| Journal of Renewable Materials |
DOI: 10.32604/jrm.2021.016301
ARTICLE
In Silico Disulfide Bond Engineering to Improve Human LEPTIN Stability
1School of Energy and Power Engineering, Jiangsu University, Zhenjiang, 212013, China
2Naghshe Jahan-Iliya Chemistry, Research&Development Centre, Isfahan, Iran
3Department of Microbiology, Qom Branch, Islamic Azad University, Qom, Iran
*Corresponding Author: Shuang Wang. Email: alexjuven@ujs.edu.cn
Received: 24 February 2021; Accepted: 25 March 2021
Abstract: Enhancing the stability of biomolecules is one of the hot topics in industry. In this study, we enhanced the stability of an important protein called LEPTIN. LEPTIN is a hormone secreted by fat cells playing an essential role in body weight and composition, and its deficiency can result in several disorders. The treatment of related LEPTIN dysfunctions is often available in the form of injection. To decrease the cost and the frequency of its applications can be achieved by increasing its lifetime through engineering LEPTIN. In this study, to engineer LEPTIN, we have introduced disulfide bonds. Disulfide By Design server was used to predict the suitable nominate pairs, which suggested three pairs of amino acids to be mutated to cysteine for disulfide bond formation. Additionally, to further evaluate the effect of combined mutations, we combined these three nominated pairs to produce three more mutants. In order to assess the effect of introduced mutations, molecular dynamic (MD) simulation was performed. The result suggests that Mutant-1 is more stable in comparison to wild-type and the other mutants. Moreover, docking results showed that the introduced mutation does not affect the receptor binding performance; therefore, it can be considered a suitable choice for future protein engineering.
Keywords: Insilco protein engineering; LEPTIN; disulfide bond prediction; molecular dynamic simulation; docking
Historically LEPTIN was discovered in 1994; the word LEPTIN, meaning thin, refers to the Greek word LEPTOS. The primary producer of LEPTIN are adipocytes (fat cells); however, a lower level of production occurs in the gastric fundic epithelium, skeletal muscle, mammary epithelium, intestine, placenta, and brain [1]. Stored body fat directly controls the LEPTIN ratio; as body fat increases, LEPTIN concentration in blood plasma levels up, and oppositely fasting leads to declined LEPTIN secretion [2]. LEPTIN is known as a critical control in body composition, food consumption, and, more importantly, body weight [3]. LEPTIN plays a more critical role in acute (e.g., fasting) and chronic energy-deficient states (e.g., diet or exercise-induced hypothalamic amenorrhea and lipoatrophy) than in energy-replete states (e.g., obesity). These energy-deficient circumstances are linked to relative LEPTIN deficiency, which, in turn, is related to neuroendocrine abnormalities such as metabolic dysfunction, infertility, depressed immune function, and bone loss [4,5]. Human recombinant LEPTIN may aid as a treatment option in these circumstances [6]. The therapeutic option is available as an injection for both children and adults suffering from severe obesity, and other dysfunctions resulted from LEPTIN deficiency. Daily injections result in weight loss by reducing fat mass due to reduced food intake [7]. In recent years LEPTIN replacement therapy has been introduced to normalize phenotype caused by LEPTIN deficiency [8] and remarkable positive changes observed in patients [9].
LEPTIN is a secreted protein that consists of 146 residues and encompasses only one native disulfide bond [10]. LEPTIN three-dimensional structure contains two long crossover link four antiparallel α-helices, and a two-layer packing formed by a short loop located in the left-handed helical bundle. A vital naturally disulfide bond exists between residue Cys96 and Cys146, which probably plays a role in the folding and receptor binding of the molecule [11]. LEPTIN binds to a membrane protein, LepR, composed of four cytokine receptor homologous domains (CRH), an Ig-like domain, a transmembrane segment, and a C-terminal cytoplasmic domain in the long isoform. Initial work on identifying the LEPTIN binding domain (LBD) of LepR showed amino acids 323–640 in binding [12], which consists of the Ig-like and the second CRH (CRH2) domains. Various groups have addressed specific molecular interactions between Lep and LepR through the site-directed mutagenesis and modeling approaches [13–15]. Recently, the structure of the CRH2 of LepR (PDB: 3v6o) structure of the human obesity receptor LEPTIN-binding domain revealed the mechanism of LEPTIN antagonism by a monoclonal antibody. It was proposed that LEPTIN binds tightly to the CRH2 domain, while also interacting with a second LepR through the Ig-like domain [16].
As mentioned earlier, LEPTIN hormone (protein) is used in the form of injection to cure the dysfunctions resulted from LEPTIN deficiencies. However, there is a high chance that it becomes unfolded and lose its natural structure upon exposure to uncontrolled conditions [17]. Temperature is the primary environmental factor affecting protein structure and functionality or even poses a severe risk to consumers’ health [18,19]. This led pharmaceutical company face considerable challenges in the production and logistics of protein therapeutics. To retain a constant temperature, cold chain instruments have become vital to the biopharma industry globally, projected to spend beyond 18 billion USD by 2022 for the temperature-controlled shipment [20]. Therefore, enhancing the stability of proteins to tolerate high temperatures is critical to elucidate the challenges in efficacy, economy, and safety of therapeutics [20]. Besides, considering the economic reasons, the increase of stability and consequently longer half-life can reduce doses and the number of uses. Therefore, protein engineering can be employed to enhance protein efficiency and stability [21].
There are several methods to enhance protein stability, such as disulfide bridges [22], hydrogen bonds [23], ion pairs [24]. Disulfide bonds exist in many proteins, where they make the conformation more stable by reducing the entropy of the unfolded conformation [25]. The introduction of disulfide bonds has been proposed as a promising stabilizing strategy, potentially leading to more stable protein conformation by decreasing conformational entropy compared with their unfolded state [26]. Fundamentally, loops are known as the most fluctuating region of the protein. The disulfide bonds that bring longer loops closer are more effective than the shorter ones [27]. In protein rational design, amino acid replacement is challenging due to the high number of possible substitutions. In silico techniques can predict stability changes after mutations quickly and with reasonable accuracy [28]. In recent years, progress in computers and the development of simulation software have significantly contributed to advancing biological science. It is now possible to generate natural components, mimic environmental conditions in computers, and perform experiments in this simulated condition. One of the methods that can be used for simulation is molecular dynamics (MD), which applies physics-based formulas to model protein dynamics. It offers atomistic data of the molecular interactions that participate in protein stability or protein function [29]. Moreover, it is possible to assess the interaction of protein with its receptor or ligand via docking study [30]. Hence, in this study, several mutants were created by the addition of novel disulfide bonds; further, the effect of introduced mutations on protein stability were evaluated using molecular dynamic simulation. Furthermore, the effect of LEPTIN on its receptor was studied using molecular docking.
2.1 Mutation Site Prediction and Generation
LEPTIN protein tertiary structure was downloaded from Protein Data Bank (PDB) (http://www.rcsb.org/pdb) with the entry (1AX8) and examined to remove water molecules. In order to find potential mutation sites to form disulfide bonds, Disulfide by Design server [31] was used. Based on the server calculations, six candidates were predicted and listed; then, according to the described criteria by the server, the first three potential mutation sites were selected to create the mutant files (Fig. 1).

Figure 1: Result of disulfide by design server and generated mutations. initially, the potential mutation sites were suggested and ranked, and then mutations were generated
Accordingly, the residues involved in the formation of both novel and native disulfide bonds (C96 and C146) are highlighted in using PyMOL (The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC) software. Four mutation sites are located in a loop, including LEU 39, ALA 101, GLY 44 and PRO 47; two other sites are also located in the helix, including SER 127 and ALA 59 (Fig. 2).

Figure 2: Mutation site position on LEPTIN structure: the LEU 39, ALA 101, GLY 44, PRO 47, SER 127 and ALA 59 are candidate mutation sites selected to be replaced with cys
2.2 Molecular Dynamic Simulation
The molecular dynamic simulation was performed using the Gromacs 4.6.3 program [32] by the OPLS-AA force field [33] implemented on a LINUX operating system in triplicate. Prepared PDB files of six mutants and a native structure were used for MD simulation. The structures were solvated with 32702 simple point charges (SPC) water model, and the system was neutralized with six Na+ ions. The cubic type of box by 10 nm size was used for MD simulation. The solvated structure was minimized by the steepest descent method for 10000 steps at 300°K and constant pressure. Then the structure's equilibration was conducted in two phases. The first phase is conducted under an NVT ensemble (constant number of particles, volume, and temperature), and in the second phase, equilibration of pressure is conducted under an NPT ensemble, wherein the number of particles, pressure, and temperature are all constant, both for the length of 2500 ps. After equilibration production, MD was run for 50000 ps at constant temperature and pressure. The LINCS algorithm [34] was used to constrain the bond length. Periodic boundary conditions were applied to the system. The electrostatic interactions were calculated using a PME algorithm with 0.9 Å cut off [35]. During the simulation, every 1.000 ps of the actual frame was stored. The integration time step was 2 fs, with the neighbor list being updated every fifth step by using the grid option and a cut-off distance of 12 A˚. The isotropic Parrinello–Rahman [36] protocol was used for pressure (1 bar), and the velocity-rescaling thermostat was used for temperature coupling. The trajectory data were calculated by GROMACS tools, including g_rms, g_rmsd, g_gyrate, and do_dssp. To understand the effect of the mutations, molecular dynamics simulations were successfully performed for 50 ns on six structures of in silico engineered LEPTIN and wild-type LEPTIN (1AX8). The standard GROMACS routines were employed in the analysis of trajectory files such as root mean square deviation (RMSD), root mean square fluctuations (RMSF) and radius of gyration (Rg). Also, the secondary structures were assigned by using the DSSP program [37].
In this study, molecular docking calculations were carried out to visualize the binding mechanism of all mutants and wild-type of LEPTIN with the receptor to investigate the effect of the mutations on the performance of LEPTIN. The average structures of LEPTIN and its mutants after 40 ns simulation were used for docking calculations. Docking calculations were accomplished using the HADDOCK interface [38], which is one of the best protein-protein docking packages and directly uses experimental information. Experimental data that are feed into this docking engine are active residues of proteins that have central importance for the interactions. These data are transformed by HADDOCK into ambiguous interaction restraints (AIRs) and then used to drive the docking. The docking procedure with HADDOCK consists of three phases: a rigid body energy minimization, a semi-flexible refinement in torsion angle space, and a final refinement in explicit solvent. After each of these stages, structures are scored and ranked, and the best structures are implemented for the next stage. After a successful docking run, clustered results are displayed, and for every cluster, various energies are presented that make up the HADDOCK score. The HADDOCK score is a linear weighted sum of van der Waals, electrostatic, desolvation, and restraint violation energies together with the buried surface area.
This program calculates the secondary structure evolution of a protein over time and represents the β-sheet, α-helix contents, and other secondary structures. The analysis of secondary structures helps further understand how mutations are linked to conformational and functional changes [39]. In general, three main regions, including the residue between 15 to 25, 50 to 55, and 85 to 105, were observed to differ in the mutants compared to wild type. Indicating the introduction of disulfide bonds has led to the relocation of the main structures.
In mutant-1, in the region between the residues 15 to 25, after 27000 ps, the residue was shown to change their conformation from bend to turn, and turn to α-helix. Although this transition enhanced the stability of the structure, the transition of the residues between 50 and 55 (α-helix to bend) together with the residues from 90 to 105 (α-helix to 3-helix and bend), could diminish this effect and unstablize the structure. In mutant-2, in the region between the residues 15 to 25, conformation changed from the bend and turn to coil. Also, the residues between 50 and 55 had a transition from the bend and the turn to coil. While, for the residues from 90 to 105, the secondary structure displayed a change from α-helix to 3-helix and bend. All the mentioned changes in conformation caused structure alteration to a less stable state. Similarly, in mutant-3, the conformation changed to a lesser stable state. In the region between residues 15 and 25, the conformation altered from the bend and turn to coil. While residue 30 shifted the conformation from the coil to bend through the simulation time, which is slightly more stable. Also, the residues between 50 and 55 had a transition from bend and turn to coil, and also the secondary structure of the residues from 90 to 105 tended to change from α-helix to 3-helix and bend.
In mutant-4, after 23000 ps of simulation, the region between the residues 1 to 5 altered from α-helix to bend and turn, which may reduce the structure’s stability. While, in the region between the residues 15 and 20, the conformation transformed from the bend and turn to α-helix, which confer more stability in this region. Also, residue 30 transformed from the coil to bend throughout the simulation time. While, for the residues from 90 to 105, the secondary structure was changed from α-helix to turn, which substantially reduces the stability of the structure. In mutant-5, in the region between the residues 15 and 20, it tended to change from bend and turn to coil. The residues between 50 and 55, exhibited no significant changes in comparison to the wild-type. While, the residues from 90 to 105, a significant transition of α-helix to mostly bends was observed, which contributing to the reduction of structural stability. In mutant-6, the region between the residues 15 to 20, the residue exhibited the conformation changes by altering bend and turn to coil, which could lessen the stability of the structure. Whereas residue 30 shifted the conformation from the coil to bend through the simulation time. The residue between 50 and 55 shown only some transition from bend to turn during the simulation. While, the residues from 90 to 105 presented the tendency to transit from α-helix to mostly turn, which destabilizes the structures (Fig. 3).
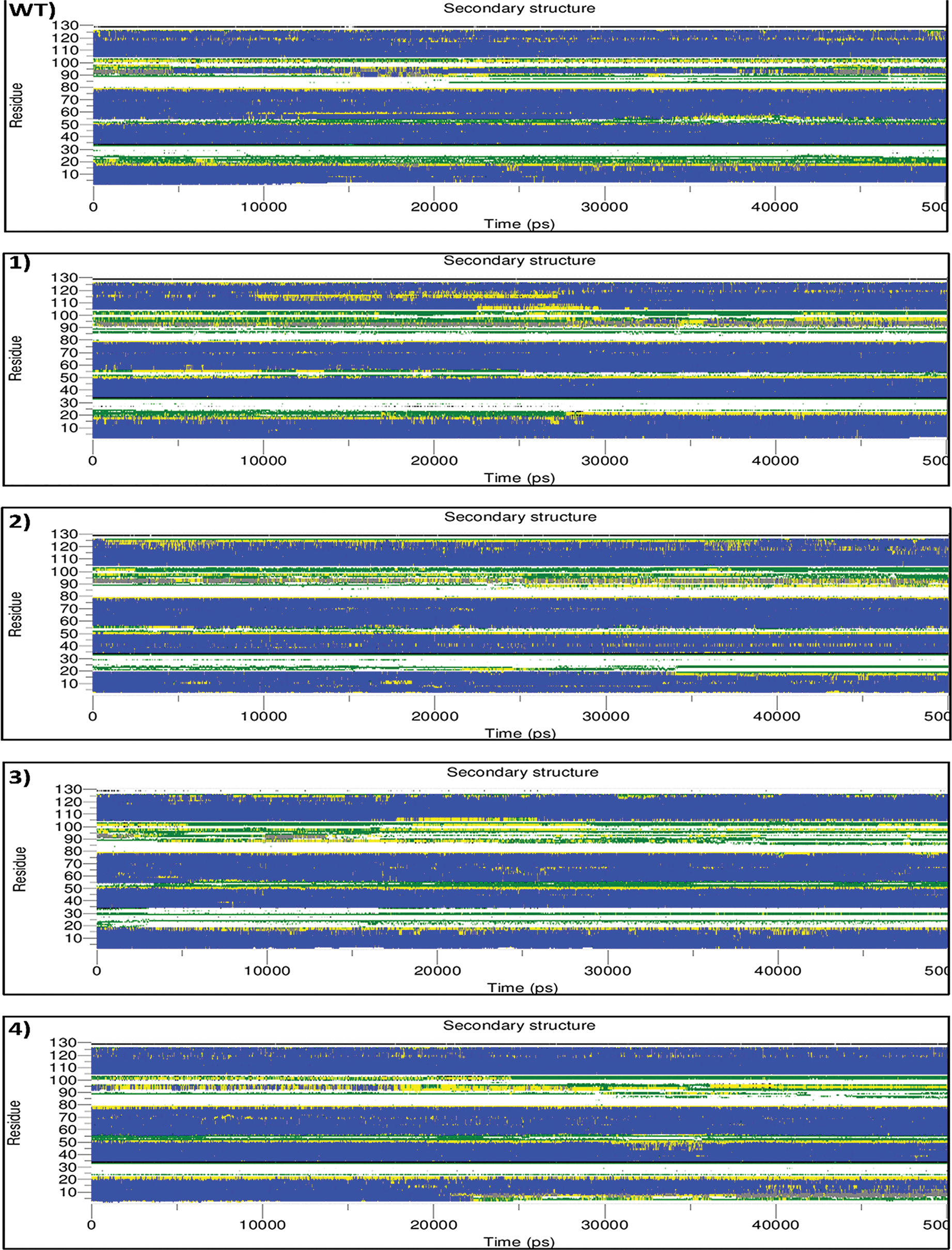

Figure 3: Analysis of the secondary structure elements vs. time. color codes are used to represent secondarystructure elements, coil by white, B-bridge by black, bend by green, turn by yellow, α-helix by blue and 3-helix (3/10) by gray
Root Mean Square Deviations (RMSD) for mutants and wild type to their initial minimized structures were calculated and plotted. As shown in Fig. 4, the RMSDs of each system tends to be stabilized at different time points during the simulation. In order to elucidate the stability of protein structure in the period of simulation, an analysis of backbone atom deviation in comparison to original crystal conformation was performed. RMSD value is widely applied for evaluating structural similarity and protein stability during MD simulation [40]. These results indicate that mutants have undergone a different pattern of fluctuations. For example, in mutant-5, the structure significantly became unstable after the middle of the simulation time; as observed in the previous section, the alpha-helix structure loses its stability and turns into a bend. Except for mutant-1, the rest of the structures showing an increasing trend, at least for the first 5 ns.

Figure 4: RMSD graphs of structures during 50000 ps simulation time. in all graphs, the black line is wild-type RMSD and mutated protein highlighted by different colors
The wild-type structure showed equilibration at 25000 ps around the value of 0.3 nm. In mutant-1, the equilibration time was observed to be at first 5000 ps and remain stable until the end of simulation by around 0.2 nm. In mutant-2, the increasing trend at the first 30000 ps was observed, and then becomes stable around the value of 0.3 nm by the end of simulation time, adjacent to the wild-type value. In mutant-3, the RMSD increased and reached 0.35 nm at the first 10000 ps, then remained stable by the end of the period. In mutant-4, RMSD increased moderately and reached its peak of 0.38 at 33000 ps then began to decrease and fluctuated around 0.3 nm; the pattern of fluctuations showed more instability compared to wild-type. In mutant-5, RMSD dramatically increased in the first 26000 ps, then showed some fluctuations around 0.5 nm until the end of the simulation. It can be interoperated the mutation caused a significant disturbance in structure stability. In mutant-6, RMSD began to increase during the first 5000 ps and reached slightly more than 0.3 nm. Then they dramatically decreased to 0.2 nm at 20000 ps and remained stable until 38000 ps. At 38000 ps of simulation to the rest of the simulation, it followed a similar fluctuation pattern compared to wild-type until the end of the simulation.
3.3 Root-Mean-Square Fluctuation (RMSF)
To summarize, in all mutants, mutations caused the increase in the RMSF value of all residue, specifically in the flexible regions, except for the mutant-1, in which the mutations decreased in the RMSF value of all residue (Fig. 5). To address flexibility of LEPTIN and its mutants, RMSF per residue was calculated from 0 to 50 ns simulation studies. RMSF graphs were plotted for these seven structures, as shown in Fig. 5. Similar RMSF distribution was observed in the fluctuations of these structures, which could be attributed to the secondary structure, including loops and helix. The RMSF is another tool to monitor the dynamic stability of the systems. RMSF can reflect the mobility of the residue around its mean position.

Figure 5: RMSF graphs of mutant and wild-type per residue number during 50 ns simulation time
In wild-type, three central regions are presenting significant fluctuation in RMSF during the simulation, including the residue between 23 and 43, 65 and 80, and 92 and 120. However, a significant increase in the fluctuation was observed in the region between residues 90 and 120. In mutant-1, a significant decrease in the flexibility of residue 105 was observed, which could be due to the introduced mutations, as the linked disulfide bond between residue 39 and 127 might cause both the mutation sites to become closer. In mutant-2, the flexibility of all the residues increased compared to the wild-type. However, the residue's flexibility between 105 and 125 amplified more extensively, with the 117 being the most fluctuating reissue. In mutant-3, the residues 46 and 48 shown significant fluctuation during simulation, which was inconsistent with other mutant and wild-type. Also, in the region between residue 105 and 125, a significant fluctuation was observed. In mutant-4, residues showed the same pattern of flexibility; while, in the region between the residues 105 and 120, the flexibility increased compared to wild-type. In mutant-5, overall, the residue flexibility increased. However, in the region between reidue95 and 120, the RMSF value considerably increased. In mutant-6, the RMSF fluctuation was almost similar to the wild-type, while, in the region between 105 and 125, the residues’ flexibility noticeably increased.
Analysis of Rg of a protein helps to understand the protein compactness during the simulation time. The movements in the secondary structure might contribute to variations in Rg and may influence the required time to attain a compact structure. As shown in Fig. 6, in wild-type, the Rg values during the first 10000 ps fluctuated around 1.45, then slightly increased to around 1.47 for the rest of the simulation time. In mutant-1, in the first 25000 ps of simulation time, the Rg value fluctuated around 1.448, then after, the Rg decreased to around 1.445 nm by the end of the time. In mutant-2, the RG values were fluctuating slightly lower than wild-type around 1.448 while mimicking the same pattern of fluctuation. In mutant-3, the Rg value began to decrease from the beginning and reached 1.442 nm at 25000 ps and maintained the same value by the end of the simulation. In mutant-4, Rg fluctuated very close to the values of Rg in wild-type, although, after the middle of the simulation, Rg fluctuations slightly increased, it followed the same pattern of changes as wild-type. In mutant-5, initially, the Rg value was around 1.448 while it increased and reached 1.455 at 250000 ps, then after it showed intensive fluctuation ranging from 1.445 to 1.555. In mutant-6, the Rg value significantly reduced and fluctuated around 1.445, which is considerably lower than the wild-type.

Figure 6: Radius of gyration graphs of mutant and wild-type
The active residues PHE41, SER93, LYS94, ASP135, LEU142 and SER143, which are located on the surface of all mutants and wild-type of LEPTIN and the active residues ASN431, ASN433, ILE 434, SER435, ASN567, TYR589, LYS614 and GLY618 belonging to CRH2 domain of LepR have central importance for the interactions [41] and were feed into the HADDOCK interface. HADDOCK clustered 137, 140, 141, 131, 138, 139, 130 structures for complex of LEPTIN and Mutants 1, 2, 3, 4, 5 and 6 in the interaction with the receptor in 7, 9, 10, 5, 8, 6, 10 clusters, respectively. Tab. 1 represents the results of the docking run for the best clusters produced by HADDOCK. The negative values of Z-score and small values of RMSD for docking experiments suggested that the complex structures of LEPTIN and its mutants with LBD are acceptable models.
Table 1: Protein-protein docking results of the best cluster produced by HADDOCK for all mutants and wild-type of LEPTIN in the interaction with the receptor

As shown in Fig. 7a the best-ranked pose in the best cluster of LEPTIN/LepR was found to involve four H-bond interactions between the two docking partners. Good global energy scores of docking results showed that residues located at CRH2 domain of LepR have a suitable binding affinity for residues located at the surface of LEPTIN and all mutants; this means that the mutations do not have any effect on the performance of LEPTIN. Fig. 7c shows the superposed structures of the complex between wild-type of LEPTIN and all mutants with the LEPTIN receptor. Also, structural superposition shows similar interaction interfaces in all complexes, and there is no observable difference between the wild type and the mutant protein structures.

Figure 7: a) superposed structures of the complex between wild-type of LEPTIN and all mutants with LEPTIN receptor; b) LEPTIN structure obtained from PDB (3V6O); c) top-ranked poses of the best cluster of LEPTIN/lepR
The first evidence of Cys residue in forming disulfide bond was demonstrated in mice [42]. A LEPTIN variant that is incapable of forming the disulfide bonds exhibited a reduced biological response once injected into LEPTIN-deficient mice. A study performed by a point mutation replacing Cys with Ser disturbed the natural folding of LEPTIN [43], which resulted in dysfunctionality and lack of secretion of LEPTIN. This indicates that preserving the natural folding of LEPTIN is the critical factor for its activity. The addition of disulfide bonds has been introduced as a promising approach to enhance the stability of proteins, and finding the suitable residue to replace with Cys is the critical aspect [29]. In a remarkable study, point mutation of a residue (Lys237) of Homotetrameric β-xylosidase introduced a disulfide bond in Selenomonas ruminantium, which resulted in significant thermal stability [44]. In addition, in the simulation results, it was observed that the RMSF of the mutation site was reduced considerably, which is in agreement with the mutant-1. The engineering disulfide bond in Bacillus subtilis 168/pMA5 enhanced the half-life of when T181C was introduced to 4-hydroxyisoleucine, which also increased the specific enzyme activity by 3.56-fold [45]. Therefore, the addition of disulfide bond not only enhances protein stability, it also increases enzyme activity. In a recent study, it was observed that the introduction of a disulfide bond in GH11 xylanase of Bacilus firmus Strain K-1 at S100C/N147C, accompanied with K40L mutation, enhanced the enzymatic activity at room temperature [46]. Therefore, engineering disulfide bonds can improve both enzyme stability and activity. There are many processes in biofuel production from renewable materials [47–49] in which enzymes can be engineered by disulfide bond addition. In recent years, few studies have applied this approach [50–52] that suggest its suitability for future enzyme engineering studies for important medical and industrial enzymes and proteins, particularly in biofuel production processes [53–55].
The analysis suggests that mutation 1, which encompasses mutation in the residues Lue 39 and Ser 127, exhibited the most stable structure. The mutation made a disulfide bond between loop and alpha-helix structure, which caused more stability in RMSD, reduced flexibility of structure (RMSF analysis), and growth in compactness (RG analysis). Moreover, the docking study indicated that the introduction of disulfide bonds did not alter the performance of the mutant-1. Engineering disulfide bonds to proteins can increase their stability without affecting their performance using algorithms provided by Disulfide by Design. However, in vivo experiments are required for the final validation of this result.
Funding Statement: This work was supported by China Postdoctoral Science Foundation Grant (2019M661742).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Yadav, A., Kataria, M. A., Saini, V., Yadav, A. (2013). Role of leptin and adiponectin in insulin resistance. Clinica Chimica Acta, 417, 80–84. DOI 10.1016/j.cca.2012.12.007. [Google Scholar] [CrossRef]
2. Pénicaud, L., Meillon, S., Brondel, L. (2012). Leptin and the central control of feeding behavior. Biochimie, 94, 2069–2074. DOI 10.1016/j.biochi.2012.04.013. [Google Scholar] [CrossRef]
3. Baak, J. P., Mutter, G. L., Robboy, S., van Diest, P. J., Uyterlinde, A. M. et al. (2005). Food for thought: The role of appetitive peptides in age-related cognitive decline. Cancer, 103, 2304–2312. DOI 10.1002/(ISSN)1097-0142. [Google Scholar] [CrossRef]
4. Friedman, J. M. (2019). Leptin and the endocrine control of energy balance. Nature Metabolism, 1, 754–764. DOI 10.1038/s42255-019-0095-y. [Google Scholar] [CrossRef]
5. do Carmo, J. M., da Silva, A. A., Gava, F. N., Moak, S. P., Dai, X. et al. (2019). Impact of leptin deficiency compared with neuronal-specific leptin receptor deletion on cardiometabolic regulation. American Journal of Physiology-Regulatory Integrative, 317, R552–R562. DOI 10.1152/ajpregu.00077.2019. [Google Scholar] [CrossRef]
6. Dardeno, T. A., Chou, S. H., Moon, H. S., Chamberland, J. P., Fiorenza, C. G. et al. (2010). Leptin in human physiology and therapeutics. Frontiers in Neuroendocrinology, 31, 377–393. DOI 10.1016/j.yfrne.2010.06.002. [Google Scholar] [CrossRef]
7. Heymsfield, S. B., Greenberg, A. S., Fujioka, K., Dixon, R. M., Kushner, R. et al. (1999). Recombinant leptin for weight loss in obese and lean adults. Journal of the American Medical Association, 282, 1568. DOI 10.1001/jama.282.16.1568. [Google Scholar] [CrossRef]
8. Oral, E. A., Simha, V., Ruiz, E., Andewelt, A., Premkumar, A. et al. (2002). Leptin-replacement therapy for lipodystrophy. New England Journal of Medicine, 346, 570–578. DOI 10.1056/NEJMoa012437. [Google Scholar] [CrossRef]
9. Paz-Filho, G., Mastronardi, C. A., Licinio, J. (2015). Leptin treatment: Facts and expectations. Metabolism, 64, 146–156. DOI 10.1016/j.metabol.2014.07.014. [Google Scholar] [CrossRef]
10. Kline, A. D., Becker, G. W., Churgay, L. M., Landen, B. E., Martin, D. K. et al. (1997). Leptin is a four-helix bundle: Secondary structure by NMR. Federation of European Biochemical Societies Letters, 407, 239–242. DOI 10.1016/S0014-5793(97)00353-0. [Google Scholar] [CrossRef]
11. Imagawa, K., Numata, Y., Katsuura, G., Sakaguchi, I., Morita, A. et al. (1998). Structure-function studies of human leptin. Journal of Biological Chemistry, 273, 35245–35249. DOI 10.1074/jbc.273.52.35245. [Google Scholar] [CrossRef]
12. Fong, T. M., Huang, R. R. C., Tota, M. R., Mao, C., Smith, T. et al. (2018). Localization of leptin binding domain in the leptin receptor. Molecular Pharmacology, 53, 234–240. DOI 10.1124/mol.53.2.234. [Google Scholar] [CrossRef]
13. Iserentant, H. (2005). Mapping of the interface between leptin and the leptin receptor CRH2 domain. Journal of Cell Science, 118, 2519–2527. DOI 10.1242/jcs.02386. [Google Scholar] [CrossRef]
14. Niv-Spector, L., Gonen-Berger, D., Gourdou, I., Biener, E., Gussakovsky, E. E. et al. (2006). Identification of the hydrophobic strand in the a–B loop of leptin as major binding site III: Implications for large-scale preparation of potent recombinant human and ovine leptin antagonists. Biochemical Journal, 391, 221–230. DOI 10.1042/BJ20050457. [Google Scholar] [CrossRef]
15. Peelman, F., van Beneden, K., Zabeau, L., Iserentant, H., Ulrichts, P. et al. (2004). Mapping of the leptin binding sites and design of a leptin antagonist. Journal of Biological Chemistry, 279, 41038–41046. DOI 10.1074/jbc.M404962200. [Google Scholar] [CrossRef]
16. Prokop, J. W., Duff, R. J., Ball, H. C., Copeland, D. L., Londraville, R. L. (2012). Leptin and leptin receptor: Analysis of a structure to function relationship in interaction and evolution from humans to fish. Peptides, 38, 326–336. DOI 10.1016/j.peptides.2012.10.002. [Google Scholar] [CrossRef]
17. Bisht, M., Jha, I., Venkatesu, P. (2016). Comprehensive evaluation of biomolecular interactions between protein and amino acid based-ionic liquids: A comparable study between [Bmim][Br] and [Bmim][Gly] ionic liquids. ChemistrySelect, 1, 3510–3519. DOI 10.1002/slct.201600524. [Google Scholar] [CrossRef]
18. Wang, W. (2005). Protein aggregation and its inhibition in biopharmaceutics. International Journal of Pharmaceutics, 289, 1–30. DOI 10.1016/j.ijpharm.2004.11.014. [Google Scholar] [CrossRef]
19. Sauna, Z. E., Lagassé, H. A. D., Alexaki, A., Simhadri, V. L., Katagiri, N. H. et al. (2017). Recent advances in (therapeutic protein) drug development. F1000 Research, 6, 1–17. DOI 10.12688/f1000research.9970.1. [Google Scholar] [CrossRef]
20. Sundaram, V., Ramanan, R. N., Selvaraj, M., Vijayaraghavan, R., MacFarlane, D. R. et al. (2021). Structural stability of insulin aspart in aqueous cholinium aminoate ionic liquids based on molecular dynamics simulation studies. Journal of Molecular Liquids, 322, 114501. DOI 10.1016/j.molliq.2020.114501. [Google Scholar] [CrossRef]
21. Marshall, S. A., Lazar, G. A., Chirino, A. J., Desjarlais, J. R. (2003). Rational design and engineering of therapeutic proteins. Drug Discovery Today, 8, 212–221. DOI 10.1016/S1359-6446(03)02610-2. [Google Scholar] [CrossRef]
22. Barati, B., Sadegh Amiri, I. (2015). In silico engineering of disulphide bonds to produce stable cellulase, springerbriefs in applied sciences and technology. Springer, Singapore. DOI 10.1007/978-981-287-432-0 [Google Scholar] [CrossRef]
23. Chen, A., Xu, T., Ge, Y., Wang, L., Tang, W. et al. (2019). Hydrogen-bond-based protein engineering for the acidic adaptation of bacillus acidopullulyticus pullulanase. Enzyme and Microbial Technology, 124, 79–83. DOI 10.1016/j.enzmictec.2019.01.010. [Google Scholar] [CrossRef]
24. Huang, S. M., Chou, W. Y., Lin, S. I., Chang, G. G. (1998). Engineering of a stable mutant malic enzyme by introducing an extra ion-pair to the protein. Proteins: Structure. Function and Genetics, 31, 61–73. DOI 10.1002/(sici)1097-0134(19980401)31:1<61::aid-prot6>3.0.co;2-k. [Google Scholar] [CrossRef]
25. Siadat, O. R., Lougarre, A., Lamouroux, L., Ladurantie, C., Fournier, D. (2006). The effect of engineered disulfide bonds on the stability of drosophila melanogaster acetylcholinesterase. BMC Biochemistry, 7, 1–7. DOI 10.1186/1471-2091-7-12. [Google Scholar] [CrossRef]
26. Ásgeirsson, B., Adalbjörnsson, B. V., Gylfason, G. A. (2007). Engineered disulfide bonds increase active-site local stability and reduce catalytic activity of a cold-adapted alkaline phosphatase. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics, 1774, 679–687. DOI 10.1016/j.bbapap.2007.03.016. [Google Scholar] [CrossRef]
27. Zhang, L., Chou, C. P., Moo-Young, M. (2011). Disulfide bond formation and its impact on the biological activity and stability of recombinant therapeutic proteins produced by escherichia coli expression system. Biotechnology Advances, 29, 923–929. DOI 10.1016/j.biotechadv.2011.07.013. [Google Scholar] [CrossRef]
28. Dehouck, Y., Grosfils, A., Folch, B., Gilis, D., Bogaerts, P. et al. (2009). Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks: PoPMuSiC−2.0. Bioinformatics, 25, 2537–2543. DOI 10.1093/bioinformatics/btp445. [Google Scholar] [CrossRef]
29. Childers, M. C., Daggett, V. (2017). Insights from molecular dynamics simulations for computational protein design. Molecular Systems Design & Engineering, 2, 9–33. DOI 10.1039/C6ME00083E. [Google Scholar] [CrossRef]
30. Dhandare, B. C., Rather, M. A., Bhosale, B. P., Pawar, R., Guttula, P. K. et al. (2020). Molecular modeling, docking and dynamic simulations of growth hormone receptor (GHR) of labeo rohita. Journal of Biomolecular Structure and Dynamics, 1–14. DOI 10.1080/07391102.2020.1844063. [Google Scholar] [CrossRef]
31. Dombkowski, A. (2003). Disulfide by DesignTM: A computational method for the rational design of disulfide bonds in proteins. Bioinformatics, 19, 1852–1853. DOI 10.1093/bioinformatics/btg231. [Google Scholar] [CrossRef]
32. Pronk, S., Páll, S., Schulz, R., Larsson, P., Bjelkmar, P. et al. (2013). GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics, 29, 845–854. DOI 10.1093/bioinformatics/btt055. [Google Scholar] [CrossRef]
33. Siu, S. W. I., Pluhackova, K., Böckmann, R. A. (2012). Optimization of the OPLS-aA force field for long hydrocarbons. Journal of Chemical Theory and Computation, 8, 1459–1470. DOI 10.1021/ct200908r. [Google Scholar] [CrossRef]
34. Hess, B., Bekker, H., Berendsen, H. J. C., Fraaije, J. G. E. M. (1997). LINCS: A linear constraint solver for molecular simulations. Journal of Computational Chemistry, 18, 1463–1472. DOI 10.1002/(ISSN)1096-987X. [Google Scholar] [CrossRef]
35. Darden, T., York, D., Pedersen, L. (1993). Particle mesh ewald: An N⋅log(N) method for ewald sums in large systems. The Journal of Chemical Physics, 98, 10089–10092. DOI 10.1063/1.464397. [Google Scholar] [CrossRef]
36. Parrinello, M., Rahman, A. (1981). Polymorphic transitions in single crystals: A new molecular dynamics method. Journal of Applied Physics, 52, 7182–7190. DOI 10.1063/1.328693. [Google Scholar] [CrossRef]
37. Kabsch, W., Sander, C. (1983). Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers, 22, 2577–2637. DOI 10.1002/(ISSN)1097-0282. [Google Scholar] [CrossRef]
38. Van Zundert, G. C. P., Rodrigues, J. P. G. L. M., Trellet, M., Schmitz, C., Kastritis, P. L. et al. (2016). The HADDOCK2.2 Web server: User-friendly integrative modeling of biomolecular complexes. Journal of Molecular Biology, 428, 720–725. DOI 10.1016/j.jmb.2015.09.014. [Google Scholar] [CrossRef]
39. Hirschberger, T., Stork, M., Schropp, B., Winklhofer, K. F., Tatzelt, J. et al. (2006). Structural instability of the prion protein upon m205s/R mutations revealed by molecular dynamics simulations. Biophysical Journal, 90, 3908–3918. DOI 10.1529/biophysj.105.075341. [Google Scholar] [CrossRef]
40. Zhang, Y., Skolnick, J. (2005). The protein structure prediction problem could be solved using the current PDB library. Proceedings of the National Academy of Sciences of the United States of America, 102, 1029–1034. DOI 10.1073/pnas.0407152101. [Google Scholar] [CrossRef]
41. Sudhakar, M., Silambanan, S., Chandran, A. S., Prabhakaran, A. A., Ramakrishnan, R. (2018). C-reactive protein (CRP) and leptin receptor in obesity: Binding of monomeric CRP to leptin receptor. Frontiers in Immunology, 9, 1–13. DOI 10.3389/fimmu.2018.01167. [Google Scholar] [CrossRef]
42. Rock, F. L., Altmann, S. W., van Heek, M., Kastelein, R. A., Bazan, J. F. (1996). The leptin haemopoietic cytokine fold is stabilized by an intrachain disulfide bond. Hormone and Metabolic Research, 28, 649–652. DOI 10.1055/s-2007-979871. [Google Scholar] [CrossRef]
43. Boute, N., Zilberfarb, V., Camoin, L., Bonnafous, S., Le Marchand-Brustel, Y. et al. (2004). The formation of an intrachain disulfide bond in the leptin protein is necessary for efficient leptin secretion. Biochimie, 86, 351–356. DOI 10.1016/j.biochi.2004.06.005. [Google Scholar] [CrossRef]
44. Dehnavi, E., Fathi-Roudsari, M., Mirzaie, S., Arab, S. S., Ranaei Siadat, S. O. et al. (2017). Engineering disulfide bonds in selenomonas ruminantium β-xylosidase by experimental and computational methods. International Journal of Biological Macromolecules, 95, 248–255. DOI 10.1016/j.ijbiomac.2016.10.104. [Google Scholar] [CrossRef]
45. Qiao, Z., Xu, M., Shao, M., Zhao, Y., Long, M. et al. (2020). Engineered disulfide bonds improve thermostability and activity of L-isoleucine hydroxylase for efficient 4-hIL production in bacillus subtilis 168. Engineering in Life Sciences, 20, 7–16. DOI 10.1002/elsc.201900090. [Google Scholar] [CrossRef]
46. Boonyaputthikul, H., Muhammad, A., Roekring, S., Rattanarojpong, T., Khunrae, P. et al. (2019). Synergistic effects between the additions of a disulphide bridge and an N-terminal hydrophobic sidechain on the binding pocket tilting and enhanced xyn11a activity. Archives of Biochemistry and Biophysics, 672, 108068. DOI 10.1016/j.abb.2019.108068. [Google Scholar] [CrossRef]
47. Barati, B., Zafar, F. F., Rupani, P. F., Wang, S. (2021). Bacterial pretreatment of microalgae and the potential of novel nature hydrolytic sources. Environmental Technology & Innovation, 21, 101362. DOI 10.1016/j.eti.2021.101362. [Google Scholar] [CrossRef]
48. Cao, B., Yuan, J., Jiang, D., Wang, S., Barati, B. et al. (2021). Seaweed-derived biochar with multiple active sites as a heterogeneous catalyst for converting macroalgae into acid-free biooil containing abundant ester and sugar substances. Fuel, 285, 119164. DOI 10.1016/j.fuel.2020.119164. [Google Scholar] [CrossRef]
49. Wang, S., Zhao, S., Uzoejinwa, B. B., Zheng, A., Wang, Q. et al. (2020). A state-of-the-art review on dual purpose seaweeds utilization for wastewater treatment and crude bio-oil production. Energy Conversion and Management, 222, 113253. DOI 10.1016/j.enconman.2020.113253. [Google Scholar] [CrossRef]
50. Xie, D. F., Fang, H., Mei, J. Q., Gong, J. Y., Wang, H. P. et al. (2018). Improving thermostability of (R)-selective amine transaminase from aspergillus terreus through introduction of disulfide bonds. Biotechnology and Applied Biochemistry, 65, 255–262. DOI 10.1002/bab.1572. [Google Scholar] [CrossRef]
51. Nakhaee, N., Asad, S., Khajeh, K., Arab, S. S., Amoozegar, M. A. (2018). Improving the thermal stability of azoreductase from halomonas elongata by introducing a disulfide bond via site-directed mutagenesis. Biotechnology and Applied Biochemistry, 65, 883–891. DOI 10.1002/bab.1688. [Google Scholar] [CrossRef]
52. Wang, R., Wang, S., Xu, Y., Yu, X. (2020). Engineering of a thermo-alkali-stable lipase from rhizopus chinensis by rational design of a buried disulfide bond and combinatorial mutagenesis. Journal of Industrial Microbiology & Biotechnology, 47, 1019–1030. DOI 10.1007/s10295-020-02324-1. [Google Scholar] [CrossRef]
53. Shuang, W., Zhao, S., Uzoejinwa, B. B., Zheng, A., Wang, Q. et al. (2020). A state-of-the-art review on dual purpose seaweeds utilization for wastewater treatment and crude bio-oil production. Energy Conversion and Management, 222, 113253. DOI 10.1016/j.enconman.2020.113253. [Google Scholar] [CrossRef]
54. Lakshmikandan, M., Murugesan A. G., Wang, S., Abomohra, A. E. F. et al. (2020). Sustainable biomass production under CO2 conditions and effective wet microalgae lipid extraction for biodiesel production. Journal of Cleaner Production, 247, 119398. DOI 10.1016/j.jclepro.2019.119398. [Google Scholar] [CrossRef]
55. Xu, S., Cao, B., Uzoejinwa, B. B., Odey, E. A., Wang, S. et al. (2020). Synergistic effects of catalytic co-pyrolysis of macroalgae with waste plastics. Process Safety and Environmental Protection, 137, 34–48. DOI 10.1016/j.psep.2020.02.001. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |