
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

The Oxyalkylation of Hydrophilic Black Alder Bark Extractives with Propylene Carbonate with a Focus on Green Polyols Synthesis
Laboratory of Lignin Chemistry, Latvian State Institute of Wood Chemistry, Riga, 1006, Latvia
* Corresponding Author: Alexandr Arshanitsa. Email:
(This article belongs to the Special Issue: Advances in Biorefinery Technologies and Products – 2024)
Journal of Renewable Materials 2024, 12(11), 1927-1948. https://doi.org/10.32604/jrm.2024.056466
Received 23 July 2024; Accepted 23 August 2024; Issue published 22 November 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
The isolated hydrophilic black alder (Alnus glutinosa) bark extractives were characterized in terms of component and functional composition and converted at 150°C–170°C into liquid green polyols using solvent-free and low-toxic base-catalyzed modification with propylene carbonate (PC). FTIR spectroscopy, HP-LC, GC, GPC, and wet chemistry methods were used to characterize the starting constituents, intermediate and final products of the reaction and to monitor the different pathways of PC conversion. The reaction of extractives as well as the model compounds, including catechol, xylose, PEG 400, and benzoic acid, with PC indicated the ability of OH groups of different origins present in the extractives to condense with equivalent amounts of PC. The polyols obtained consist of a copolymer fraction with one oxypropyl unit grafted per OH functionality of extractive components on average and oligo oxypropyl diols with a small number of carbonate linkages in the chain, obtained as a result of remaining PC homopolymerization. The domination of the oxypropylation mechanism vs. transcarbonation for PC ring opening was observed for both copolymerization and homopolymerization processes, making the process of oxypropylation with PC similar to that of conventional oxypropylation. At optimal reaction conditions, including a PC/OH ratio of 3.0 and a 24-h duration at 150°C, uniform polyols with low viscosity of ~900 mPa·s−1, a biomass content of ~27%, and an OHV of ~500 mg KOH·g−1 were obtained. Increasing the temperature of modification allows shortening the process but drastically increases the polyol viscosity. At fixed temperature values, increasing the PC/OH ratio not only decreases the biomass content but also strongly prolongs the processing. The significantly increased duration of the process using PC as an alternative oxyalkylation agent compared to that of oxyalkylation with propylene oxide is a reasonable trade-off for using a safer and more environmentally friendly technology.Graphic Abstract
Keywords
Supplementary Material
Supplementary Material FileNomenclature
| Mw | Mass-average molecular weight |
| Mn | Number-average molecular weight |
| Đ | Dispersity |
| EW | Equivalent weight |
| HPLC | High performance liquid chromatography |
| GC | Gas chromatography |
| FTIR | Fourier-transform infrared spectroscopy |
| GPC | Gel permeation chromatography |
Today, plastic materials are indispensable in the life of modern human society, with a continuously growing market. In 2021, the global production of plastic was about 390 million tonnes (MT), a twenty-fold increase since 1950, and it is expected to triple by 2060 [1]. More than 97% of plastics are derived from fossil feedstocks, which are not renewable or biodegradable [2]. Considering that less than 30% of the 25.8 MT of plastic waste produced annually in Europe is collected for recycling, it can be concluded that further increasing the plastic market without developing novel approaches toward plastic production will exacerbate global environmental problems. Therefore, the plastics strategy is a key element of Europe’s transition towards a carbon-neutral and circular economy. It will contribute to reaching the 2030 Sustainable Development Goals, the Paris Climate Agreement objectives, and the EU’s industrial policy objectives.
Using biomass as an alternative feedstock for the development of bio-renewable monomers, which are available for the sustainable production of biobased plastics, can be considered a key element of this strategy, thus reducing the industry’s dependence on fossil-based resources [3]. Among other renewables, lignocellulosic biomass, including wood and cereal crops, the deposits of which are renewed by photosynthesis, as well as the wastes from their processing, are the most available alternative feedstocks for the chemical industry. The primary metabolites of lignocellulosic biomass, such as cellulose, and hemicellulose, and the secondary metabolites, such as lignin, extractives, despite their differences in chemical structure and composition, are rich in hydroxyl groups available for condensation reactions [4]. Therefore, they are considered natural precursors for the development of bio-polyols for conventional polyurethane (PU) plastics, the most versatile class of polymers applicable in practically all branches of modern industry [5].
In general, PU bonds are formed as a result of the condensation of OH and NCO groups. Obtaining isocyanate-free PU is a highly promising step towards creating environmentally friendly materials, considering the harmful process of producing fossil-derived isocyanates and their toxic properties. While the synthesis of isocyanate-free PU by reacting cyclic carbonates with diamines is attracting increasing attention [6–8], the substitution of fossil-based polyols with renewable alternatives in conventional PU systems is recognized as the most viable pathway for the development of bio-based PU plastics [9].
The most studied precursors of bio-polyols for PU production are various lignocellulose-originated multi-tonnage wastes of the pulping and timber industries like tree bark, lignin, and extractives [10–13]. The application of bark extractives as PU precursors can be explained by the comparatively low labor and energy costs of their isolation using green solvents, the low molecular weight of constituents, in comparison with parent biomass, and the high content of OH groups [10–13].
The transition of lignocellulosic biomass from a solid to a liquid state via their chemical modification or liquefaction/solubilization in inert (aprotic) solvents or polyols used in PU synthesis serves as the basis for developing approaches to their use as macromonomers rather than fillers in PU systems, including elastomers, adhesives, and rigid PU foams [11,14–16].
The highly effective method for modification/liquefaction of lignin by its oxypropylation with propylene oxide (PO) in the presence of alkali as a catalyst was proposed by Glasser and co-workers [17,18]. This approach was developed further for different lignins [19,20], tree bark, and tannin-enriched bark extractives [21–23].
The main advantage of biomass modification with PO over other methods of liquefaction is the production of polyols with uniform functionality. This is attributed to the grafting of oxypropyl units (OPU) onto the OH groups of biomass components, including phenolic and acidic groups, transforming them into secondary aliphatic groups that are free from electronic and steric constraints. Consequently, these polyols exhibit high reactivity toward isocyanates [16]. It has been shown that polyols based on oxypropylated lignin can be substituted for conventional polyether polyols in PU synthesis, improving characteristics such as compressive and tensile strength, thermal conductivity, and flame-retardant properties [14,24]. However, the high flammability, low explosive limit in air, toxicity, and carcinogenicity of PO, as well as the high temperatures (200°C–250°C) and pressures (15–30 bars) needed for processing [19], present challenges for biomass modification with PO. Using cyclic carbonates for biomass oxyalkylation is considered a much more eco-friendly and safer alternative to biomass processing with PO [25,26].
The special interest in this class of organic compounds arises from their biodegradability and the use of greenhouse gas CO2 as one of the feedstocks for their production [26]. A well-known and commercially available representative of this compound class is propylene carbonate (PC), with a boiling point of 248°C and a vapor pressure of 0.04 hPa at 20°C, in comparison with 34°C and 588 hPa for PO, respectively [27]. The oxypropylation of organosolv and kraft lignins with PC in the presence of a base catalyst, focusing on the characteristics of the copolymers, was studied at the PC/OH ratio 10–50 and a temperature of 100°C–170°C [16,24,27–29].
In this case, the major part of the PC remains unreacted and acts as a solvent. It was shown that the phenolic groups of lignin reacted faster with PC compared to aliphatic OH groups [27,29]. At 160°C–170°C, 24 h were needed to oxypropylate all OH groups in lignin, including phenolic and aliphatic groups, with an average grafted chain length of 2.2–4.6 units. 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) promoted longer grafted chains compared to potassium carbonate. Nevertheless, despite the different nucleophilicity of phenolic and aliphatic OH groups, their oxypropylation proceeded via nucleophilic attack on the propylene carbon of PC. Only 0.3% of the PC was attached via carbonate linkage [27]. Unlike PO, which has a high ability for homopolymerization [19], only 3%–15% of PC was converted into propylene glycols. The full derivatization of only phenolic groups of lignin into aliphatic groups during lignin interaction with ethylene carbonate (EC) was determined at 140°C–170°C in the presence of potassium carbonate [30]. The absence of multichain grafting and EC homopolymerization led to a high amount of unreacted EC in the products, which was converted into ethylene glycol by the addition of an equivalent amount of water. PEG 400, as a non-reacted co-solvent, was added to the oxyalkylation product to tune the hydroxyl values and viscosity of polyols tested in PU foam compositions [31].
The oxypropylation of condensed tannins by PC revealed some differences compared to the oxypropylation of lignin. The grafted chains of up to six units were achieved, which is close to conventional oxypropylation with PO [32]. About 25% of them are represented by carbonate linkages, which appeared as a result of the oxypropylation exclusively of aliphatic OH groups. This confirms the data indicating that the combination of two alkylene carbon attacks and one carbonyl attack by the active chain end is necessary to make the pathway of aliphatic cyclocarbonate polymerization via carbonate carbon attack thermodynamically possible [31]. The chain coupling reactions in the presence of carbonate units in grafted chains can lead to an unexpectedly high molecular mass of PC copolymers compared to products obtained by conventional oxypropylation [33,34].
The brief overview of the state of the art presented above indicates that the process of biomass oxypropylation with PC can differ from conventional oxypropylation with PO due to the ability of two pathways for reactions via nucleophilic attacks on one carbonyl carbon and two alkylene carbons. Both oxypropylation methods and biomass composition will influence the composition and properties of the products obtained.
Black alder (Alnus glutinosa) is a widespread, fast-growing, and adaptive tree species in Europe, with high beneficial silvicultural impact for purification of waters. Black alder wood is used for furniture manufacturing, veneer and plywood production and has high potential for timber production [35]. The bark of black alder is a known source of diarylheptanoids, namely oregonin [36]. In contrast to tannins, well-known phenolic extractives consisting of combinations of rigid aromatic and heterocyclic structures, diarylheptanoids, including oregonin, consist of aromatic rings connected by a flexible seven-carbon chain. This could positively influence the mechanical properties of the PU polymer matrix, and avoid the excessive brittleness of the materials.
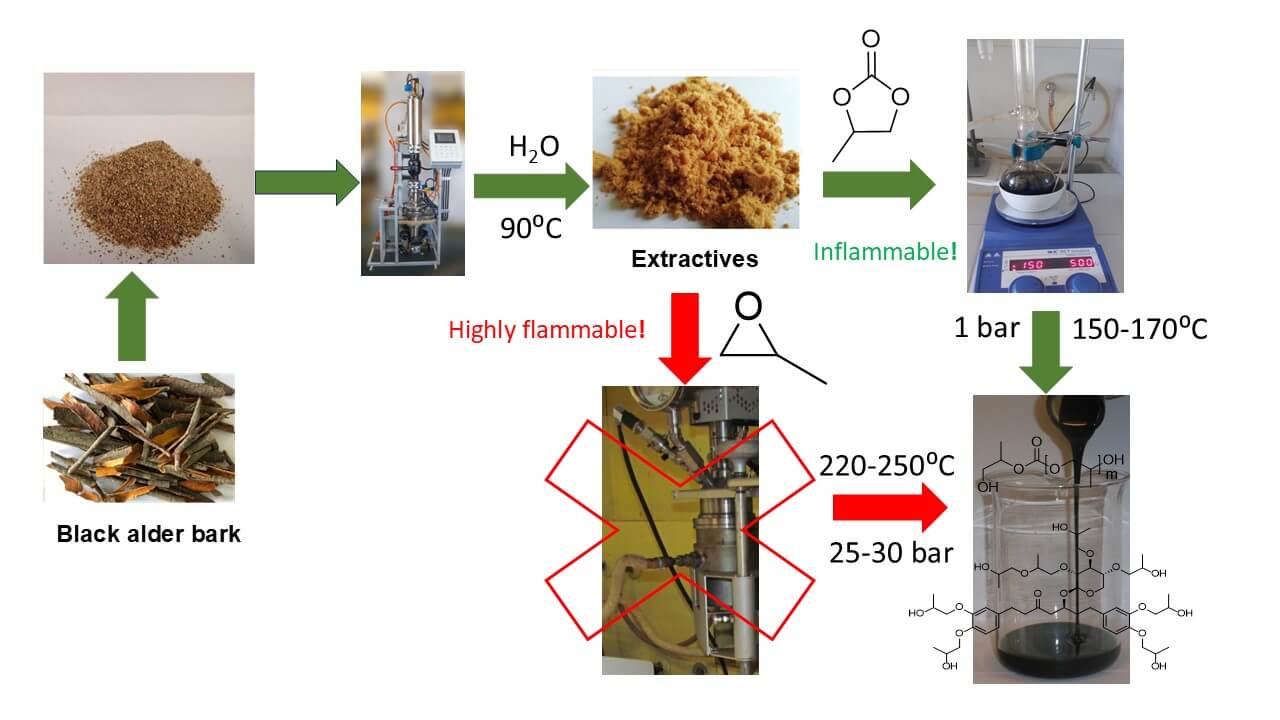
In this paper, the oxypropylation of black alder (Alnus glutinosa) (BA) bark hydrophilic extracts, consisting primarily of oregonin, which is the xyloside form of diarylheptanoid enriched with phenolic and aliphatic OH groups in similar proportions, was studied using PC in the presence of 1,8-Diazabicyclo [5.4.0]undec-7-ene as a catalyst. The oxypropylation of pure catechol, xylose, benzoic acid, and polyethylene glycol, as compounds modeling OH groups of different origins to predict their ability to condense with PC in the real extracts, was also studied. The weight loss of the reaction blends, dependent on the reaction temperature, duration, and the PC/OH ratio, was monitored, along with the FTIR spectra of the products. The molecular mass distribution (MMD), OH group content, and viscosity of the polyols obtained were analyzed based on oxypropylation regimes with a focus on obtaining low-viscosity green polyols enriched with biomass and possessing characteristics similar to those of commercial polyols usable for producing rigid PU foam.
Extract isolated from black alder (BA) bark by microwave-assisted extraction with water, was used as a precursor of polyol polyethers obtained by oxypropylation with PC.
The other reagents including, 1,2-dihydroxybenzene (catechol), ≥99%, D-(+)-xylose, ≥99%, benzoic acid, ≥99.5%, polyethylene glycol 400 (PEG 400), ≥99%, tetrahydrofuran (THF) anhydrous containing 250 ppm of butylated hydroxytoluene (BHT) as inhibitor, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), ≥99%, 1,4-diazabicyclo[2.2.2]octane, ≥99%, propylene carbonate (PC), oregonin with a purity of ≥95% (LC/MS-ELSD), were supplied from Merck (Darmstadt, Germany) and were used without additional purification.
Extractives from BA bark were isolated by a short-time and low-energy water extraction performed at 90°C using an innovative microwave extractor, as described in [37]. The yield of extractives after lyophilized drying was about 15% on a dry matter basis of the bark. The lyophilized extractives, without additional purification, were used for oxypropylation by PC.
2.3 Composition and Functional Analysis of BA Bark Extractives
2.3.1 HPLC Chromatography with PDA/ELS Detectors
The composition of extractives was determined using a Waters HPLC (Waters Corp., Singapore, Singapore) system equipped with photodiode array (PDA) and evaporative light scattering (ELS) detectors. The analysis regimes were described in detail in [37]. Oregonin was used as an external standard for its quantification in the extractives.
2.3.2 Total Polyphenolic Content
The total polyphenolic content (TPC) in the extractives was determined using the Folin–Ciocalteu method as described by [38] and expressed as grams of gallic acid equivalent (GAE) per gram of produced bio-polyol.
The total monomeric sugar content in the extractives was determined both without and with complete carbohydrate hydrolysis using an alditol acetate procedure with the GC-FID quantification as described by [39]. Glucose, galactose, mannose, xylose, arabinose, and rhamnose were used for calibration. 1-O-Methyl-D-glucose was used as internal standard.
The OH functionality of the extract was examined in duplicate using 31P NMR. The 2-chloro-4,4,5,5-tetramethyl-1,3-2-dioxaphospholan (TMDP) and the mix of N-hydroxy-5-norbornene-2,3-dicarboximide and chromium acetylacetonate were used as phosphorylation agent and internal standard correspondingly. Signal assignment was performed according to [40].
2.4 Oxypropylation of Model Compounds, Water, and Extractives
A mixture of 25 g, consisting of one hydroxyl-containing compound or water, PC, and DBU, was loaded into a 50 mL round-bottom flask equipped with a magnetic stirrer and a reflux condenser cooled with cold water. The weight of each component was adjusted to vary the PC/OH molar ratio from 1 to 7 while maintaining a constant DBU/OH molar ratio of 0.1. The flask was placed in a silicone bath with automatic temperature control and allowed to react at a temperature range of 125°C–170°C under constant stirring and argon flow. After 1, 2, 3, and 5 h, the weight loss of the reaction mixture was determined by weighing the flask, removed from the bath, with a precision of 1 mg, followed by the continuation of the reaction. After 24 h, the flask was weighed again, cooled to room temperature, and the reaction product was stored for further analysis. The idle experiments, including the heating of separetely PC and propylene glycol under the same reactional conditions, were performed. No weight loss was observed in this case after 24 h of heating at 170°C.
The total amount of OH groups in the initial extract and in the oxypropylated products (polyols) was quantified by acetylation methods using pyridine as a catalyst. The unreacted acetic anhydride was hydrolyzed by water, followed by potentiometric titration of acetic acid with 0.1 N NaOH [41]. In the case of the initial extract, conductometric titration of the alkali solution of extracts with 0.1 N HCl and calcium-acetate-based chemisorption were additionally used to separately quantify aliphatic, phenolic, and carboxylic groups [41].
The rheological measurements were made at 25°C using the Anton Paar Modular Compact Rheometer MCR 92 (Anton Paar, Graz, Austria) with a cone-plate measuring system and a gap of 48 μm. Shear rate ramps were carried out from 1 to 100 s−1.
2.7 Gel Permeation Chromatography
The molecular mass distribution (MMD) of extractives and polyols was studied using an Agilent 1260 Infinity HPLC system equipped with an isocratic pump, degasser, auto-sampler, PLGEL Mixed-E 300 mm × 8 mm column, and Optilab refractive index (RI) detector. The sample concentration in the THF solution and injection volume were 5 mg∙L−1 and 100 μL, respectively. THF was used as the mobile phase with a flow rate of 1.0 mL∙min−1. Sodium-polystyrene sulfonates were used as the calibration standard for MMD, with a specified molecular weight range of 5007–20,000 Da [42].
2.8 Non-Isothermal TG/DTG of biomass
Non-isothermal thermal analysis of BA bark extractives was performed in argon using a Seteram Setline device (France) in the temperature range from 25°C to 700°C with a heating rate of 5°C∙min−1.
FTIR spectra of polyols were studied by the attenuated total reflectance (ATR) method. FTIR spectra of BA bark extractives, catechol, and xylose were determined using the KBr technique. Spectrum One FTIR spectrometer (Perkin Elmer) equipped with an ATR diamond-ZnSe top plate was used. The spectral range was 4000–500 cm−1, scan resolution and the number of scans were 4 cm−1 and 64 scans, respectively.
3.1 Composition of BA Bark Extractives
As was shown by the HPLC experiments the isolated BA bark extractives consist of different classes of organic compounds, including major non-lignin phenolics: diarylheptanoids, flavonoids in oligomeric form, and carbohydrates with organic acids admixture (Fig. S1) [37]. Oregonin, which is the xyloside form of a diarylheptanoid (Fig. 1), is a dominant polyphenolic constituent in extractives and comprises 31.0% ± 1.8% on the organic basis of extractives that was determined using commercial oregonin for external calibration.
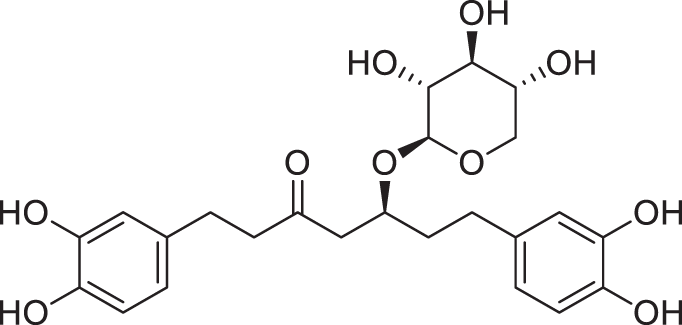
Figure 1: The structural form of oregonin
In total, 38% of monomeric sugars content were determined in the extracts after complete hydrolysis, compared to 6.7% in the raw extract (Table 1). This indicates that the majority of carbohydrates present in the extract are in oligomeric form or as glucosides. The portion of xylose liberated after complete hydrolysis of biomass was 15.1% on a dry matter basis. The yield of xylose after complete hydrolysis of the glucoside bond of oregonin should be 31%. This means that about 64% of monomeric xylose and 25% of all sugars in the hydrolyzed extract are the xylose moiety of oregonin.

The analysis of physical-chemical characteristics indicates that isolated BA bark extractives can be described as a natural solid polyol, rich in both phenolic and aliphatic OH groups, with a low number of inorganic admixtures and low dispersity in terms of MMD. The Mn of extractives was more than twice that of pure oregonin (478 Da), which can be attributed to higher molecular fractions of carbohydrates and admixtures of high-molecular-weight procyanidins that are unavailable for detection by HPLC-ELSD (Table 2; Fig. S4) [15].
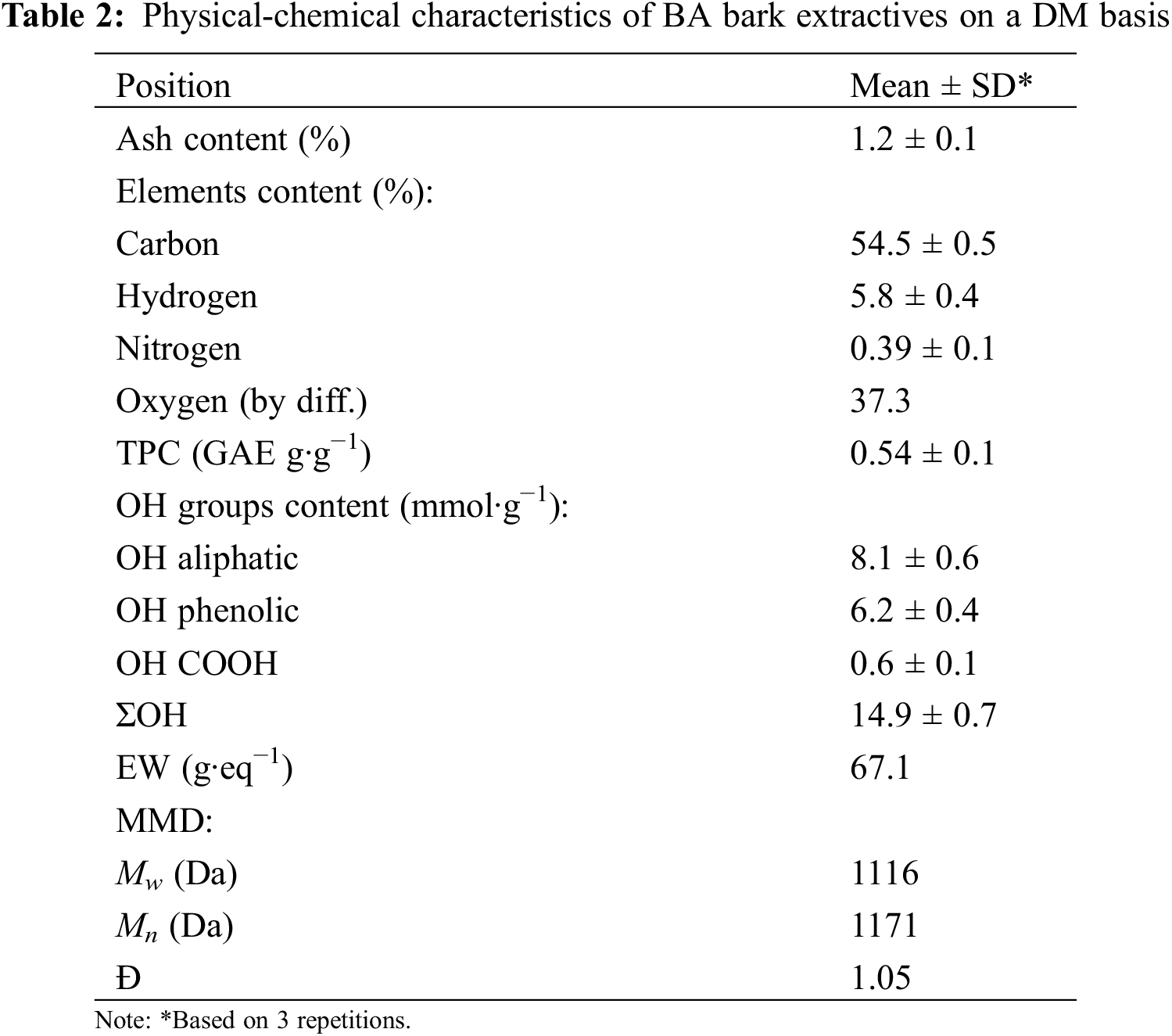
The TPC value of 0.54 GAE g∙g−1 indicates that the BA bark extract is enriched with plant polyphenolics. This was confirmed by the phenolic group content, which comprises about 42% of the total OH functionality of the extract (Table 2). The majority of this functionality is aliphatic OH at 55%, while the least is carboxylic OH at 3%. The 31P NMR analysis showed that catechol groups made up 90% of all phenolic groups in the extract (Fig. 2). This suggests that, in addition to oregonin, other unidentified diarylheptanoids are present in the extracts [43]. The content of other classes of polyphenolics is insignificant.
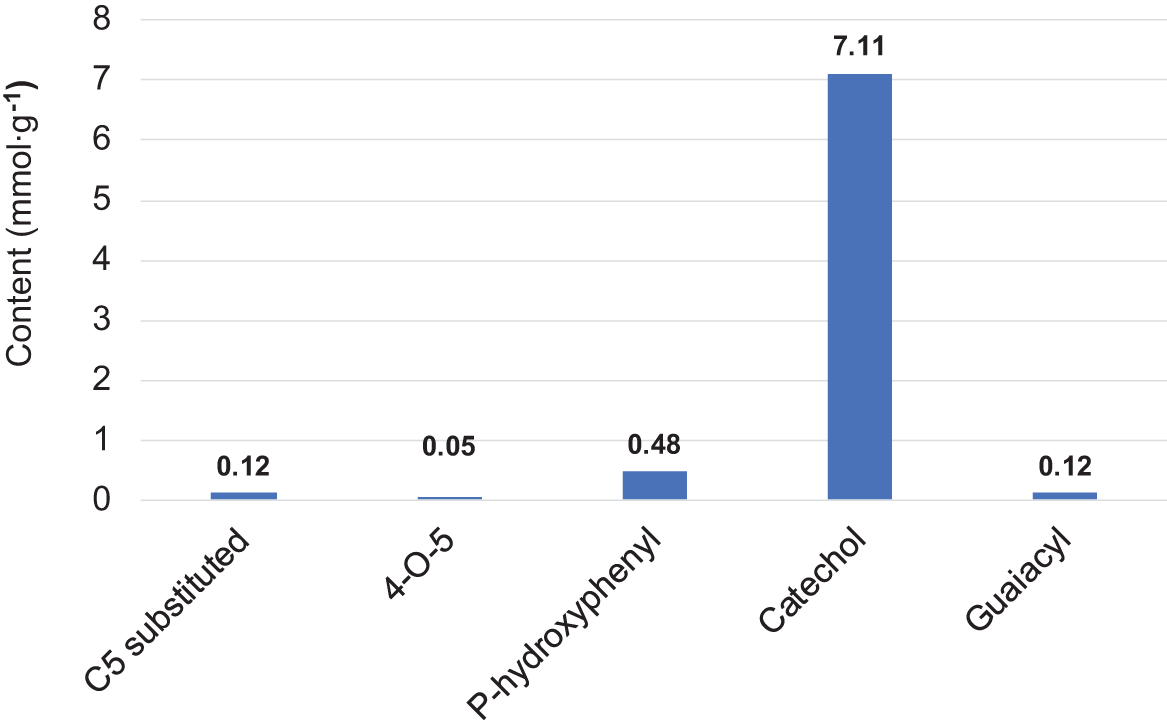
Figure 2: The content of phenolic groups of different originations in BA bark extract according to 31P NMR
3.2 Conversion of PC in Reactions with Model Hydroxyl-Containing Compounds and Water: Dependence on Reaction Conditions
The different acidity of extractives bearing OH groups—aliphatic OH, phenolic groups, and carboxylic groups suggests that they interact differently with PC during the oxypropylation process [30].
To separately estimate the conversion of PC in the presence of phenolic, aliphatic, and carboxylic groups as initiators of PC polymerization, catechol, xylose, and benzoic acid were used as model compounds. Additionally, the interaction of PC with water and PEG 400 was investigated. In the first case, it was justified that trace amounts of water could promote the formation of PC homopolymers, which generally compete with copolymer grafting. In the second case, PEG 400 was considered due to its potential application as a co-solvent in combination with cyclic carbonate for biomass liquefaction, as described by Zhang et al. [30] (Figs. 3 and S2).
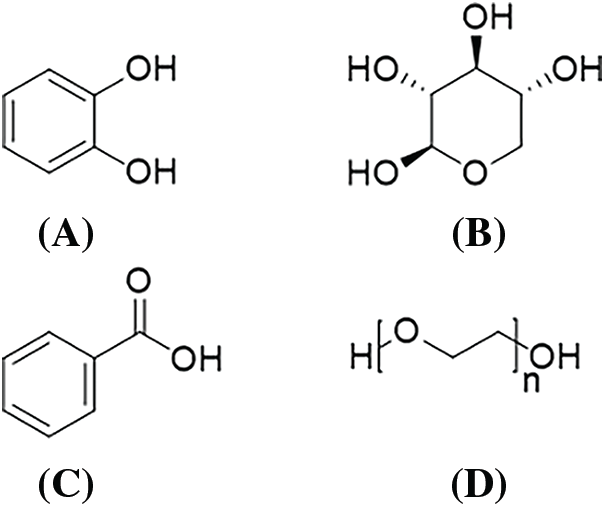
Figure 3: The formulas of model compounds: catechol (A); xylose (B); benzoic acid (C) and PEG 400 (D)
Additionally, to the two electrophilic centers available for the PO molecule, PC has one more site at the carbonyl carbon. Therefore, in general, depending on the nucleophiles used, two pathways for the opening of the PC cycle are available. These include transcarbonation with the formation of a carbonate linkage and oxypropylation with the release of CO2 (Fig. 4) [31].
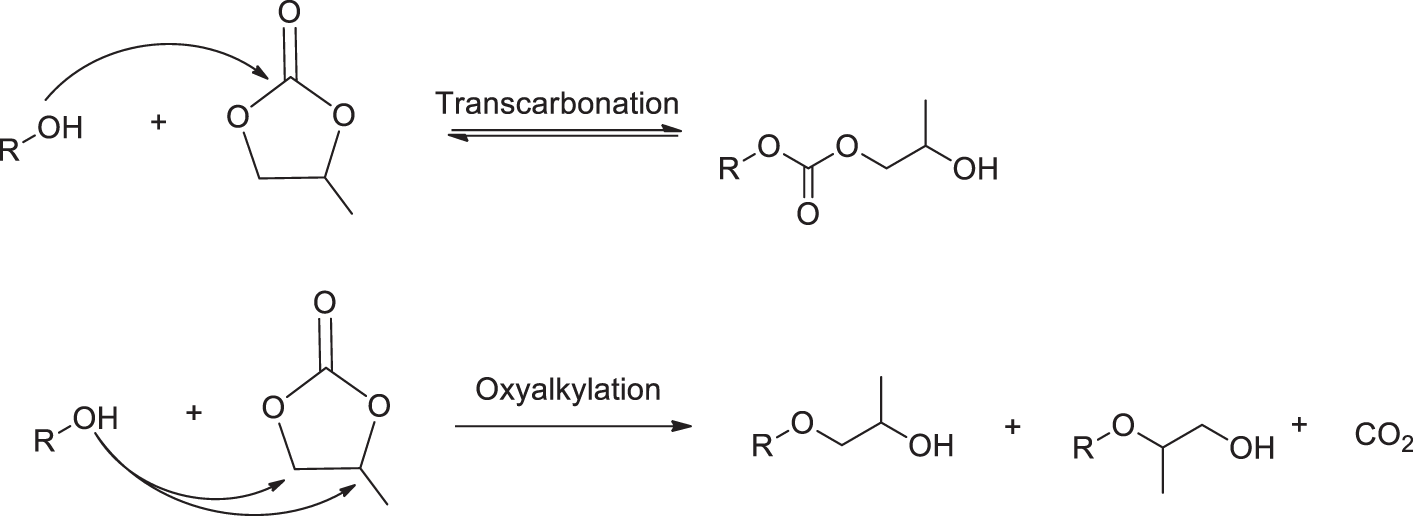
Figure 4: Two pathways of the PC ring opening in its reaction with hydroxyl-containing compounds
It is recognized that weak nucleophiles such as phenols and carboxylic acids react with PC via an oxyalkylation mechanism, whereas aliphatic hydroxyls, as strong nucleophiles, must react via a transcarbonation mechanism [25]. It should be mentioned that one unit grafting of PC transforms acidic groups of the substrate into aliphatic ones, and therefore, further PC grafting has to be accompanied by carbonate linkage formation without CO2 release. However, this mechanism is not thermodynamically favorable. Theoretically, a combination of one carbonyl carbon atom attack and two alkylene carbon atom attacks is the most probable mechanism of cyclic carbonate polymerization [31]. In agreement with this, about 25 mol% of PC was grafted via carbonate linkage in the product of oxypropylation of condensed tannins, as reported [32]. In contrast, it was reported that in the case of oxypropylation of lignin, the etherification of both acidic and aliphatic OH groups is the favored mechanism of PC conversion in the presence of basic catalysts and enhanced temperature [27].
The contradictory data presented above indicate that the oxypropylation of biomass with PC is a more complicated process compared to conventional oxypropylation. In addition to the two pathways of ring opening, the main reaction resulting in the grafting of oxypropyl units (OPU) along the chain initiated by OH groups of biomasses can be accompanied by hydrolysis of PC by water, forming propylene glycol (PG) followed by homopolymer formation in a similar manner as in the case of oxypropylation using conventional PO [29] (Fig. 5).
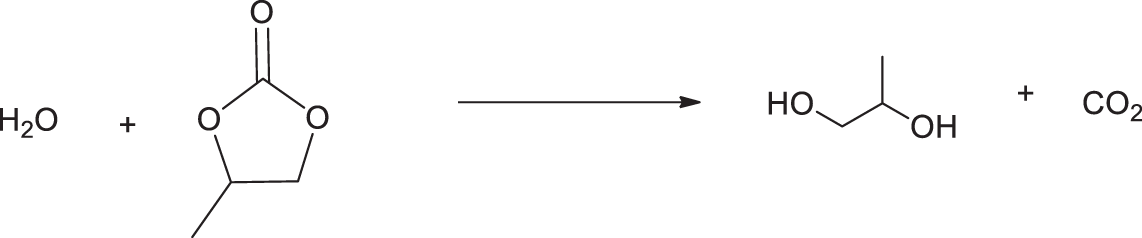
Figure 5: The scheme of PC hydrolysis
In real conditions, the occurrence of this reaction is high, probably due to the presence of traces of physically bonded water in the biomass as well as its partial dehydration during thermal treatment (Fig. S2). In this work, the monitoring of weight loss of reaction blends due to CO2 elimination during the oxypropylation of model compounds, water, PEG 400, and extracts under different conditions was performed to explain which of two the ring opening mechanisms will be realized. The values of PC conversion (X) were calculated using Eq. (1), assuming that oxypropylation is the dominant mechanism of reaction compared to transcarbonation.
where:
In this study, PC acts as both a solvent and a reagent, with no other solvents used. It was established that solubilization of xylose in PC at an equivalent PC/OH ratio is not feasible. Solubilization was achieved at a higher concentration of solvent (PC/OH = 3.0). It was shown that, at an equivalent PC content, water exhibited the highest reactivity among the compounds tested (Fig. 6A). After 1 h of reaction, about 86% of PC was converted into PG and CO2 according to the reaction scheme shown in Fig. 5. At 125°C, the other compounds exhibited significantly lower reactivity compared to water. These results indicate that, in the presence of trace amounts of water, the formation of PG will be a favorable process compared to the copolymerization of PC with hydroxyl-containing compounds. Therefore, in further reactions, PC homopolymerization will compete with the copolymer chain grafting.
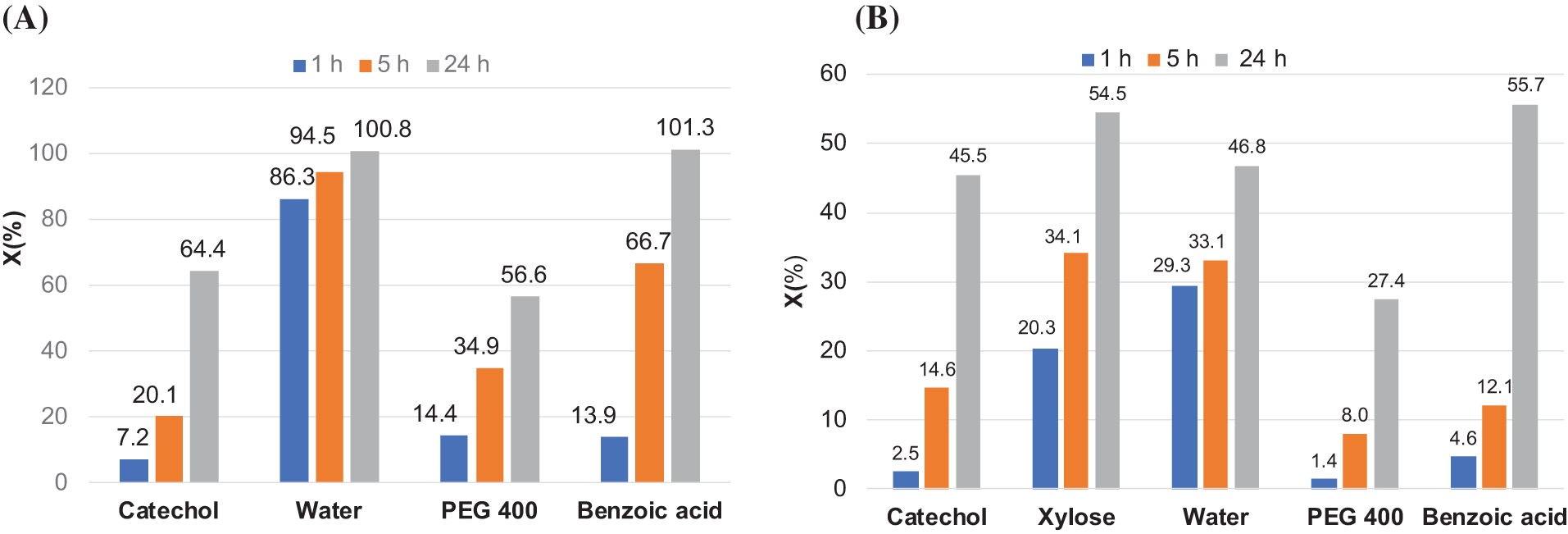
Figure 6: The PC conversion (X) at 125°C in dependence of the duration of its reaction with model compounds, PEG 400, and water at different PC/OH ratio: 1.0 (A); 3.0 (B)
After 24 h of reaction at 125°C, complete PC conversion was achieved only in the cases of water and benzoic acid, which has the highest acidity (pKa = 4.2) and thus the highest deprotonation ability. The conversion values slightly higher than 100% can be explained by the negligible loss of PC due to its evaporation at the moment of disassembly from the condenser before weighing.
With an increasing the PC/OH ratio up to 3.0, the conversion of PC at a given time was significantly decreased for all compounds. No complete conversion was achieved after 24 h of reactions (Fig. 6B). This can be explained by a combination of factors. Firstly, since PC is used as both a solvent and reagent, its concentration in solution remains the same, ~12 mol−1 at 20°C, regardless of the PC/OH ratio. However, the concentration of hydroxyl-containing compounds and the catalyst DBU in the solution decreases steadily as the PC/OH ratio increases. Therefore, the reaction rate, which depends on the concentrations of reagents and catalyst, must decrease. Secondly, the rate of single-unit grafting is higher compared to multi-unit’s chain grafting. For example, the rate of PC conversion in reactions with catechol and benzoic acid should decrease after single-unit grafting due to the transformation of acidic and phenolic groups into secondary aliphatic groups with lower proton-donor ability.
Water revealed the highest activity compared to OH-containing substances at the initial stage of the reaction. After 1 h, the conversion was about 30%, indicating almost complete consumption of water in the fast reaction of PC hydrolysis. However, the further steps of PC conversion through propagation reaction with PG proceed at a significantly lower rate. In total, less than 50% of PC was converted over 24 h. In both cases, including equivalent content of reagents and an excess of PC, PEG 400 reached with primary OH groups showed the lowest activity toward oxypropylation with PC.
For all studied compounds at a PC/OH ratio of 1.0, the PC conversion steadily increased as the reaction temperature rose in the range of 125°C to 170°C (Fig. 7A). After 1 h of interaction at the PC conversion at 170°C, compared to that at 125°C, increased mostly for the weak nucleophiles catechol and benzoic acid, by 7.0 and 6.7 times, respectively, and by only 2.3 times for PEG 400, which bears aliphatic OH groups. As shown by the experiments, slight evaporation of water from the open reactor occurred despite using a condenser when the temperature in the reactor was up to 150°C. Therefore, water was oxypropylated only at 125°C.
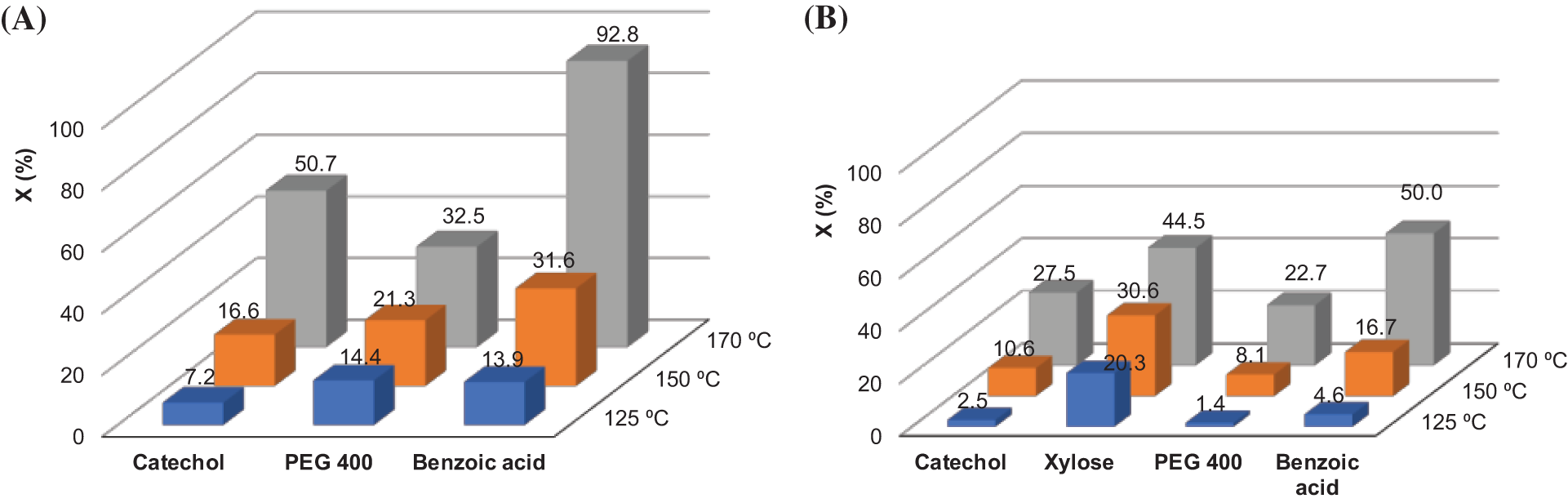
Figure 7: The PC conversion (X) after a 1-h reaction with model compounds and PEG 400 in dependence of reaction temperature at different PC/OH ratios: 1.0 (A); 3.0 (B)
PC conversion closed to complete (92.8%) in 1 h of reaction at 170°C was achieved for benzoic acid, whose pKa value is rather lower compared to the phenolic groups in catechol (pKa1 = 9.3; pKa2 = 13.0) [44]. For catechol, complete PC conversion was established after 5 h of reaction based on the weight loss of the reaction mixture. 24 h were needed for the complete oxypropylation of PEG 400 at 170°C. With an excess of PC in the reaction system (PC/OH = 3.0), a similar trend in the increase of PC conversion with increasing temperature was observed. However, the extent of PC conversion was lower compared to when a stoichiometric ratio of reactive sites was used (Fig. 7A,B).
3.2.1 The Characterization of Intermediate and Final Products of Oxypropylation by FTIR Spectroscopy and GPC Methods
The results of PC conversion were confirmed by monitoring the FTIR spectra of reaction products (Fig. 8A,B). It should be mentioned that the presence of a DBU catalyst at concentrations of 4.8% and 8% (w/w) in the starting catechol and PEG 400-based compositions, respectively, resulted in the appearance of FTIR absorbance in the region of 1615 cm−1 stretching vibration bonds of C=N (Fig. 8A,B; Fig. S3).
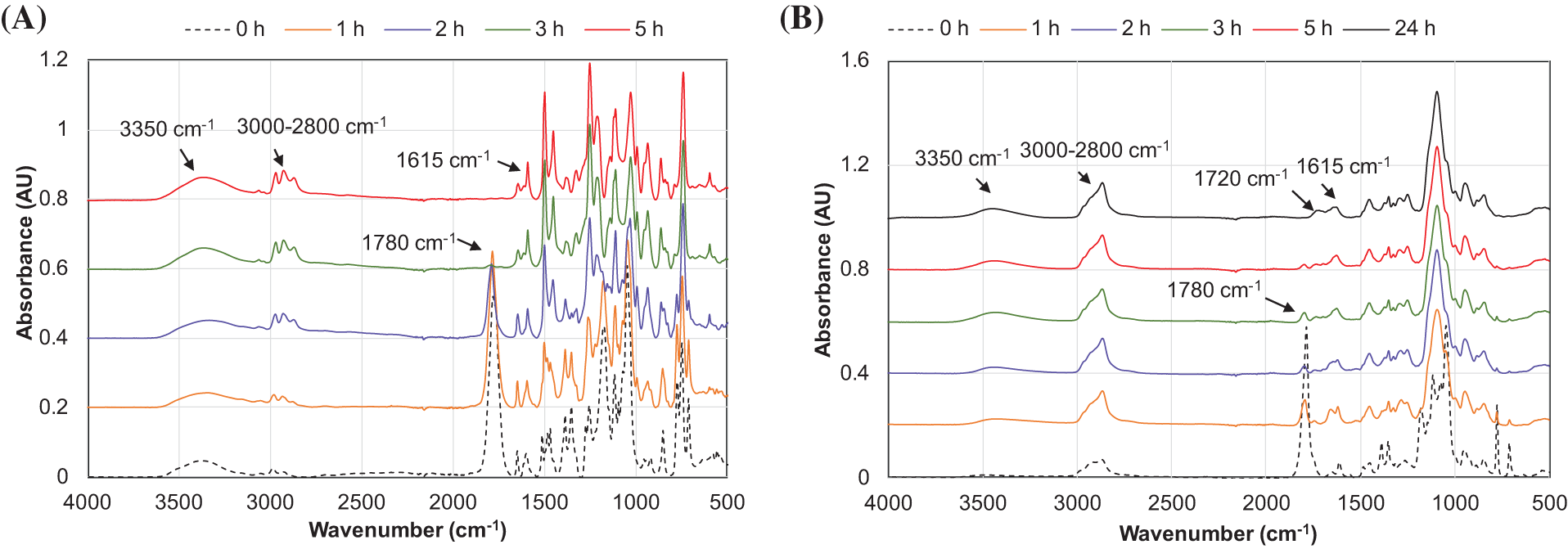
Figure 8: The FTIR spectra of reaction products of catechol (A) and PEG 400 (B) with PC performed at 170°C and PC/OH = 1.0 in dependence of the reaction duration
The continuous decrease in absorbance intensity at ~1780 cm−1, attributed to the carbonyl group in PC (Fig. S3), alongside the simultaneous increase in absorbance in the aliphatic hydrocarbon region (symmetric and asymmetric stretching of -CH2- in the region 2950–2850 cm−1), confirms the grafting of a single PC unit onto the OH groups of catechol and PEG 400, respectively (Fig. 8A). According to FTIR analysis, the final products of catechol oxypropylation can be characterized as polyol ethers without the incorporation of carbonyl linkages (Fig. 8A). The FTIR spectra of the final products from the interaction of PEG 400 with PC show a low-intensity absorbance at ~1720 cm−1, which can be explained by the presence of a negligible number of carbonate units grafted via an esterification mechanism. Therefore, we can conclude that under the studied conditions, the copolymerization of both catechol and PEG 400 with PC proceeded predominantly via an oxyalkylation mechanism, despite the different nucleophilicity of the formed anions.
A more systematic study of the effect of the PC/OH ratio on the structure and composition of the obtained polyols was conducted using products obtained at 150°C. For catechol, PEG 400, and water, the PC/OH ratios were 1.0, 3.0, 5.0, and 7.0. For xylose, the ratios were 3.0, 5.0, and 7.0. The duration of the process was 24 h, after which complete conversion according to Eq. (1) was confirmed. For water and PEG 400 based systems at PC/OH = 5–7, 48 h were needed for complete PC conversion.
The complete disappearance of FTIR absorbance in the region of 1780 cm−1 in the products of the reaction between catechol and PC was observed independently of the PC/OH ratio (Fig. 9A). Small absorbance intensities at 1780 cm−1 were observed in the FTIR spectra of PEG 400- and xylose-based products at PC/OH = 3, which disappeared at the higher PC/OH ratios (Fig. 9B,C). For water-based products, unreacted PC was present only at the highest amount of PC (Fig. 9D).
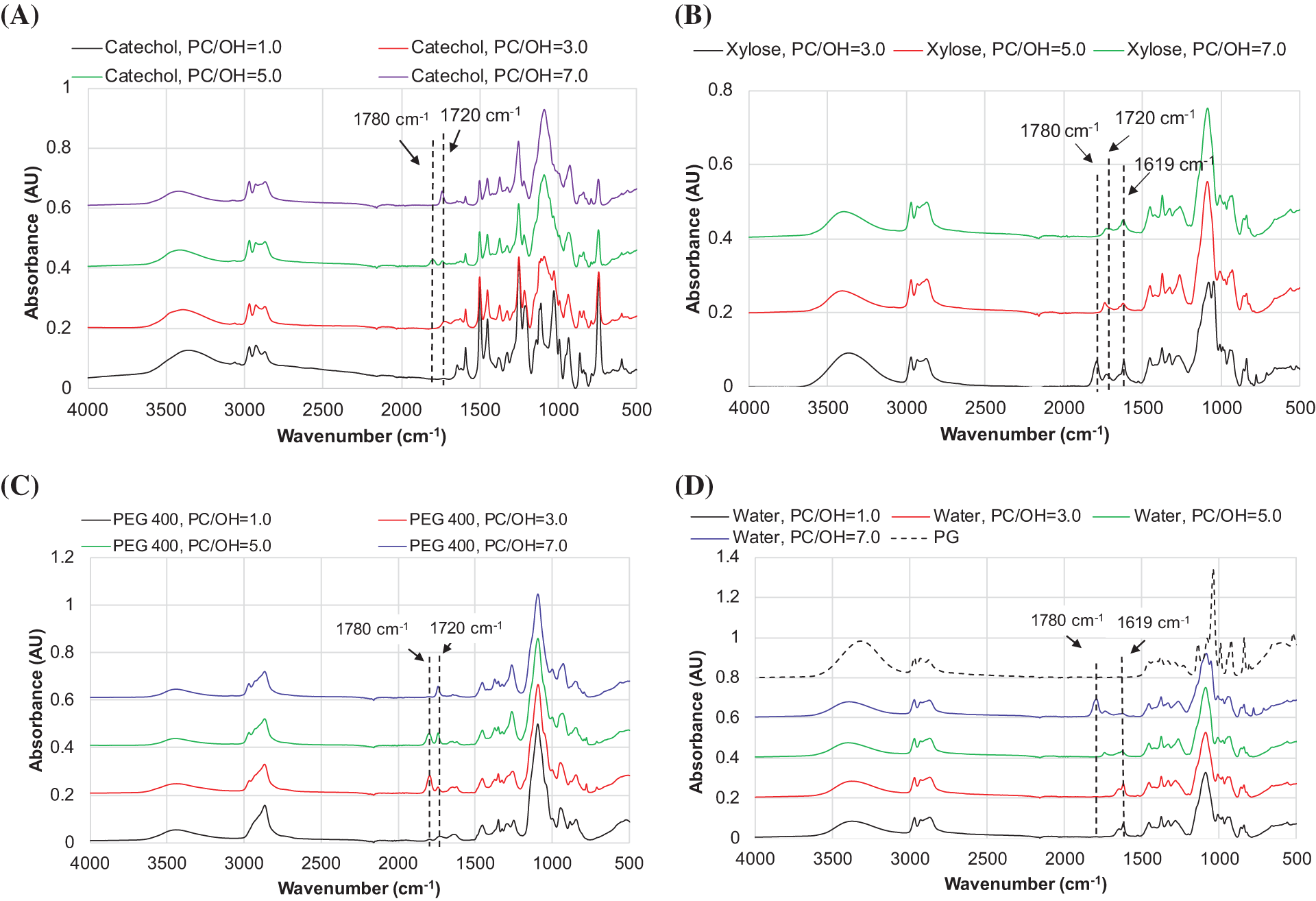
Figure 9: The FTIR spectra of products obtained by the reaction of catechol (A), xylose (B), PEG 400 (C), and water (D) with PC at different PC/OH ratios
With an increasing PC/OH ratio, the intensity at ~1720 cm−1 in the FTIR spectra of all catechol-derived products increased, indicating that a small portion of PC was attached via a carbonate linkage in this case. A similar increase in the absorbance band at ~1720 cm−1 with an increasing PC amount was observed in the FTIR spectra of PEG 400-derived products (Fig. 9C) and oligo oxypropyl diols (Fig. 9D).
The results presented above confirm that the reaction of catechol with PC proceeded exclusively via the oxyalkylation mechanism. One-unit grafting transforms the phenolic groups into aliphatic OH groups. The attachment of PC to aliphatic groups can occur via oxyalkylation and transcarbonation pathways, with the former dominating due to the use of basic catalysts.
In general, an increase in the C–H stretching intensity of methyl, methylene, and methane groups at 2800–3000 cm−1 relative to that of OH groups at 3500 cm−1 indicates the growing length of oxypropyl chains in the products with increasing PC content. Two processes could be responsible for this: the grafting of oxypropyl units onto the OH groups of the starter and the formation of propylene glycol and its oligomers as a result of PC homopolymerization.
The GPC results of catechol-based products have shown the appearance of high molecular fractions compared to catechol in products of its oxypropylation, clearly indicating copolymer formation. It was established that the low molecular weight peak present in all chromatograms is attributed to the DBU catalyst, which exhibited high refractive intensity (Fig. 10A–D). In the case of catechol the lower molecular peak is attributed to BHT presented in THF solvent. In the GPC of reaction products this peak is overlapped with peaks of DBU catalyst and therefore is not observed separately (Fig. 10A).
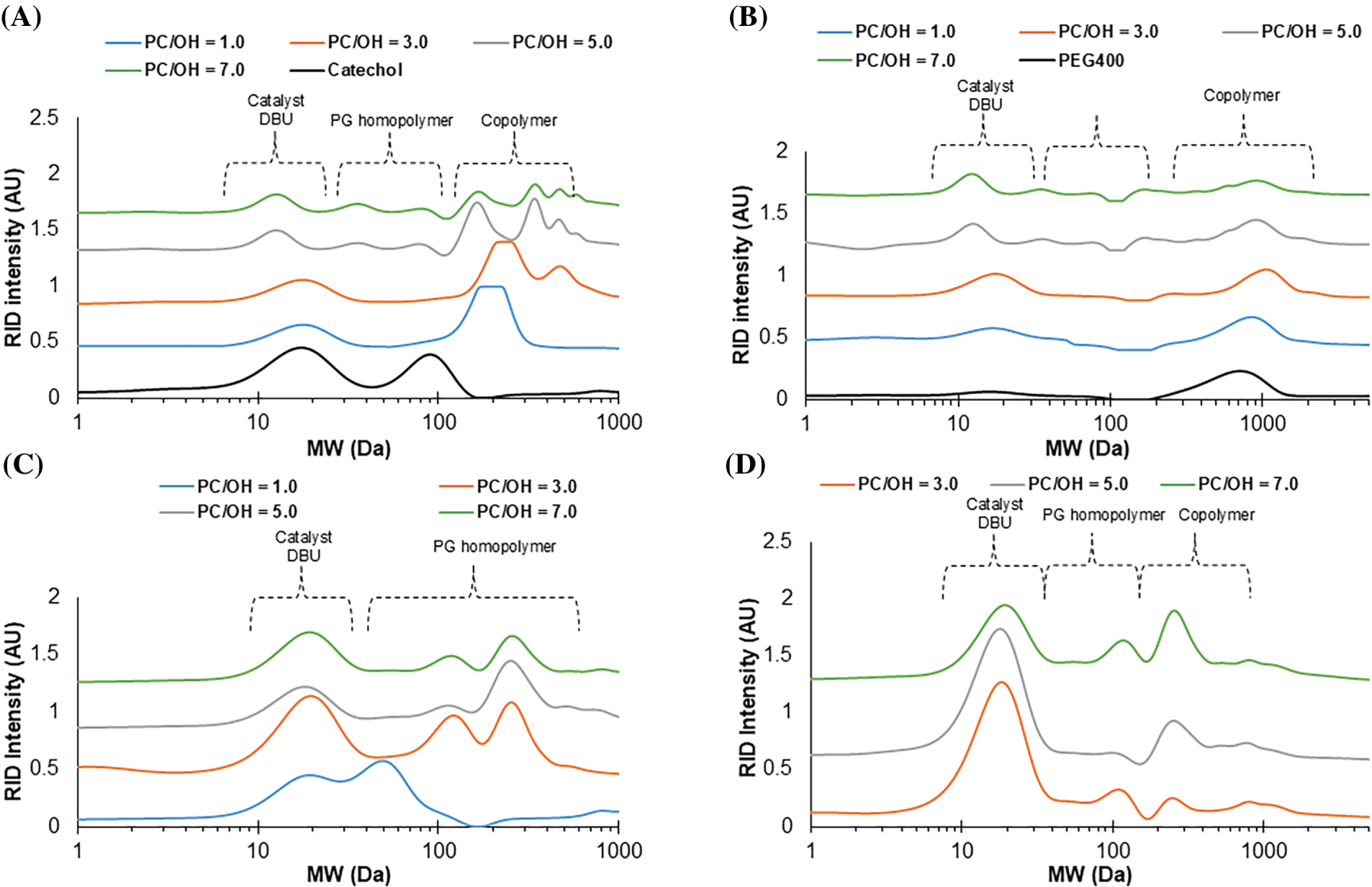
Figure 10: The effect of PC/OH ratio on GPC of oxypropylation products of catechol (A), PEG 400 (B), water (C), xylose (D)
At the PC/OH ratios of 5 and 7, the monomodal MMD distribution typical for initial catechol and products of its oxypropylation at PC/OH = 3 changes to multimodal (Fig. 10A). This suggests that the process becomes more complex and that both copolymerization and homopolymer formation take place. A slight shift of the MMD towards higher molecular mass with an increasing PC/OH ratio was also observed for products of PEG 400 oxypropylation (Fig. 10B). With an increasing PC/water ratio, the MMD of the products became bimodal and shifted to higher molecular weights typical of oligo oxypropyl diols compared to that of propylene glycol (PG), which is the product of PC hydrolysis (Fig. 10C).
For xylose-derived products, the Mw values of the copolymer determined by GPC were in the range of 310–320 Da, which is double the absolute molecular weight of pure xylose. This fact and the solubility of xylose-derived products in THF confirm their condensation with PC (Fig. 10D). The GPC test for pure xylose was not available due to its practical insolubility in THF. Therefore, the relative molecular weights of xylose and the copolymers obtained were unfortunately not compared using GPC data.
3.3 The Oxypropylation of BA Bark Extractives by PC, Composition, and Properties of Ensuing Green Polyols
The results presented above indicate that all functional groups of extractives, including phenolic groups of catechol units, and aliphatic and carboxylic groups, could be available for condensation with PC. Aliphatic OH is the least reactive; therefore, more time is needed for its interaction with PC in comparison with phenolic and carboxylic OH. Additionally, the process of forming oligo oxypropyl diols initiated by PC hydrolysis will compete with the copolymerization of biomass with PC. The relative content of copolymer and homopolymer will predetermine the major properties of the polyols including OH groups content, viscosity, and functionality [19].
As in the case of xylose oxypropylation, a uniform suspension of extractives in PC was obtained at a PC/OH ratio of not less than 3.0. The study of the interaction of BA extractives with PC at an equivalent ratio of components is possible only in the presence of aprotonic co-solvents and, therefore, was not performed in this study.
For extractives derived composition at the PC/OH ratio equal to 3, the conversion values were strongly dependent on temperature. At 125°C, the X values increased continuously with reaction time, but complete conversion was not achieved after 24 h of interaction (Fig. 11A). 48 h was needed for that. With an increase in temperature to 150°C and 170°C, complete conversion was achieved after 24 and 5 h of reaction, respectively (Fig. 11A).
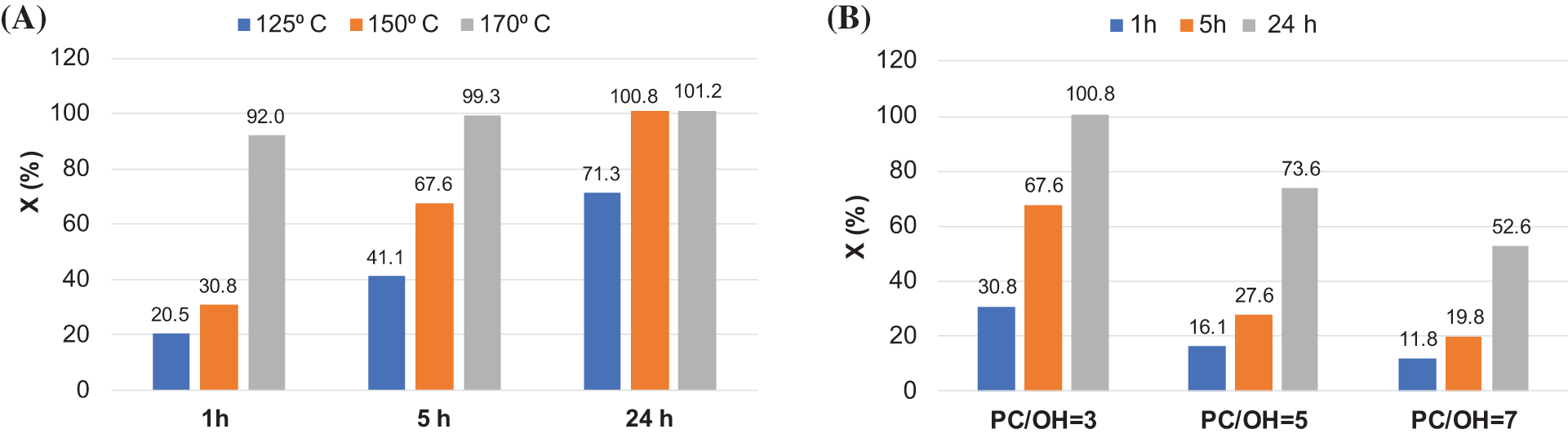
Figure 11: The PC conversion (X%) in the reaction of BA bark extractives oxypropylation vs.: (A) reaction time at different temperatures with a fixed PC/OH ratio of 3.0; (B) PC/OH ratio at different reaction times with a fixed temperature of 150°C
The effect of PC/OH ratio values on the process of biomass oxypropylation was studied using a moderate temperature of 150°C (Fig. 11B). The motivation for choosing a moderate temperature is based firstly on the desire to minimize the thermal degradation of biomass during processing, secondly on the safety of processing, and thirdly on decreasing energy consumption, which is an urgent task for any technology.
A decrease in the conversion rate was observed as the amount of PC in the starting suspension increased. For example, complete conversion of PC after 24 h of interaction was established for the system with the PC/OH ratio 3, while only 74% and 53% conversion was determined for compositions with the PC/OH ratios of 5 and 7, respectively (Fig. 11A,B).
The FTIR spectra of polyols obtained confirmed the results presented above (Fig. 12A,B).
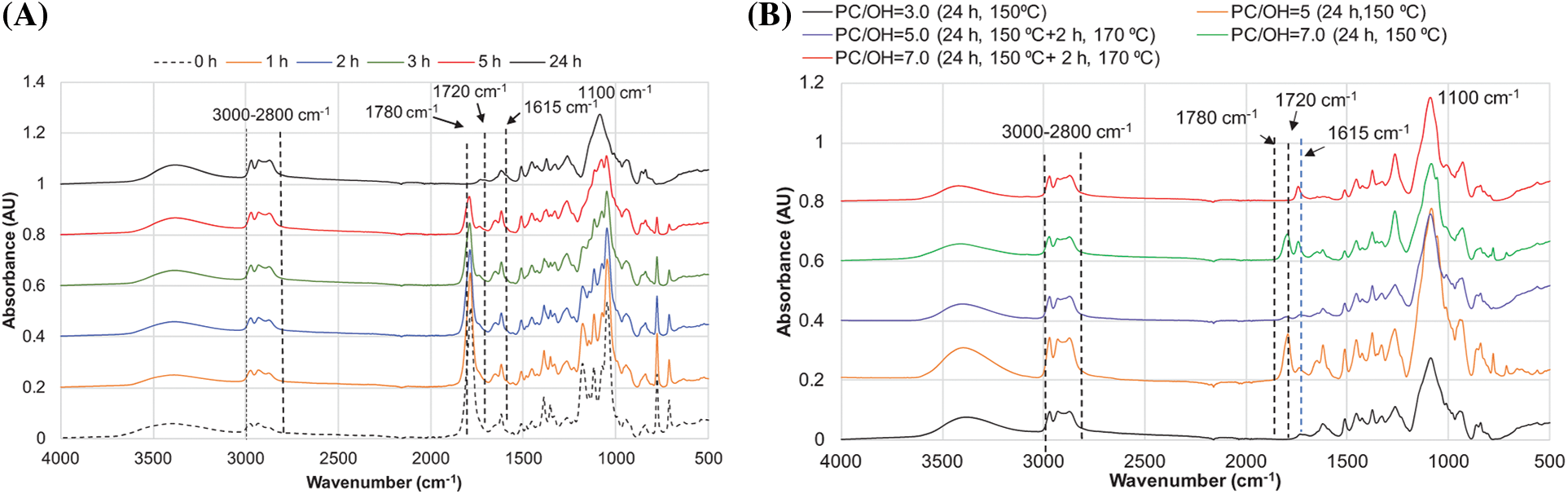
Figure 12: FTIR spectra of polyols obtained by reacting BA extractives with PC at 150°C, showing: the effect of reaction duration with a fixed PC/OH ratio of 3.0 (A), and (the effect of varying PC/OH ratios after 24 h of reaction (B)
The interaction of BA bark extractives with PC was accompanied by a continuous decrease in absorbance intensity at 1780 cm−1 in the extractives-based polyols, along with a simultaneous increase in the intensity of aliphatic group stretching (3000–2800 cm−1), similar to that observed in the products of model compound oxypropylation (Fig. 12A; Fig. 8A). The FTIR data confirm the results of weighing experiments, indicating that complete PC conversion in the reaction with BA extractives performed at 150°C and a PC/OH ratio of 3 was achieved after 24 h. With an increasing PC/OH ratio, additional treatment of the reaction mixture at 170°C for 2 h was needed to complete the reaction (Fig. 12B).
According to the FTIR spectra, at complete PC conversion the ratio of maximal absorbance intensities attributed to aliphatic hydrocarbons (3000–2800 cm−1) to those of OH groups (3350 cm−1) was 1.27, 1.42, and 1.78 for polyols with PC/OH ratios of 3.0, 5.0, and 7.0, respectively. This clearly indicates an increase in oxypropyl unit concentration in polyols in this order, achievable through both copolymer and homopolymer formation pathways. Therefore, regardless of the PC/OH ratio, the FTIR spectra of the products of BA bark extractives oxypropylation show a strong intensity of around 1100 cm−1, attributed to secondary OH groups and ether bonds. This testifies that oxyalkylation is the dominant mechanism in the reaction of BA extractives with PC vs. transcarbonation (Fig. 12B).
The appearance of low-intensity absorbance bands at ~1710 cm−1, typical for unconjugated C=O in the FTIR spectra, indicates that a negligible number of products of PC conversion is present in polyols as carbonate linkages, with a trend towards increasing with the PC/OH ratio. These results are in good agreement with the data on model compound oxypropylation presented in Section 3.2.
The GPC analysis of polyols compared to the initial extractives indicates the presence of fractions with differing molecular weights in the obtained polyols. The appearance of higher molecular weight fractions compared to the initial extractives can be explained by the copolymerization of extractives through the condensation of OH groups with PC (Fig. 13).
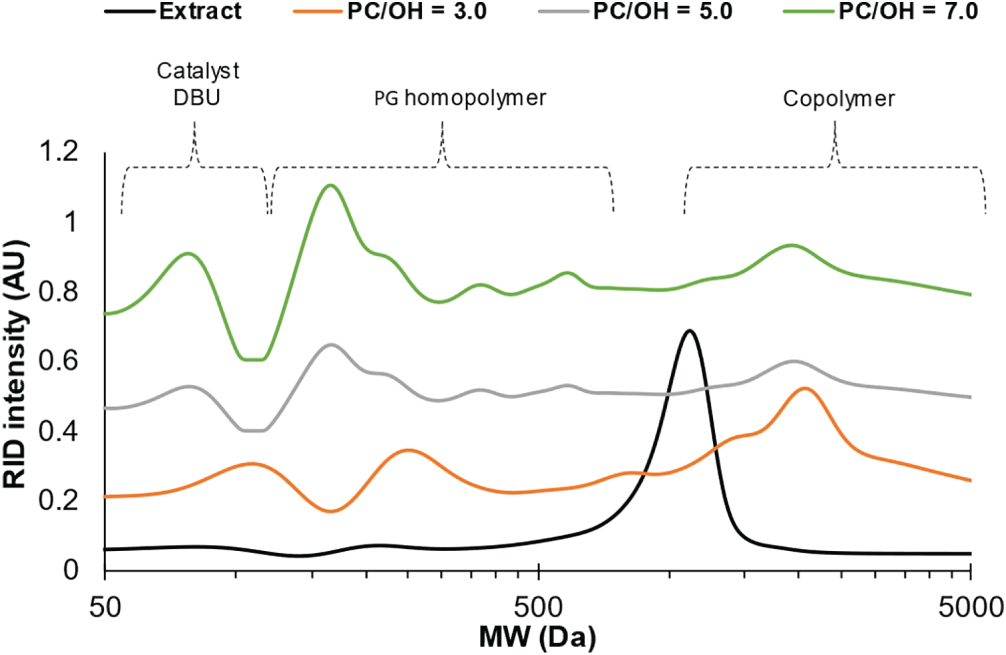
Figure 13: The GPC of BA bark extract and their based polyols synthesized at different PC/OH ratios
Together, the existence of lower molecular fractions is attributed to PG and their oligomeric forms obtained as the result of PC homopolymerization that could be initiated by traces of water avoid less presented in hydrophilic extractives, as well as removed in the results of partial biomass thermal dehydration (Fig. S5). The MMD characteristics of copolymer fractions of polyols obtained in different conditions as well as other the main characteristics of whole polyols are presented in Table 3.
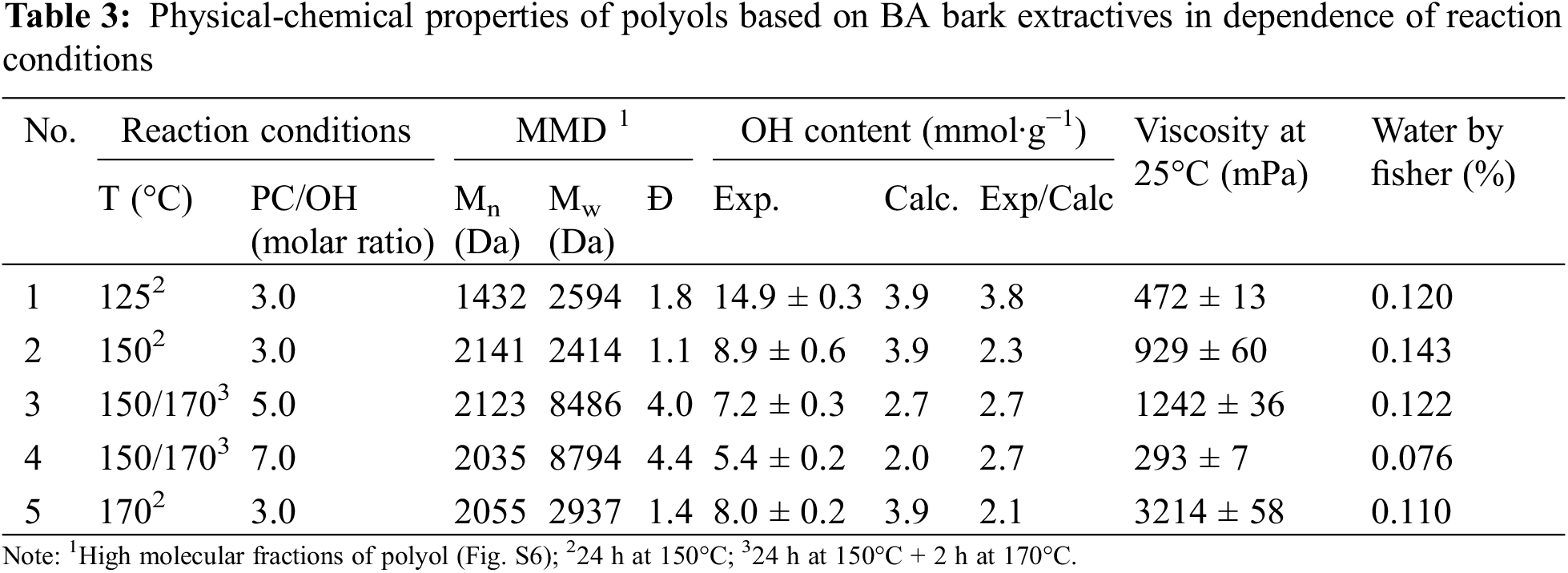
The experimentally determined OH content in polyols, ranging from 5.1 to 14.9 mmol∙g−1 (286–836 mgKOH∙g−1), showed a 2.1- to 3.8-fold increase compared to the calculated values, assuming that oxypropyl unit grafting proceeds only as a result of copolymerization with extractive components. These data, along with the experimentally verified increase in the molecular weight of copolymers compared to the initial extractives, clearly indicate that both the attachment of OPU to the OH groups of extractives and the conversion of propylene carbonate (PC) through homopolymerization, resulting in the formation of propylene glycol (PG) and its oligomers, occurred in the reaction of extractives with PC (Table 3). Therefore, the obtained polyols can be defined as a blend of relatively high molecular weight copolymer fractions of extractives compounds with attached OPU and lower molecular weight oligo oxypropyl diols with small number of carbonate linkages in the chain, both enriched with aliphatic OH groups reactive with isocyanate [19]. It should be noted that the presence of PG oligomers helps decrease the viscosity of polyols and allows for the enhancement of the OH content in polyols to the desired concentration.
The Mn values of the high molecular fractions were shown to increase approximately twofold compared to the initial extracts and remained similar for all compositions synthesized at 150°C and 170°C (Table 3). The most uniform MMD (Đ = 1.1) of the copolymer fraction was determined in the polyol synthesized at 150°C with a PC/OH ratio of 3.0. The significant increase in dispersity with the increasing amount of PC in the compositions can be explained by the appearance of a small amount of high molecular fractions. This explanation is available because the Mw value is more sensitive to the molecular mass of long-chain molecules than to their number, while the Mn of the copolymer fractions remains very close. Probably, the formation of such high molecular fractions can be attributed to the presence of carbonate linkages in polyols synthesized at the high PC/OH ratios (Fig. 12B). As a result, the coupling reaction via the interaction of the carbonate anion with OH-terminated chains proceeds [32]. The increase in temperature from 150°C to 170°C at the fixed PC/OH ratio equal to 3 led to a less significant increase in the Mw of the copolymer (Table 3).
Based on the Mn values of high molecular fractions and initial extractives, along with the OH group content in them and the amount of PC used, the average length of grafting and the composition of polyol in terms of copolymer and homopolymer content were calculated. It was shown that, on average and independent of the amount of PC, only about one OPU per OH group of extractives was attached as a result of the reaction performed at 150°C–170°C (Table 4). Therefore, we can conclude that the grafting of one OPU to the OH groups of extractives, resulting in the transformation of acidic groups into aliphatic ones, is followed by competitive reactions forming oligo oxypropyl diols. These reactions become dominant, leading to the complete conversion of PC present in the reaction mixture.

This explanation is approved by the results of model compounds and water interaction with PC presented in Section 3.2 and coincides with the data presented in the manuscript by Zhang et al. [30].
At 125°C, the number of grafts was only 0.34 OPU per OH group, and the remaining PC was converted into homopolymers via the oxypropylation mechanism.
Correspondingly, the maximum portions of copolymer were calculated for polyols synthesized at the PC/OH ratio of 3.0. The highest biomass content in the resulting polyol, 26.9%, was also achieved at this ratio, decreasing continuously to 14.2% as the PC/OH ratio increased to 7.0 (Table 3).
Viscosity is one of the important parameters of polyols used for PU synthesis [45]. The low viscosity is a technological advantage of polyols that expands the area of their practical application. As shown by Vieira et al. [16]. Increasing the reaction temperature and decreasing the amount of PC promote an increase in the viscosity of polyols obtained by the oxypropylation of lignin with PC. In our opinion, the relative portion of copolymer and its Mw are the major factors that have a rather complex influence on the viscosity of polyols. In our case, the viscosity of polyols synthesized at 170°C is significantly higher than those synthesized at 150°C (Table 3). This can be explained by the higher Mw value of the copolymer fraction in polyol No. 5 compared to that of No. 2, despite both having a similar content of homopolymers in composition. (Tables 3 and 4). The lowest viscosity was determined for the polyol synthesized at a PC/OH ratio of 7.0. We propose that in this case, the high content of oligomeric oxypropyl diols was the dominant factor, outweighing the Mw value of the copolymers due to its low content (25.9%) in the polyol. Together with the increasing copolymer content in polyol up to 35%, its higher Mw became the dominant factor responsible for the higher viscosity of composition No. 3 compared to that of No. 2 (Tables 3 and 4).
The results presented in the manuscript indicates that BA bark extractives were successfully converted into the liquid polyol polyether with OHV = 286–500 mg KOH∙g−1 and viscosity of 593–3200 mPa∙s−1 by reactions with PC performed at temperatures of 150°C–170°C and PC/OH ratio in the range of 3–7. These characteristics meet the requirements of commercial polyether polyols with variable OHV and viscosity values in the ranges of 300–800 mg KOH∙g−1 and 300–30000 mPa∙s−1 respectively which are used for rigid PU foam producing [45].
Was established that the PC/OH ratio equal to 3.0 is optimal in terms of the highest content of biomass in polyol and its properties. In this case, the reaction was performed at a moderate temperature of 150°C for 24 h and a low viscous polyol (929 mPa∙s−1) was obtained in the result (Table 3). Increasing of temperature up to 170°C allowed us to shorten the duration of the reaction to 5 h but the viscosity of the polyol obtained was three times higher (Fig. 11A; Table 3). However, it is about 1.5 times lower in comparison with that of polyols obtained by oxypropylation of lignin at a similar content of biomass [16].
After 24 h of reaction at 150°C with a higher amount of PC, an additional 2-h heating at 170°C was necessary to complete the conversion of PC.
Besides copolymer and homopolymer, DBU is another important component of the polyols obtained. The presence of tertiary amine, which can be used repeatedly for catalysis of polyol interaction with isocyanate, can be considered as a favorable factor from a technological point of view because simplify the preparation of A system. However, the inspection of polyol activity in the composition of PUR foam is necessary. The polyols obtained are under testing as substituents of commercial polyol polyether Lupranol 3300 (oxypropylated glycerol) in compositions of rigid PU foams.
Finally, it should be mentioned that the experiments performed indicate that the alternative process of biomass oxypropylation using cyclic carbonate is significantly longer compared to conventional oxypropylation of biomass. Using PO and a high-pressure reactor only a few minutes were needed for the conversion of solid lignin into liquid polyols suitable for PU synthesis [19]. We can conclude that the increased duration of the process using cyclic carbonates as an alternative oxyalkylation agent is a reasonable trade-off for using a safer and more environmentally friendly technology. The possible speed-up of processing by increasing temperature will decrease the desirable safety of technology.
The solid hydrophilic extractives isolated through short-time and low-energy water extraction of BA bark were transformed into liquid green polyol polyethers using 8-diazabicyclo[5.4.0]undec-7-ene as a basic catalyst and propylene carbonate as an alternative oxyalkylation agent instead of environmentally harmful propylene oxide.
The composition of the extractives, predominantly consisting of diarylheptanoids and hemicelluloses, determines their functionality. The phenolic and aliphatic OH groups, originating from catechol units and carbohydrates, respectively, were present in the extractives in a 1:1.3 ratio, with small admixtures of COOH groups, resulting in a total OH group concentration of 14.9 mmol∙g−1.
Research on the reaction of model compounds, including catechol, xylose, benzoic acid, and PEG-400 with propylene carbonate demonstrated that all functional groups of the extracts undergo condensation with PC. It was shown that, at 150°C–170°C, the extractive compounds reacted with an equivalent amount of PC via an etherification mechanism, producing, a copolymer with on average, close to one oxypropyl unit grafting independently of the initial PC/OH ratio in the reaction blend. The remaining PC was transformed into oligo oxypropylene diols, containing a negligible amount of carbonate linkages in the chain, and acted as an active co-reagent in the extractive-based polyols while simultaneously reducing the viscosity of the polyols.
The PC/OH ratio of 3, a temperature of 150°C, and a reaction duration of 24 h can be considered optimal conditions. These conditions allow for the synthesis of a polyol with approximately 26.9% biomass content, a low viscosity of 930 mPa∙s at 25°C, and an OHV of 500 mg KOH∙g−1, meeting the requirements for commercial polyols use in the production of rigid PU foam. Increasing the synthesis temperature to 170°C shortens the reaction but increases the viscosity of the polyol threefold to 3200 mPa∙s, slightly decreasing the OHV values. Increasing the PC/OH ratio above 3 decreases the biomass content in the polyols and requires more time to achieve complete conversion of PC.
Acknowledgement: The financial support from the Latvian Budget—the State Research Program No. lzp-2021/1–0207 and the comprehensive study of the viscosity by Dr. Maris Lauberts from Latvian State Institute of Wood Chemistry are gratefully acknowledged.
Funding Statement: The article was published with financial support from the Latvian Council of Science, Project No. lzp-2021/1-0207.
Author Contributions: The authors confirm their contribution to the paper as follows: study conception and design: Alexandr Arshanitsa; data collection: Matiss Pals, Daniela Godina, Oskars Bikovens; analysis and interpretation of results: Alexandr Arshanitsa, Matiss Pals; draft manuscript preparation: Alexandr Arshanitsa, Matiss Pals. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The data presented in this study are available on request from the corresponding authors.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Supplementary Materials: The supplementary material is available online at https://doi.org/10.32604/jrm.2024.056466.
References
1. Statista. Market volume of polyurethane worldwide from 2015 to 2025, with a forecast for 2022 to 2029. Available from: https://www.statista.com/statistics/720341/global-polyurethane-market-size-forecast/. [Accessed 2023]. [Google Scholar]
2. NOVA. European bioplastics. 2023. Available from: https://www.european-bioplastics.org/tag/nova-institut/. [Accessed 2023]. [Google Scholar]
3. Coppola G, Gaudio MT, Lopresto CG, Calabro V, Curcio S, Chakraborty S. Bioplastic from renewable biomass: a facile solution for a greener environment. Earth Syst Environ. 2021 Jun 29;5(2):231–51. doi:10.1007/s41748-021-00208-7. [Google Scholar] [CrossRef]
4. Phung Hai TA, Tessman M, Neelakantan N, Samoylov AA, Ito Y, Rajput BS, et al. Renewable polyurethanes from sustainable biological precursors. Biomacromolecules. 2021 May 10;22(5):1770–94. doi:10.1021/acs.biomac.0c01610. [Google Scholar] [PubMed] [CrossRef]
5. Ma Y, Xiao Y, Zhao Y, Bei Y, Hu L, Zhou Y, et al. Biomass based polyols and biomass based polyurethane materials as a route towards sustainability. React Funct Polym. 2022 Jun;175:105285. doi:10.1016/j.reactfunctpolym.2022.105285. [Google Scholar] [CrossRef]
6. Gholami M, Shakeri A, Zolghadr M, Yamini G. Non-isocyanate polyurethane from the extracted tannin of sumac leaves: synthesis, characterization, and optimization of the reaction parameters. Ind Crops Prod. 2021 Mar;161:113195. doi:10.1016/j.indcrop.2020.113195. [Google Scholar] [CrossRef]
7. Kathalewar MS, Joshi PB, Sabnis AS, Malshe VC. Non-isocyanate polyurethanes: from chemistry to applications. RSC Adv. 2013;3(13):4110. doi:10.1039/c2ra21938g. [Google Scholar] [CrossRef]
8. Aristri MA, Lubis MAR, Yadav SM, Antov P, Papadopoulos AN, Pizzi A, et al. Recent developments in lignin- and tannin-based non-isocyanate polyurethane resins for wood adhesives—a review. Appl Sci. 2021 May 7;11(9):4242. doi:10.3390/app11094242. [Google Scholar] [CrossRef]
9. Rubens M, Wesemael MV, Feghali E, Lufungula LL, Blockhuys F, Vanbroekhoven K, et al. Exploring the reactivity of aliphatic and phenolic hydroxyl groups in lignin hydrogenolysis oil towards urethane bond formation. Ind Crops Prod. 2022 Jun;180:114703. doi:10.1016/j.indcrop.2022.114703. [Google Scholar] [CrossRef]
10. Ajao O, Benali M, Faye A, Li H, Maillard D, Ton-That MT. Multi-product biorefinery system for wood-barks valorization into tannins extracts, lignin-based polyurethane foam and cellulose-based composites: techno-economic evaluation. Ind Crops Prod. 2021 Sep;167:113435. doi:10.1016/j.indcrop.2021.113435. [Google Scholar] [CrossRef]
11. Arshanitsa A, Ponomarenko J, Pals M, Jashina L, Lauberts M. Impact of bark-sourced building blocks as substitutes for fossil-derived polyols on the structural, thermal, and mechanical properties of polyurethane networks. Polymers. 2023 Aug 22;15(17):3503. doi:10.3390/polym15173503. [Google Scholar] [PubMed] [CrossRef]
12. Pals M, Lauberte L, Ponomarenko J, Lauberts M, Arshanitsa A. Microwave-assisted water extraction of aspen (Populus tremula) and pine (Pinus sylvestris L.) barks as a tool for their valorization. Plants. 2022 Jun 9;11(12):1544. doi:10.3390/plants11121544. [Google Scholar] [PubMed] [CrossRef]
13. Md Salim R, Asik J, Sarjadi MS. Chemical functional groups of extractives, cellulose and lignin extracted from native Leucaena leucocephala bark. Wood Sci Technol. 2021 Mar 23;55(2):295–313. doi:10.1007/s00226-020-01258-2. [Google Scholar] [CrossRef]
14. Alinejad M, Henry C, Nikafshar S, Gondaliya A, Bagheri S, Chen N, et al. Lignin-based polyurethanes: opportunities for bio-based foams, elastomers, coatings and adhesives. Polymers. 2019 Jul 18;11(7):1202. doi:10.3390/polym11071202. [Google Scholar] [PubMed] [CrossRef]
15. Arshanitsa A, Ponomarenko J, Pals M, Jashina L. Controlling the reactivity of hydrophilic bark extractives as biopolyols in urethane-formation reactions using various catalysts. Ind Crops Prod. 2023 Nov;204:117385. doi:10.1016/j.indcrop.2023.117385. [Google Scholar] [CrossRef]
16. Vieira FR, Magina S, Evtuguin DV, Barros-Timmons A. Lignin as a renewable building block for sustainable polyurethanes. Materials. 2022 Sep 5;15(17):6182. doi:10.3390/ma15176182. [Google Scholar] [PubMed] [CrossRef]
17. Kelley SS, Glasser WG, Ward TC. Engineering plastics from lignin XIV. Characterization of chain-extended hydroxypropyl lignins. J Wood Chem Technol. 1988 Sep;8(3):341–59. doi:10.1080/02773818808070689. [Google Scholar] [CrossRef]
18. Oliveira WD, Glasser WG. Engineering plastics from lignin. XVI. Starlike macromers with propylene oxide. J Appl Polym Sci. 1989 Jun 5;37(11):3119–35. doi:10.1002/app.07.v37:11. [Google Scholar] [CrossRef]
19. Cateto CA, Barreiro MF, Rodrigues AE, Belgacem MN. Optimization study of lignin oxypropylation in view of the preparation of polyurethane rigid foams. Ind Eng Chem Res. 2009 Mar 4;48(5):2583–9. doi:10.1021/ie801251r. [Google Scholar] [CrossRef]
20. Li Y, Ragauskas AJ. Kraft lignin-based rigid polyurethane foam. J Wood Chem Technol. 2012 Jul;32(3):210–24. doi:10.1080/02773813.2011.652795. [Google Scholar] [CrossRef]
21. García DE, Glasser WG, Pizzi A, Paczkowski S, Laborie MP. Hydroxypropyl tannin from Pinus pinaster bark as polyol source in urethane chemistry. Eur Polym J. 2015 Jun;67:152–65. doi:10.1016/j.eurpolymj.2015.03.039. [Google Scholar] [CrossRef]
22. D’Souza J, Camargo R, Yan N. Polyurethane foams made from liquefied bark-based polyols. J Appl Polym Sci. 2014 Aug 15;131(16). doi:10.1002/app.40599. [Google Scholar] [CrossRef]
23. Evtiouguina M, Barros-Timmons A, Cruz-Pinto JJ, Neto CP, Belgacem MN, Gandini A. Oxypropylation of cork and the use of the ensuing polyols in polyurethane formulations. Biomacromolecules. 2002 Jan 1;3(1):57–62. doi:10.1021/bm010100c. [Google Scholar] [PubMed] [CrossRef]
24. Kühnel I, Saake B, Lehnen R. Comparison of different cyclic organic carbonates in the oxyalkylation of various types of lignin. React Funct Polym. 2017 Nov;120:83–91. doi:10.1016/j.reactfunctpolym.2017.09.011. [Google Scholar] [CrossRef]
25. Clements JH. Reactive applications of cyclic alkylene carbonates. Ind Eng Chem Res. 2003 Feb 1;42(4):663–74. doi:10.1021/ie020678i. [Google Scholar] [CrossRef]
26. Taherimehr M, Pescarmona PP. Green polycarbonates prepared by the copolymerization of CO2 with epoxides. J Appl Polym Sci. 2014 Nov 5;131(21):1–17. [Google Scholar]
27. Kühnel I, Saake B, Lehnen R. Oxyalkylation of lignin with propylene carbonate: influence of reaction parameters on the ensuing bio-based polyols. Ind Crops Prod. 2017 Jul;101:75–83. doi:10.1016/j.indcrop.2017.03.002. [Google Scholar] [CrossRef]
28. Kühnel I, Podschun J, Saake B, Lehnen R. Synthesis of lignin polyols via oxyalkylation with propylene carbonate. Holzforschung. 2015 Jul 1;69(5):531–8. doi:10.1515/hf-2014-0068. [Google Scholar] [CrossRef]
29. Vieira FR, Barros-Timmons A, Evtuguin DV, Pinto PCR. Effect of different catalysts on the oxyalkylation of eucalyptus Lignoboost® kraft lignin. Holzforschung. 2020 Jun 25;74(6):567–76. doi:10.1515/hf-2019-0274. [Google Scholar] [CrossRef]
30. Zhang X, Kim Y, Elsayed I, Taylor M, Eberhardt TL, Hassan EB, et al. Rigid polyurethane foams containing lignin oxyalkylated with ethylene carbonate and polyethylene glycol. Ind Crops Prod. 2019 Dec;141:111797. doi:10.1016/j.indcrop.2019.111797. [Google Scholar] [CrossRef]
31. Lee JC, Litt MH. Ring-opening polymerization of ethylene carbonate and depolymerization of poly(ethylene oxide-co-ethylene carbonate). Macromolecules. 2000 Mar 1;33(5):1618–27. doi:10.1021/ma9914321. [Google Scholar] [CrossRef]
32. Duval A, Avérous L. Oxyalkylation of condensed tannin with propylene carbonate as an alternative to propylene oxide. ACS Sustain Chem Eng. 2016 Jun 6;4(6):3103–12. doi:10.1021/acssuschemeng.6b00081. [Google Scholar] [CrossRef]
33. Harris RF. Molecular weight advancement of poly(ethylene ether carbonate) polyols. J Appl Polym Sci. 1989 Aug 5;38(3):463–76. doi:10.1002/app.07.v38:3. [Google Scholar] [CrossRef]
34. Kéki S, Török J, Deák G, Zsuga M. Mechanism of the anionic ring-opening oligomerization of propylene carbonate initiated by the tert-Butylphenol/KHCO3 system. Macromol Symp. 2004 Aug 27;215(1):141–50. doi:10.1002/masy.v215:1. [Google Scholar] [CrossRef]
35. Salca EA. Black alder (Alnus glutinosa L.)—a resource for value-added products in furniture industry under european screening. Curr For Rep. 2019 Mar 26;5(1):41–54. doi:10.1007/s40725-019-00086-3. [Google Scholar] [CrossRef]
36. Smeriglio A, D’Angelo V, Cacciola A, Ingegneri M, Raimondo FM, Trombetta D, et al. New insights on phytochemical features and biological properties of alnus glutinosa stem bark. Plants. 2022 Sep 23;11(19):2499. doi:10.3390/plants11192499. [Google Scholar] [PubMed] [CrossRef]
37. Arshanitsa A, Ponomarenko J, Lauberte L, Jurkjane V, Pals M, Akishin Y, et al. Advantages of MW-assisted water extraction, combined with steam explosion, of black alder bark in terms of isolating valuable compounds and energy efficiency. Ind Crops Prod. 2022 Jul;181:114832. doi:10.1016/j.indcrop.2022.114832. [Google Scholar] [CrossRef]
38. Singleton VL, Orthofer R, Lamuela-Raventós RM. Analysis of total phenols and other oxidation substrates and antioxidants by means of folin-ciocalteu reagent. In: Methods in Enzymology. USA: Academic Press; 1999. vol. 299, p. 152–78. doi:10.1016/S0076-6879(99)99017-1. [Google Scholar] [CrossRef]
39. Blakeney AB, Harris PJ, Henry RJ, Stone BA. A simple and rapid preparation of alditol acetates for monosaccharide analysis. Carbohydr Res. 1983 Mar;113(2):291–9. doi:10.1016/0008-6215(83)88244-5. [Google Scholar] [CrossRef]
40. Argyropoulos DS, Pajer N, Crestini C. Quantitative 31P NMR analysis of lignins and tannins. J Vis Exp. 2021 Aug 2;174:1–21. [Google Scholar]
41. Zakis GF. Functional analysis of lignins and their derivatives. Atlanta, GA, USA: TAPPI Press; 1994. [Google Scholar]
42. Godina D, Makars R, Paze A, Rizhikovs J. Analytical method cluster development for comprehensive characterisation of suberinic acids derived from birch outer bark. Molecules. 2023 Feb 27;28(5):2227. doi:10.3390/molecules28052227. [Google Scholar] [PubMed] [CrossRef]
43. Ganapathy G, Preethi R, Moses JA, Anandharamakrishnan C. Diarylheptanoids as nutraceutical: a review. Biocatal Agric Biotechnol. 2019 May;9:101109. [Google Scholar]
44. Schweigert N, Zehnder AJB, Eggen RIL. Chemical properties of catechols and their molecular modes of toxic action in cells, from microorganisms to mammals. Environ Microbiol. 2001 Feb 20;3(2):81–91. doi:10.1046/j.1462-2920.2001.00176.x. [Google Scholar] [PubMed] [CrossRef]
45. Ionescu M. Chemistry and technology of polyols for polyurethanes. Shriopshire: Rapra Technology; 2005. vol. 1. [Google Scholar]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools