| Journal of Renewable Materials |
DOI: 10.32604/jrm.2021.015047
ARTICLE
Organosolv Lignin for Non-Isocyanate Based Polyurethanes (NIPU) as Wood Adhesive
1Biotechnical Faculty, University of Ljubljana, Ljubljana, 1000, Slovenia
2LERMAB-ENSTIB, Université de Lorraine, Epinal, 88000, France
3Fraunhofer Institute for Chemical Technology, Pfinztal, 76327, Germany
*Corresponding Author: Milan Šernek. Email: milan.sernek@bf.uni-lj.si
Received: 18 November 2020; Accepted: 19 December 2020
Abstract: A non-isocyanate-based polyurethane (NIPU) wood adhesive was produced from organosolv lignin, which is a bio-sourced raw material, available in large quantities and produced as a by-product of the paper industry. The formulation of this new lignin-based NIPU adhesive, which is presented, was chemically characterised by Matrix-Assisted Laser Desorption Ionization Time of Flight (MALDI ToF) mass spectrometry and by Fourier Transform Infra-Red (FTIR) spectrometry analyses. The oligomers formed were determined and showed that the three species involved in the NIPU adhesive preparation were formed by the co-reaction of the three reagents used: lignin, dimethyl carbonate, and hexamethylene diamine. Linear and branched structures were both identified. Mechanical properties of the adhesive were determined using the Automated Bonding Evaluation System (ABES) and internal bond (IB) strength test of the laboratory particleboard bonded with it. The adhesive has shown satisfactory mechanical properties after hot pressing at 230°C. Such a temperature is used industrially in the most modern particleboard factories, but since it is hardly feasible for more conventional wood bonding equipment, the reactivity of the NIPU adhesive was successfully increased with the addition of a small percentage of a silane coupling agent. With the addition of the silane, the proposed NIPU adhesive could also be used at a hot-pressing temperature lower than 200°C.
Keywords: Organosolv lignin; NIPU; MALDI ToF; FTIR; ABES
Lignins are natural phenolic compounds, very common in the vegetable world, particularly in trees, for which they constitute one of the two main fundamental structural constituents of wood. They are widely available from the pulp and paper industry, and they are the second most abundant polymers on earth. Different lignin production processes lead to their different chemical structures. Organsolv lignins (OL) are produced by the chemical reaction between lignin and organic solvents, while the most commonly produced technical lignin with inorganic reagent is the Kraft lignin [1].
Many approaches to upgrading these materials to bring them to the level of industrial utilisation conditions as wood panel adhesives have been taken [2−5]; some are also used as commercial plastics [6]. Their use as commercial wood adhesives has been less common, due to some of their drawbacks, such as lower reactivity and consequently longer hot press times. Some methods have been reported for lignin modification for application in wood adhesives, such as demethylation [7−10], hydroxy-methylation [11,12], phenolation [13−16], oxidation [17], and others more recent ones [18], which can effectively improve the reactivity of lignin. Lignins have also been used as partial substitutes for phenol in phenol-formaldehyde resins for plywood [17−19].
Bio-sourced polyols have already been used to manufacture polyurethanes for a variety of applications [20−26]. Nonetheless, notwithstanding the bio-sourced polyols, the industrial need to use isocyanates to manufacture polyurethanes is still present.
Non-isocyanate based polyurethanes (NIPU) have been and can be prepared by a number of routes. Poly-hydroxy-urethanes are generally prepared by the reaction of five-terms cyclic carbonates [27] with diamines [28]. This type of approach is well represented in the relevant literature [29−45]. The preparation of cyclic carbonates for such an application is, however, a technical drawback. Work to eliminate this drawback by employing approaches based on intermediates of glycerine carbonate or by using transformed epoxidized plant oils have been published. As regards the needed amines, an approach based on fatty diacids to obtain polyamines has also been proposed [46].
The first carbonation has been proven to be effective by also using a much simpler non-cyclic aliphatic carbonate, namely dimethyl carbonate. Dimethylcarbonate is easily available, inexpensive, reacts well and is also a good reaction solvent [47,48]. It has a pleasant smell and neither mutagenic, nor irritating effects are reported whatsoever, neither on the skin nor on respiration (Merck index) [48].
Dimethyl carbonate does carbonate hydroxygroups in general around 90°C [48], thus near to its boiling point. The reaction is a base-catalysed two molecules nucleophile substitution by acyl cleavage [48].
Preparation from condensed and hydrolysable tannins of non-isocyanates poly-hydroxy-urethanes has recently been reported, these resins presenting good wood surface coats and wood adhesives performances [49−53]. Even more notable was the inclusion of pre-aminated tannin as a bio-sourced polyamine [51] bringing the bio-content of these NIPU polyurethanes to around 90%.
The reactivity of NIPU has already been successfully increased by adding silane coupling agents. Silanes can modify the chemical formulation of adhesives [54] or substrates [55], which are then better compatible with each other. This leads to shorter pressing times at lower pressing temperatures.
The development of a new adhesive formulation and its successful application in the wood industry requires a characterisation of the curing process, which enables the determination of the optimal pressing parameters, which are essential for the economical production of wood-based composites. For this reason, several methods have been developed to provide insight into the curing process of the adhesives.
The shear strength development of the adhesive bond is the direct measure of the curing process. The majority of those methods measure various parameters that indirectly approximate the shear strength development of the adhesive. Only a few of those directly measure the adhesive bond strength during (or immediately after) pressing. The automated bonding evaluation system (ABES) that was used in this research is one of those [56−58].
This paper deals with the development, the preparation and the characterisation of NIPU starting from organosolv lignin, their analysis, and their applicability and performance as thermosetting wood adhesives for wood panels.
For the preparation of the organosolv lignin NIPU, the following chemicals were used: Organosolv lignin (OL) (generated via the acid-catalysed ethanol/water organosolv-cooking of standard beechwood chips, supplied by Fraunhofer CBP, Leuna, Germany); Dimethyl carbonate (99%, CAS Number 616-38-6, supplied by Sigma-Aldrich); Hexamethylenediamine (70%, CAS Number 124-09-4, supplied by Sigma-Aldrich); silane coupling agent (HK560, 98%, CAS Number 2530-83-8, supplied by Sigma-Aldrich), with the structural formula
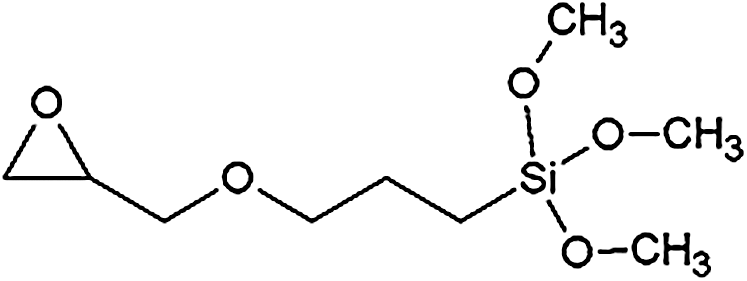
2.2 Organosolv Lignin Characterisation
Exactly 20.00 mg of the organosolv lignin (vacuum dried, 10 mbars, 55°C, 96 hrs) was weighed in a High-Performance Liquid Chromatography (HPLC) vial. 600 µL of a freshly prepared solvent–mix I, containing pyridine/CDCl3 (1.6/1 v/v), 100 µl of solvent mix I, containing 0.5 mg Cr(III) acetylacetonate as well as 100 µL solvent mix I, containing 1.085 mg cyclohexanol (internal standard) were added to the sample, and the vial was capped. The sample was mixed by vortex mixing at 21°C. 10 min before the Phosphorus-31 nuclear magnetic resonance (31P-NMR) analysis 50 µL of in situ labelling reagent 2-Chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (Cl-TMDP) were added to the vial and homogenised again by vortex mixing at 21°C. The sample was completely dissolved. The final sample solution was transferred to an NMR tube. The 31P-NMR analysis was done on a Bruker AVANCE 300 MHz. Chemical shifts are reported relative to the sharp signal (132.2 ppm) originating from the reaction between traces of water and Cl-TMDP. The following NMR parameters were used: scans = 1024, pulse delay = 5 s, 90° pulse and line broadening = 2 and default baseline correction. 31P-NMR measurement is based on the method by Granata and Argyropoulos [59]. 31P-NMR-spectra were processed on ACD Lab 12.0, 1D-processor software. Integration for quantitative calculation was done according to published data of Pu et al. [60].
The determination of weight-average molecular weight (MW) and number-average molecular weight (MN) was carried out by gel permeation chromatography (GPC) on two GPC systems.
GPC system I used an HPLC-system with a Diode Array Detector (Agilent Technologies, LC 1100, DAD: 280 nm) with three columns (PSS SDV 50 Å, 5 µ, guard column, and three analytical columns PSS SDV 5 µ, 102 Å, 5 µ, 103 Å and 5 µ, 105 Å), according to Baumberger et al. [61]; 2–3 mg of acetylated organosolv lignin (detailed description is in Appendix A of Supplementary Materials) were dissolved in 1 mL of THF. The mobile phase was THF with a flow rate of 1 mL min−1 and an oven temperature of 35°C. Sample injection volume was 100 µl. Conventional polystyrene standards (266 g mol−1 to 1.252.000 g mol−1, PSS Mainz, Germany) were used for calibration. Therefore, the results regarding weight-average molecular weight (MW) and number-average molecular weight (MN) are presented versus polystyrene.
A GPC system II using an HPLC-system with a refractive index detector (Agilent Technologies, LC 1200, RID cell temperature: 35°C) with three columns (PSS MCX 10 µm, guard column, analytical columns 10 µ, 100 Å and 10 µ, 1000 Å) according to Baumberger et al. [61]; 2–3 mg of the samples were dissolved in 1 mL of 0.1 M sodium hydroxide solution. The mobile phase was 0.1 M sodium hydroxide solution with a flow rate of 1 mL min−1 and an oven temperature of 35°C. Sample injection volume was 100 µl. Conventional pullulan standards (342–805.000 g mol−1, PSS Mainz, Germany) were used for calibration. Therefore, results regarding weight-average molecular weight (MW) and number-average molecular weight (MN) are presented versus pullulan.
2.2.3 Elemental Analysis (CHONS)
The elemental composition of the organosolv lignin was analysed using an automatic elemental analyser Flash EA 1112 from Thermo Scientific, which was equipped with a MAS 200R auto-sampler. The calibration-standard methionine and vanadium pentoxide, tin capsules, and silver containers were purchased from IVA Analysentechnik e. K., Germany. Helium 5.0 for the GC–TCD analysis was purchased from Linde, Germany. The calibration and the determination of the elements C, H, O, N and S were done according to Nebhani et al. [62].
Determination of ash in organosolv lignin was done as follows. 10 g of OL were exactly weighed and vacuum dried (10 mbars, 55°C, 96 hrs). The determination of ash was exactly done according to the technical report NREL/TP-510-42622 [63].
2.3 Preparation of Lignin NIPU
In the first stage, 25.9 weight units of organosolv lignin powder were poured into a three-necked flask equipped with a reflux condenser and thermometer. Then 17.5 weight units of dimethyl carbonate and 21.6 weight units of distilled water were added and continuously stirred with a magnetic stirrer for 40 min at 50°C, then cooled to room temperature. After that, a total of 35.1 weight units hexamethylenediamine was added to the mixture and continuously stirred with a magnetic stirrer for 120 min at 90°C. Next, the resin was cooled to room temperature and ready to use. The viscosity of the prepared adhesive was determined to 64 mPa·s, the pH value to 11 and the solid content to 46%.
To increase the curing process of the OL NIPU, the silane coupling agent (KH560) was used. The silane was added to the prepared NIPU adhesive on the basis of a percentage of the dry NIPU weigh.
Samples for matrix-assisted laser desorption ionisation time-of-flight (MALDI-ToF) analysis were prepared first by dissolving 5 mg of sample powder in 10 mL of a 50:50 v/v acetone/water solution. Then 10 mg of this solution was added to 10 μL of a 2,5-dihydroxy benzoic acid (DHB) matrix to obtain a homogeneous solution. The locations dedicated to the samples on the analysis plaque were first covered with 2 μL of a NaCl solution 0.1 M in 2:1 v/v methanol/water, and predried. Then, 1 μL of the sample solution was placed on its dedicated location, and the plaque was dried again. The reference substance used for the equipment calibration was red phosphorus. MALDI-ToF spectra were obtained using an Axima-Performance mass spectrometer from Shimadzu Biotech (Kratos Analytical Shimadzu Europe Ltd., Manchester, UK) using a linear polarity positive tuning mode. The measurements were carried out, making 1000 profiles per sample with two shots accumulated per profile. The spectra precision is of +1 Da.
Fourier Transform Infra-Red (FTIR) analysis was carried out using a Shimadzu IR Affinity-1 (Shimadzu Europe Ltd., Manchester, UK) spectroscopy. A blank sample tablet of potassium bromide, ACS reagent from ACROS Organics (Geel, Belgium), was prepared for the reference spectra. Similar tablets were prepared by mixing potassium bromide with 5% weight on the weight of the sample powders to be analysed. The spectra were obtained in transmittance by combining 32 scans with a resolution of 2.0 cm−1 in the 400–4000 cm−1 range.
2.6 Adhesion Development of NIPU Adhesive with ABES
Adhesion development of NIPU adhesive was determined with ABES (Adhesive Evaluation Systems Inc., Oregon, USA), following the ASTM [64] standard. ABES is a combination of small hot press and tensile testing machine.
Beech (Fagus sylvatica) veneers, 0.84 mm thick, were bonded in the hot press at different temperatures and for different pressing times. The geometry of the bonded area of the veneer lap joints was 5 mm × 20 mm (100 mm2), as presented on Fig. 1. However, the actual area of each tested sample was measured.

Figure 1: Schematic of the Bonding and Testing Concept [64]
Selected temperatures of the hot press were 180°C, 200°C, and 230°C. The real temperature transition to the bond line was observed with K-type thermocouple for three samples at each of the tested temperatures.
Ten different press times between 15 s and 600 s (15 s, 30 s, 60 s, 90 s, 120 s, 180 s, 240 s, 300 s, 420 s, and 600 s) were chosen to present the building of the bond shear strength in time. After the opening of the hot press, the adhesive bond was cooled with the compressed air for 5 s.
The last step was the shear strength test of the adhesive bond. The lap joint was pulled until the rupture occurred (approx. 11 s after the hot press opening). The maximum achieved force was compared with the bonded area, and the resulting shear strength was expressed in N/mm2.
2.7 Dry Internal Bond Strength Test
For a dry internal bond strength test, different monolayer particleboards were prepared. The adhesive solids load was 10% on the weight of the bone-dry wood. The panels were pressed with a three-stage hot pressing cycle for 10 min at a maximum pressure of 2.8 N/mm2, decreasing to 1.5 N/mm2 and then 0.6 N/mm2 for respectively 2, 3.5, and 4.5 minutes. Two different hot-pressing temperatures (180°C and 230°C), and densities (0.7 and 0.76 g/cm3) were used. The dry internal bond (IB) strength was tested according to European Norm EN 319 [65].
3.1 Organosolv Lignin Characterisation
In Tab. 1 and Fig. 2, the GPC and 31P-NMR results according to structural features of OL are presented. Both used GPC methods illustrate a lower molecular weight and a small molecular weight distribution of the used OL compared to technical Kraft lignin.
Table 1: Structural features of organosolv lignin (OL) weight-average molecular weight (MW) and number-average molecular weight, determined by two GPC-methods and number of phenolic, aliphatic and carboxylic OH groups in mmol/g, determined by 31P-NMR analysis, after in-situ labelling with Cl-TDMP, internal standard: Cyclohexanol


Figure 2: Result of the 31P-NMR-analysis of OL, after in-situ labelling with 2-Chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (Cl-TMDP), region of aliphatic OH: 145.4-150.0 ppm; region of C5-substituted and syringyl OH: 140.0-144.5 ppm, region of guaiacyl OH: 139.0-140.2 ppm, region of carboxylic OH: 133.6–136.0 ppm, ISTD Cyclohexanol: 144.75–145.39 ppm according to published data of Pu et al. [60]. Chemical shifts are reported relative to the sharp signal (132.2 ppm) originating from the reaction between traces of water and Cl-TMDP
Additional, the functionality within the total OH number, aliphatic OH number as well as the phenolic OH number is smaller than expected. Compared to Kraft lignin, the used OL does not contain carboxylic OH groups as additional difference.
Detailed data regarding the structural features of Kraft lignin were published by Crestini et al. [66].
A target driven chemical functionalisation, such as illustrated in the paper, is essential to increase the chemical reactivity of Kraft lignins as well as of OL for the development of material applications.
3.1.2 Elemental Composition (CHONS) Ash and Humidity
The elemental composition (CHONS) of the organosolv lignin was as follows: C: 64.13%, H: 5.69%, O: 28.99%, N: 0.53%, S: 0.00% with an elemental balance of 99.3%.
The ash content was 0.01%. The analysis was done on vacuum-dried (10 mbars, 55°C, 96 hrs) materials. The humidity of the original OL was 1.55%, determined by vacuum drying (10 mbars, 55°C, 96 hrs) of 10 g powdered sample.
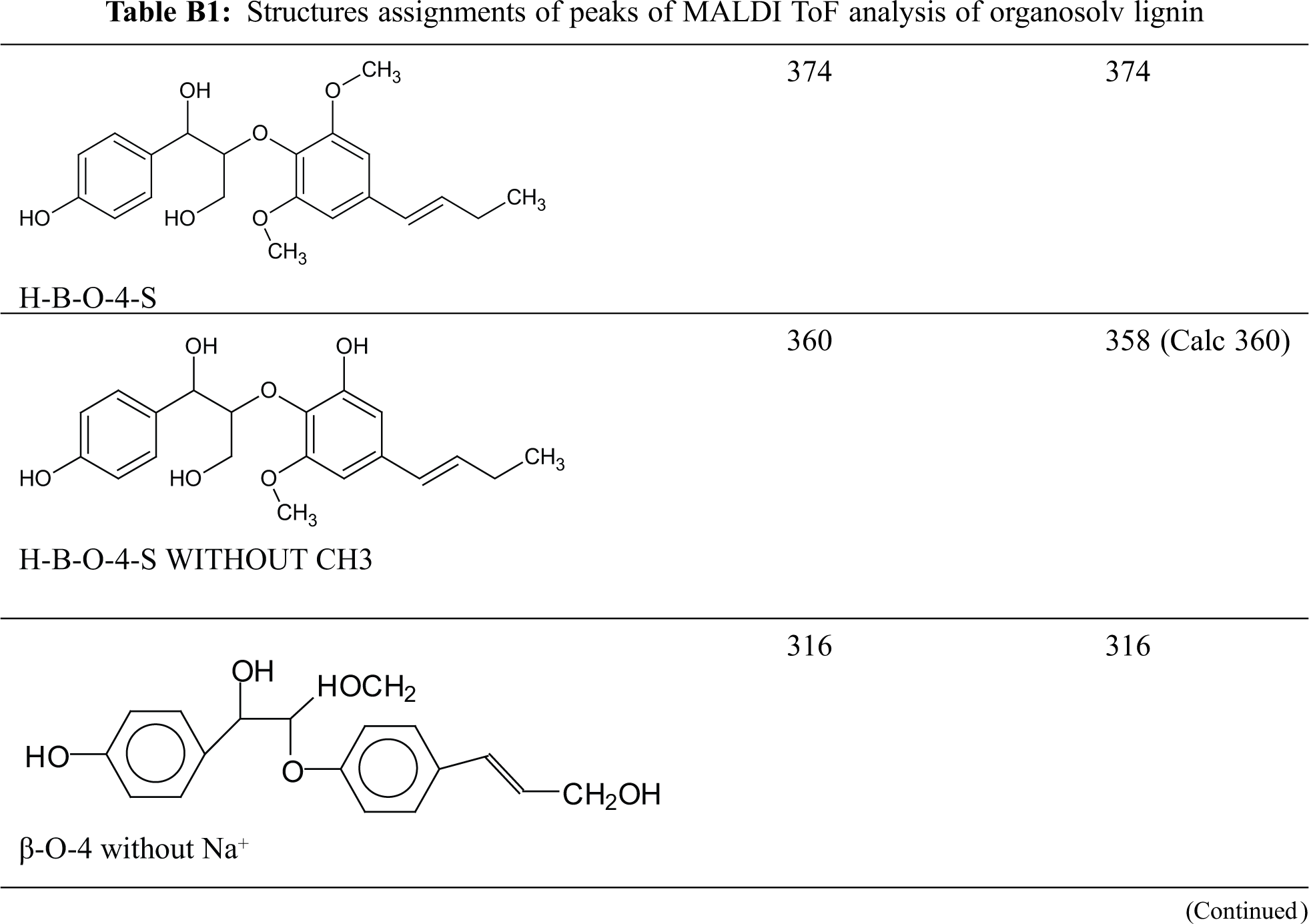
MALDI ToF analyses of both the organosolv lignin and of the NIPU obtained from the same lignin after reaction with dimethyl carbonate and hexamethylene diamine were done to understand the structures involved and formed in the reaction. In Figs. 3a–3c, the MALDI spectra of the organosolv lignin, are shown. The peaks in these spectra are those of monomeric lignin units and different lignan dimers derived from conyferyl, synapyl, and cynnammyl alcohols and their couplings. The structures assigned to the main peaks are shown and listed in Appendix B in the Supplementary Material. It was of interest to first determine the oligomers present in the original lignin. This was necessary to identify the new peaks and to identify the oligomers formed in the preparation of the organosolv lignin NIPU resin. The MALDI spectra of the lignin NIPU resin are shown in Figs. 4a–4f and the list and structures assigned are shown in Appendix C in the Supplementary Material. In Appendix C, none of the peaks and structures of the pure organosolv lignin in Appendix B has been reported in order that only some of the relevant peaks and structures from the reaction can be considered. The list of compounds is long, but here only structures that are of interest will be discussed.

Figure 3: (a) MALDI ToF spectrum of organosolv lignin. 0 to 1000 Da range (b) MALDI ToF spectrum of organosolv lignin. 0 to 450 Da range (c) MALDI ToF spectrum of organosolv lignin. 0 to 1000 Da range


Figure 4: (a) MALDI ToF spectrum of organosolv lignin NIPU resin. 0 to 500 Da range (b) MALDI ToF spectrum of organosolv lignin NIPU resin. 400 to 800 Da range (c) MALDI ToF spectrum of organosolv lignin NIPU resin. 600 to 800 Da range (d) MALDI ToF spectrum of organosolv lignin NIPU resin. 800 to 1000 Da range (e) MALDI ToF spectrum of organosolv lignin NIPU resin. 1000 to 1200 Da range (f) MALDI ToF spectrum of organosolv lignin NIPU resin. 1200 to 1500 Da range
Several types of reaction and a number of rather different oligomers are formed by the NIPU preparation method used. Intermediate structures derived from the reaction of monomeric lignin units, such as conyferyl and synapyl alcohol reacted with dimethyl carbonate (DMC), are present such as structure (I) at 320–321 Da and (II) at 329 Da as well as dimers of these alcohols linked exclusively through a carbonate bridge, such as structure (III) at 409 Da.
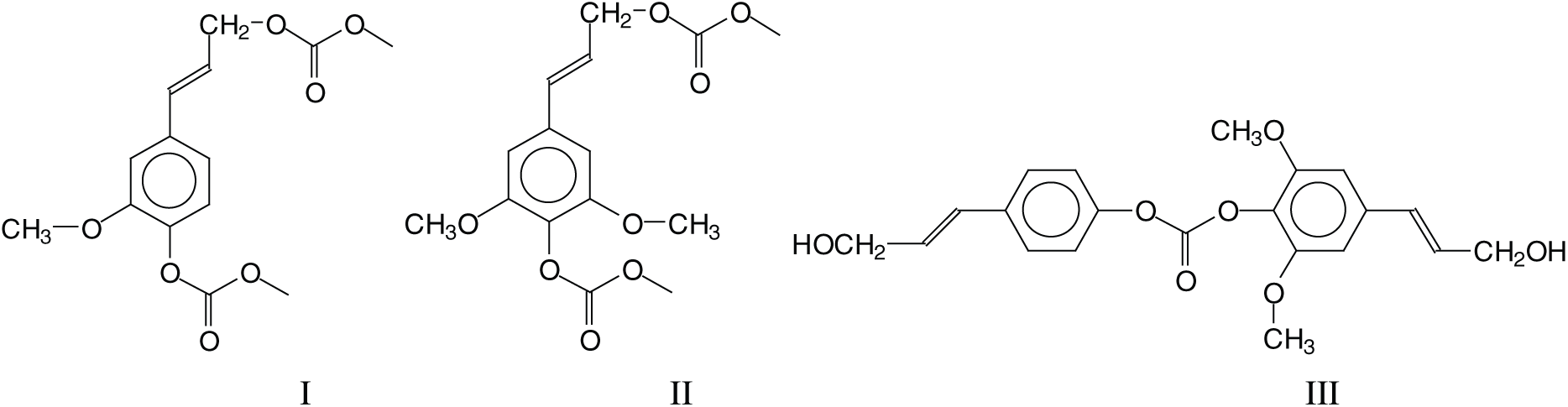
Structure (III) is the result of a wasteful side reaction as two lignin units that could form urethane linkages are blocked through this side reaction. There are not many structures such as (III), only the ones at 451 Da and at 469–471 Da. Thus, due to their low proportion, structures of type III do not detract to the preparation of NIPU resins. The next type of structure of interest (IV) at 457 Da indicates that all the –OH groups of lignin, both aromatic and aliphatic, do react with DMC and then with the diamine to form urethane linkages.
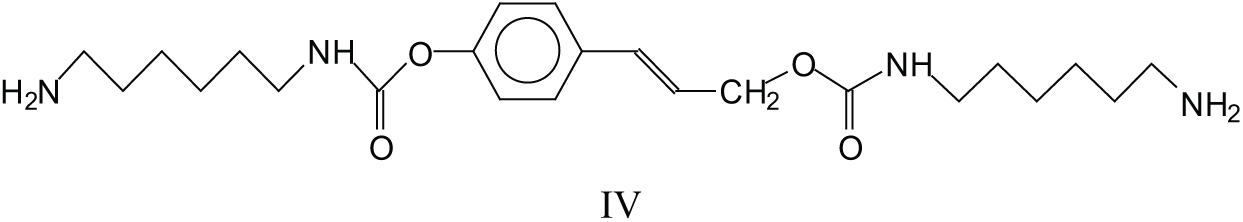
Structures like these lead to the formation of double urethane bridges between lignin units. There are several of these at 591 Da, 607 Da, 637 Da, 646-647 Da, 666 Da, 691 Da, 722 Da, 727 Da, 782 Da, 792 Da, 864 Da, 872-873 Da, 902 Da, 933 Da, 955 Da, 979Da, 1016 Da and many others at higher molecular weights. One example is species (V) at 873 Da.

The long series of these species leads to a series of linear oligomers that respond to the general structural formula (VI).
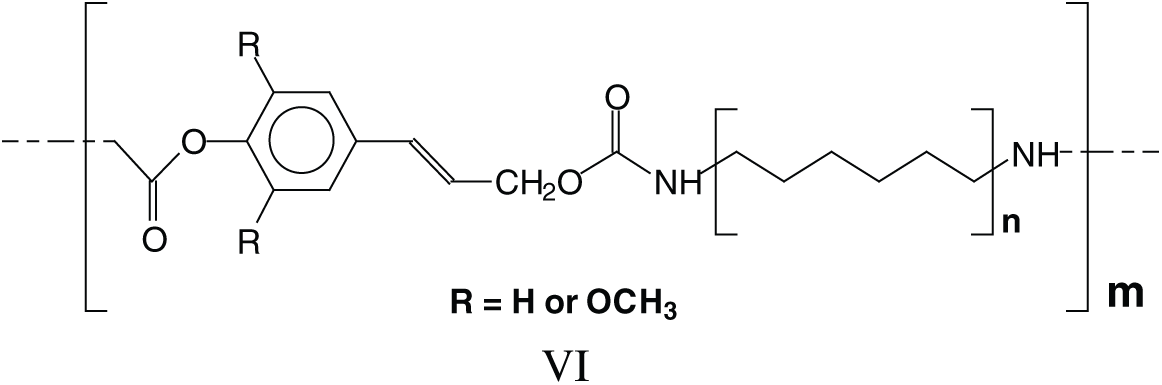
The cross-linking that is obtained with hardening these NIPU adhesives should entail a mechanism in which tridimensional networking is formed, as in effect these NIPU resins cure just with heat. Many species in which tridimensional branching has occurred, explaining the NIPU resins cross-linking, were detected with the MALDI Analysis. Thus, branching is evident in a considerable number of structures as those assigned to the peaks at 787 Da, 902 Da, 930–931 Da, 963 Da, 986 Da, 995 Da, 1027 Da, 1062 Da, 1095 Da, 1128–1131 Da, 1341 Da, 1403 Da and several other at higher molecular weights. These species can be divided into two categories: (i) Species in which branching has occurred just with the DMC and the diamine, without reaction with further lignin units, thus branching without cross-linking, an example being structure VII at 787 Da, and (ii) Species in which branching has cross-linked several lignin units in the structure, explaining how these resins can cross-link and harden with heat, such as structure VIII at 1403 Da.
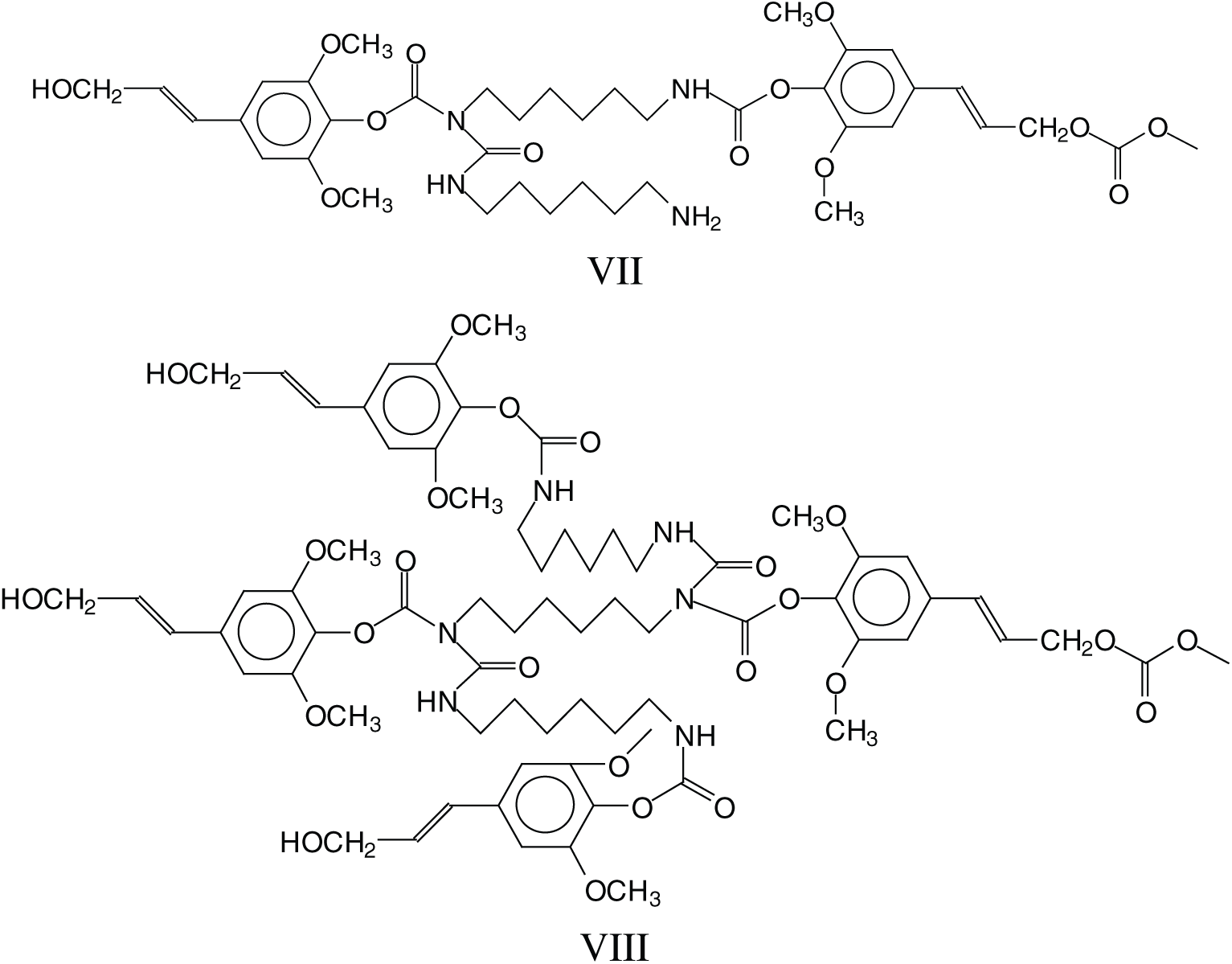
The above indicates that a generalised formula for the type of NIPU resins formed is more as structure IX rather than just structure VI.
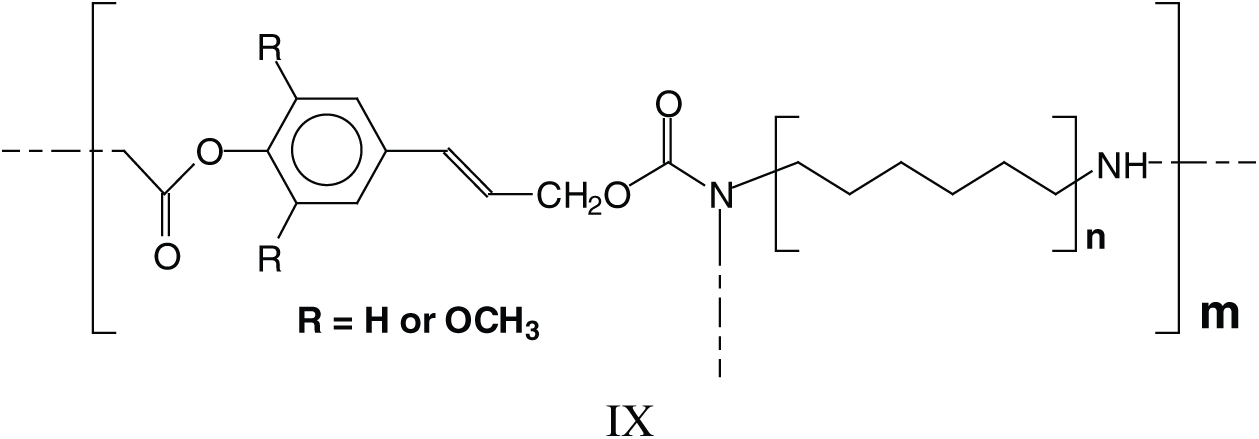
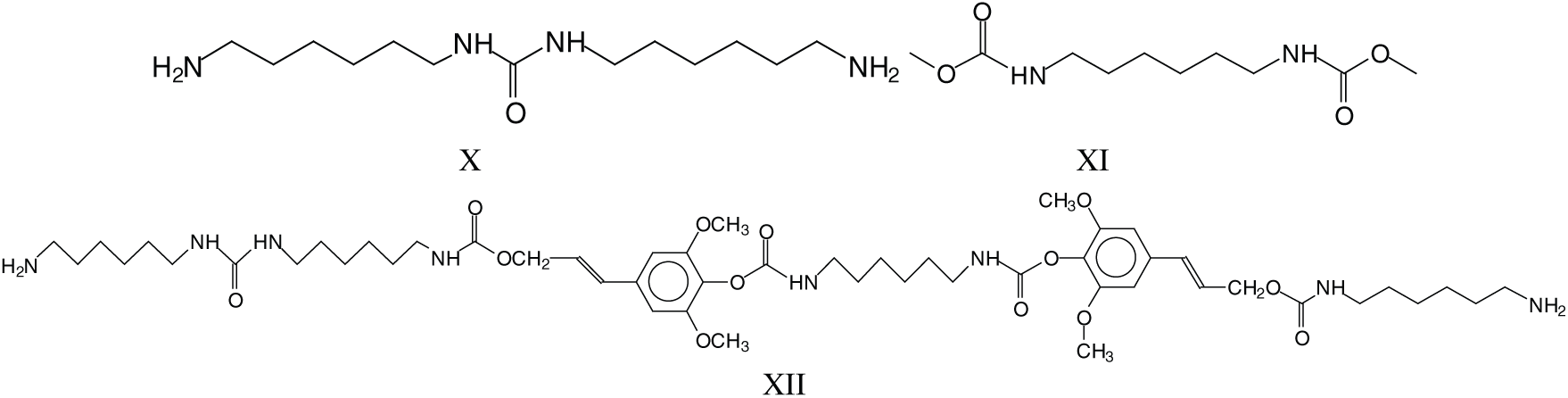
Parasite side reactions do occur. The one leading to structure III has already been presented, and a second one leads to the reaction of DMC with amine only, hence linking two amines such as structures X and XI at 255 Da. This reaction can also occur on groups linked to established structures involving lignin units, such as those at 979–980 Da and 1016 Da (XII).
The C=O group of urethanes at 1635 cm–1 and at 1557 cm–1 appears in the NIPU spectrum and is not present in the lignin spectrum (Fig. 5). These two peaks both belong to NH-CO urethane bridges. Confirming the presence of the urethane linkages is the peak at 1240 cm−1, also belonging to a urethane. This latter peak can also belong to an amide again indicating a urethane linkage but is also superposed masking a carbonate signal, confirmed by the small shoulder peak at 1744 cm−1. The rest of the peaks are assigned as follows: the much larger peaks at 2929 and 2856 cm−1 and the peak at 1455 cm−1 to the asymmetric stretching of the to –CH2- chain belonging to the diamine used to prepare the NIPU resin. These peaks also occur in the pure lignin spectrum, but their intensity is much smaller, belonging to the aliphatic side chains of lignin units. The small peaks at 912 and 834 cm−1 in the pure lignin spectrum are describing the different multi-substituted aromatic nuclei of the lignin. The peak at 1031 cm−1 and the broad peak at 3330–3400 cm−1 which remain unaltered when comparing the two spectra belongs to the hydrogen-bonded stretching of aliphatic alcohols, thus of the side chain of lignin units. This latter is then indicative that the urethane NIPU linkage either forms mainly on the free phenolic –OH groups of the lignin, or that on opening the C=C double bonds in the alpha and beta lignin units aliphatic chains an equal amount of –OH groups are formed than that are consumed by the reaction. With the data available, it is not possible to ascertain more on this.
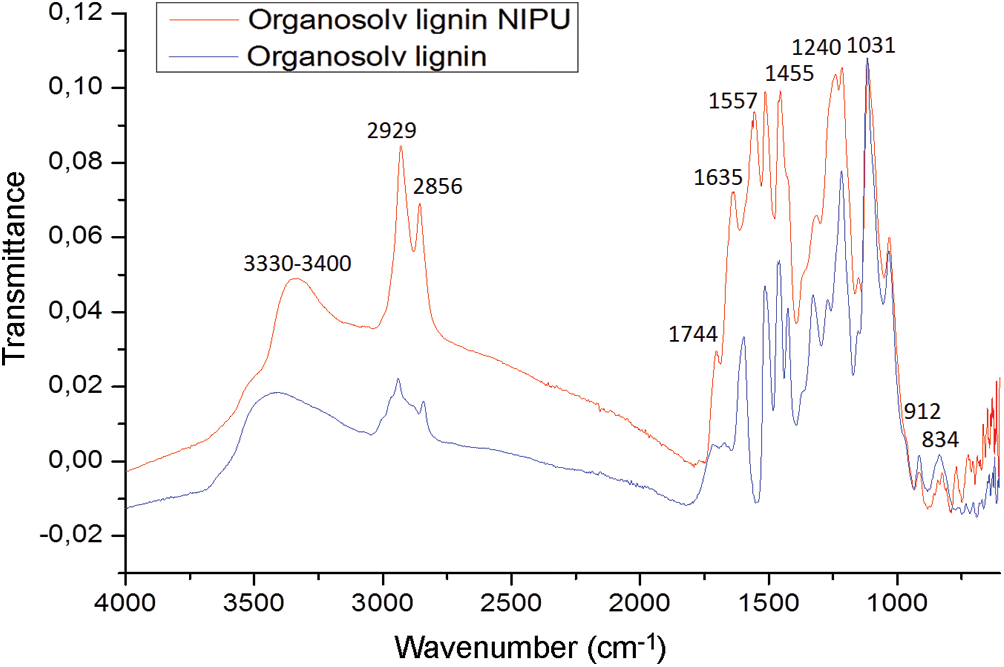
Figure 5: FTIR spectrum comparison of organosolv lignin and organosolv lignin NIPU
3.4 Adhesion Development of NIPU Adhesive with ABES
Curing of the adhesive was carried out at three press temperatures. Temperature transition to the adhesive bond line (an example for 60 s of selected pressing time) is shown in Fig. 6. Each curve represents an average value of 3 measurements. After 15 s in the hot press, the average temperatures reached 168°C, 190°C, and 223°C at different hot press temperatures (180°C, 200°C, and 230°C, respectively); however, ±5°C of set temperature was reached in 20 ± 2 s. After 5 s cooling step, all lap joints were tested in a similar temperature range between 70°C and 85°C, approximately 11 s after hot press opening.
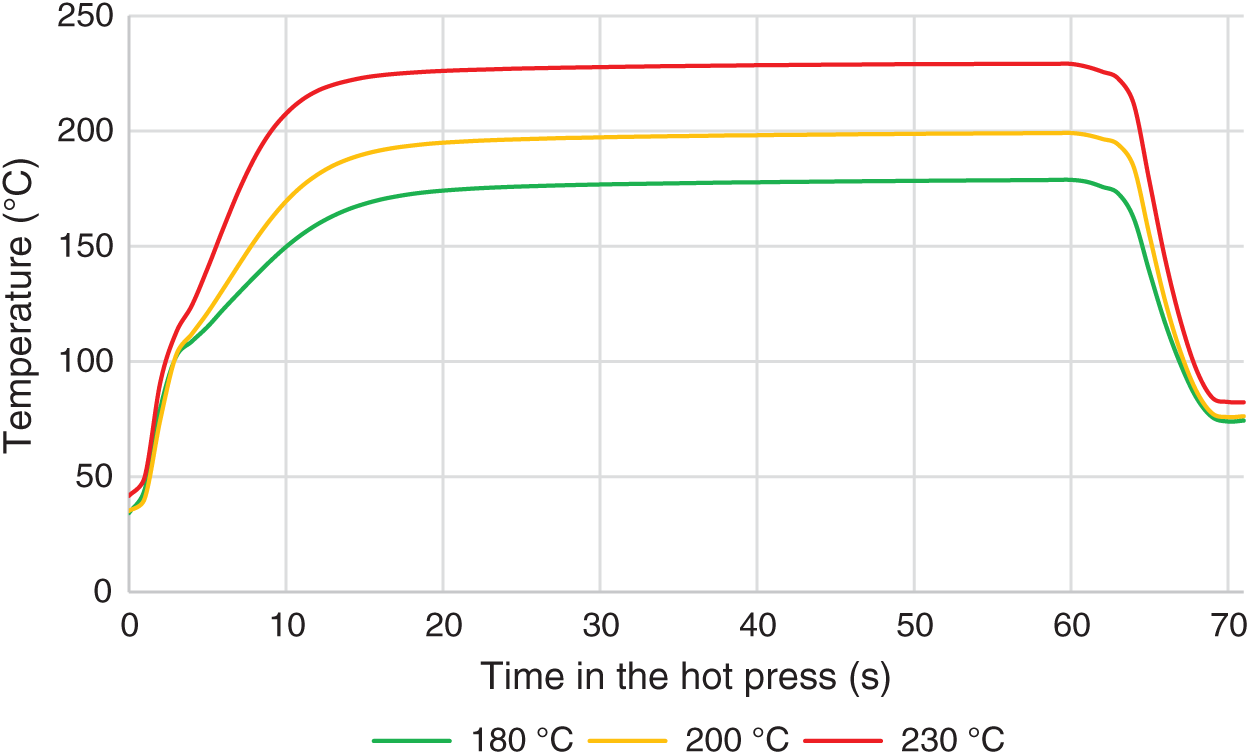
Figure 6: Temperature transition to the bond line in the hot press, during 60 s heating + cooling segment
Average shear strength test results are shown in Fig. 7. OL NIPU adhesive achieved the final strength (above 7 N/mm2), with high wood failure only at the highest temperature (230°C). Since the beech veneer loses its tensional strength at such a high temperature, this can be seen in the results as loss of the shear strength after 3 min. At lower temperatures, the adhesive did not achieve the final strength in the first 10 min. Similar results were also obtained after performing preliminary tests with kraft lignin NIPU.
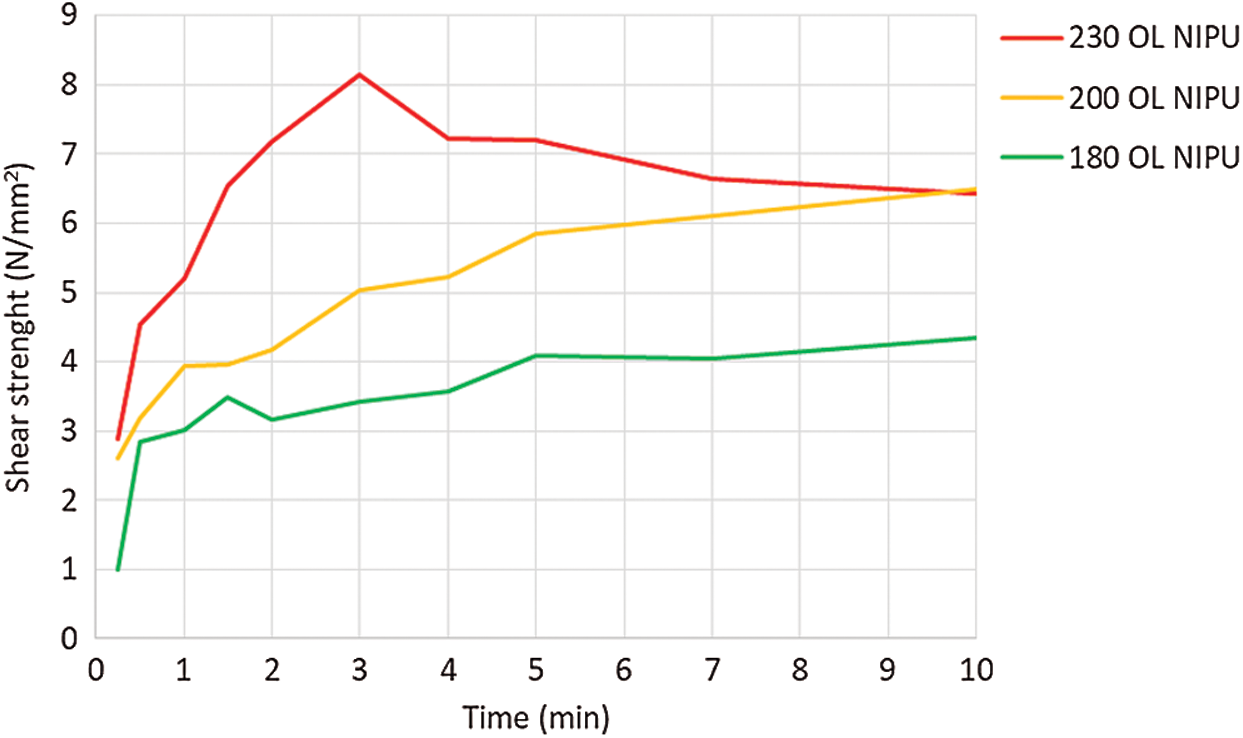
Figure 7: Shear strength development of organosolv lignin NIPU on ABES at different pressing temperatures (180°C, 200°C and 230°C)
A temperature of 230°C is used for fast press times in modern particleboard factories, which why panels were tried at this temperature. However, since hot pressing at 230°C is not feasible for all wood bonding equipment in older factories, as well as to try to save precious energy, we aimed to try and lower the hot-pressing temperature. That is why the impact of silane coupling agent on NIPU curing was observed in the next step. The results are presented in Fig. 8. Increasing the percentage of the silane in the mixture accelerated the curing process of OL NIPU. The best results, with final high wood failure, were obtained at 200°C with 22% silane addition.
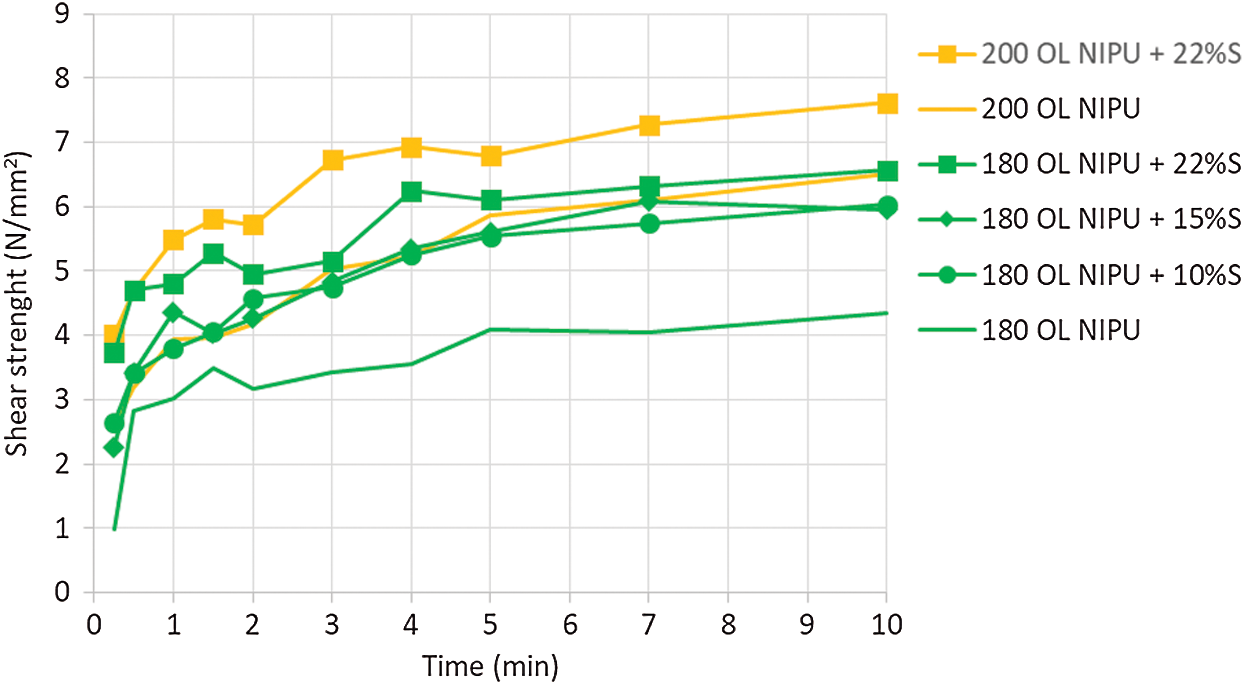
Figure 8: Impact of silane coupling agent (S) on shear strength development of organosolv lignin NIPU on ABES at different pressing temperatures (180°C and 200°C)
To summarise, the observed lignin NIPU could be used as a wood adhesive with pressing parameters of 3 min hot press at 230°C, or for 10 min at 200°C with the addition of silane coupling agent.
3.5 Dry Internal Bond Strength
In Tab. 2, the results of a dry internal bond (IB) strength test are shown. The European Norm EN 319 standard requires for particleboard a dry IB strength equal to or higher than 0.35 MPa [65]. From the results in Tab. 2, it is evident that if the organosolv lignin NIPU adhesive is used alone, it can satisfy the dry IB strength result only if the panels are pressed at 230°C for an average density of 0.70 g/cm3, and this IB strength value is also on the allowed minimum limit envisaged by the EN standard. The results become much better if the density of the panel is increased to 0.76 g/cm3. This density and temperature would indeed be suitable to pass the standard requirements for medium density fiberboard (MDF). To lower the needed energy for board production, silane coupling agent was used. Addition of 15% on resins solids of a silane satisfied the relevant requirements of the standard (it obtained a dry IB strength of 0.60 MPa), while pressing 0.70 g/cm3 dense particleboard at 200°C.
Table 2: Dry internal bond strength of different OL NIPU particleboards

A novel NIPU wood adhesive was produced from organosolv lignin, dimethyl carbonate, and hexamethylene diamine.
The formulation of this new lignin-based NIPU adhesive was chemically characterised using MALDI ToF and FTIR spectrometry analyses. The oligomers formed were determined, and they showed that the three species involved in the NIPU adhesive preparation were formed by the co-reaction of the three reagents used: Lignin, dimethyl carbonate, and hexamethylene diamine. Both linear and branched structures were identified.
The mechanical properties of the adhesive were determined by ABES and IB strength test. Both methods confirmed that the adhesive had satisfactory mechanical properties after short-time hot pressing at 230°C. Such a temperature is currently used in the most modern particleboard factories, but since it is hardly feasible for more conventional wood bonding equipment, the reactivity of the NIPU adhesive successfully increased by the addition of a silane coupling agent. The observed influence of silane differs between both research methods. The results of the ABES method shown that for the goal shear strength of the adhesive bond above 7 N/mm2 the pressing parameters of at least 200°C for 10 min, with a 22% addition of silane coupling agent were needed. However, at dry IB strength test, the prepared particleboard superseded the goal strength of 0.35 N/mm2 already after hot pressing at 180°C for 10 min, with 15% addition of silane coupling agent.
Funding Statement: This research was financed by the ERA-CoBioTech project WooBAdh (Environmentally-friendly bioadhesives from renewable resources). The University of Ljubljana, Biotechnical Faculty was financed by the Slovenian Ministry of Education, Science and Sport and the Slovenian Research Agency within the framework of program P4-0015. The LERMAB was financed by the French Agence Nationale de la Recherche (ANR) as part of the laboratory of excellence (LABEX) ARBRE.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Fengel, D., Wegener, G. (1989). Wood: chemistry, ultrastructure, reactions. Berlin: De Gruyter. [Google Scholar]
2. Nimz, H. (1983). Lignin-based adhesives. In: Pizzi, A. (eds.Wood adhesives chemistry and technology. New York: Marcel Dekker. [Google Scholar]
3. Pizzi, A. (1994). Advanced wood adhesives technology. New York: Marcel Dekker.
4. Pizzi, A. (2003). Natural phenolic adhesives 2: lignin. In: Pizzi, A., Mittal, K. L. (eds.Handbook of adhesive technology. 2nd edition. New York: Marcel Dekker.
5. Pizzi, A. (2017). Natural phenolic adhesives derived from Tannins and Lignin. In: Pizzi, A., Mittal, K. L. (eds.Handbook of adhesive technology. 3rd edition. New York: Marcel Dekker. [Google Scholar]
6. TECNARO GmBH (2020). Ilsfeld, Germany. https://www.tecnaro.de/. [Google Scholar]
7. Li, J., Zhang, J., Zhang, S., Gao, Q., Li, J. et al. (2017). Fast curing bio-based phenolic resins via lignin demethylated under mild reaction condition. Polymers, 9(12), 428. DOI 10.3390/polym9090428. [Google Scholar] [CrossRef]
8. Wang, F., Kuai, J., Pan, H., Wang, N., Zhu, X. (2018). Study on the demethylation of enzymatic hydrolysis lignin and the properties of lignin–epoxy resin blends. Wood Science and Technology, 52(5), 1343–1357. DOI 10.1007/s00226-018-1024-z.
9. Zhou, H., Qiu, X., Yang, D., Xie, S. (2015). Laccase and xylanase incubation enhanced the sulfomethylation reactivity of alkali lignin. ACS Sustainable Chemistry & Engineering, 4(3), 1248–1254. DOI 10.1021/acssuschemeng.5b01291.
10. Song, Y., Wang, Z., Yan, N., Zhang, R., Li, J. (2016). Demethylation of wheat straw alkali lignin for application in phenol formaldehyde adhesives. Polymers (Basel), 8(6), 209. DOI 10.3390/polym8060209. [Google Scholar] [CrossRef]
11. Upton, B. M., Kasko, A. M. (2016). Strategies for the conversion of lignin to high-value polymeric materials: Review and perspective. Chemical Reviews, 116(4), 2275–2306. DOI 10.1021/acs.chemrev.5b00345. [Google Scholar] [CrossRef]
12. Naseem, A., Tabasum, S., Zia, K. M., Zuber, M., Ali, M. et al. (2016). Lignin-derivatives based polymers, blends and composites: A review. International Journal of Biological Macromolecules, 93, 296–313. DOI 10.1016/j.ijbiomac.2016.08.030. [Google Scholar] [CrossRef]
13. Podschun, J., Stücker, A., Buchholz, R. I., Heitmann, M., Schreiber, A. et al. (2016). Phenolated lignins as reactive precursors in wood veneer and particleboard adhesion. Industrial & Engineering Chemistry Research, 55(18), 5231–5237. DOI 10.1021/acs.iecr.6b00594. [Google Scholar] [CrossRef]
14. Zhao, M., Jing, J., Zhu, Y., Yang, X., Wang, X. et al. (2016). Preparation and performance of lignin–phenol–formaldehyde adhesives. International Journal of Adhesion and Adhesives, 64, 163–167. DOI 10.1016/j.ijadhadh.2015.10.010.
15. Stewart, D. (2008). Lignin as a base material for materials applications: Chemistry, application and economics. Industrial Crops and Products, 27(2), 202–207. DOI 10.1016/j.indcrop.2007.07.008.
16. Gonçalves, A. R., Benar, P. (2001). Hydroxymethylation and oxidation of organosolv lignins and utilization of the products. Bioresource Technology, 79(2), 103–111. DOI 10.1016/S0960-8524(01)00056-6. [Google Scholar] [CrossRef]
17. Calvé, L. R. (1999). Fast-cure and pre-cure resistant cross-linked phenol-formaldehyde adhesives and methods of making the same. Canada Patent 2042476. [Google Scholar]
18. Chen, X., Xi, X., Pizzi, A., Fredon, E., Du, G. et al. (2020). Oxidized demethylated lignin as a bio-based adhesive for wood bonding. Journal of Adhesion, 1–18. DOI 10.1080/00218464.2019.1710830. [Google Scholar] [CrossRef]
19. Valkonen, S., Hübsch, C. (2017). Lignin based binders: An industrial reality, latest developments. Wood Adhesives 2017, 11th International Conference on Wood Adhesives. Atlanta, GE: UPM Biochemicals. [Google Scholar]
20. Peng, Y., Zheng, Z., Sun, P., Wang, X., Zhang, T. (2013). Synthesis and characterization of polyphenol-based polyurethane. New Journal of Chemistry, 37(3), 729–734. DOI 10.1039/c2nj41079f. [Google Scholar] [CrossRef]
21. Ge, J. J., Sakai, K. (1993). Compressive properties and biodegradabilities of polyurethane foams derived from condensed tannins. Mokusai Gakkaishi, 39, 801–806.
22. Ge, J. J., Sakai, K. (1998). Decomposition of polyurethane foams derived from condensed tannin II: Hydrolysis and aminolysis of polyurethane foams. Journal of Wood Science, 44(2), 103–105. DOI 10.1007/BF00526253.
23. Ge, J. J., Shi, X., Cai, M., Wu, R., Wang, M. (2003). A novel biodegradable antimicrobial PU foam from wattle tannin. Journal of Applied Polymer Science, 90(10), 2756–2763. DOI 10.1002/app.12928.
24. Faruk, O., Sain, M. (2013). Continuous extrusion foaming of lignin enhanced thermoplastic polyurethane (TPU). Journal of Biobased Materials and Bioenergy, 7(3), 309–314. DOI 10.1166/jbmb.2013.1365.
25. Hatakeyama, H., Hatakeyama, T. (2013). Advances of polyurethane foams derived from lignin. Journal of Renewable Materials, 1(2), 113–123. DOI 10.7569/JRM.2012.634111.
26. Liu, J., Liu, H. F., Deng, L., Liao, B., Guo, Q. X. (2013). Improving aging resistance and mechanical properties of waterborne polyurethanes modified by lignin amines. Journal of Applied Polymer Science, 130(3), 1736–1742. DOI 10.1002/app.39267. [Google Scholar] [CrossRef]
27. Whelan, J. M. Jr., Hill, M., Cotter, R. J. (1963). Multiple cyclic carbonate polymers. US Patent 3072613. [Google Scholar]
28. Rokicki, G., Piotrowska, A. (2002). A new route to polyurethanes from ethylene carbonate, diamines and diols. Polymer, 43(10), 2927–2935. DOI 10.1016/S0032-3861(02)00071-X. [Google Scholar] [CrossRef]
29. Kihara, N., Endo, T. (1993). Synthesis and properties of poly(hydroxyurethane)s. Journal of Polymer Science, Part A: Polymer Chemistry, 31(11), 2765–2773. DOI 10.1002/pola.1993.080311113. [Google Scholar] [CrossRef]
30. Kihara, N., Kushida, Y., Endo, T. (1996). Optically active poly(hydroxyurethane)s derived from cyclic carbonate and L-lysine derivatives. Journal of Polymer Science, Part A: Polymer Chemistry, 34, 2173–2179.
31. Tomita, H., Sanda, F., Endo, T. (2001). Structural analysis of polyhydroxyurethane obtained by polyaddition of bifunctional five-membered cyclic carbonate and diamine based on the model reaction. Journal of Polymer Science, Part A: Polymer Chemistry, 39, 851–859.
32. Tomita, H., Sanda, F., Endo, T. (2001). Polyaddition behavior of bis(five- and six-membered cyclic carbonate)s with diamine. Journal of Polymer Science, Part A: Polymer Chemistry, 39, 860–867.
33. Tomita, H., Sanda, F., Endo, T. (2001). Model reaction for the synthesis of polyhydroxyurethanes from cyclic carbonates with amines: Substituent effect on the reactivity and selectivity of ring-opening direction in the reaction of five-membered cyclic carbonates with amine. Journal of Polymer Science, Part A: Polymer Chemistry, 39(21), 3678–3685. DOI 10.1002/pola.10009.
34. Birukov, O., Potashnikova, R., Leykin, A., Figovsky, O., Shapovalov, L. (2009). Advantages in chemistry and technology of non-isocyanate polyurethane. Scientific Israel-Technological Advantages, 11, 160–167.
35. Figovsky, O., Shapovalov, L. (2002). Features of reaction amino-cyclocarbonate for production of new type polyurethanes. Macromolecular Symposia, 187, 325–332.
36. Camara, F., Benyahya, S., Besse, V., Boutevin, G., Auvergne, R. et al. (2014). Reactivity of secondary amines for the synthesis of non-isocyanate polyurethanes. European Polymer Journal, 55, 17–26. DOI 10.1016/j.eurpolymj.2014.03.011.
37. Blattmann, H., Fleischer, M., Bähr, M., Mülhaupt, R. (2014). Isocyanate- and phosgene-free routes to polyfunctional cyclic carbonates and green polyurethanes by fixation of carbon dioxide. Macromolecular Rapid Communications, 35(14), 1238–1254. DOI 10.1002/marc.201400209.
38. Boyer, A., Cloutet, E., Tassaing, T., Gadenne, B., Alfos, C. et al. (2010). Solubility in CO2 and carbonation studies of epoxidized fatty acid diesters: Towards novel precursors for polyurethane synthesis. Green Chemistry, 12(12), 2205–2213. DOI 10.1039/c0gc00371a.
39. Kim, M. R., Kim, H. S., Ha, C. S., Park, D. W., Lee, J. K. (2001). Syntheses and thermal properties of poly(hydroxy)urethanes by polyaddition reaction of bis(cyclic carbonate) and diamines. Journal of Applied Polymer Science, 81(11), 2735–2743. DOI 10.1002/app.1719.
40. Ochiai, B., Inoue, S., Endo, T. (2005). Salt effect on polyaddition of bifunctional cyclic carbonate and diamine. Journal of Polymer Science, Part A: Polymer Chemistry, 43(24), 6282–6286. DOI 10.1002/pola.21081.
41. Ubaghs, L., Fricke, N., Keul, H., Höcker, H. (2004). Polyurethanes with pendant hydroxyl groups: Synthesis and characterization. Macromolecular Rapid Communications, 25(3), 517–521. DOI 10.1002/marc.200300064.
42. Fleischer, M., Blattmann, H., Mülhaupt, R. (2013). Glycerol-, pentaerythritol- and trimethylolpropane-based polyurethanes and their cellulose carbonate composites prepared via the non-isocyanate route with catalytic carbon dioxide fixation. Green Chemistry, 15(4), 934–942. DOI 10.1039/c3gc00078h.
43. Besse, V., Auvergne, R., Carlotti, S., Boutevin, G., Otazaghine, B. et al. (2013). Synthesis of isosorbide based polyurethanes: An isocyanate free method. Reactive and Functional Polymers, 73(3), 588–594. DOI 10.1016/j.reactfunctpolym.2013.01.002.
44. Camara, F., Benyahyaa, S., Besse, V., Boutevin, G., Auvergne, R. et al. (2014). Reactivity of secondary amines for the synthesis of non-isocyanate polyurethanes. European Polymer Journal, 55, 17–26. DOI 10.1016/j.eurpolymj.2014.03.011.
45. Cornille, A., Auvergne, R., Figovsky, O., Boutevin, B., Caillol, S. (2017). A perspective approach to sustainable routes for non-isocyanate polyurethanes. European Polymer Journal, 87, 535–552. DOI 10.1016/j.eurpolymj.2016.11.027. [Google Scholar] [CrossRef]
46. Nohra, B., Candy, L., Blanco, J. F., Guerin, C., Raoul, Y. et al. (2013). From petrochemical polyurethanes to biobased polyhydroxyurethanes. Macromolecules, 46(10), 3771–3792. DOI 10.1021/ma400197c. [Google Scholar] [CrossRef]
47. Annunziata, L., Diallo, A. K., Fouquay, S., Michaud, G., Simon, F. et al. (2014). α,ω-Di(glycerol carbonate) telechelic polyesters and polyolefins as precursors to polyhydroxyurethanes: An isocyanate-free approach. Green Chemistry, 16, 1947–1956. [Google Scholar]
48. Tundo, P., Selva, M. (2002). The chemistry of dimethyl carbonate. Accounts of Chemical Research, 35(9), 706–716. DOI 10.1021/ar010076f. [Google Scholar] [CrossRef]
49. Thébault, M., Pizzi, A., Dumarçay, S., Gerardin, P., Fredon, E. et al. (2014). Polyurethanes from hydrolysable tannins obtained without using isocyanates. Industrial Crops and Products, 59, 329–336. DOI 10.1016/j.indcrop.2014.05.036. [Google Scholar] [CrossRef]
50. Thébault, M., Pizzi, A., Essawy, H., Baroum, A., van Assche, G. (2015). Isocyanate free condensed tannin-based polyurethanes. European Polymer Journal, 67, 513–526. DOI 10.1016/j.eurpolymj.2014.10.022.
51. Thébault, M., Pizzi, A., Santiago-Medina, F. J., Al-Marzouki, F. M., Abdalla, S. (2017). Isocyanate-free polyurethanes by coreaction of condensed tannins with aminated tannins. Journal of Renewable Materials, 5(1), 21–29. DOI 10.7569/JRM.2016.634116. [Google Scholar] [CrossRef]
52. Xi, X., Wu, Z., Pizzi, A., Gerardin, C., Lei, H. et al. (2019). Non-isocyanate polyurethane adhesive from sucrose used for particleboard. Wood Science and Technology, 53(2), 393–405. DOI 10.1007/s00226-019-01083-2.
53. Xi, X., Pizzi, A., Delmotte, L. (2018). Isocyanate-free polyurethane coatings and adhesives from mono- and di-saccharides. Polymers, 10(4), 402. DOI 10.3390/polym10040402. [Google Scholar] [CrossRef]
54. Xi, X., Wu, Z., Pizzi, A., Gerardin, C., Lei, H. et al. (2019). Non‐isocyanate polyurethane adhesive from sucrose used for particleboard. Wood Science and Technology, 53(2), 393–405. DOI 10.1007/s00226-019-01083-2. [Google Scholar] [CrossRef]
55. Vovk, M., Sernek, M. (2020). Aluminium trihydrate-filled poly(methyl methacrylate) (PMMA/ATH) waste powder utilization in wood-plastic composite Boards bonded by MUF resin. BioResources, 15(2), 3252–3269. [Google Scholar]
56. Humphrey, P. (1993). Device for testing adhesive bonds. US Patent 5176028. [Google Scholar]
57. Jošt, M., Šernek, M. (2009). Shear strength development of the phenol–formaldehyde adhesive bond during cure. Wood Science and Technology, 43(1–2), 153–166. DOI 10.1007/s00226-008-0217-2.
58. Šernek, M., Dunky, M. (2010). Adhesive bond strength development. In: Thoemen, H., Irle, M., Sernek, M. (eds.Wood-Based panels: an introduction for specialists. pp. 203–224. London: Brunel University Press. [Google Scholar]
59. Granata, A., Argyropoulos, D. S. (1995). 2-Chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane, a reagent for the accurate determination of the uncondensed and condensed phenolic moieties in lignins. Journal of Agricultural and Food Chemistry, 43(6), 1538–1544. DOI 10.1021/jf00054a023. [Google Scholar] [CrossRef]
60. Pu, Y., Cao, S., Ragauskas, A. J. (2011). Application of quantitative 31P NMR in biomass lignin and biofuel precursors characterization. Energy & Environmental Science, 4(9), 3154–3166. DOI 10.1039/c1ee01201k. [Google Scholar] [CrossRef]
61. Baumberger, S., Abaecherli, A., Fasching, M., Gellerstedt, G., Gosselink, R. et al. (2007). Molar mass determination of lignins by size-exclusion chromatography: Towards standardisation of the method. Holzforschung, 61(4), 459–468. DOI 10.1515/HF.2007.074. [Google Scholar] [CrossRef]
62. Nebhani, L., Schmiedl, D., Barner, L., Barner-Kowollik, C. (2010). Quantification of grafting densities achieved via modular ‘‘grafting-to’’ approaches onto divinylbenzene microspheres. Advanced Functional Materials, 20(12), 2010–2020. DOI 10.1002/adfm.200902330. [Google Scholar] [CrossRef]
63. Sluiter, A., Hames, B., Ruiz, R., Scarlata, C., Sluiter, J. et al. (2008). Determination of ash in biomass-laboratory analytical procedure (LAP). Technical Report NREL/TP-510-42622, Golden, Colorado, USA: NREL National Renewable Energy Laboratory. [Google Scholar]
64. ASTM D7998–19. (2019). Standard test method for measuring the effect of temperature on the cohesive strength development of adhesives using lap shear bonds under tensile loading. West Conshohocken, PA: ASTM International. www.astm.org. [Google Scholar]
65. European Norm EN 319. (1993). Perpendicular tensile strength on particleboards and fibreboards. [Google Scholar]
66. Crestini, C., Lange, H., Sette, M., Argyropoulos, D. S. (2017). On the structure of softwood kraft lignin. Green Chemistry, 19(17), 4104–4121. DOI 10.1039/C7GC01812F. [Google Scholar] [CrossRef]
Appendix A
Lignin Acetylation
The organosolv lignin powder (OL) was vacuum dried at a temperature of 45°C for 3 days at a pressure of 10 mbars. 200 mg of vacuum dried OL powder were solved in 4 ml of acetic anhydride/pyridine (1/1, v/v) at room temperature by vortex mixing. Subsequently, the mixture was stirred at room temperature for 5 days in a 10 ml flask with cap. The concentration of the lignin in this solution was approximately 50 mg/ml. After 5 days, the solution was diluted with approx. 5 ml of ethanol and stirred for an additional 30 minutes. The solvents were removed by rotation evaporation at 60°C in a pressure range from 800 to 5 mbars. The addition of ethanol and the rotation evaporation were repeated twice to remove the solvents. Subsequently, the acetylated lignin was dried in a vacuum oven at 55°C at a pressure of 10 mbars for additional 3 days.
Appendix B
Table B1: Structures assignments of peaks of MALDI ToF analysis of organosolv lignin
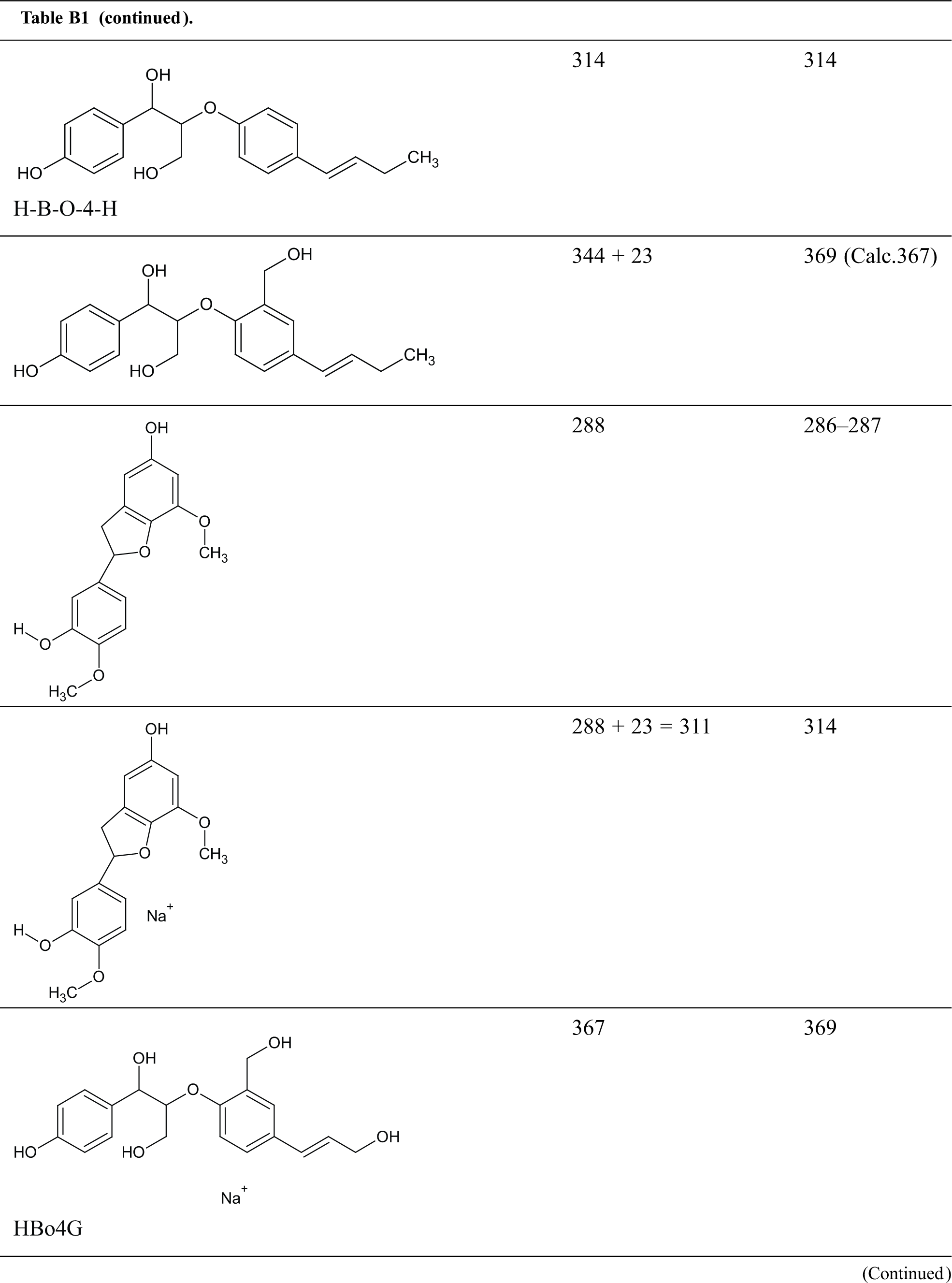
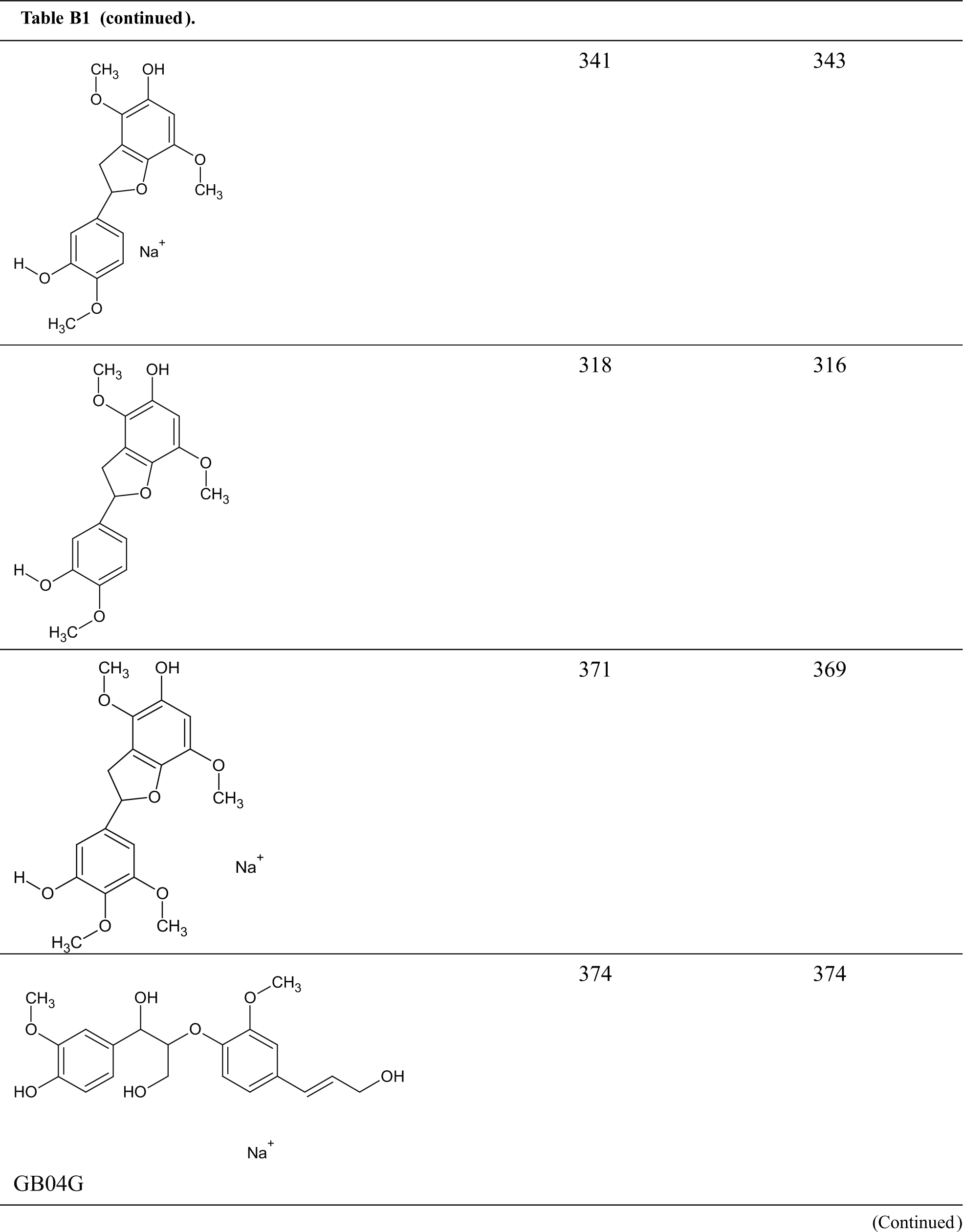
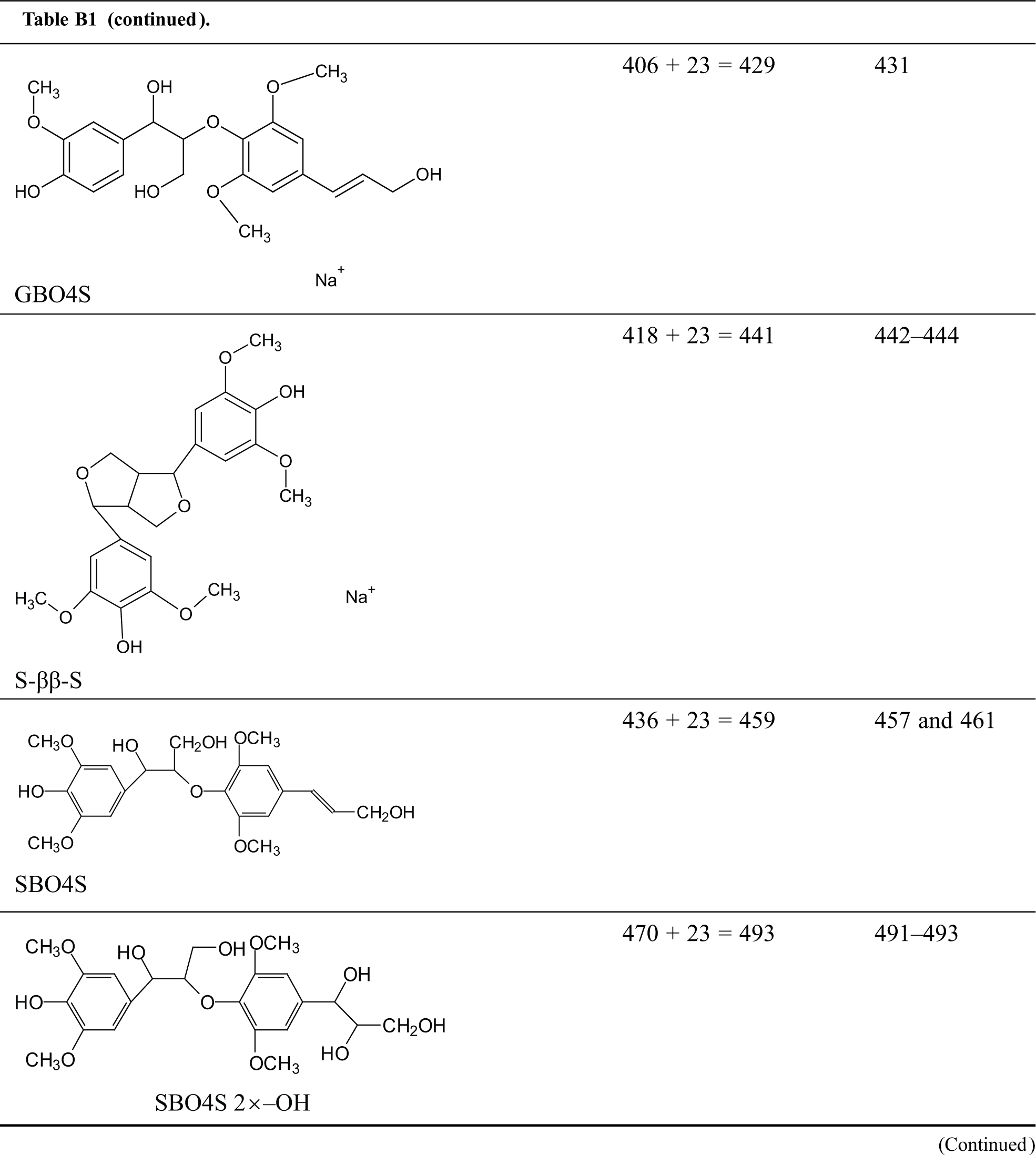
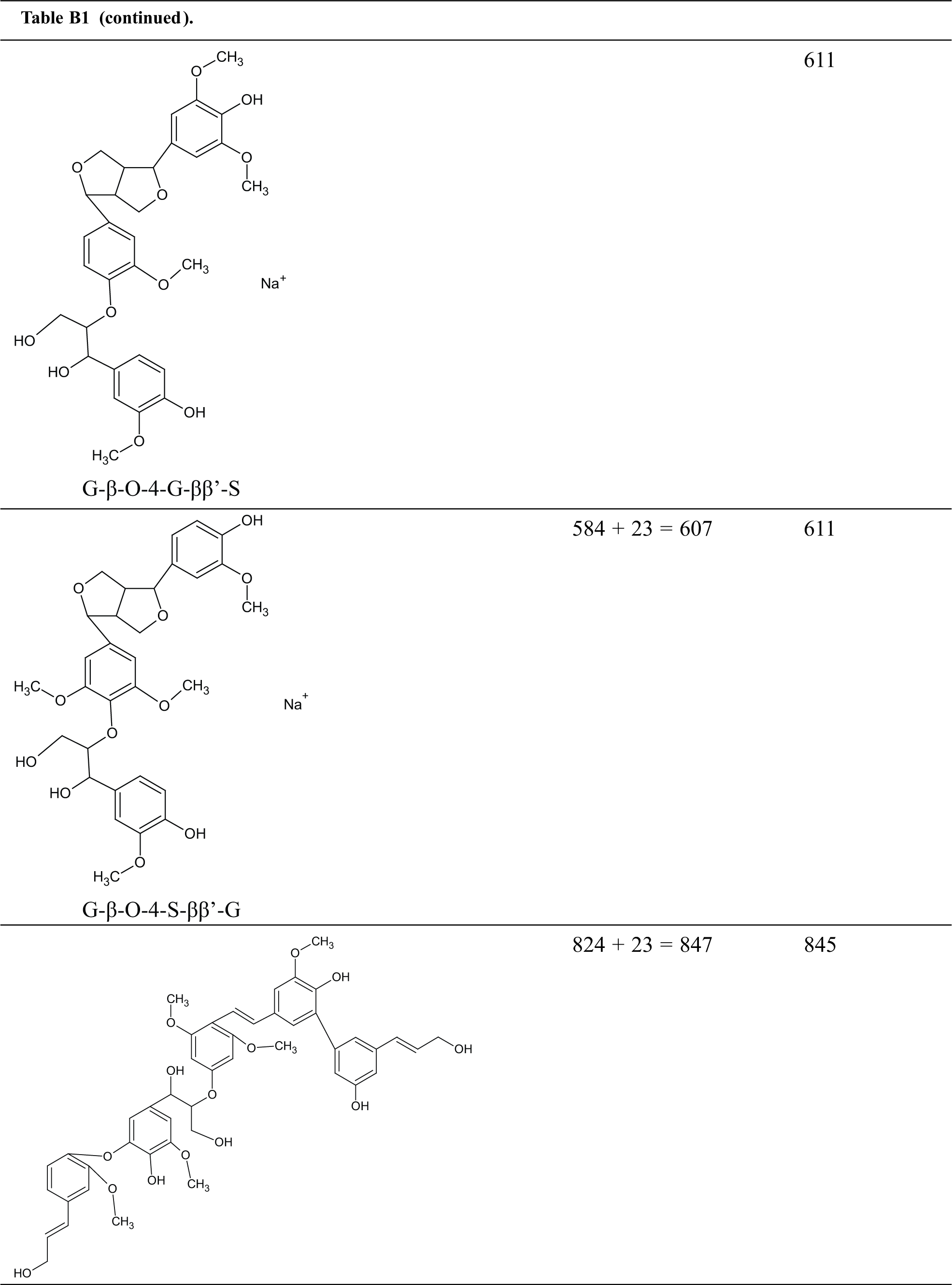
Appendix C
Structures assignments of peaks of MALDI ToF analysis of organosolv lignin NIPU Resin.




 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |