| Journal of Renewable Materials |
DOI: 10.32604/jrm.2021.015604
ARTICLE
Improving Polylactide Toughness by Plasticizing with Low Molecular Weight Polylactide-Poly(Butylene Succinate) Copolymer
1Manufacturing and Materials Research Unit, Department of Manufacturing Engineering, Faculty of Engineering, Mahasarakham University, Mahasarakham, 44150, Thailand
2Department of Chemistry, Faculty of Science and Technology, Rajabhat Maha Sarakham University, Mahasarakham, 44000, Thailand
3Polymer Engineering Center, Department of Mechanical Engineering, University of Wisconsin-Madison, Madison, 53705, USA
4King Mongkut Institute of Technology Ladkrabang, Bangkok, 10520, Thailand
*Corresponding Author: Yottha Srithep. Email: yottha.s@msu.ac.th
Received: 31 December 2020; Accepted: 03 February 2021
Abstract: A low-molecular-weight polylactide-poly(butylene succinate) (PLA-PBS) copolymer was synthesized and incorporated into polylactide (PLA) as a novel toughening agent by solvent casting. The copolymer had the same chemical structure and function as PLA and it was used as a plasticizer to PLA. The copolymer was blended with PLA at a weight ratio from 2 to 10 wt%. Phase separation between PLA and PLA-PBS was not observed from their scanning electron microscopy (SEM) images and the crystal structure of PLA almost remained unchanged based on the X-ray diffraction (XRD) measurement. The melt flow index (MFI) of the blends was higher as the amount of PLA-PBS increased, indicating that the block copolymer did improve the mobility of the PLA chains. Moreover, tensile tests revealed that PLA with greater PLA-PBS copolymer exhibited higher elongation at break and it reached the maximum at 8 wt% of PLA-PBS in PLA, which was around 6 times higher than that of pure PLA. Furthermore, the glass transition temperature, measured by differential scanning calorimetry (DSC), markedly decreased with an increasing amount of the copolymer as it decreased from 61.2°C for pure PLA to 41.3°C when it was blended with 10 wt% PLA-PBS copolymer. Therefore, the PLA-PBS copolymer was shown to be a promising plasticizer for fully biobased and toughened PLA.
Keywords: Polylactide; poly (butylene succinate); low molecular weight copolymer; biodegradability; plasticizer
Polylactide (PLA) is a biodegradable polymer, which is derived from renewable resources such as corn, potato, cassava, cane, etc. It is now widely used in many applications, e.g., medical uses, films, and packaging [1]. The mechanical and thermal properties of PLA are mostly influenced by the monomer composition and degree of polymerization. One of the essential polymer properties is molecular weight, which dictates the material strength and brittleness that are essential factors for quality control and product development. PLA can be synthesized either by direct polycondensation of lactic or by ring opening polymerization (ROP) of lactides. In direct condensation, solvent is used, and higher reaction times are required. The resulting polymer has low molecular weight and poor mechanical properties. On the other hand, ring opening polymerization needs a catalyst, but the molecular weight can be controlled by selecting the monomer and reaction conditions [2].
PLA is relatively brittle, has low elongation-at-break and crystallizes slowly. Therefore, its applications in some areas, such as films for the packaging industry, are limited [1,3]. These limitations can be overcome by several techniques, such as copolymerization, plasticization, and polymer blending [4,5]. Li et al. [6] reported that blending PLA with other soft and biodegradable polymers, e.g., poly(butylene succinate) (PBS), polybutylene(adipate terephthalate) (PBAT), and poly(vinyl alcohol) (PVA), circumvented the brittleness of PLA. They showed that as the PBAT content increased in the PLA matrix, the elongation-at-break and toughness of PLA/PBAT blends improved considerably, while tensile strength and tensile modulus reduced [7]. Nevertheless, these soft polymers are not miscible with PLA, leading to a weak interfacial bond between PLA and the elastic polymers, due to inadequate chain entanglement across the interface [8]. To reduce that problem, PLA and PBS were used to form a single chain of block copolymer via bulk ring opening polymerization from L-lactide and still maintain biodegradability [9,10].
Further, additives, such as plasticizers, are commonly added to the PLA to further improve its flexibility. Plasticizers are low-to-medium molecular weight compounds, up to a few thousand monomers. In some applications, e.g., film formation, an appropriate plasticizer greatly improves processing. However, the choice of plasticizer, especially in biobased applications, may be limited due to required safety, environmental conditions, and chemical and physical properties that dictate their miscibility [11]. A wide variety of plasticizers have been used, including the widely studied poly(ethylene glycol) (PEG), but also many others, e.g., oligomeric lactic acid (OLA), poly(propylene glycol) (PPG), glycerol, tributyl citrate, citrate ester, triacetine, acetyltriethyl citrate, tributyl citrate oligomers, glucosemonoesters, diethyl bishydroxymethyl malonateoligomers, partial fatty acid esters, and triphenyl phosphate [12]. Biodegradable plasticizers are particulary relevant and include triethyl citrate (TEC) and acetyl tributyl citrate (ATBC) [11,13], and recycled poly(3-hydroxybutyrate) (PHB) [14]. Migration of the plasticizer in contact with aqueous solutions is a key problem [14]. At high TEC concentrations, while they successfully increased the MFI of PLA by factors of 6, up to 20% of the plasticizer was lost at 100°C. However, the loss of ATBC was much lower—up to 5% [13]. This migration increases rigidity over extended periods, e.g., in storage, and limits PLA’s applications where flexibility is required. Efforts to retain the plasticizer in the polymer structure include using reactive end groups to create covalent bonding and anchor the plasticizer [14] and adding fillers, such as CaCO3 and chitin nanofibrils [11]. Long chain bio-derived compounds have also been used. For example, cardanol(m-pentadecenyl phenol), which is derived from cashew nut shells and exhibits a 15 C chain with a phenol end group that allows interaction with the polymer, has been found to provide flexibility to the PLA [15].
In this study, we aimed to meet broader application requirements, by improving the toughness of PLA with no plasticizer migration, while maintaining full biodegradability. The idea is to use additives that have the same chemical structure and function as PLA as a plasticizer, such as low-molecular-weight PLA-PBS copolymers.
Polylactide (Luminy L175, Total Corbion Co., Ltd., Thailand), with a density of 1,250 kg/m3 and a melt flow index (MFI) of 8 g/10 min at 210°C, contained ≥ 99% L-lactic acid. The macroinitiator, biobased poly(butylene succinate) (PBS, grade FZ71PM) was purchased from PTT MCC Biochem Co., Ltd., Thailand. It had an MFI of 22 g/10 min at 190°C and a melting temperature 115°C. Stannous octoate (Sn(Oct)2) was obtained from Sigma-Aldrich. Methanol, tetrahydrofuran (THF), and chloroform (CHCl3), were acquired from RCI Labscan Limited, Thailand.
2.2 Synthesis of Low Molecular Weight PLA-PBS Block Copolymer
Low-molecular-weight polylactide-poly(butylene succinate) (PLA-PBS) block copolymer was prepared from L-lactide. Pellets were dried for 7 h at 60°C under vacuum before use. The PLA-PBS block copolymers were synthesized by ring-opening polymerization under nitrogen for 4 h at 120°C. L-lactide 1 mol (144.1 g), PBS 0.002 mol (60 g) was used as a macroinitiator, and Sn(Oct)2 3.2 mL (1% mol of L-lactide) was used as a catalyst. After the reaction was complete, the polymers were dissolved in CHCl3 and then precipitated by adding cold methanol. Finally, PLA-PBS block copolymer was dried overnight in an oven at 45°C. The synthesis of low-molecular-weight polylactide-poly(butylene succinate) is shown in Sch. 1.

Schematic 1: Synthesis of low-molecular-weight polylactide-poly(butylene succinate)
2.3 Preparation of Polylactide/PLA-PBS Block Copolymer Blends
Blends of PLA with low-molecular-weight PLA-PBS block copolymer were prepared by solution casting. The block copolymer was blended with PLA at 2, 4, 6, 8, and 10 wt%, by dissolving in chloroform using a magnetic stirrer (3 h, ~25°C). The resulting mixed solution was poured onto glass petri dishes. Films were dried for 3 days at ~25°C to remove the solvent, and the sample was further vacuum dried to a constant weight. Final films were ~1 mm thick.
2.4.1 Gel Permeation Chromatography (GPC)
GPC was used to determine number average molecular weights (Mn), weight average molecular weights (Mw), and polydispersity index (PDI) of the PLA-PBS block copolymer, PLA, and PBS. Approximately 5 mg specimens were dissolved in 3 ml tetrahydrofuran (THF). A Waters 2414 refractive index (RI) detector was used. The GPC columns were eluted using THF at a flow rate of 1.0 mL/min. The columns were calibrated with polystyrene standards.
2.4.2 Proton Nuclear Magnetic Resonance (1H-NMR)
The functional groups in the PLA-PBS block copolymer were determined from 1H-NMR spectra (Bruker Advanced DPX at 300 MHz using CDCl3 solvent at room temperature) and compared with the PLA backbone signal.
2.4.3 Fourier Transform Infrared Spectroscopy (FTIR)
The chemical structures of PLA and PLA-PBS block copolymer were analyzed by Fourier transform infrared spectra (FTIR) using the attenuated total reflection (ATR) mode, from 400 to 4000 cm–1 at 2 cm–1 resolution.
2.4.4 Scanning Electron Microscope (SEM)
The sample cross section surface was recorded with a scanning electron microscope (TM4000Plus Tabletop Microscope, HITACHI) operated at 10 kV. Samples were frozen in liquid nitrogen and quickly impacted fractured. Specimens were coated with a thin layer of gold (20 nm in thickness) before the examination.
Melt flow indices were obtained with an extrusion plastometer (melt flow indexer), model MP1200 polymer tester (Tinius Olsen, Horsham, PA, USA) following ASTM D1238. Samples were measured at 210°C with a load of 2.16 kgs. The MFI (g/10 min) is the rate of extrusion of the sample, under a static load pressure through an orifice.
Mechanical properties of the PLA/PLA-PBS blends were tested by a tensile tester (Texture Analyzer Stable Micro System Model TA.XT Plus) at a cross speed of 2 mm/min. Thin 15 × 50 mm samples were cut from the cast films. Young’s modulus, tensile strengths, and elongation at break were measured at a relative humidity of 50 ± 5% and 25°C. Five samples of each group were tested and averaged.
2.4.7 Differential Scanning Calorimetry (DSC)
The thermal properties of the mixtures were evaluated by differential scanning calorimetry (DSC) (PerkinElmer DSC PyrisDimond). About 5 mg samples were cut from the films and placed in aluminum pans. They were then heated from 25°C to 200°C at a rate of 10 °C/min and held for 5 min at 200°C to remove previous thermal history before cooling at 10 °C/min to 25°C. The specimens were then reheated to 200°C, using the same heating rate. The cold crystallization temperature (Tcc), and its enthalpy (ΔHcc), the melting temperature (Tm), and its enthalpy (ΔHm), as well as glass transition temperature (Tg), were recorded. The absolute degree of crystallinity (χc) of the PLA and blended PLA was calculated by
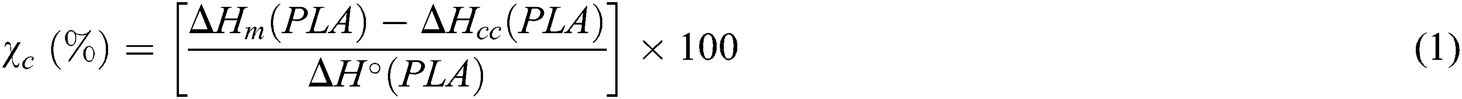
where ΔH°(PLA) is the melting enthalpy per gram with 100% crystals (i.e., perfect crystalline structure) (93 J/g) [16].
2.4.8 Wide-Angle X-Ray Diffraction (WAXD)
WAXD was used to probe the crystallinity of the neat PLA and PLA blended with PLA-PBS copolymer. The films were analyzed using X-ray diffractometry (Bruker/D8 Advance Bruker-Biospin Ag, Bruker, Waltham, MA, USA). A 2θ range from 5° to 40° was scanned at 2 °/min at room temperature. The source was Cu Kα under a thin Ni filter.
2.4.9 Thermogravimetric Analysis (TGA)
Thermal properties were measured with a thermogravimetric analyzer (PerkinElmer TGA4000). About 10 mg samples were heated from 10°C to 600°C with a heating rate of 10°C/min under N2 flowing at 20 ml/min. The weight loss was recorded and referred to the initial weight. The thermal degradation temperatures were specified from the decomposition temperatures at 20% weight loss (T20%). Samples were dried at 60°C overnight prior to the test.
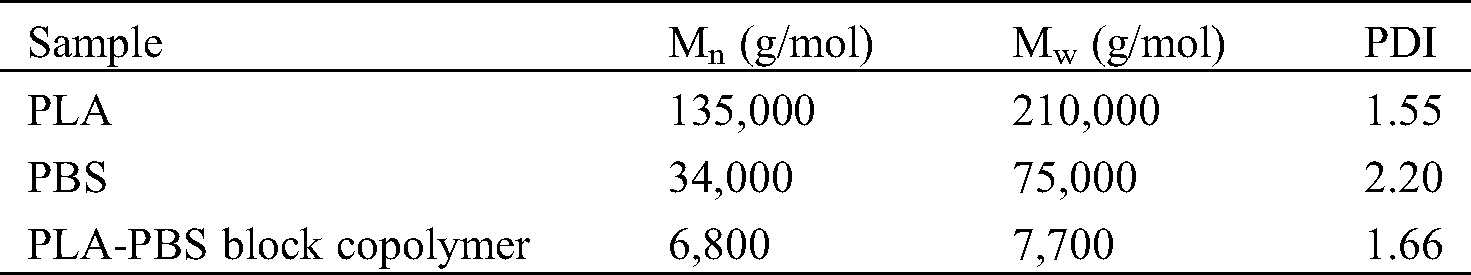
The number average molecular weights (Mn), weight average molecular weights (Mw), and polydispersity indices (PDI) of PLA, PBS, and PLA-PBS block copolymer as measured by GPC are shown in Tab. 1.
Table 1: Molecular weights of PLA, PBS, and PLA-PBS block copolymer
1H-NMR spectra of PLA, PBS, and PLA-PBS block copolymer are shown in Fig. 1. The spectrum of pure PLA (spectrum a) had two 1H peaks at 1.53 ppm (Peak 1) and 5.18 ppm (Peak 2). Peak 1 was assigned to the methyl H (–OCH3) and Peak 2 to the methane H (–OCH) of the PLA [9]. For pure PBS (spectrum b), the peak at 4.10 ppm (Peak 4) and 1.69 ppm (Peak 5) were attributed to H atoms in the end and central CH2 groups in the 1,4-butanediol segments, while the chemical shift of methylene H (–C=OCH2CH2) in succinic acid appeared at 2.61 ppm (Peak 3) [17]. For the PLA-PBS block copolymer (spectrum (c)), the signals at 1.56 ppm (Peak 1') were assigned to the methyl H (-OCH3), and the signals at 5.16 ppm (Peak 2') assigned to methane H (–OCH) in the PLA unit. Additional signals were observed in the copolymer spectra at 1.69 ppm (Peak 5') and 4.10 ppm (Peak 4'), which were assigned to H atoms in the 1,4-butanediol segments. Moreover, the 2.6 ppm signal (Peak 3') was assigned to methylene H (–C=OCH2CH2) in the butylene succinate monomer. This showed that the PBS hydroxyl group had started the L-lactide ring-opening polymerization, and the polymerization was completed by linking through the carbonyl group of the L-lactide [18], showing that the PLA-PBS copolymer had been formed.

Figure 1: 1H-NMR spectra of (a) PLA, (b) PBS and (c) PLA-PBS block copolymer
Fig. 2 shows the FTIR spectra from 400 to 4000 cm–1 of (a) PLA, (b) PBS, and (c) PLA-PBS block copolymer. The FTIR spectra clearly indicated that PLA and PLA-PBS block copolymer had comparable chemical bonding because of identical functional groups. Two distinctive peaks in the 2850–2980 cm–1 region were related to the symmetrical and asymmetrical stretching modes of the CH3 groups. In each spectrum, the C=O group occurred in the area of 1751–1758 cm–1, whereas that of C–O–C appeared at 1050–1200 cm–1. Peaks in the 1430–1480 cm–1 area were C–H deformation, whereas those at 733–756 cm–1 corresponded to the methylene groups [19]. In PBS, the group at 917 cm–1 was related to the stretching vibration of the carboxylic acid groups (C–OH). The peak around 1044–1046 cm–1 was assigned to the O–C–C bending, and the peak in the range 1144–1264 cm–1 was caused by the C–O–C groups in the ester linkage of PBS. The band at 1325 cm–1 was created from the asymmetric vibration of the CH2 group in the PBS backbone. The group at 1710–1713 cm–1 was formed from the bending of the PBS ester group (C=O) [20].
The FTIR spectra of PLA-PBS block copolymer were distinct from those of PLA, because a peak at ~800 cm–1 was observed: This peak is a rocking of the CH2 group in PBS, indicating that PLA-PBS block copolymer had formed.

Figure 2: FTIR spectra for (a) PLA, (b) PBS, and (c) PLA-PBS block copolymer
Mechanical properties of PLA and the PLA/PLA-PBS blends were evaluated by tensile test. Stress-strain curves are shown in Fig. 3 and the derived properties are listed in Tab. 2. An increase in PLA-PBS block copolymer content led to a gradual decline of tensile modulus and tensile strength, but an increase in elongation-at-break. The reductions of tensile modulus and tensile strength with increasing copolymer were attributed to block copolymer interference with the PLA chains, allowing enhanced chain movement. Therefore, the elongation-at-break of the blend was better than that of pure PLA. The strain for the PLA blended with 8 wt% PLA-PBS block copolymer increased from 10% to 60% (Tab. 2). However, when the amount of block copolymer increased to 10%, the elongation at break started to decrease. This suggested that the copolymer, which had a low molecular weight, was dispersed well at lower concentration levels (8 wt%) [21], but started to aggregate when a further amount was added so that the material became brittle again.

Figure 3: Stress-strain curve of PLA and PLA/PLA-PBS block copolymer blends
Table 2: Tensile properties of PLA and PLA/PLA-PBS block copolymer blends

3.5 Differential Scanning Calorimetry (DSC)
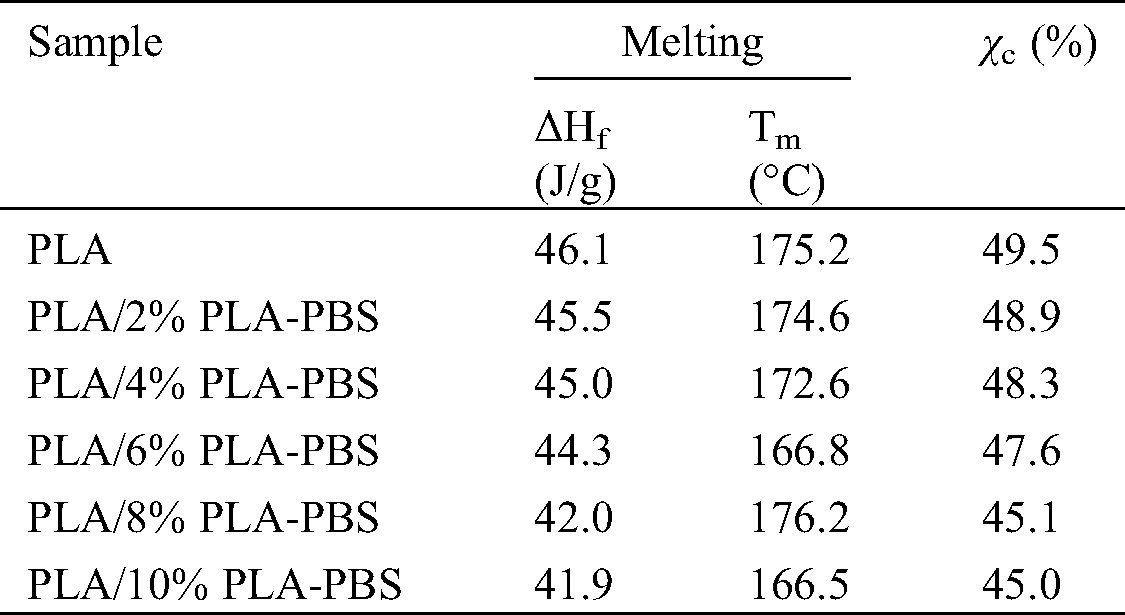
DSC curves (Fig. 4) of PLA and PLA/PLA-PBS blends show the heating thermograms and melting peak temperatures for varying amounts of the block copolymer. The derived thermal parameters are listed in Tab. 3. The endothermic peaks observed at 166.5°C to 175.2°C can be assigned to the melting peak of PLA. As the amount of PLA-PBS copolymer increased, the melting point and enthalpy of melting became smaller. The decreased melting point was due to enhanced mobility of segments of the PLA chains, caused by the presence of PLA-PBS copolymer as plasticizer. That is, increasing the PLA-PBS content led to higher polymer flexibility [22].

Figure 4: First heating scan DSC thermograms for PLA and PLA/PLA-PBS blends
Table 3: Temperatures and melting enthalpies obtained from the first heating cycle
PLA exhibits slow crystallization rates [16]. Tab. 4 and Fig. 5 show the effect of the copolymer on the crystallization temperature (Tc). As the content of PLA-PBS copolymer increased, Tc steadily declined. The copolymer showed robust polar and hydrogen bond interactions with PLA. It acted as a polymer diluent and reduced the Tc of PLA. Moreover, the area underneath the peak, which corresponded to crystallization enthalpy (ΔHc), increased with the content of copolymer. That is, the copolymer enhanced the PLA mobility, leading to a higher degree of crystallization as reflected by an increase of ΔHc from 4.9 J/g for pure PLA to 17.8 J/g when 10%PLA-PBS was added.

Figure 5: Cooling scan thermograms of PLA and PLA/PLA-PBS block copolymer blends
Table 4: Crystallization temperatures and enthalpies obtained from the cooling cycle
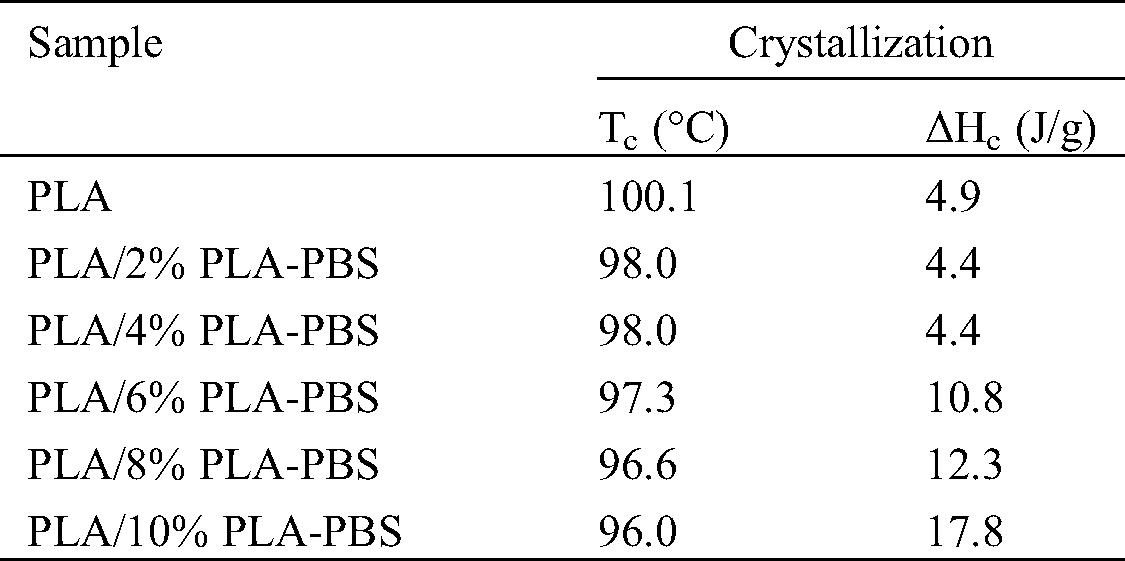
Fig. 6 and Tab. 5 show the thermograms and calculated data of the PLA/PLA-PBS films from the second heating cycle. Unlike the first heating cycle, glass transition and cold crystallization peaks were observed because the samples were cooled at a significantly faster rate (10 °C/min) during the previous cooling cycle than when the films were dried during the solution casting for the chloroform to evaporate. This is consistent with the assumption that, at lower cooling rates, the polymer chains would have more time to relax and combine into an organized crystalline structure, facilitating crystallization and leading to higher degrees of crystallinity [23].

Figure 6: Second heating scan DSC thermograms for PLA and PLA/PLA-PBS blends
Table 5: Temperatures and crystallization enthalpies obtained from the second heating cycle
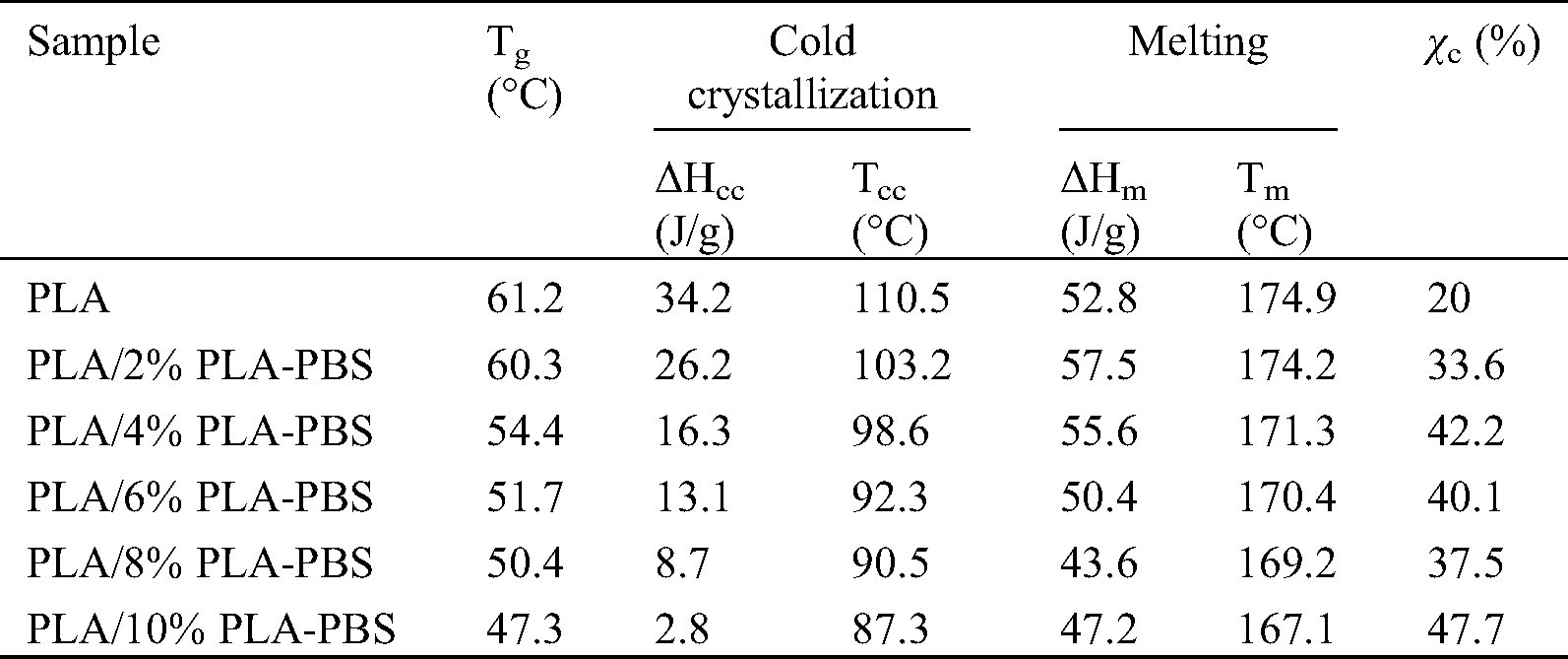
As shown in Fig. 6 and Tab. 5, as the amount of the block copolymer increased, Tg,, Tcc, and Tm of PLA decreased. This further demonstrated that the block copolymer increased the segmental mobility of PLA. The Tg of PLA was 61.2°C and it was consistently reduced to 47.3°C, when 10 wt% PLA-PBS copolymer was blended.
The cold crystallization temperatures (Tcc) for the blends appeared in the range, from 87.3°C for PLA/10%PLA-PBS to the maximum at 110.5°C for pure PLA, and enthalpies (ΔHcc) of the PLA/PLA-PBS block copolymer blends decreased following the trend similar to the shift in Tg. This can be related to two different phenomena. The first one was the reduced crystallization induction period, due to the presence of crystal nuclei, already formed during cooling [12]. The second phenomenon was the increased chain mobility at lower temperatures, associated with the higher Tg depression. This Tg reduction enabled crystallization to begin at a lower temperature.
Neat PLA melted at ~174°C, and when the copolymer was added at 10 wt%, the melting point decreased to 167°C. The small molecular size of the PLA-PBS block copolymer permitted it to occupy intermolecular spaces among polymer chains, reducing the energy for molecular motion and the creation of hydrogen bonding between the polymer chains, which sequentially increased free volume and molecular flexibility. Accordingly, the material was more prone to soften when heated. The lower melting point suggested that PLA/PLA-PBS block copolymer blends were more readily processable than neat PLA.
Fig. 7 shows the MFI values of PLA and the copolymer blends under a 2.16 kg load at 210°C. MFI for PLA was 8.34 g/10 min. As the quantity of the copolymer increased, MFI steadily increased. The 10 wt% PLA-PBS blend had the highest MFI 30.1 g/10 min. When PLA-PBS was added in PLA, the small block copolymer molecule could diffuse into the PLA matrix and make the PLA molecules slip and flow more easily [24], thereby increasing the polymer chain movement. This led to a decrease in viscosity and an increase in the MFI as compared to PLA [13].

Figure 7: MFI of PLA and PLA/PLA-PBS blends at various PLA-PBS copolymer contents
Fig. 8 shows SEM micrographs of cryo-fractured surfaces of neat PLA and the copolymer blends. As shown in Fig. 8a, the neat PLA showed a fairly smooth fracture surface, exhibiting a typical brittle fracture behavior and low impact strength [25]. Meanwhile, fractured PLA with 2–6 wt% PLA-PBS block copolymer (Figs. 8b–8c) showed craggy surfaces and exhibited more ductile fracture behavior than the neat PLA. Whereas the blends with 8 wt% PLA-PBS block copolymer (Fig. 8e) was rougher, implying local ductile regions were generated during fracture. This showed that the block copolymers improved the interfacial adhesion of PLA. However, when the amount of copolymer increased to 10 wt% (Fig. 8f), the copolymer dispersed phases were similar to the fracture surfaces of the 6 wt% copolymer blends. However, there was no distinct phase between PLA and PLA-PBS block copolymer even at 10 wt% PLA-PBS, at which the material started to aggregate, indicating that both materials were miscible.

Figure 8: SEM micrographs of fractured surfaces of (a) neat PLA, (b) PLA/2% PLA-PBS block copolymer, (c) PLA/4% PLA-PBS block copolymer, (d) PLA/6% PLA-PBS block copolymer, (e) PLA/8% PLA-PBS block copolymer, and (f) PLA/10% PLA-PBS block copolymer (Scale bars represent 50 µm)
Fig. 9 shows WAXD profiles of neat PLA and PLA/PLA-PBS block copolymer films at room temperature. Neat PLA displayed three sharp diffraction peaks at around 16.7°, 18.8°, and 23.4°, respectively [12]. For the PLA/PLA-PBS blends, they included all the diffraction peaks corresponding to neat PLA, and the intensity of the diffraction peaks of PLA diminished with increasing PLA-PBS content. This suggested that PLA/PLA-PBS blends did not alter PLA’s crystal structure but reduced the diffraction peak intensity. The lower intensity of the diffraction peak indicated that the PLA-PBS block copolymer decreased the crystallinity-similar behavior was reported in Sub-section 3.5.1: First heating cycle.

Figure 9: XRD patterns of the neat PLA and PLA/PLA-PBS blends
The thermal stability of the neat PLA and PLA/PLA-PBS blend samples, measured using TGA, is shown in Fig. 10 and Tab. 6. The initial PLA decomposition temperature moved systematically to a lower temperature when copolymer was added. As copolymer was added, the 20% weight loss decomposition temperature, T20%, moved from 385°C for pure PLA to 383°C and 381°C when 6% wt and 10% wt of copolymer was added, respectively. This further confirmed that the copolymer improved the PLA molecule mobility and the van der Waals interactions between the molecules reduced, leading to decreased thermal stability [26].

Figure 10: TGA curves for PLA and PLA/PLA-PBS block copolymer blends
Table 6: Degradation temperatures, T20%, of neat PLA and PLA/PLA-PBS blends

PLA films can be made tougher by adding small amounts of PLA-PBS copolymer as plasticizer and remain fully biodegradable. SEM micrographs showed that the two polymers were miscible and WAXD spectra showed that the crystal structure remained substantially unchanged. Melting points and glass transition temperatures of the blends were reduced with increasing amounts of the copolymer. When adding 8%PLA-PBS to PLA, elongation-at-break of blended material improved approximately 6 times compared to the pure PLA. These measurements demonstrated that the copolymer interfered with the PLA chains, allowing enhanced chain mobility. However, toughness diminished as the amount of copolymer exceeded 8 wt%, due to aggregation of the PLA-PBS copolymer.
Funding Statement: This research project was financially supported by Mahasarakham University (Fast Track 2020).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Lim, L. T., Auras, R., Rubino, M. (2008). Processing technologies for poly(lactic acid). Progress in Polymer Science, 33(8), 820–852. DOI 10.1016/j.progpolymsci.2008.05.004. [Google Scholar] [CrossRef]
2. Pholharn, D., Cheerarot, O., Baimark, Y. (2017). Stereocomplexation and mechanical properties of polylactide-b-poly(propylene glycol)-b-polylactide blend films: Effects of polylactide block length and blend ratio. Chinese Journal of Polymer Science, 35(11), 1391–1401. DOI 10.1007/s10118-017-1989-3. [Google Scholar] [CrossRef]
3. Hassouna, F., Raquez, J. M., Addiego, F., Toniazzo, V., Dubois, P. et al. (2012). New development on plasticized poly (lactideChemical grafting of citrate on PLA by reactive extrusion. European Polymer Journal, 48(2), 404–415. DOI 10.1016/j.eurpolymj.2011.12.001. [Google Scholar] [CrossRef]
4. Peng, Q., Mahmood, K., Wu, Y., Wang, L., Liang, Y. et al. (2014). A facile route to realize the copolymerization of l-lactic acid and ε-caprolactone: Sulfonic acid-functionalized Brønsted acidic ionic liquids as both solvents and catalysts. Green Chemistry, 16(4), 2234–2241. DOI 10.1039/C3GC42477D. [Google Scholar] [CrossRef]
5. Xiu, H., Huang, C., Bai, H., Jiang, J., Chen, F. et al. (2014). Improving impact toughness of polylactide/poly (ether) urethane blends via designing the phase morphology assisted by hydrophilic silica nanoparticles. Polymer, 55(6), 1593–1600. DOI 10.1016/j.polymer.2014.01.057. [Google Scholar] [CrossRef]
6. Li, Y., Shimizu, H. (2007). Toughening of polylactide by melt blending with a biodegradable poly(ether) urethane elastomer. Macromolecular Bioscience, 7(7), 921–928. DOI 10.1002/mabi.200700027. [Google Scholar] [CrossRef]
7. Jiang, L., Wolcott, M. P., Zhang, J. (2006). Study of biodegradable polylactide/poly(butylene adipate-co-terephthalate) blends. Biomacromolecules, 7(1), 199–207. DOI 10.1021/bm050581q. [Google Scholar] [CrossRef]
8. Bai, H., Bai, D., Xiu, H., Liu, H., Zhang, Q. et al. (2014). Towards high-performance poly(L-lactide)/elastomer blends with tunable interfacial adhesion and matrix crystallization via constructing stereocomplex crystallites at the interface. RSC Advances, 4(90), 49374–49385. DOI 10.1039/C4RA08823A. [Google Scholar] [CrossRef]
9. Supthanyakul, R., Kaabbuathong, N., Chirachanchai, S. (2017). Poly(l-lactide-b-butylene succinate-bl-lactide) triblock copolymer: A multi-functional additive for PLA/PBS blend with a key performance on film clarity. Polymer Degradation and Stability, 142(8), 160–168. DOI 10.1016/j.polymdegradstab.2017.05.029. [Google Scholar] [CrossRef]
10. Al Hosni, A. S., Pittman, J. K., Robson, G. D. (2019). Microbial degradation of four biodegradable polymers in soil and compost demonstrating polycaprolactone as an ideal compostable plastic. Waste Management, 97, 105–114. DOI 10.1016/j.wasman.2019.07.042. [Google Scholar] [CrossRef]
11. Aliotta, L., Vannozzi, A., Panariello, L., Gigante, V., Coltelli, M. B. et al. (2020). Sustainable micro and nano additives for controlling the migration of a biobased plasticizer from PLA-based flexible films. Polymers, 12(6), 1366. DOI 10.3390/polym12061366. [Google Scholar] [CrossRef]
12. Xiao, H., Lu, W., Yeh, J. T. (2009). Effect of plasticizer on the crystallization behavior of poly(lactic acid). Journal of Applied Polymer Science, 113(1), 112–121. DOI 10.1002/app.29955. [Google Scholar] [CrossRef]
13. Maiza, M., Benaniba, M. T., Quintard, G., Massardier-Nageotte, V. (2015). Biobased additive plasticizing Polylactic acid (PLA). Polimeros-Ciencia e Tecnologia, 25(6), 581–590. DOI 10.1590/0104-1428.1986. [Google Scholar] [CrossRef]
14. Yang, X., Clénet, J., Xu, H., Odelius, K., Hakkarainen, M. (2015). Two step extrusion process: From thermal recycling of PHB to plasticized PLA by reactive extrusion grafting of PHB degradation products onto PLA chains. Macromolecules, 48(8), 2509–2518. DOI 10.1021/acs.macromol.5b00235. [Google Scholar] [CrossRef]
15. Kang, H., Li, Y., Gong, M., Guo, Y., Guo, Z. et al. (2018). An environmentally sustainable plasticizer toughened polylactide. RSC Advances, 8(21), 11643–11651. DOI 10.1039/C7RA13448G. [Google Scholar] [CrossRef]
16. Srithep, Y., Nealey, P., Turng, L. S. (2013). Effects of annealing time and temperature on the crystallinity and heat resistance behavior of injection-molded poly(lactic acid). Polymer Engineering & Science, 53(3), 580–588. DOI 10.1002/pen.23304. [Google Scholar] [CrossRef]
17. Phua, Y., Chow, W., Mohd Ishak, Z. (2013). Reactive processing of maleic anhydride-grafted poly(butylene succinate) and the compatibilizing effect on poly(butylene succinate) nanocomposites. Express Polymer Letters, 7(4), 340–354. DOI 10.3144/expresspolymlett.2013.31. [Google Scholar] [CrossRef]
18. Ba, C., Yang, J., Hao, Q., Liu, X., Cao, A. (2003). Syntheses and physical characterization of new aliphatic triblock poly(L-lactide-b-butylene succinate-b-L-lactide)s bearing soft and hard biodegradable building blocks. Biomacromolecules, 4(6), 1827–1834. DOI 10.1021/bm034235p. [Google Scholar] [CrossRef]
19. Vilay, V., Mariatti, M., Ahmad, Z., Pasomsouk, K., Todo, M. (2009). Characterization of the mechanical and thermal properties and morphological behavior of biodegradable poly(L-lactide)/poly(ε-caprolactone) and poly(L-lactide)/poly(butylene succinate-co-L-lactate) polymeric blends. Journal of Applied Polymer Science, 114(3), 1784–1792. DOI 10.1002/app.30683. [Google Scholar] [CrossRef]
20. Muthuraj, R., Misra, M., Mohanty, A. (2015). Hydrolytic degradation of biodegradable polyesters under simulated environmental conditions. Journal of Applied Polymer Science, 132(27), 1–13. DOI 10.1002/app.42189. [Google Scholar] [CrossRef]
21. Deng, Y., Thomas, N. L. (2015). Blending poly(butylene succinate) with poly(lactic acidDuctility and phase inversion effects. European Polymer Journal, 71(9), 534–546. DOI 10.1016/j.eurpolymj.2015.08.029. [Google Scholar] [CrossRef]
22. Srithep, Y., Pholharn, D. (2017). Plasticizer effect on melt blending of polylactide stereocomplex. e-Polymers, 17(5), 409–416. DOI 10.1515/epoly-2016-0331. [Google Scholar] [CrossRef]
23. Srithep, Y., Javadi, A., Pilla, S., Turng, L. S., Gong, S. et al. (2011). Processing and characterization of recycled poly(ethylene terephthalate) blends with chain extenders, thermoplastic elastomer, and/or poly(butylene adipate-co-terephthalate). Polymer Engineering & Science, 51(6), 1023–1032. DOI 10.1002/pen.21916. [Google Scholar] [CrossRef]
24. Burgos, N., Tolaguera, D., Fiori, S., Jiménez, A. (2014). Synthesis and characterization of lactic acid oligomers: Evaluation of performance as poly(lactic acid) plasticizers. Journal of Polymers and the Environment, 22(2), 227–235. DOI 10.1007/s10924-013-0628-5. [Google Scholar] [CrossRef]
25. Homklin, R., Hongsriphan, N. (2013). Mechanical and thermal properties of PLA/PBS co-continuous blends adding nucleating agent. Energy Procedia, 34(4), 871–879. DOI 10.1016/j.egypro.2013.06.824. [Google Scholar] [CrossRef]
26. Roudaut, G., Simatos, D., Champion, D., Contreras-Lopez, E., Le Meste, M. (2004). Molecular mobility around the glass transition temperature: A mini review. Innovative Food Science & Emerging Technologies, 5(2), 127–134. DOI 10.1016/j.ifset.2003.12.003. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |