fv1) (E) MHC-II (PDB Id: 1h15) (F) MHC-II (PDB Id: 1i4f) (G) MHC-II (PDB Id: 1j8h) (H) MHC-II (PDB Id: 1jk8) (I) MHC-II (PDB Id: 3lqz) (J) TCR αβ (PDB Id: 3mff) (K) HER2 and ECD (PDB Id: 6j71)
2.5 Docking Complex Visualization
Accelyra’s computational tools for protein-protein docking and protein domain development are used to develop a modified structural model [25]. The Discovery Studio 2.5 is used to analyze the docked complex [26].
In this study, the toxicity and anticancer properties of the peptide are investigated using the web server based on machine learning approaches. The AntiCp server determined that only Pep11 was toxic, as shown in Table 1. The negative SVM score predicted the possibility of a non-toxic peptide sequence in nature. All other peptides may be regarded as possible bioactive peptides against cancer for additional docking study, except Pep11. Consequently, we have predicted 20 antimicrobial peptides based on physicochemical properties (Protein length, peptide mass, hydrophobicity, steric hindrance, side bulk, hydropathicity, amphipathicity, hydrophilicity, net hydrogen, charge, pI, boman index, hydropathy index, aliphatic index, instability index, GRAVY (grand average of hydropathicity) from the proteins of medicinal plants, i.e., Curcuma longa, which can also act as an anticancer peptide. So, we consider these peptides under investigation for anticancer peptides.

Physicochemical properties of 20 predicted Antimicrobial Peptides (AMPs) from Curcuma longa have been computed and compared with the AMP patent database shown in Table 2. The comparison included the frequency and composition (%) of the amino acids in each of the 20 anticipated AMPs. The most frequent amino acids found in AMP are 10 which cover more than 66% in the proteolytic cleavage of Curcuma longa and 77% of the overall amino acid composition, i.e., GAVLIKNSTE in the patent database. On the other hand, 44% & 23% of amino acid composition cover M P W Q Y C F D R H, respectively. The content of amino acids in the proteolytic peptides was calculated and compared with the patent peptide database. The frequency of specific amino acids is more common in the AMPs and can influence their biological activity. For docking and interaction analyses, only 20 peptides were considered. The main goal of this study was to figure out which amino acids are involved in MHC classes I, II, and TCR binding to Pep1 at Pep20. Finally, compare the binding affinity of the energy state of the peptide-receptor complex. Subdomain III of Human Epidermal Growth Factor Receptor (HER2) and sub-domain III of the extracellular domain (ECD), HER 2 (Fig. 2K 6j71) is overexpressing SK-BR-3 breast cancer cells, which is one of the best crucial targets in the cancer research for the development of immunotherapeutic strategy because the cell surface proteins are overexpressed under the clinical condition like breast, gastric and ovarian cancer.

Table 3 shows that most antimicrobial peptides are cationic amphipathic peptides, also containing one or more hydrophobic residues. Hydrophobic amino acids such as I G L account for more than 30 percent of analyzed AMPs from Curcuma longa and play an important role in secondary structure formation and interactions with bacterial membrane. The cationic and hydrophobic characters peptides affect the antimicrobial activity. The analysis of amino acid content of antimicrobial peptides revealed many fragments with the predominance of one or several amino acids. In comparison with all AMPs listed in the patent database, amino acids such as V & S are less frequently encountered from Curcuma longa. On average, the evaluated peptides in both cases have a higher content of I, K & G. In both cases, the result shows the significant frequency of serine to be antimicrobial.
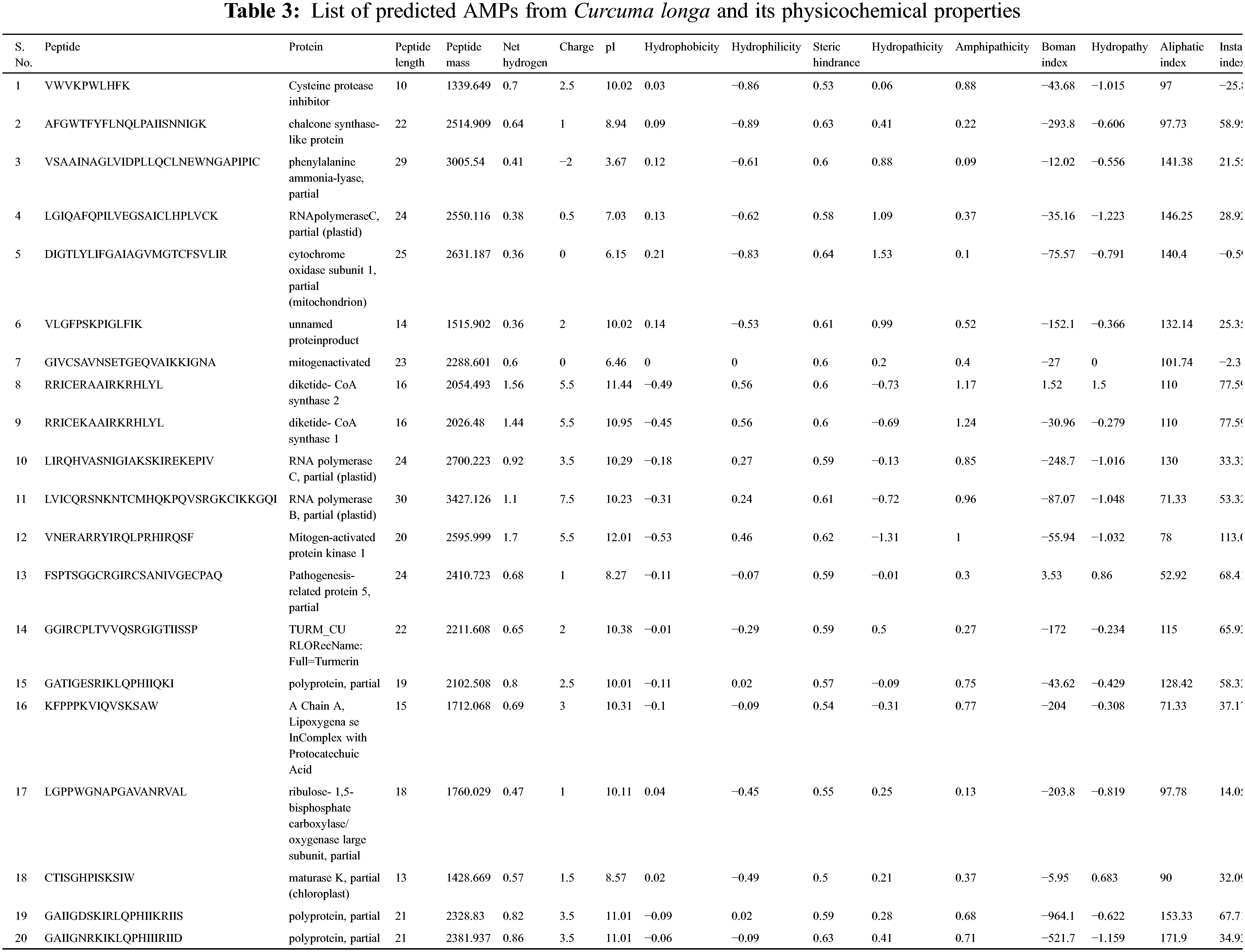
The selected peptides in our work are potentially antimicrobial AMPs with 6 to 30 amino acids, corresponding to a peptide mass of 117.175 to 3427.126 Da. The general classification of AMPs includes cationic peptides divided into three subclasses: linear peptides forming helical structures, cysteine-rich open-ended peptides containing single or several disulfide bridges and molecules rich in specific amino acids, such as proline, glycine, histidine and tryptophan. The majority of antimicrobial peptides with positive charges are known as cationic antimicrobial peptides (CAPs). In our study, 85.71%, 3.57% & 7.14% of peptides are found to have a cationic, anionic and neutral charge.
MHC involved in breast cancer interacts with predicted peptides from Curcuma longa. The highest binding energy molecules further selected and interacted with TCR to determine its affinity is shown in Table 4.

In the breast cancer, 1i4f, 1a07 MHC I, MHC II 1bx2, 1fv1, 1h15, 1a6a, 1j8h, 1jk8, 31qz and TCR 3mff. Pep5, DIGTLYLIFGAIAGVMGTCFSVLIR with MHC class I with binding global energy −210.97. Pep13, FSPTSGGCRGIRCSANIVGECPAQ with MHC class I with binding global energy −521.58. Pep3, VSAAINAGLVIDPLLQCLNEWNGAPIPIC with MHC class II binding global energy 1jk8 with −566.98 and 3Iq2 with −587.34. Pep20, GAIIGNRKIKLQPHIIIRIID with MHC class II with ng global energy of 1bx2 and 1hI5 is −747.85 and −728.14 kj/Mol, respectively. Finally, Pep10, LIRQHVASNIGIAKSKIREKEPIV with 3mff TCR has the highest binding energy −695.61 compared with Pep20 GAIIGNRKIKLQPHIIIRIID, which is −600.97 kj/Mol, is the second-highest. Binding interaction site for each receptors represents in terms of hydrogen binding HLA-A0201, HLA-DR2, DRB50101, HLA-DR3, HLA-DR4, HLA-DQ8 and HLA-DP2 MHC receptor. Pep10 also have moderate binding affinity with MHC classes I and II but further it achieves the highest binding affinity with TCR, i.e., −695.61 which is highest among all other peptides. Fig. 3A shows the interaction sites between peptide 10 with TCR are B: GLN 139: HE 21 – LEU 11: O, B: THR 138: O – GLY 1: H 1, C: PRO 90: O – GLY 5: H and C: GLN 45: O – ARG 7: HH 21. The binding interaction site for each receptor represents hydrogen binding HLA-A0201, HLA-DR2, DRB50101, HLA-DR3, HLA-DR4, HLA-DQ8, and HLA-DP2 MHC receptor. Pep10 also has a moderate binding affinity with MHC Classes I and II, but it achieves the highest binding affinity with TCR, i.e., −695.61, the highest among all other peptides. Overall, all the receptors have the highest binding energy with the peptide 20 and have the highest binding energy of −747.85 with HLA-DR 2 and −728.14 kj/Mol with DRB50101. Further, Pep20 docked to TCR and achieved a −600.97 kj/Mol global energy level, the second-highest energy among all other predicted peptides shown in Fig. 3B. The interaction sites between peptide 10 with TCR are GLN 123 : HE Z1 – PRO 22: O, LYS 140 : HZ 1 – GLU 21 : OE 1, LYS 140 : HZ 3 – GLU 21 : OE 1, LYS 140 : HZ 3 – GLU 21 : OE 2, PRO 174 : O – ARG 3 : HH 12, HIS 167 : NE2 – ASN 9 : HD 22, GLN 139: OE 1 – SER 15: HG and GLN 123 : OE 1 – ARG 18 : HH 11, ARG 18 (see Appendix Tables 1A and 1B in the supplemental material).

Figure 3: Docking complex with interaction (A) Pep10 with TCR (B) Pep20 with TCR
These findings indicated that Pep10 and 20 could strongly bind to MHC and TCR. This interaction leads to only four hydrogen bonds, which have effective binding. Thus, it could generate an effective immune response against HER 2 receptors to prevent the growth of human breast cancer cells.
Our knowledge of the significance of peptides may be used directly as a cytotoxic agent or as a transporter of cytotoxic chemicals and radioisotopes to cancer cells. Peptide-based hormone treatment has been thoroughly studied and treated with breast and prostate cancers. In this research work, two peptides, Pep10 and Pep20, have been identified as having a significant impact on stopping cancer progression. The Pep10 has moderate binding energy with eight hydrogen bonds, and Pep20 achieves the highest binding energy with four hydrogen bonds on the MHC classes I and II. Both peptides are harmless, according to computational methods based on experiments. It could ultimately serve as a novel essential therapeutic target as a cancer peptide vaccine in the future. This method might aid researchers in achieving better disease regulation and predicting treatment targets of disease.
Funding Statement: The authors received no specific funding for this study.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Garrett, D., Yoder, L. H. (2007). An overview of stem cell transplant as a treatment for cancer. MedSurg Nursing, 16(3), 183. [Google Scholar]
2. Garrett, J. T. (2007). Peptide-based B-cell epitope vaccines targeting HER-2/neu (Ph.D. Thesis). The Ohio State University. [Google Scholar]
3. Calabrich, A., Fernandes, G. D. S., Katz, A. (2008). Trastuzumab: Mechanisms of resistance and therapeutic opportunities. Oncology, 22(11), 1250–1258. [Google Scholar]
4. Xiang, B., Snook, A. E., Magee, M. S., Waldman, S. A. (2013). Colorectal cancer immunotherapy. Discovery Medicine, 15(84), 301–308. [Google Scholar]
5. Li, W., Joshi, M. D., Singhania, S., Ramsey, K. H., Murthy, A. K. (2014). Peptide vaccine: Progress and challenges. Vaccines, 2(3), 515–536. [Google Scholar]
6. Reche, P. A., Fernandez-Caldas, E., Flower, D. R., Fridkis-Hareli, M., Hoshino, Y. (2014). Peptide-based immunotherapeutics and vaccines. Journal of Immunology Research, 2014, 256784. DOI 10.1155/2014/256784. [Google Scholar] [CrossRef]
7. SlingluffJr., C. L. (2011). The present and future of peptide vaccines for cancer: Single or multiple, long or short, alone or in combination? Cancer Journal, 17(5), 343–350. [Google Scholar]
8. Zhang, X. W. (2013). A combination of epitope prediction and molecular docking allows for good identification of MHC class I restricted T-cell epitopes. Computational Biology and Chemistry, 45(23–24), 30–35. DOI 10.1016/j.compbiolchem.2013.03.003. [Google Scholar] [CrossRef]
9. Bian, H., Reidhaar-Olson, J. F., Hammer, J. (2003). The use of bioinformatics for identifying class II-restricted T-cell epitopes. Methods, 29(3), 299–309. DOI 10.1016/S1046-2023(02)00352-3. [Google Scholar] [CrossRef]
10. Martin, W., Sbai, H., de Groot, A. S. (2003). Bioinformatics tools for identifying class I-restricted epitopes. Methods, 29(3), 289–298. DOI 10.1016/S1046-2023(02)00351-1. [Google Scholar] [CrossRef]
11. Garboczi, D. N., Ghosh, P., Utz, U., Fan, Q. R., Biddison, W. E. et al. (1996). Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature, 384(6605), 134–141. DOI 10.1038/384134a0. [Google Scholar] [CrossRef]
12. Jardetzky, T. S., Brown, J. H., Gorga, J. C., Stern, L. J., Urban, R. G. et al. (1996). Crystallographic analysis of endogenous peptides associated with HLA-DR1 suggests a common, polyproline II-like conformation for bound peptides. Proceedings of the National Academy of Sciences, 93(2), 734–738. DOI 10.1073/pnas.93.2.734. [Google Scholar] [CrossRef]
13. Tong, J. C., Tan, T. W., Ranganathan, S. (2004). Modeling the structure of bound peptide ligands to major histocompatibility complex. Protein Science: A Publication of the Protein Society, 13(9), 2523–2532. DOI 10.1110/(ISSN)1469-896X. [Google Scholar] [CrossRef]
14. Dakappagari, N. K., Douglas, D. B., Triozzi, P. L., Stevens, V. C., Kaumaya, P. T. (2000). Prevention of mammary tumors with a chimeric HER-2 B-cell epitope peptide vaccine. Cancer Research, 60(14), 3782–3789. [Google Scholar]
15. Manijeh, M., Mehrnaz, K., Violaine, M., Hassan, M., Abbas, J. et al. (2013). In silico design of discontinuous peptides representative of B and T-cell epitopes from HER2-ECD as potential novel cancer peptide vaccines. Asian Pacific Journal of Cancer Prevention, 14(10), 5973–5981. DOI 10.7314/APJCP.2013.14.10.5973. [Google Scholar] [CrossRef]
16. Wang, B., Kaumaya, P. T., Cohn, D. E. (2010). Immunization with synthetic VEGF peptides in ovarian cancer. Gynecologic Oncology, 119(3), 564–570. DOI 10.1016/j.ygyno.2010.07.037. [Google Scholar] [CrossRef]
17. Flower, D. R. (2003). Databases and data mining for computational vaccinology. Current Opinion in Drug Discovery & Development, 6(3), 396–400. [Google Scholar]
18. Patronov, A., Dimitrov, I., Flower, D. R., Doytchinova, I. (2011). Peptide binding prediction for the human class II MHC allele HLA-DP2: A molecular docking approach. BMC Structural Biology, 11(1), 1–10. DOI 10.1186/1472-6807-11-32. [Google Scholar] [CrossRef]
19. Young, D. C. (2009). Computational drug design: A guide for computational and medicinal chemists. USA: John Wiley & Sons. DOI 10.1002/9780470451854. [Google Scholar] [CrossRef]
20. D’Incalci, M., Steward, W. P., Gescher, A. J. (2005). Use of cancer chemopreventive phytochemicals as antineoplastic agents. The Lancet Oncology, 6(11), 899–904. DOI 10.1016/S1470-2045(05)70425-3. [Google Scholar] [CrossRef]
21. Maupetit, J., Derreumaux, P., Tuffery, P. (2009). PEP-FOLD: An online resource for de novo peptide structure prediction. Nucleic Acids Research, 37(suppl_2), W498–W503. DOI 10.1093/nar/gkp323. [Google Scholar] [CrossRef]
22. Guex, N., Peitsch, M. C. (1997). SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis, 18(15), 2714–2723. DOI 10.1002/(ISSN)1522-2683. [Google Scholar] [CrossRef]
23. Scott, W. R., Hünenberger, P. H., Tironi, I. G., Mark, A. E., Billeter, S. R. et al. (1999). The GROMOS biomolecular simulation program package. The Journal of Physical Chemistry A, 103(19), 3596–3607. DOI 10.1021/jp984217f. [Google Scholar] [CrossRef]
24. Gupta, S., Kapoor, P., Chaudhary, K., Gautam, A., Kumar, R. et al. (2013). In silico approach for predicting toxicity of peptides and proteins. PLoS One, 8(9), e73957. DOI 10.1371/journal.pone.0073957. [Google Scholar] [CrossRef]
25. Huang, H. J., Lee, K. J., Yu, H. W., Chen, H. Y., Tsai, F. J. et al. (2010). A novel strategy for designing the selective PPAR agonist by the “Sum of Activity” model. Journal of Biomolecular Structure and Dynamics, 28(2), 187–200. DOI 10.1080/07391102.2010.10507352. [Google Scholar] [CrossRef]
26. Accelrys Software Inc. (2009). Discovery studio 2.5. San Diego: Accelrys Software Inc. [Google Scholar]

Cite This Article
 Copyright © 2022 The Author(s). Published by Tech Science Press.
Copyright © 2022 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools