
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Effect of Concentration and Residence Time of Joncryl®ADR4368 on Melt Processability of Poly(3-hydroxybutyrate)
1 Post-Graduation Program in Materials Engineering (PPCEM), Federal University of Paraíba (UFPB), João Pessoa, PB 58051-900, Brazil
2 Laboratory of Materials and Biosystems (LAMAB), Federal University of Paraíba (UFPB), João Pessoa, PB 58051-900, Brazil
3 Department of Materials Engineering, Federal University of Sergipe (UFS), Jardim Rosa Elze, São Cristóvão, SE 49100-000, Brazil
4 Institute of Science and Technology, Federal University of São Paulo (UNIFESP), São José dos Campos, SP 12231-280, Brazil
5 Department of Chemical Engineering, Federal University of Pernambuco (UFPE), Cidade Universitária, Recife, PE 50670-901, Brazil
* Corresponding Author: Amélia S. F. Santos. Email:
Journal of Renewable Materials 2024, 12(12), 2079-2094. https://doi.org/10.32604/jrm.2024.055361
Received 25 June 2024; Accepted 23 September 2024; Issue published 20 December 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Poly(3-hydroxybutyrate) (PHB) is a biothechnological and biodegradable thermoplastic polymer from the polyhydroxyalkanoates (PHAs) family, whose chain regularity, high molecular weight, and physical and mechanical properties comparable to polypropylene (PP) are characteristics that have made PHB a prominent commercial bioplastic. Nevertheless, its susceptibility to thermal degradation and hydrolysis has limited many applications. To address the challenges associated with processing, a random copolymer of 95.86 mol% 3-hydroxybutyrate and 4.14 mol% 3-hydroxyvalerate (referred as PHB) was compounded without (neat PHB) and with 0.25, 0.5, 1, and 1.5 wt% of chain extender (Joncryl®ADR 4368), consisting of multifunctional epoxy groups, and melt mixed in an internal mixer at 170°C, 50 rpm, and residence times of 5, 10, and 15 min. The effect of chain extender concentration and residence time on the effectiveness of Joncryl®ADR 4368 to recover the pristine PHB properties were investigated through torque rheometry, melt flow index (MFI), differential scanning calorimetry (DSC), tensile testing, and thermogravimetric analysis. The results indicated that an increase in residence time strongly influenced the prevalence of thermomechanical degradation reactions over chain extension reactions of PHB during processing in the molten state. Shorter residence times favored chain extension, while longer times favored chain scission. This trend led to no enhancement of PHB mechanical properties processed for 10 and 15 min, regardless of additive content used. Melt flow index, thermogravimetric analysis and tensile strength exhibited an asymptotic behavior with the increase in additive concentration, exhibiting a noteworthy improvement in tensile strength and temperature at the highest rate of thermal degradation (TMAX), and a decrease in melt flow index of neat PHB with only 0.25 wt% of additive mixed for 5 min. The results found in this work allow additional data for processing PHB by manipulating the ratios of multifunctional chain extender and mixing time, which can create new approach in various applications.Keywords
The increased utilization of plastics and the significant amount of waste generated, that is not properly disposed, have made scientists and technologists around the world search for new synthetic routes and monomers from renewable sources to produce or modify biodegradable polymers so that they can compete and replace conventional ones. This interest arises from the urgent need for sustainable alternatives to traditional polymers that minimize plastic pollution, reduce carbon emissions from petroleum extraction and refinement or, preferably, meet the carbon neutrality requirement [1,2]. Biodegradable polymers offer the advantage of undergoing decomposition under the influence of environmental microorganisms shortly after being discarded [3,4]. This degradation process produces carbon dioxide (CO2) or methane (CH4), depending on the oxygen availability, cellular components, as well as several other by-products [2]. Technologies for their production have been developed, utilizing locally available and renewable raw materials such as corn, agricultural waste, dairy effluent, sugar cane, among others [5–7]. Therefore, biobased and biodegradable polymers can be an alternative to combat the environmental impact of plastics disposed improperly, mainly, where there are problems related to solid waste management, including products not suitable for recycling, and also lack of people’s or industry engagement and concern about the environment.
Poly(3-hydroxybutyrate) is a thermoplastic, biodegradable, and biocompatible polymer belonging to the polyhydroxyalkanoates (PHAs) family, produced by gram-negative and gram-positive bacteria, which constitutes a unique class of bacterial storage polyesters composed of 3-hydroxyalkanoic acids (3HAs or β-hydroxyalkanoics). One of PHB challenges is to reduce its production cost thus, in the last few years, many studies have arisen exploring microorganisms that accumulate poly-b-hydroxyalkanoates from several sterile and cheap carbon sources, like: sugar cane molasses, sago molasses, rice bran, potato (Solanum tuberosum), and tobacco (Nicotiana tabacum) [8–12]. PHB exhibits a high degree of chain regularity, high molecular weight, and physical and mechanical properties akin to traditional polymers such as polyethylene (PE) and polypropylene (PP), being potentially capable of replacing fossil-based plastics [10]. It possesses a glass transition temperature (Tg) close to 5°C and a crystallinity ranging from 55% to 80% [11–14]. Because of these attributes, PHB enjoys a strong foothold in the market and holds significant potential for the production of short-lifecycle items, including disposables, as well as other applications such as in cosmetics, food packaging, personal hygiene products, medical devices for both human and veterinary uses, automotive industry products, enhanced efficiency fertilizers (EEFs), among others [13–17].
The use of PHB with wide technological applications is still somewhat limited due to, for example, its fragility to be used in flexible packaging applications, increase in crystallinity when stored at room temperature, low thermal stability, narrow processing window that makes PHB processing difficult [9,10,18], and combinations of these factors. Among these, thermal degradation has been identified as the primary mechanism affecting PHB, and involves random chain scission, resulting in the formation of both carboxylic end group and unsaturated ester groups. At temperatures between 225°C–300°C, complete degradation occurs in a single step, leading to the depolymerization of PHB to crotonic acid. Consequently, numerous studies in the literature [11–13,19] highlight the significant property loss during first extrusion cycles, as a result of the decrease in molecular weight above 170°C.
In this way, other alternatives for its general waste stream have been proposed, in order to return PHB waste to the industrial production cycle, thus achieving circularity. Development of additives to improve melt processability, blends to reduce melting temperature of PHB, and biological or chemical 4 recycling routes are some of them [20–24]. Parodi et al. [7], for example, studied chemical recycling of PHB to produce methyl-3-hydroxybutyrate (MHB) and methyl-3-methoxybutyrate (MMB) molecules that can replace toxic chlorinated solvents, for example, to recover poly(hydroxy-alkanoates) (PHAs) from single strain cultures (SSC).
The use of chain extenders has been reported in some studies to control the thermal degradation of polymers [25–27], thus bringing new perspective to PHB melt processability and recyclability [17]. The chain extension reaction aims to improve the physical properties of polymers. Additionally, small amounts of chain-extending agents, usually, induce significant changes in polymers at a low cost [23,28]. The increase in polymer average molecular weight and improvement in physical properties are achieved through the formation of primary bonds between chains [29–31].
Compounds containing multifunctional epoxide groups serve as significant agents for extending polymer chains. Joncryl®ADR-4368 is a multifunctional chain extender, featuring a total of nine epoxy groups. The epoxy groups found in this additive can undergo reactions involving the opening of the epoxy rings, facilitating the formation of covalent bonds and the generation of hydroxyl groups. This enables them to react with functional end groups of condensation-derived polymers, counteracting the thermal degradation and recovering their molecular weight [23,31]. Poly(ethylene terephthalate) (PET), poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV), polycarbonate (PC), nylon, poly(butylene adipate-co-terephthalate) (PBAT), poly(propylene carbonate) (PPC), poly(lactic acid) (PLA), poly(butylene succinate-co-L-lactate) (PBSL) are examples of condensation-derived polymers [23,29–31].
Successful and significant increase in the molecular weight of PLA and PET has already been achieved with the use of Joncryl®ADR-4368 [17]. Chen [31] used an internal chamber of Brabender plastograph to mixture PHB and Joncryl®ADR-4368 at 30 rpm and 170°C for 7 min and, surprisingly, obtained torque values of additivated PHB lower than neat PHB. Bousfield [17] evaluated the incorporation of 1.0 and 4.0 wt% Joncryl®ADR 4368 in PHB pre-melted for about 150 s at 175°C, 60 rpm and 10 min of total residence time in a Haake internal mixer. As pointed out by these authors, the concern about using a longer time of melt mixing relies on the quality of PHB mixture with the additive, mainly at a reduced temperature, like 170°C. Duangphet et al. [22] also evaluated the incorporation of Joncryl®ADR 4368 up to 1 wt%, since US FDA Food Contact Notification 429 suggested a maximum level of 0.5 wt% for food contact safety. Their study was carried out in a HAKKE Polylab twin screw extruder at 180°C and residence time around 8 min.
Conversely, chain extenders have been reported to hinder the readily biodegradability of PHB in a variety of aerobic environments, such as soil, compost, and marine [2], more specifically for compositions of PHBV with 5 wt% of Joncryl®ADR 4368 [31]. Therefore, chemical interactions of Joncryl®ADR 4368, if added up to 5 wt%, hindered the overall biodegradation rate of PHB [31], reducing its green market value. Despite the undesirable effect of chain extenders in PHB biodegradation for use in post-consumer single use plastics, the biodegradation modulation can be interesting for products applied in agriculture [2] and controlled release of drugs, for example.
Although some studies of PHB with Joncryl®ADR 4368 have already been carried out [2,17,23,31], questions related to the best processing time for temperature as low as 170°C still persist, as well as a detailed study for concentrations varying up to 1.5 wt% of Joncryl®ADR 4368 in PHB is also missing. According to the literature [17,31] concentrations greater than 2 wt% of Joncryl®ADR 4368 can be disregarded, since they did not bring any significant improvement in PHB flow properties. Doubts related to best processing time still exist because short residence times may compromise extension reactions and, conversely, long residence times may allow degradation mechanisms to overcome chain extension reactions [17]. Therefore, this study was performed, aiming to investigate the effects of Joncryl®ADR 4368’s concentration ranging from 0.25 to 1.5 wt%, and residence time on the rheological, thermal and mechanical properties of PHB melt processed in an internal mixer. The role of these variables to counteract the thermal degradation and recover molecular weight of PHB during melt processing was accessed by torque rheometry, melt flow index (MFI), differential scanning calorimetry (DSC), tensile testing and thermogravimetric analysis (TGA).
Commercial Biocycle® 1000, a statistical copolymer of poly(3-hydroxybutyrate) (PHB) with 4.14% 3-hydroxyvalerate comonomer, was provided by PHB Industrial S/A (Serrana-SP, Brazil). This polymer has a melting point in the 170°C–180°C range, weight-average molar mass (
The chain-extending agent under the trade name Joncryl®ADR 4368, was provided by BASF Brazil in the form of a white powder. Joncryl®ADR 4368 has a high average functional number (fn > 4), a specific gravity of 1.08 g.cm−3, a glass transition temperature (Tg) of ≈54°C, and an epoxy mass equivalent of 285 g.mol−1.
2.2 Melt Processing of PHB with Joncryl®ADR 4368
Neat PHB and its formulations with different concentrations of Joncryl®ADR 4368 were processed for 5, 10, and 15 min in a Rheomix HAAKETM PolyLab OS (Thermo Scientific, Germany) with a mixing chamber capacity of 69 cm3, using roller-type rotors and operating at 170°C and 50 rpm. Compositions were prepared with 0, 0.25, 0.5, 1, and 1.5 wt.% of Joncryl®ADR 4368, and were respectively designated as PHB, PHB/0.25JC, PHB/0.50JC, PHB/1.0JC, and PHB/1.5JC. Melting torque-time data for each sample was recorded by the abovementioned internal mixer to verify any improvement on PHB melt processability.
2.3 Compression Molding of Neat and Additivated PHB Films
Compression-molded films were prepared using about 3 g of each sample in a 15 kgf hydraulic press (Marconi MA098/A, São Paulo, Brazil) operating at 170°C for 1 min (30 s without pressure and 30 s at 6 kgf). The films were then quenched in an ice-water bath, in order to hinder the growth of large crystallites and assure reproducible cooling cycles. Prior to each processing step, the polymer and its melt-processed formulations, previously cut into regular shapes and sizes (Fig. 1), were dried in a vacuum oven (600 mmHg) for 3 h at 80°C.

Figure 1: PHB after melt processing in the internal mixer of Haake Rheomix 600
Melt flow index (MFI) measurements of PHB compounds were performed in triplicate using a model 7023 Ceast plastometer, according to ASTM D 1238. Testing parameters were 2.16 kg at 180°C with intervals of 6 s for melt cutting. Previously, samples were vacuum dried for 3 h at 80°C.
Tensile testing was carried out according to ISO 527-2 standard using 5A test specimens. A universal testing machine, Instron-3365, was employed at 1 mm/min. Test specimens were prepared by laser cutting from the compression molded films. The results (average and standard deviation) were reported from at least 5 test-specimens of each sample.
Differential Scanning Calorimetry (DSC) analyses were performed using a DSC-60 equipment (Shimadzu, Kyoto, Japan), operating at heating and cooling rates of 10°C/min and nitrogen flow rate of 50 mL/min in aluminum crucibles. A three-stage heating/cooling/heating protocol was utilized, i.e., samples were initially heated from 30°C to 190°C, held at 190°C for 2 min, then cooled to room temperature, maintained for 2 min at 30°C, and finally reheated to 190°C. The degree of crystallinity (Xc) was determined during the second heating stage and calculated using the following Eq. (1):
where: ΔHm represents the enthalpy of fusion for each sample, ΔH100% the enthalpy of fusion for 100% crystalline PHB, assumed to be 146 J/g [30], and w denotes the weight fraction of PHB.
Thermogravimetric analyses (TGA) were conducted under 50 mL/min of nitrogen flow, using a STA 6000 thermogravimetric analyzer (Perkin Elmer, USA). The samples were heated to 600°C at 10°C/min. The characteristic temperature at the maximum degradation rate (TMAX) was obtained from the first derivative of the thermogravimetric curve (DTG). Also the temperature at which the sample loses 10 wt% of its initial weight (T10%) was determined.
The torque curves as a function of processing time are shown in Fig. 2. All curves have similar behavior and any shift among them can be attributed to feeding reproducibility of the raw material in the internal chamber, or differences in the homogenization rate of the chain-extender agents in the melt PHB. It was possible to observe an increase in the torque values during the feed, due to friction, and plastic deformation of pellets, followed by an asymptotic decay. As the polymer heats and starts to melt, viscosity decreases and so does the torque. It was expected that the addition of chain extender would increase and/or stabilize torque of the PHB compositions, inhibiting the reduction of polymer molecular weight during melt mixing.
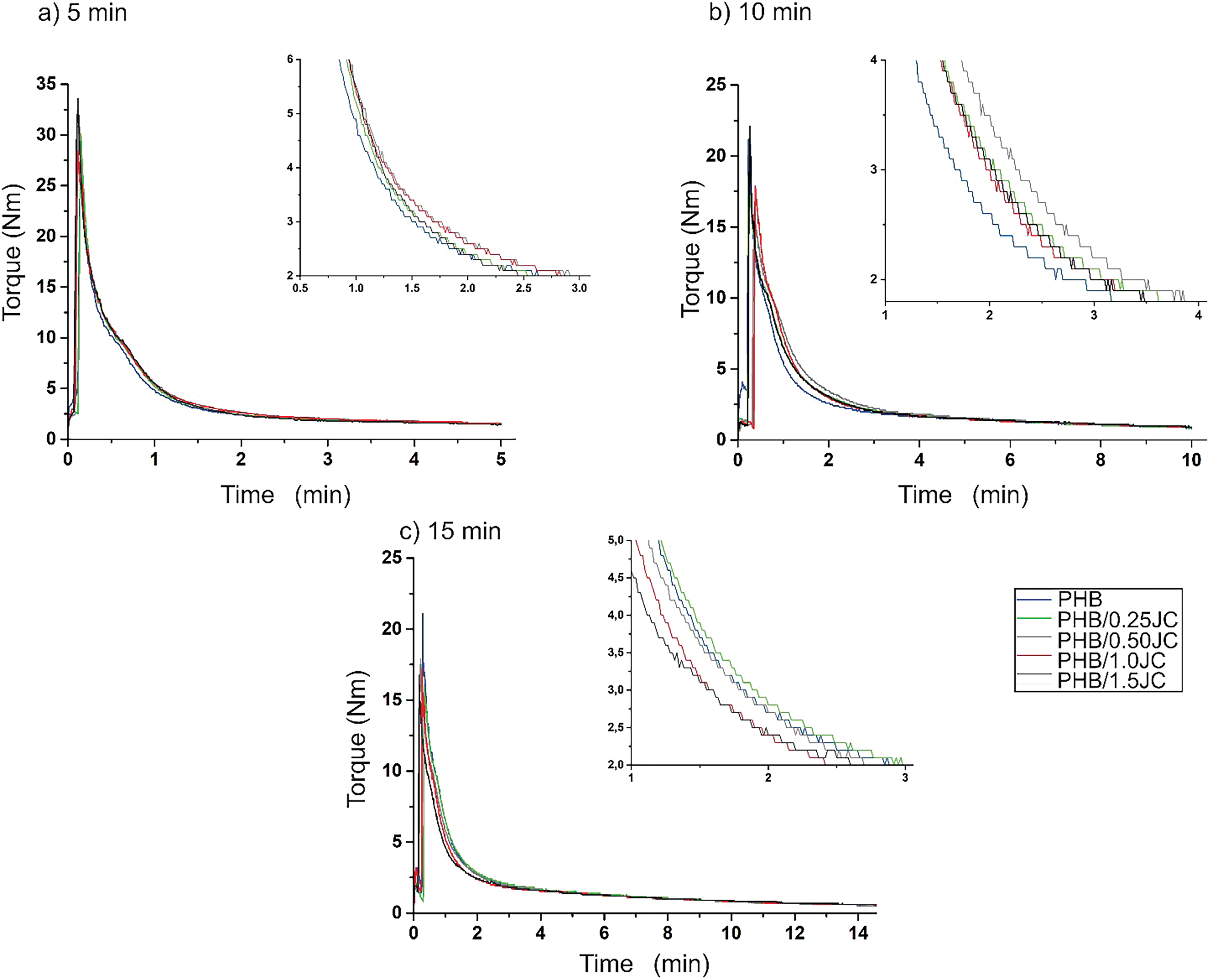
Figure 2: Torque curves and insects of PHB containing different chain extender contents processed in the molten state at different residence times
Nevertheless, the torque differences among the samples are verified up to 2–3 min of mixing, which is exactly the time required for the bulk temperature to reach the internal chamber temperature. After this period of time, the torque decay rate reduces continuously for 5, 10 and 15 min without any difference among the PHB compositions, regardless of additive content. Boushield [17] finds a similar result because the change in the complex viscosity of PHB after 30 min is only slightly less than it was after processing for 10 min, suggesting that the majority of degradation occurred in the first ten minutes of compounding. From this behavior, it can be inferred that chain extenders reacted to completion during compounding, which did not inhibit future degradation.
Among the differences observed in these 2–3 min of mixing interval, Fig. 2a shows, as expected, a relatively higher torque for the PHB compositions with the chain extender relative to neat PHB. However, for the others residence time (Fig. 2b,c), the curves up to 2–3 min of mixing indicate only the PHB/0.5CJ, as the composition with the lowest torque decay rate. The other PHB compositions presented the same torque decay rate as neat PHB, being overlapped or displaced, due to the time spent on loading the raw materials into the internal chamber.
This behavior suggests that the torque rheometer is not accurate enough to detect structural changes in PHB without and with Joncryl®ADR 4368; and/or that the chain extension reactions are fast, being the additive consumed or decomposed at the first 2–3 min of melt processing. Therefore, the additives would not be available to interfere in the thermo-oxidative and mechanical degradation mechanisms of PHB, explaining the torque decay rate of PHB with chain extender equal to that of neat PHB for residence times higher than 3 min. This last hypothesis corroborates with the supplier datasheet that reports Joncryl®ADR 4368’s reaction being over 99% complete, if exposed for at least 120 s at 200°C [32]. Another hypothesis highlighted by Chen [31] is the high molecular weight of PHB, which hinders the chain extension reaction between the reactive terminal groups from PHB with Joncryl®ADR 4368 additive. These authors found an opposite effect in the torque during the PHB processing relative to the increase in the Joncryl®ADR 4368’s concentration. Regardless, any one of the abovementioned explanations for the low efficiency of the Joncryl®ADR 4368 to enhance the PHB viscosity, competition between chain extension and thermal degradation surely is one of the most plausible explanation [33].
Fig. 3 illustrates the melt flow index (MFI) results as a function of the additive concentration and residence time. The neat PHB presented the same MFI after 10 and 15 min of melt processing, and both are values close to the neat PHB melt processed for 5 min. This behavior corroborates with the Bousfield hypothesis [17] that the majority of PHB degradation occurs in the first minutes of compounding.
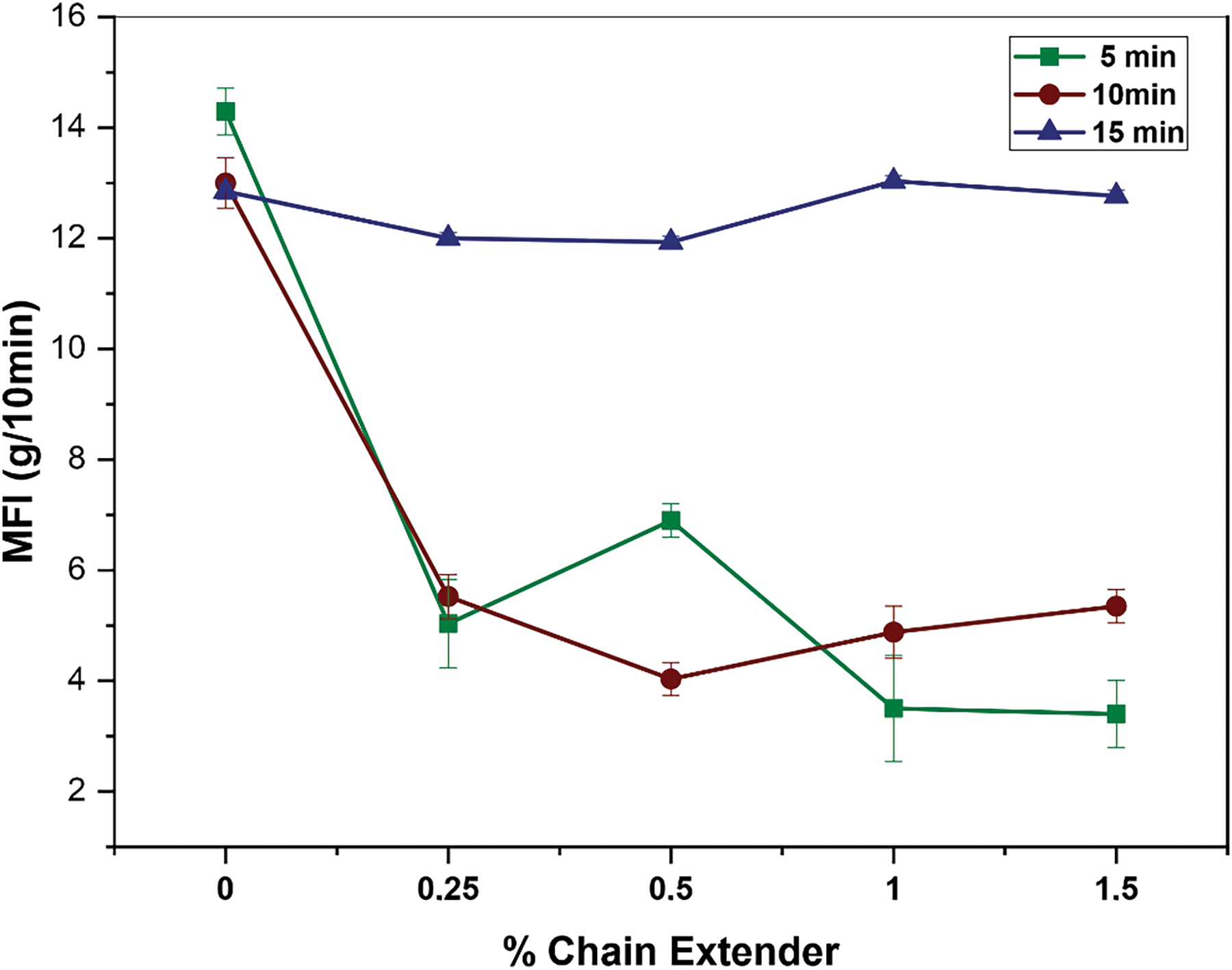
Figure 3: Melt flow index of PHB containing different chain extender contents processed in the molten state at different residence times
For residence times (RT) of 5 and 10 min, there was a substantial reduction in MFI with the addition of just 0.25 wt.% of Joncryl®ADR 4368, decreasing from 13–14 g/10 min−1 to approximately 5 g/10 min−1. For RT = 5 min, MFI values displayed an asymptotic tendency, as the additive content increased, reaching a plateau around 3.5 g/10 min for a mixture containing 1.0 wt% of Joncryl®ADR 4368. Therefore, contents up to 1.0 wt% of Joncryl®ADR 4368 chain extender are enough to decrease MFI values of PHB, facilitating the reaction of its multiple epoxy groups with the carboxyl or hydroxyl end groups of PHB [23,27–29]. This shows that the MFI reduction is maximum for 5 min and that no significant improvement was achieved when up to 1.5 wt% Joncryl®ADR 4368 was added.
For RT = 10 min, if Joncryl®ADR 4368 concentrations are equal to or less than 0.5 wt%, the MFI reduction is equivalent or slightly better than that obtained with only 5 min of mixing. On the other hand, for additive concentrations higher than 0.5 wt%, the chain extender is more effective at residence times of 5 min. This phenomenon is likely connected to the prolonged exposure to shear stress during processing within the internal mixer, where the molten material is subjected to this stress over an extended period of time, leading to degradation and a subsequent reduction in viscosity [34].
The overlapping of chain extension reactions over thermal degradation was only possible for additive content capable of counteract chain scission reactions at their short half-life. Perhaps, the additive incorporation in the melted PHB, e.g., after around 120 s of mixture, would improve these results, as already observed by Bousfield [17] even at 10 min of mixture.
This explanation corroborates the results obtained for samples processed at the maximum residence time, RT = 15 min. There was no further decrease in the MFI with increasing additive content, suggesting that the chain extender was unable to control the degradation mechanisms of PHB, even at a concentration as high as 1.5 wt% due to the heightened thermal and shear stress that the polymer underwent during the prolonged mixing. These results indicate that a residence time of 5 min was enough to promote the PHB chain extension reaction during melt processing. Longer mixing times do not bring advantages, on the contrary, they probably promote the reversal of the chain extension reaction, side reactions or simply accentuate the effects of thermal degradation reactions.
3.3 Differential Scanning Calorimetry (DSC)
The influence of the chain extender addition and the processing time inside the Haake chamber on the melting and crystallization behavior of PHB was investigated using differential scanning calorimetry (DSC). Aiming to erase the quenching influence on the PHB crystallizable parameters, DSC scans acquired during the second heating are presented in Fig. 4, whereas collected data for PHB without and with different tested chain extender concentrations are displayed. Investigated compounds showed double and asymmetric endothermic melting peaks, being the first peak characterized by a shoulder around 160°C and a main peak at a higher temperature, i.e., between 170°C–180°C. The presence of these double peaks can be attributed to the crystals melting with distinct sizes/morphologies/perfection, rearrangement/secondary crystallization improvement, as well as melting/recrystallization/remelting of crystallizable PHB [23,33].
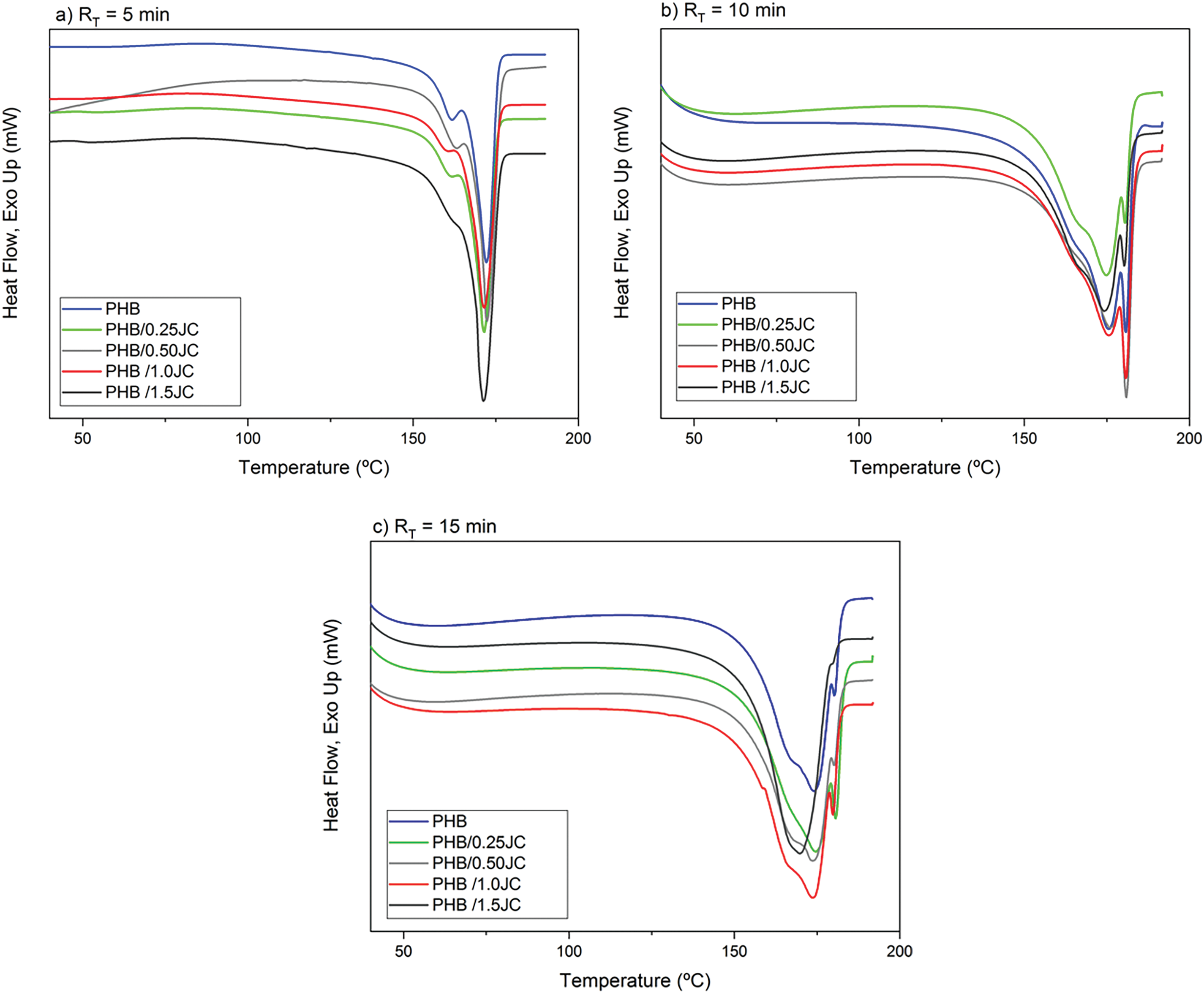
Figure 4: DSC scans acquired during the second heating cycle for PHB without and with different contents of chain extender for RT = 5 min (a), 10 min (b) and 15 min (c)
In general, the double melting peak broadened in the DSC curves, as the amount of chain extender increased. For RT =10 min and RT = 15 min, a third shoulder appeared at high temperatures, suggesting the formation of more perfect crystals, without however increasing crystallinity. Probably, the relatively lower molecular weight of neat PHB at longer mixing times in the molten state (Fig. 3) and consequently, higher molecular mobility contributed to broadening the melting peak [33] and to crystals ability to undergo continuous melting and recrystallization during heating scan. Furthermore, addition of a chain extender under these conditions may also have favored cross-linking and chain branching reactions rather than reactions between chain ends, mainly at RT = 15 min, and the packing of the polymer chains in these areas acted as nucleating sites [35–37] upon melting and recrystallization of crystals during heating scan. The epoxide functional group chain extender can react quickly and irreversibly with carboxylates present in the PHB. Simultaneously, the increase in the chain extender content turned up the first peak less evident, as already observed by Chen [31], suggesting that the presence of a greater amount of extender led to crystals developed at higher temperatures, with a heterogeneous distribution and non-uniform thickness.
Table 1 displays the melting temperature (Tm) and degree of crystallinity (Xc) of neat PHB with varying chain extender contents, processed for 5, 10, or 15 min. Neat PHB and PHB with different chain extender concentrations processed for RT = 5 min exhibit comparable Tm values (Table 1). Therefore, for RT = 5 min, the incorporation of the chain extender up to 1.5 wt% did not lead to significant changes in melting temperatures, as they remained similar to those of the neat polymer.
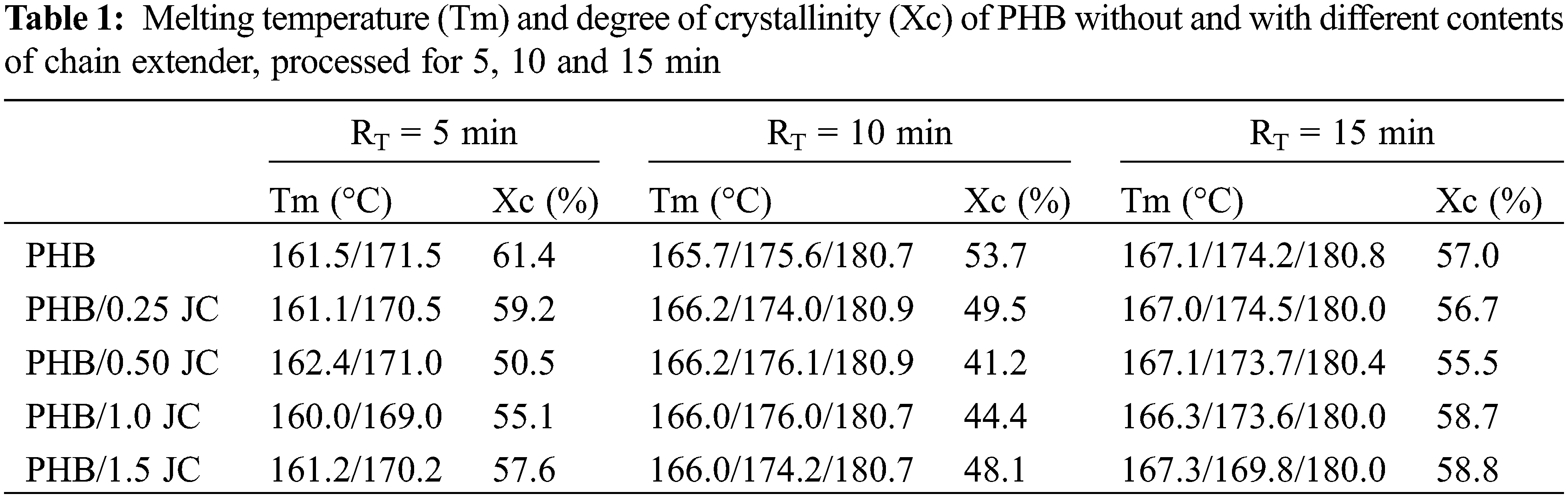
However, PHB with 0.50 wt% of the chain extender showed a decrease in the degree of crystallinity relative to neat PHB from 61.42% to 50.53%. For higher amounts of the chain extender, a slight increase in the degree of crystallinity is observed, but without exceeding neat PHB. This behavior indicates that structural/morphological changes may have taken place in PHB, reducing the crystallizable character of its compounds. This decrease in crystallinity can be attributed to the increase in the molecular weight of PHB chains, more entanglements, the restriction of chain mobility, and also possibly to the introduction of chain branching and/or crosslinking in the PHB structure [23,32,37].
The decrease in crystallinity noted for RT = 5 and 10 min reflects the interaction between PHB chains and the chain extender, primarily, due to increased viscosity (Fig. 3) due to the reactions between chain ends and/or crosslinking, hampers the crystallization kinetics and/or the ease of arrangement of the PHB chains [38], as aforementioned.
For the maximum residence time studied, RT = 15 min, the melting temperature also did not change significantly, remaining with values close to those obtained for neat PHB (Table 1). The degree of crystallinity remained almost constant and close to neat PHB even when chain extender up to 1.5 wt% was added, corroborating with the MFI results for RT = 15 min (Fig. 3). This behavior suggests that at this residence time, the additive was not efficient in changing the polymer molecular weight by crosslinking or reactions between chain ends.
Figs. 5 and 6 show results for tensile strength and elongation at break, respectively. Samples processed with RT = 5 min showed an improvement in tensile strength with the addition of only 0.25 wt% additive. Further increase in the additive content did not lead to any improvement of PHB/0.25JC’s tensile strength, reproducing the same trend observed for MFI (Fig. 3). Conversely, PHB with different contents of the chain extender processed for 10 min at 170°C showed almost the same tension strength and elongation at break of neat PHB, mainly if the experimental error of the measurements is considered. Similar result was obtained by Bousfield [17] that also processed PHB with 1 wt% of Joncryl®ADR4368 for 10 min in a torque rheometer. However, the difference in tensile strength between neat PHB processed at RT = 5 min and RT = 10 min was negligible. Therefore, the chain extension reactions that reduced MFI of additivated PHB processed for 10 min were not enough to increase of tie molecules, responsible for transferring stress between two neighboring crystalline lamellae [39].
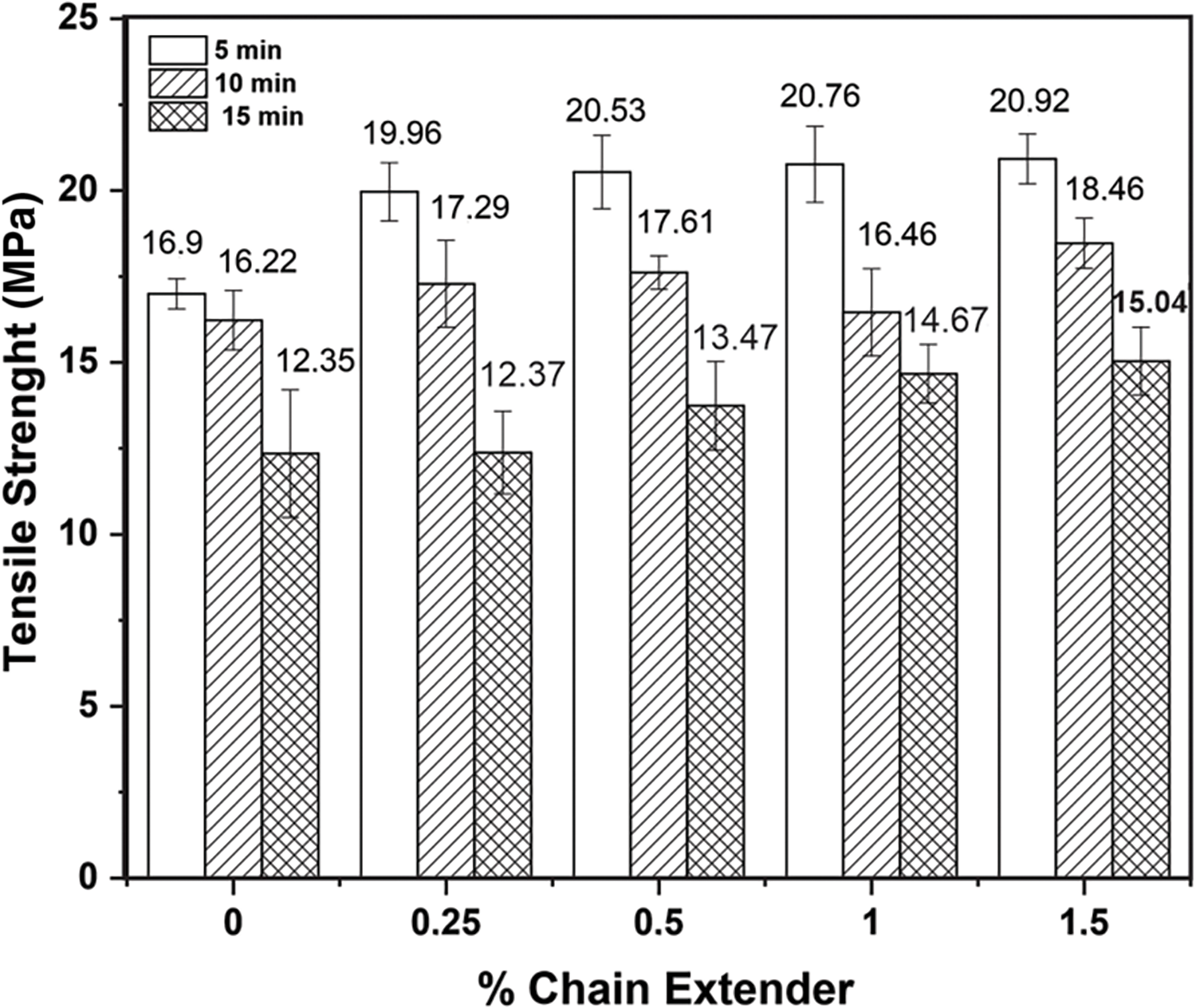
Figure 5: Tensile strength for PHB without and with different concentrations of the chain extender melt mixed for 5, 10 and 15 min in the internal mixer
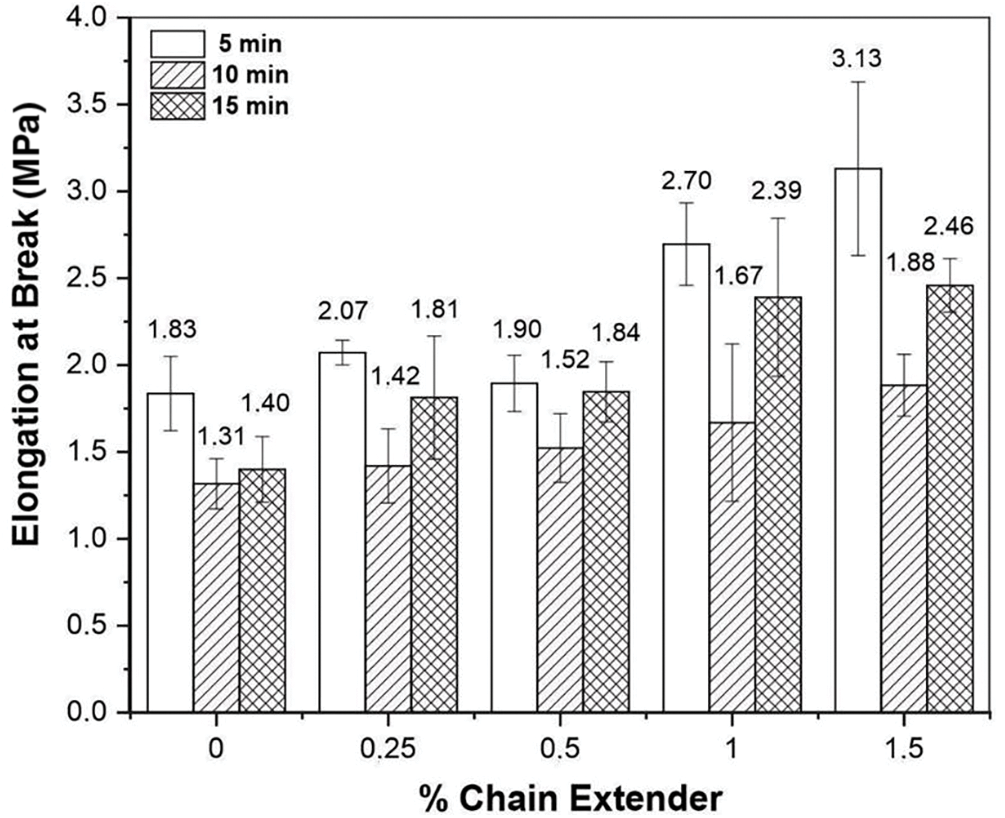
Figure 6: Elongation at break for PHB without and with different concentrations of the chain extender melt mixed for 5, 10 and 15 min in the internal mixer
Samples processed for 15 min at 170°C reproduced the trend observed in the MFI test (Fig. 3) and showed no improvement in the tension strength of PHB with increasing additive content. All of them exhibited a reduced tensile strength compared to PHB samples processed for 5 and 10 min. This behavior can be associated with the longer residence time of the material in the internal mixer, suggesting the prevalence of thermal degradation reactions [25,40,41] over chain extension reactions.
Thus, the tensile strength measurements were not sensitive to changes in polymer molecular weight with increasing content of the chain extender content. Furthermore, the slight reduction in the efficiency of the chain extender in recovering the molecular weight of PHB, when mixed for 10 min instead of just 5 min, did not improve the mechanical properties for the neat PHB. Therefore, residence times greater than 5 min for mixing the chain-extending agent are not recommended, since the increase in maximum tension with increasing additive content did not occur for RT = 10 and 15 min [25].
The increase in ductility observed for RT = 5 min and RT = 10 min with increasing additive content, corroborated with the decrease in crystallinity and reduction in MFI observed for additivated PHB samples (Table 1). This behavior is attributed to an increase of PHB’s molecular weight due to reactions between chain ends of the PHB with Joncryl®ADR 4368 over cross-linking and/or branching reactions, considering a residence time of up to 5 min. The excess of additive did not drawback extension reactions up to 5 min of residence time. Surprisingly, the rise in ductility for RT = 15 min with increasing additive content suggests that chain extender can interfere in the chain scission reactions from reverse and thermal degradation reactions favored at 15 min of residence time. Therefore, reverse reactions for mixing times above 5 min probably changed PHB’ structure justifying the ductility increment of additivated PHB melt mixed for 15 min. Probably, as already pointed out in previous analyses, chain branching reactions may have been favored, reducing the number of tie molecules and, consequently, decreasing the tensile strength of the polymer, however without significantly affecting its ductility [38–40].
These results indicate that concentrations of 0.25 wt% chain extender and mixing times of up to 5 min are enough for the tensile strength of PHB staying around 20 MPa. The increase in tensile strength is probably associated with the increase in the molecular weight of PHB and tie molecules, due to reactions between chain ends, as previously suggested [39].
3.5 Thermogravimetric Analysis (TGA)
Fig. 7 illustrates the effect of Joncryl® ADR 4368 content on the thermal degradation of PHB over the three considered residence times. As the TGA curves are all overlaid, any structural changes in PHB and its formulations were not enough to change their thermal behavior. It is evident that the weight loss of PHB, both with and without Joncryl® ADR 4368, occurs in a one-step process between 230°C and 300°C [16]. No mass loss was observed until about 230°C, and the temperature at the DTG peak (TMAX) was between 279°C and 303°C (Table 2).
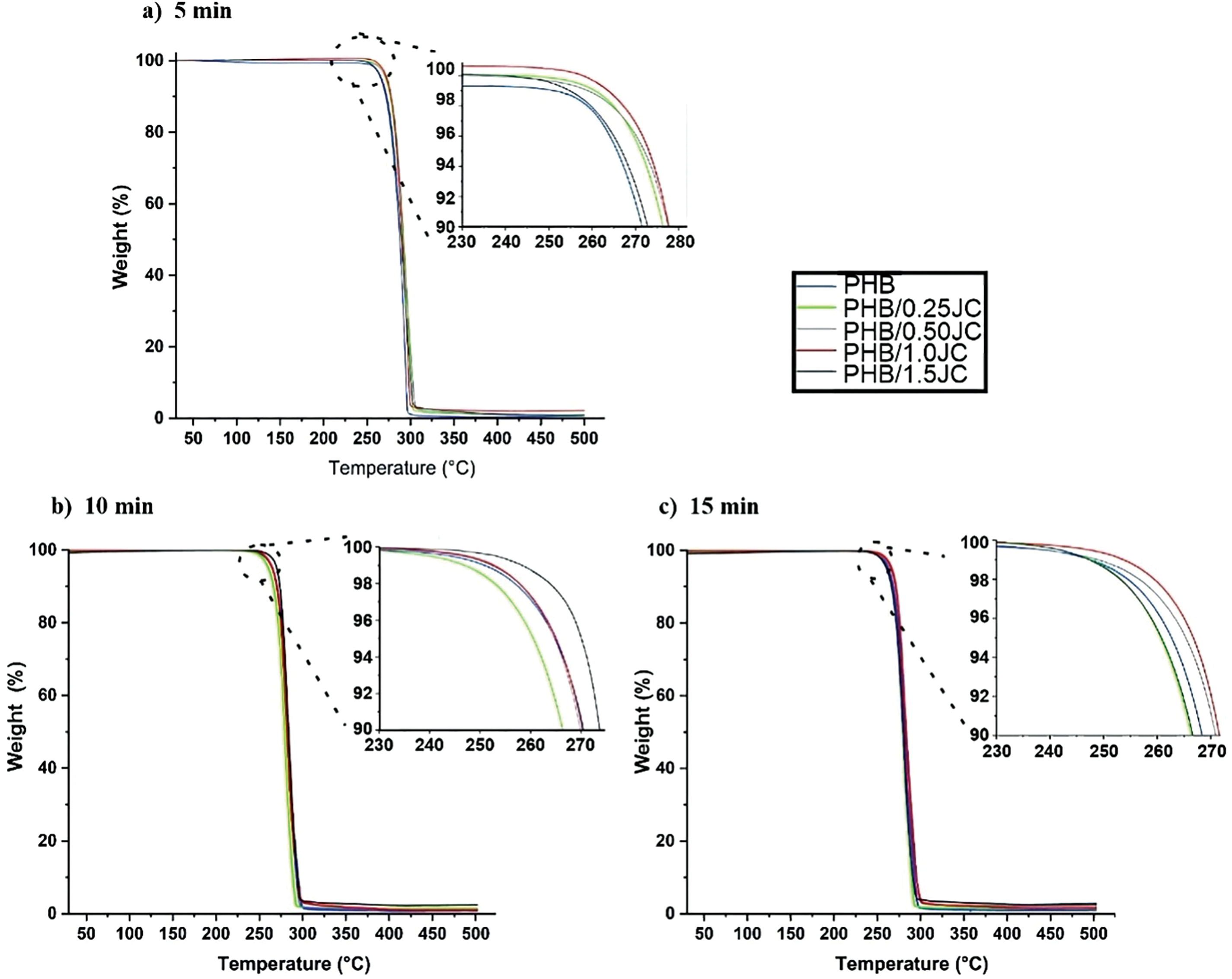
Figure 7: Thermogravimetric curves for PHB without and with different concentrations of the chain extender melt processed for 5 min (a), 10 min (b) and 15 min (c) in the internal mixer
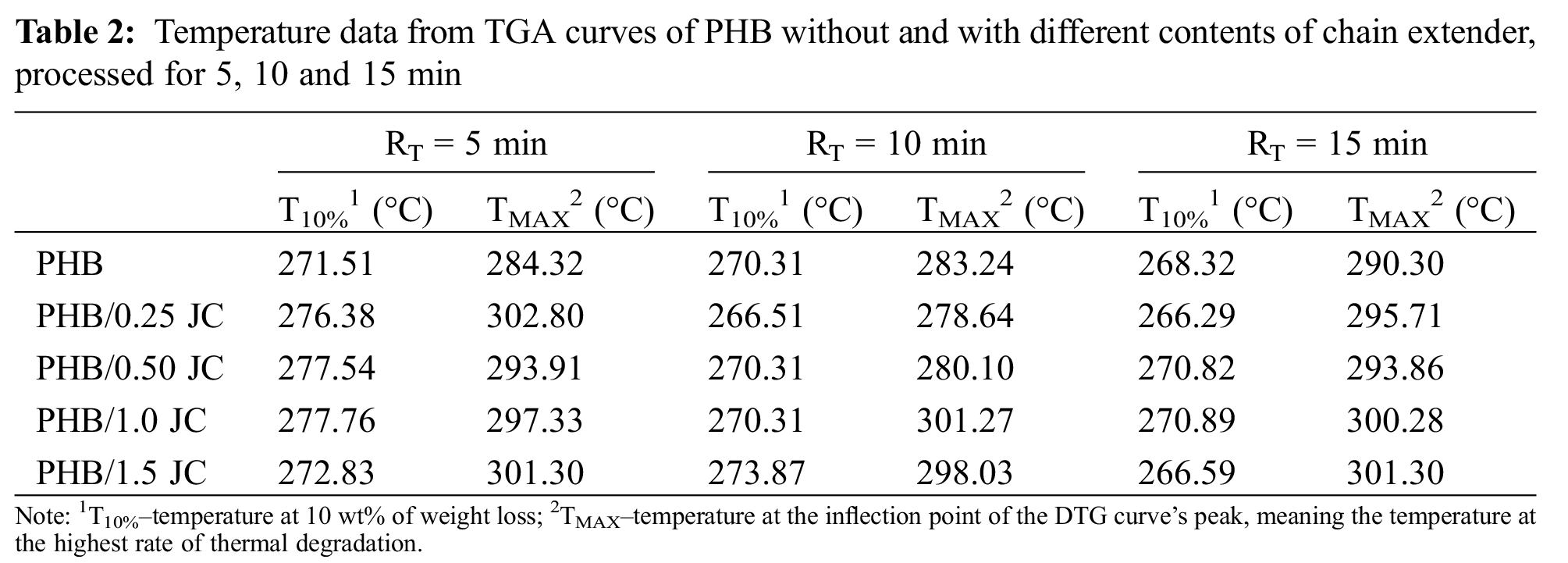
Taking a closer look at the beginning of TGA curves of PHB compounds (insets of Fig. 7), a slight shift among these curves can be observed. All formulations with Joncryl® ADR 4368 processed at the same residence time exhibited thermal stability near or higher than neat PHB. Particularly, PHB/1.0 JC and PHB/1.5 JC presented the highest increase in TMAX relative to neat PHB, which was increased by around 10°C–11°C, 13°C–17°C and 15°C–18°C after 15, 5 and 10 min of mixing, respectively. Since at RT = 15 min, MFI data did not notice any improvement in melt viscosity of PHB with increasing content of Joncryl® ADR 4368, and literature [42–44] usually describe a proportional relationship between changes in the thermal stability of polymers and changes in their molecular weight, the gain in thermal stability (Table 2) and mechanical properties of additivated PHB (Figs. 5 and 6) at RT = 15 min indicates that other reactions alongside chain extension and thermal degradation reactions are occurring, thereby changing PHB structure.
Looking at the influence of residence time on the T10% for each one of PHB compounds, the thermal stability of the samples slightly reduced with increasing residence time (Table 2), having PHB at RT = 5 min the lowest decrease. These results thoroughly corroborate with MFI data (Fig. 3), emphasizing that the majority of PHB degradation occurs in the first minutes of compounding [17] and the effectiveness of Joncryl®ADR 4368 occurs in the first minutes of melt processing due to its fast reactivity [32].
Finally, an increase of about 10°C to 18°C in TMAX of neat PHB with the incorporation of 1.0 or 1.5 wt% of Joncryl® ADR 4368, depending on the residence time, indicates that the introduction of Joncryl® ADR 4368 can broaden the processing window and enhance the thermal stability of PHB. Also, concentrations as low as 0.25 wt% of Joncryl® ADR 4368 can reach the same performance, if the residence time is up to 5 min.
The effect of Joncryl®ADR 4368 chain extender on thermal behavior, crystallization, and changes in melt viscosity of PHB was studied. Results showed that it is possible to increase the viscosity and improve the mechanical properties of PHB with the incorporation of only 0.25 wt% Joncryl®ADR 4368, without compromising polymer thermal properties for residence times up to 5 min. However, increasing the residence time up to 15 min addition of any content of Joncryl®ADR 4368 resulted in the same properties of neat PHB after processing, except for elongation at break. Incorporating up to 1.5 wt% of this additive has been demonstrated to be effective primarily in enhancing the ductility of PHB. Furthermore, this study revealed that processing times of up to 5 min are ample to facilitate the chain extension reaction without the occurrence of secondary reactions involving the reversal and/or branching of the PHB main chain. This research has yielded promising outcomes for the utilization of Joncryl®ADR 4368 as a PHB chain extender, resulting in direct and positive effects on the transformation process of this polymer. Thus, manipulation of the multifunctional chain extender contents and mixing time can create new approaches in various applications.
Acknowledgement: The authors thank the National Foundation for Scientific and Technological Development (CNPq) for financial support of this study by projects @ss_oceanos, and INCT Circularity in Polymer Materials, Authors are also grateful to the Laboratories of Materials and Biosystems (LAMAB), Laboratory of Polymeric Materials (LAMPOL) and Laboratory of Rapid Solidification (LSR) at the Federal University of Paraíba (UFPB) for experimental support.
Funding Statement: The research described in this manuscript received funding from National Foundation for Scientific and Technological Development, CNPq (grant numbers: 405428/2022-7 and 406925/2022-4).
Author Contributions: Jéssica da Silva Chagas: Experimentation, formal analysis, and writing, Natália Fernanda Inocêncio Silva and Gelsoneide da Silva Gois: Experimentation, formal analysis, José Elson Soares Filho: Writing, editing, and formatting, Eliton Souto de Medeiros and Yêda Medeiros Bastos de Almeida: Supervision, visualization, and revision, Amélia S. F. Santos: Supervision and critical revision for approval of the manuscript, Marcelo Massayoshi Ueki: Conceptualization and design Mauricio Pinheiro de Oliveira: Experimental analysis, and discussion, Renate Maria Ramos Wellen: Conceptualization, design and revision. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The data that support the findings of this study are available from the corresponding author, Amélia S. F. Santos, upon reasonable request and/or no permissions are required.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Narayanan M, Kumarasamy S, Ranganathan M, Kandasamy S, Kandasamy G, Gnanavel K, et al. Production and characterization of polyhydroxyalkanoates synthesized by E. coli isolated from sludge soil. Mater Today Proc. 2020;33:3646–53. doi:10.1016/j.matpr.2020.05.725. [Google Scholar] [CrossRef]
2. Meereboer KW, Manjusri M, Amar KM. Review of recent advances in the biodegradability of polyhydroxyalkanoate (PHA) bioplastics and their composites. Green Chem. 2020;22:5519. doi:10.1039/D0GC01647K. [Google Scholar] [CrossRef]
3. Zytner P, Pal AK, Mohanty AK, Misra M. Performance evaluation of biodegradable polymer PHBV and PBAT blends with adjustable melt flow behaviour, heat deflection temperature, and morphological transition. Can J Chem Eng. 2024;102(8):2805–17. doi:10.1002/cjce.25235. [Google Scholar] [CrossRef]
4. Coltelli MB, Gigante V, Aliotta L, Lazzeri A. Recyclability perspectives of the most diffused biobased and biodegradable plastic materials. Macromol. 2024;4(2):401–19. doi:10.3390/macromol4020023. [Google Scholar] [CrossRef]
5. Bucci DZA, Tavares LBB, Sell I. Biodegradation and physical evaluation of PHB packaging. Polym Test. 2007;26:908–15. doi:10.1016/j.polymertesting.2007.06.013. [Google Scholar] [CrossRef]
6. Mello AFM, Vandenberghe LPS, Machado CMB, Valladares-Diestra KK, Carvalho JC, Soccol CR. Polyhydroxybutyrate production by Cupriavidus necator in a corn biorefinery concept. Bioresour Technol. 2023;370:128537. doi:10.1016/j.biortech.2022.128537. [Google Scholar] [PubMed] [CrossRef]
7. Parodi A, D’Ambrosio M, Mazzocchetti L, Martinez GA, Samori C, Torri C, et al. Chemical recycling of polyhydroxybutyrate (PHB) into bio-based solvents and their use in a circular PHB extraction. ACS Sustain Chem Eng. 2021;9(37):12575–83. doi:10.1021/acssuschemeng.1c03299. [Google Scholar] [CrossRef]
8. Nygaard D, Yashchuk O, Noseda DG, Araoz B, Hermida EB. Improved fermentation strategies in a bioreactor for enhancing poly(3-hydroxybutyrate) (PHB) production by wild type Cupriavidus necator from fructose. Heliyon. 2021;7(1):e05979. doi:10.1016/j.heliyon.2021.e05979. [Google Scholar] [PubMed] [CrossRef]
9. Briassoulis D, Tserotas P, Athanasoulia IG. Alternative optimization routes for improving the performance of poly(3-hydroxybutyrate) (PHB) based plastics. J Clean Prod. Jun 2021;318:128555. doi:10.1016/j.jclepro.2021.128555. [Google Scholar] [CrossRef]
10. Raza ZA, Noor S, Khalil S. Recent developments in the synthesis of poly(hydroxybutyrate) based biocomposites. Biotechnol Prog. 2019;35(5):e2855. doi:10.1002/btpr.2855. [Google Scholar] [PubMed] [CrossRef]
11. Kavitha G, Rengasamy R, Inbakandan D. Polyhydroxybutyrate production from marine source and its application. Int J Biol Macromol. 2018;111:102–8. doi:10.1016/j.ijbiomac.2017.12.155. [Google Scholar] [PubMed] [CrossRef]
12. Lee J, Park HJ, Moon M, Lee JS, Min K. Recent progress and challenges in microbial polyhydroxybutyrate (PHB) production from CO2 as a sustainable feedstock: a state-of-the-art review. Biores Technol. 2021;339:339125616. doi:10.1016/j.biortech.2021.125616. [Google Scholar] [PubMed] [CrossRef]
13. França D, Angelo LM, Souza CF, Roselena F. Biobased poly(3-hydroxybutyrate)/starch/cellulose nanofibrils for nutrients coatings. ACS Appl Polym Mater. 2021;3(6):3227–37. [Google Scholar]
14. Silva NFI, Filho JES, Santos TGC, Chagas JS, Medeiros SAS, Santos L, et al. Biocomposites based on poly(hydroxybutyrate) and the mesocarp of babassu coconut (Orbignya phalerata Mart.effect of wax removal and maleic anhydride-modified polyethylene addition. J Mater Sci Technol. 2021;15:3161–70. doi:10.1016/j.jmrt.2021.09.008. [Google Scholar] [CrossRef]
15. Choi YH, Kim JT, Kim MH, Park WH. Biodegradable poly(3-hydroxybutyrate-co-4-hydroxybutyrate)/curcumin composite film as a smart indicator of food spoilage. Sens Actuators: B Chem. 2024;408:135511. doi:10.1016/j.snb.2024.135511. [Google Scholar] [CrossRef]
16. Ahuja S, Bains O, Mittal M, Kamal R, Aggarwal NK, Arora S. Multifunctional chromone-incorporated poly(hydroxybutyrate) luminescent film for active and intelligent food packaging. Int J Biol Macromol. 2023;246:125625. doi:10.1016/j.ijbiomac.2023.125625. [Google Scholar] [PubMed] [CrossRef]
17. Bousfield G. Effect of chain extension on rheology and tensile properties of PHB and PHB-PLA Bblends (Master’s Thesis). Université de Montréal: Montreal; 2014. [Google Scholar]
18. Brown SB, Orlando CM. Role of plasticizers on PHB/bio-TPE blends compatibilized by reactive extrusion. Encycl Polym Sci Eng. 2017;14:169. [Google Scholar]
19. Dawin TP, Ahmadi Z, Taromi FA. Bio-based solution-cast blend films based on polylactic acid and polyhydroxybutyrate: influence of pyromellitic dianhydride as chain extender on the morphology, dispersibility, and crystallinity. Prog Org Coat. 2018;119:23–30. doi:10.1016/j.porgcoat.2018.02.003. [Google Scholar] [CrossRef]
20. Rivas LF, Casarin SA, Nepomuceno NC, Alencar MI, Agnelli JAM, Medeiros ES, et al. Reprocessability of PHB in extrusion: ATR-FTIR, tensile tests and thermal studies. Polím. 2017;27(2):122–8. doi:10.1590/0104-1428.2406. [Google Scholar] [CrossRef]
21. Ramos-Hernández T, Robledo-Ortíz JM, González-López ME, del Campo ASM, González-Núñez R, Rodrigue D, et al. Mechanical recycling of PLA: effect of weathering, extrusion cycles, and chain extender. J Appl Polym Sci. 2023;140(16):e53759. doi:10.1002/app.53759. [Google Scholar] [CrossRef]
22. Duangphet S, Szegda D. The effect of chain extender on poly(3-hydroxybutyrate-co-3-hydroxyvaleratethermal degradation, crystallization, and rheological behaviours. J Polym Environ. 2014;22:1–8. doi:10.1007/s10924-012-0568-5. [Google Scholar] [CrossRef]
23. Yang Z, Li C, Liu Y, Zhang Z, Cao X. Effect of chain extension on processability and mechanical properties of poly(3-hydroxybutyrate-co-4-hydroxybutyrate). J Appl Polym Sci. 2024;141(24):1–10. doi:10.1002/app.55490. [Google Scholar] [CrossRef]
24. Standau T, Nofar M, Dörr D, Ruckdäschel H, Altstädt V. A review on multifunctional epoxy-based Joncryl® ADR chain extended thermoplastics. Polym Rev. 2022;62(2):296–350. doi:10.1080/15583724.2021.1918710. [Google Scholar] [CrossRef]
25. Celebi S, Ozkoc G, Serhatli E. Thermal, mechanical and physical properties of chain extended recycled polyamide 6 via reactive extrusion: effect of chain extender types. Polym Degrad Stab. 2019;162:76–84. doi:10.1016/j.polymdegradstab.2019.01.026. [Google Scholar] [CrossRef]
26. Kahraman Y, Özdemir B, Gümüş BE, Nofar M. Morphological, rheological, and mechanical properties of PLA/TPU/nanoclay blends compatibilized with epoxy-based Joncryl chain extender. Coll Polym Sci. 2023;301(1):51–62. doi:10.1007/s00396-022-05043-4. [Google Scholar] [CrossRef]
27. Yahyaee N, Javadi A, Garmabi H, Khaki A. Effect of two-step chain extension using Joncryl and PMDA on the rheological properties of poly (lactic acid). Macromol Mater Eng. 2020;305(2):1–13. doi:10.1002/mame.201900423. [Google Scholar] [CrossRef]
28. Oza H, Thompson MR, Hrymak AN, Liu Q. Influence of di-functional versus multi-functional chain extenders on the foamability of a potato starch-based biopolymer. Starch. 2012;64(12):944–54. doi:10.1002/star.201200079. [Google Scholar] [CrossRef]
29. Zhao Y, Li Y, Xie D. Junjia chen effect of chain extender on the compatibility, mechanical and gas barrier properties of poly(butylene adipate-co-terephthalate)/poly(propylene carbonate) bio-composites. J Appl Polym Sci. 2021;138:50487. doi:10.1002/app.50487. [Google Scholar] [CrossRef]
30. Nishida M, Liu X, Furuya S, Nishida M, Takayama, Mitsugu T, et al. Effect of chain extender on morphology and tensile properties of poly(l-lactic acid)/poly(butylene succinate-co-l-lactate) blends. Mater Today Commun. 2021;26:101852. doi:10.1016/j.mtcomm.2020.101852. [Google Scholar] [CrossRef]
31. Chen H. Assessment of biodegradation in different environmental compartments of blends and composites based on microbial poly(hydroxyalkanoate)s (Ph.D. Thesis). University of Pisa: Pisa; 2012. [Google Scholar]
32. Battegazzore D, Frache A, Abt T, Maspoch ML. Epoxy coupling agent for PLA and PHB copolymer-based cotton fabric bio-composites. Comp Part B: Eng. 2018;148(1):188–97. doi:10.1016/j.compositesb.2018.04.055. [Google Scholar] [CrossRef]
33. Hong S-G, Lin Y-C, Lin C-H. Improvement of the thermal stability of polyhydroxybutyrates by grafting with maleic anhydride by different methods: differential scanning calorimetry, thermogravimetric analysis, and gel permeation chromatography. J Appl Polym Sci. 2008;110(5):2718–26. doi:10.1002/app.28782. [Google Scholar] [CrossRef]
34. Barham PJ, Keller A, Otun EL, Holmes PA. Crystallization and morphology of a bacterial thermoplastic: poly-3-hydroxybutyrate. J Mater Sci. 1984;19(9):2781–94. doi:10.1007/BF01026954. [Google Scholar] [CrossRef]
35. Kolahchi AR, Kontopoulou M. Chain extended poly(3-hydroxybutyrate) with improved rheological properties and thermal stability, through reactive modification in the melt state. Polym Degrad Stab. 2015;121:222–9. doi:10.1016/j.polymdegradstab.2015.09.008. [Google Scholar] [CrossRef]
36. Weinmann S, Bonten C. Thermal and rheological properties of modified polyhydroxybutyrate (PHB). Polym Eng Sci. 2019;59(5):1057–64. doi:10.1002/pen.25075. [Google Scholar] [CrossRef]
37. Al-Itry R, Lamnawar K, Maazouz A. Improvement of thermal stability, rheological and mechanical properties of PLA, PBAT and their blends by reactive extrusion with functionalized epoxy. Polym Degrad Stab. 2012;97(10):1898–914. doi:10.1016/j.polymdegradstab.2012.06.028. [Google Scholar] [CrossRef]
38. Chen C, Fei BIN, Peng S, Zhuang Y, Dong L, Feng Z. The kinetics of the thermal decomposition of poly(3-hydroxybutyrate) and maleated poly(3-hydroxybutyrate). J Appl Polym Sci. 2002;84(9):1789–96. doi:10.1002/app.10463. [Google Scholar] [CrossRef]
39. Fernandes M, Andrade C, Helena L, Mei I, Fonseca G, Rita A. Mechanical recycling simulation of polylactide using a chain extender. Adv Polym Technol. 2018;2053–60. doi:10.1002/adv.21863. [Google Scholar] [CrossRef]
40. Volova T, Shishatskaya E, Sevastianov V, Efremov S, Mogilnaya O. Results of biomedical investigations of PHB and PHB/PHV fibers. Biochem Eng J. 2003;16:125–33. doi:10.1016/S1369-703X(03)00038-X. [Google Scholar] [CrossRef]
41. Meng Q, Heuzey M, Carreau PJ. Control of thermal degradation of polylactide/clay nanocomposites during melt processing by chain extension reaction. Polym Degrad Stab. 2020;97(10):2010–20. doi:10.1016/j.polymdegradstab.2012.01.030. [Google Scholar] [CrossRef]
42. Arrieta MP, López J, Rayón E, Jiménez A. Disintegrability under composting conditions of plasticized PLAePHB blend. Polym Degrad Stab. 2014;108:307–18. doi:10.1016/j.polymdegradstab.2014.01.034. [Google Scholar] [CrossRef]
43. Iggui K, Kaci1 M, Le Moigne N, Bergeret A. The effects of accelerated photooxidation on molecular weight and thermal and mechanical properties of PHBV/Cloisite 30B bionanocomposites. J Renew Mater. 2018;6(3):288–98. doi:10.7569/JRM.2017.634184. [Google Scholar] [CrossRef]
44. Iggui K, Kaci1 M, Mahlous M, Le Moigne N, Bergeret A. The effects of gamma irradiation on molecular weight, morphology and physical properties of PHBV/Cloisite 30B bionanocomposites. J Renew Mater. 2019;7(9):807–20. doi:10.32604/jrm.2019.06778. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools