
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE
Cross-Linking of Sago Starch with Furan and Bismaleimide via the Diels-Alder Reaction
Department of Chemical Engineering, Parahyangan Catholic University, Bandung, 40141, Indonesia
* Corresponding Author: Henky Muljana. Email:
Journal of Renewable Materials 2023, 11(12), 4039-4060. https://doi.org/10.32604/jrm.2023.031261
Received 26 May 2023; Accepted 07 August 2023; Issue published 10 November 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
This research paper describes the synthesis of thermo-reversible cross-linking of sago starch by grafting a furan pendant group (methyl 2-furoate) onto the starch backbone, followed by a Diels-Alder (DA) reaction of the furan functional group with 1,1′-(methylenedi-4,1-phenylene) bismaleimide (BM). The proof of principles was provided by FTIR and 1H-NMR analyses. The relevant FTIR peaks are the carbonyl peak (υ C=O sym) at 1721 cm−1 ; the two peaks appeared after DA cross-linking, i.e., at 1510 cm−1 (corresponding to υ CH=CH BM aromatic rings, stretching vibrations), and at 1173 cm−1 (assigned to cycloadduct (C-O-C, δ DA ring)) while the 1H-NMR result shows evidence for the presence of a furan ring in the starch matrices (in the range of δ 6.3–7.5 ppm). The crosslinked starch product is indeed thermally reversible, as is evident from the appearance of exothermal (DA, temperature range of 50°C–70°C) and endothermal (retro DA, temperature range of 125°C–150°C) transitions in the DSC thermograms. This paper not only proves the thermal reversibility but also demonstrates that the final product properties (chemical, morphology, and thermal stability) can be tuned by varying the annealing temperature, BM intake, and reaction time.Graphic Abstract
Keywords
The incentive to synthesize bioplastics from various polysaccharide resources such as cellulose, starch, and chitosan has arisen over the last decade [1–7]. This trend has been triggered by the drawback of most conventional plastics properties, such as being non-biodegradable and non-recyclable, for both contribute to an increase in the accumulation of worldwide plastics waste that can be harmful to the environment. As reported in 2021 by United Nations Environment Program (UNEP), 400 million tonnes (metric tons) of plastic waste are produced annually; however, only less than 10% has been recycled while the rest of the plastic waste ends up in the environment. In addition, around 75–190 million tonnes of plastic waste are found in the ocean, creating unwanted pollution problems that are harmful to the ocean [8].
Starch, one of the most abundant polysaccharides, has been chemically modified to improve its physical and chemical properties so it can be further applied as an eco-friendly adsorbent, bioplastics including the application of modified starch as natural antibacterial materials [3,5,9–13]. Among other starch chemical modification methods, cross-linking has been employed to the starch matrices using various cross-linkers such as phosphate salts, epichlorohydrin (EPI), malonic acid, adipic acid, and acetic anhydride, vinyl chloride, citric acid, glutaraldehyde, and phosphorus oxychloride [4,11,14] resulting in higher mechanical strength (tensile strength), an increase in the resistance to heat and chemicals, and lowering the water affinity of modified products compared with the ones of the native starch properties [1,4,7,9,10,14,15]. Despite these advantages, it has also been known that after cross-linking, the materials become thermoset [4]. Thus, the possibility of recycling or reusing the cross-linked products will also be reduced [1,15].
A thermo-reversible network can be introduced into the polymer matrices via Diels-Alder (DA) chemistry to increase the recyclability of the cross-linked products. A successful example is the DA reaction between furan pendant (as a diene) and bismaleimide (as a dienophile) that has been employed in various polymeric systems [16,17], including starch matrices [1,2,14,15]. The reversibility of the DA adducts between furan and bismaleimide in the polymer network can be tuned by simply changing the processing temperature (thermo-reversible). In most cases, the cross-linking (DA) between the furan pendant group and bismaleimide occurs at a temperature range of 40°C–80°C, while the de-cross-linking (retro DA) starts at a higher temperature range (120°C–150°C) [1,17,18]. Because of the tunable cross-linking network of furan-containing polymeric materials and bismaleimide with temperature, this product has enormous potential for use in a wide range of applications such as self-healing materials (coatings, adhesives), recyclable thermoset materials (rubber, polyketone), and various biomedical applications such as biomolecular immobilization, drug delivery/release, and tissue engineering [19–22].
Moreover, compared with the amount of reported research into DA thermo-reversible cross-linking in many polymer systems, not all that much research has actually been conducted on starch or its modified products, and to the best of our knowledge, only the research studies of Antunes et al. [14] and Nossa et al. [15] have been reported in the openly available literature. Nossa and co-workers reported the synthesis of thermo-reversible cross-linking starch using corn starch, furfuryl alcohol (FA), 4,4-methylene diphenyl diisocyanate (MDI), and 1,1-(methylenedi-4,1-phenylene) bismaleimide (BM) [15]. Their work employed three reaction steps: the grafting reaction between FA-MDI, starch hydroxyl via urethane linkage to form an intermediate product (starch-g-FA-MDI), and the DA reaction between starch-g-FA-MDI with BM [15]. Furthermore, Antunes and co-workers used a different reaction route to synthesize thermo-reversible cross-linking of starch compared with the one by Nossa where starch was first oxidized with (2,2,6,6-tetramethylpiperidine-1-yl)oxyl (TEMPO), followed with the esterification of the oxidized starch with FA, and finally the cross-linking with self-made water-soluble BM at neutral pH and at body temperature. The final product was claimed to be a promising candidate for cell encapsulation and delivery within the body [14].
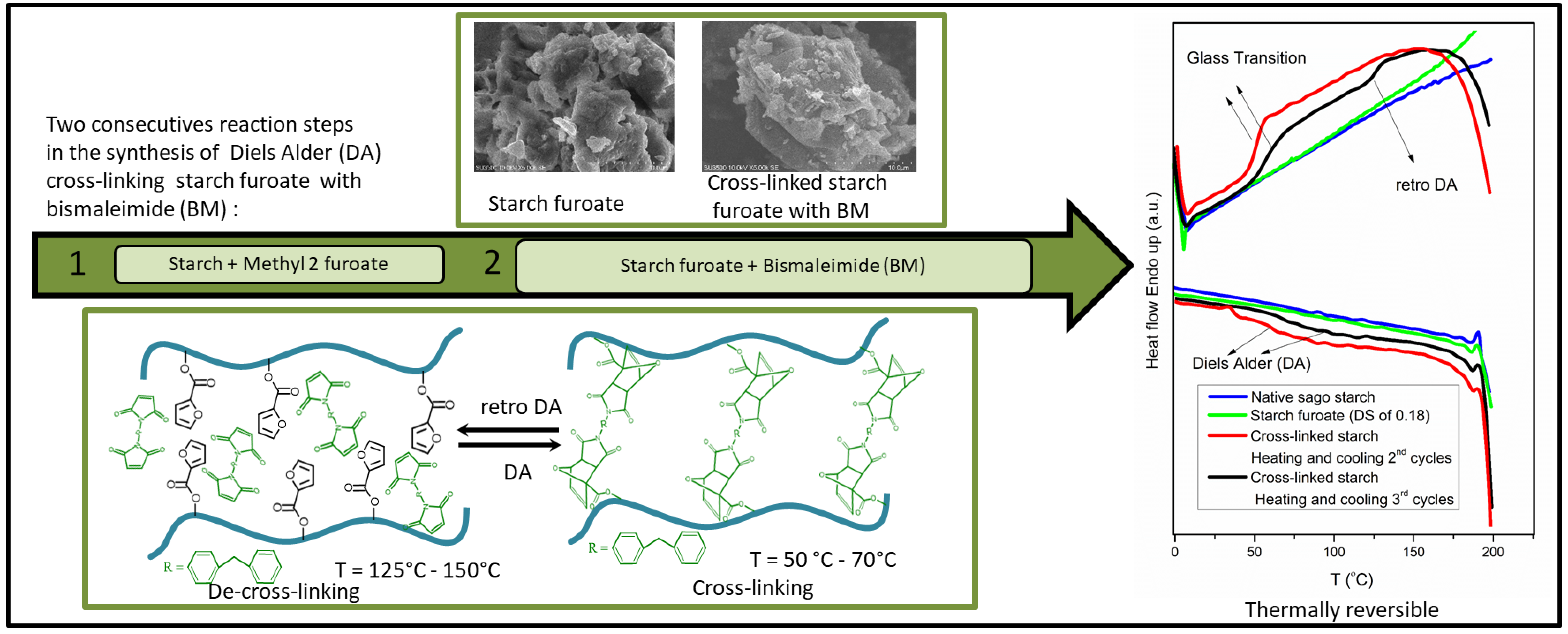
The pioneering work of Nossa et al. [15] followed by the promising results of thermo-reversible starch on the biomedical application as reported by Antunes et al. [14] not only shows the potential application of DA chemistry in the thermo-reversible cross-linking of starch synthesis (involving furan and BM) but also gives more incentive to explore another possible reaction pathway to graft furan pendant onto starch matrices and further cross-linking it with BM. Despite the potential applications of DA chemistry demonstrated by Nossa et al. [15] and Antunes et al. [14], the reaction route used in their work to produce thermo-reversible cross-linking of starch required three reaction steps (see above) and a relatively long reaction time (18 h–10 days) [14,15]. Therefore, in this research, we intend to study the DA cross-linking of starch that proceeds via a different route from the one reported in the literature, which involves fewer reaction steps and, as a result, a faster reaction time. The novel pathway consists of two consecutive reactions steps which are the transesterification of starch with methyl-2-furoate (MF) followed by the DA reaction with BM (see Scheme 1).

Scheme 1: Possible reaction scheme of transesterification of starch (a) with methyl-2-furoate and (b) Diels-Alder reaction of the starch ester (starch furoate) with BM (c). Depending on the DS values, the furoate functional group may be present in the primary or secondary alcohol of the anhydroglucose unit (AGU)
This research investigates the possibility of synthesizing thermo-reversible cross-linking of sago starch using MF and BM as the reagent (Scheme 1). Here, a different type of starch compared with the ones reported by Antunnes et al. and Nossa et al. [14,15] was used. This research provides proof of principle derived from the successful reaction. In addition, an in-depth study was made to evaluate the influence of several essential process parameters (such as catalyst intake, reagent intake, reaction time, and temperature) on the degree of substitution, the degree of cross-linking, and the relevant properties (chemical, morphology, thermal stability, and reversibility) of the final product. The latter were evaluated to gain insight into the relationship between the process and product properties of the novel product.
Sago starch (starch content of 84.6% wt/wt, water content of 16.9% wt/wt) was purchased from Bina Sago Lestari (Jakarta, Indonesia). The estimated amylose and amylopection content of typical Indonesia sago starch are 27% wt/wt and 73% wt/wt, respectively [23]. Analytical grade methyl 2-furoate (MF, >98%) was purchased from Alfa Aesar (Shanghai, China). Analytical grade 1,1′-(methylenedi-4,1-phenylene) bismaleimide (BM, 95%), dimethyl sulfoxide (DMSO, >99.5%), chloroform (CHCl3, >99%), and potassium carbonate (K2CO3, >99%), hydrochloric acid (HCl, 37%), and sodium hydroxide (NaOH, >97%) were purchased from Sigma Aldrich (Singapore).
2.2.1 Functionalization of Sago Starch with Methyl 2-Furoate
Sago starch (3 g) and DMSO (30 mL) were charged inside the 100 ml round bottom flask. The mixture was dissolved at 80°C (for approximately one hour) until a homogeneous and transparent mixture was formed. Afterward, methyl 2-furoate (0.3 mol/mol Anhydroglucose Unit, AGU−4 mol/mol AGU) and potassium carbonate (K2CO3) as catalyst (0.1 mol/mol AGU–0.4 mol/mol AGU) were added into the homogeneous mixture and stirred at 120°C for six hours. The mixture was cooled until room temperature was reached and precipitated using methanol (100 ml). The solid product was filtrated and washed with methanol (100 mL) and reverse osmosis (RO) water (100 ml). The final product was dried in a vacuum oven (70°C) to constant weight.
2.2.2 Bismaleimide Crosslinking with Furan Functionalized Sago Starch Furoate
Sago starch furoate (1 g) and BM (0.5–2 mol/mol AGU) were mixed with CHCl3 (20 g) in a round bottom flask (100 mL). The mixture was heated at 50°C in a water bath and the reaction took place for three or six hours (depending on the experimental condition). Subsequently, the method of slow evaporation of CHCl3 was employed inside the fume cabinet for 24 h to separate the solvent from the solid product. The solid product was subsequently annealed at a temperature range of 50°C–150°C for 24 h.
2.2.3 Determination of Degree of Substitution (DS)
The DS values were determined based on the procedure that has been described in the literature for typical starch ester product with slight modifications [24]. The starch furoate samples (1 g) was hydrolyzed with NaOH (0.5 M, 15 mL) in a shaker water-bath (at temperature of 30°C for 4 h), and the excess of the alkali was neutralized (pH = 7) with HCl (0.1 M). The DS value was calculated using the following Eq. (1).
where: VNaOH = volume of NaOH (ml), VHCl = volume of HCl required to neutralize excess alkali, MNaOH = concentration of NaOH (M), MHCl = concentration of HCl (M), W = weight of starch furoate sample (g), Mw = molecular weight of furoate ester = 111.07.
2.2.4 Determination of the Degree of Cross-Linking (DC)
The degree of cross-linking in the cross-linked starch products was qualitatively determined according to a previously reported procedure with a slight modification [25]. The cross-linked starch product (0.1 g, Wo)) was inserted into a round bottom flask and mixed with 10 ml of DMSO inside the flask. Subsequently, the mixture was heated in a water bath at 80°C for 24 h to dissolve the soluble part of the cross-linked products. After that, the insoluble starch samples were filtrated and dried in a vacuum oven (70°C) to constant weight. The weight of the insoluble sample was subsequently measured (denoted as W1). The degree of cross-linking was calculated by using the following equation:
where: DC = degree of cross-linking, Wo = 0.1 g, the amount of cross-linked starch product, W1 = the amount of insoluble starch cross-linked product.
Fourier transform infrared (FTIR) measurements were taken using an FTIR Prestige 21 Shimadzu (Kyoto, Japan). The spectra were acquired using potassium bromide (KBR) pellets in the absorption range of 4000 to 400 cm−1 a resolution of 4 cm−1 and scan number of 50. The deconvolution was applied on all spectra to calculate the intensity change of the relevant peaks. PeakFit software was used for deconvolution, with Savitzky Golay filtering and Gaussian fitting. The number of fitting curves was chosen at a smoothing percentage (%Sm) such that the R2 reach the minimum of 0.95 Proton nuclear magnetic resonance (1H-NMR) spectra were acquired using an Agilent NMR 500 MHz (Santa Clara, CA, USA) equipped with DD2 console system spectrometer operating at room temperature with 64 scans and a relaxation time of 1 s. 1H-NMR samples were dissolved in DMSO-d6 at 80°C and immediately inserted inside the 1H-NMR tube for direct measurement.
The thermal properties of starch furoate and the cross-linked products were investigated using thermal gravimetry analysis (TGA) and differential scanning calorimetry (DSC). TGA thermograms were obtained using a Hitachi STA 7300 (Hitachi, Japan). TGA samples (10 mg) were heated with a heating rate of 10°C min−1 to 900°C in a nitrogen atmosphere. Furthermore, DSC was measured on a Netzsch 214 Polyma (Netzsch-Geratebau, GMBH, Germany) under a nitrogen atmosphere. The samples (10 mg) were placed inside a sealed aluminum pan. The DSC thermogram was acquired with two cycles and three cycles of heating from 0°C to 200°C (heating rate of 10°C/min) and cooling to 0°C (cooling rate of 10°C) for the starch ester product and cross-linked product, respectively. The first heating and cooling run was performed to erase the material thermal history. The DSC was employed to investigate the thermal reversibility of the cross-linked product.
X-ray diffraction measurements were taken on a Bruker D8 Advance XRD System (Bruker AXS, Germany). XRD data were acquired from 5° to 40° (2θ) with an angular scanning velocity of 1°/min and a measurement frequency of 1 s−1. Morphological changes of the native and modified starch (starch ester and cross-linked starch) were recorded using a Scanning Electron Microscope (SEM Hitachi SU 3500, Hitachi, Japan). The modified starch samples were dried according to the procedure from the experiments (Sections 2.2.1 and 2.2.2). The dried powdered/granule samples were mounted on aluminum specimen stubs with adhesive tape and sputtered with a layer of gold (20 to 30 nm) using a Sputter Coater (MC1000, Hitachi, Japan).
In this research, the thermo-reversible cross-linked starch was synthesized via two consecutive reaction steps: the functionalization of starch with MF through transesterification and the DA cross-linking with a furan pendant with BM (Scheme 1). A systematic study was conducted for both reaction steps to investigate the reactivity of both reactions as correlated with the degree of substitution (DS) for transesterification and the degree of cross-linking (DC) for the DA reaction. An overview of the experimental conditions for transesterification and DA reaction is provided in Tables 1 and 2, respectively. The discussion of the obtained experimental results, including proof of principles, is shown in the following sub-section.
3.1 Transesterification of Sago Starch with Methyl 2-Furoate (MF)
After transesterification with MF, the change in the chemical structure of native sago starch was verified by FTIR. Fig. 1 shows the FTIR spectra of the native sago starch, starch furoate (DS of 0.21), and cross-linked starch with BM within two different absorption ranges (500–2000 cm−1 and 2000–3500 cm−1).Typical spectra for native sago starch are shown in Fig. 1, where the broad OH stretching (υ OH, 3400–3402 cm−1), OH water bending (δ OH water, 1647 cm−1), CH2 bending (δ CH2, 1423 cm−1), COH bending (δ COH, 1365 cm−1) and CH stretching (υ CH) at 2921–2925 cm−1) are present [3,7,14,24].

Figure 1: FTIR spectra of native sago starch, starch furoate with DS of 0.21 and cross-linked starch products with DS of 0.61 in the absorption range of 2000–3500 cm−1 (a), and 500–2000 cm−1 (b)
By comparing the spectra of the starch ester product (Fig. 1) with the spectra of the native sago starch, additional peaks at 1721 and 1579 cm−1 which corresponds to the symmetric stretching from the carbonyl group (υ C=O sym) of the ester product and aromatic furan ring absorption (see Supplementary Materials, Fig. S1, the FTIR spectra of methyl-2 furoate), appears in the IR spectra of the starch furoate product [3,26,27]. The presence of the two additional peaks in the spectra of modified starch suggests that the transesterification of starch with the MF step was successful.
In addition, the changes in the chemical structure of starch after the insertion of furan pendant in the starch backbone were observed from the 1H-NMR spectra of native sago starch (Fig. 2a) and starch furoate (with DS of 0.18, Fig. 2b). It is evident that in both spectra, the typical peaks of starch protons (six protons, label: 1S–6S, see Figs. 2a and 2b) occurred in the region of δ 3.6–5.5 ppm, while additional peaks (three in number), especially in the range between δ 6.3–7.5 ppm appear in the starch furoate spectra (Fig. 2b). The latter is assigned to the furan ring’s three protons (α and β aromatic, label: 1F–3F, Fig. 2b) [28]. This finding agrees with the FTIR results and confirms the presence of the furan group in the starch matrices suggesting the successful functionalization of starch with the furan pendant reaction step.

Figure 2: 1H-NMR spectra of native sago starch (a) and sago starch furoate with DS of 0.18 (b)
3.1.1 The Effect of Methyl 2-Furoate (MF) Intake, Temperature, and Catalyst (K2CO3) Intake on the DS Values
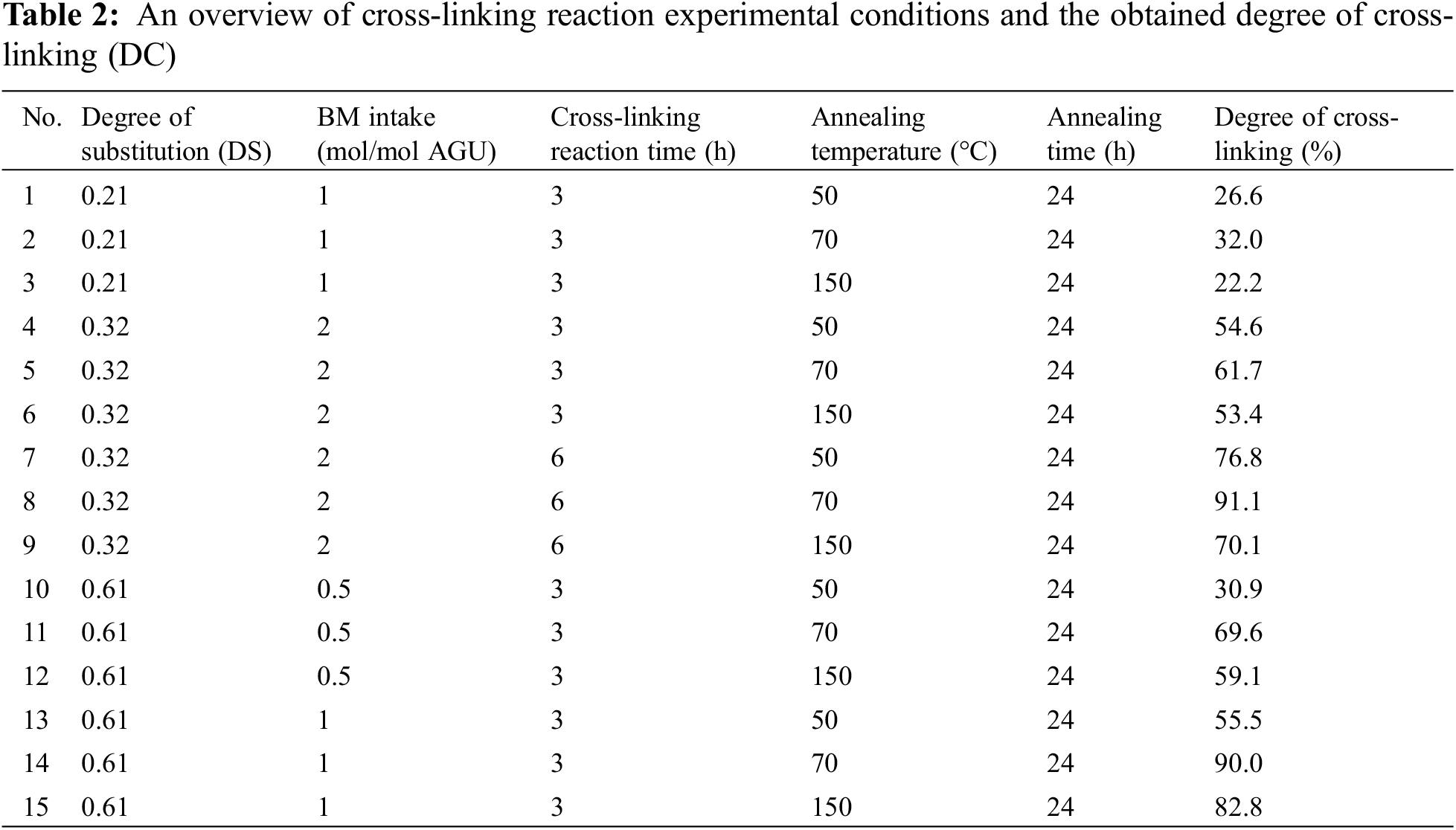
The significant influence of three reaction parameters (MF intake, reaction temperature, and K2CO3 intake) on the transesterification reaction was determined from the changes in DS values of the starch ester products (Figs. 3 and 4). The DS values obtained by varying the MF intake (0.3–4 mol/mol AGU) and reaction temperature (100°C and 120°C) are given in Table 1, while the trendlines are shown in Fig. 3. In all cases, the DS values increase with temperature from 0.055 to 0.07 for the reaction using an MF intake of 0.3 mol/mol AGU, from 0.064 to 0.11 for an MF intake of 2 mol/mol AGU and pronounced changes are observed at an MF intake of 4 mol/mol AGU where DS increases from 0.21 to 0.61 (Fig. 3).

Figure 3: DS values at different MF intakes and temperatures. The data presented in this figure were obtained from experiments using K2CO3 intake of 0.1 mol/mol AGU and reacted for six hours

Figure 4: DS values as a function of different K2CO3 intakes. The data presented in this figure were obtained from experiments using an MF intake of 2 mol/mol AGU, a temperature of 120°C and a reaction time of six hours
The positive effect of temperature on reactivity is expected as the reaction rate increases at higher temperatures, eventually leading to a higher DS value. This observation agrees with the one reported in the literature, although with different types of reagents such as fatty acid methyl and vinyl esters [29,30].
An increase in DS value with MF intake was also observed in Fig. 3. The maximum DS of 0.61 is accessible at the highest MF intake used in the experiments (MF intake of 4 mol/mol AGU). The results can be rationalized by the fact that an increase in MF intake not only increases the amount of reagent that may react with the hydroxyl group of sago starch to form the desired starch furoate, but it also enhances the reaction rate (similar to the effect of temperature) due to the higher concentration of the reagent in the vicinity of starch [24].
Thus far, the experimental results derived from MF intake and temperature variation show a positive effect on the DS values, while this is not the case for the K2CO3 intake results, as seen in Fig. 4. The role of the alkaline salt base catalyst on the reactivity may be explained with the proposed reaction scheme (see Supporting Information Scheme S1), where initially K2CO3 deprotonates the OH group of starch and produce an active starch alkoxide (see Scheme S1) [3]. Furthermore, the alkoxide reacts with methyl 2-furoate through the nucleophilic substitution reaction (SN-2) mechanism to finally form the final product starch furoate by releasing methanol as a by-product [31]. Therefore, if only based on this explanation, it is reasonable to assume that a higher amount of K2CO3 increases the formation of starch alkoxide, enhances the reaction rate, and eventually leads to higher DS values. Unfortunately, this is not the case in our results as, indeed, the DS value increases with the catalyst intake and reaches a maximum value (DS of 0.32) at a catalyst intake of 0.3 mol/mol AGU; however, it decreases at a K2CO3 intake of 0.4 mol/mol AGU and 0.5 mol/mol AGU.
These results indicate not only the positive effect on the reaction rate, but the changes in the amount of catalyst have a negative impact on the transesterification reaction as well. The latter may be explained by the possible occurrence of side reactions, including hydrolysis of methyl 2-furoate to furoic acid and methanol using K2CO3 as a catalyst [32] and possible de-esterification of the starch ester product due to the formation of methanol (Scheme S1.b).
Hydrolysis of methyl 2-furoate may happen with water available in sago starch raw materials (16.9% w/w, see above). In addition, the methanol produced in the hydrolysis of methyl 2-furoate reaction may disturb the equilibrium of the transesterification reaction. The de-esterification rate may increase, resulting in less starch furoate product (lower DS value) (Scheme S1.b). To ensure this explanation, an additional study of the kinetics of the hydrolysis of methyl 2-furoate and the influence of methanol in starch transesterification reaction is required.
3.2 Crosslinking of Furan-Bismaleimide via Diels-Alder (DA) Chemistry
After successfully obtaining the intermediate product (starch furoate), the experiment continued with the cross-linking of starch furoate via DA reaction with BM using CHCl3 as the solvent. The cross-linked product was analyzed with FTIR, and the results are shown in Fig. 1a (absorption range of 2000–3500 cm−1) and Fig. 1b (absorption range of 500–2000 cm−1). By comparing with the spectra of starch furoate, it is evident that additional peaks at the absorption band of 1598, and 1182 cm−1 are present in the spectra of the starch cross-linked product (see Supplementary Materials, Fig. S2, for the detailed FTIR spectra of BM). The broad peak at 1598 cm−1 corresponds to the CH=CH of the BM aromatic ring’s stretching vibrations (υ CH=CH), that may overlap with the peak of aromatic furan ring absorption. Moreover, the peak at 1182 cm−1 is assigned to the appearance of the cycloadduct (C-O-C, δ DA ring) after DA. The appearance of those peaks confirms the changes in the chemical structure of the modified starch products after the DA cross-linking reaction with BM [21,27,33].
In addition, 1H-NMR analysis was conducted on the cross-linked product; however, the proper measurement was unfortunately impossible due to the low solubility of the final product. The latter indicates the presence of the cross-linking network after the Diels-Alder reaction of starch furoate and BM and agrees with the findings derived from the FTIR spectra.
3.2.1 The Thermal Reversibility of the Cross-Linked Starch Furoate with BM at Different Annealing Temperatures
The reversibility of the cross-linked product after annealing at different temperatures (50°C, 70°C, and 150°C) was examined based on the FTIR spectra. The changes in the intensity of the cycloadduct peak (at 1174–1178 cm−1) were observed, as seen in Fig. S3 (Supporting Information Fig. S3). To quantify these changes, the FTIR spectra of the annealed products were deconvoluted (see Fig. 5), and the intensities of relevant peaks were determined from the deconvoluted spectra.

Figure 5: Examples of deconvoluted FTIR spectra of cross-linked starch with DS of 0.32 annealed at a temperature of 50°C for 24 h in the absorption range of 1000–1800 cm−1
Furthermore, the intensity of the peak at 1174–1178 cm−1 was normalized to the intensity of the absorption peak at 1707–1710 cm−1 to ensure the intensity comparison for each annealed product is accurate and valid. The absorption peak at 1707–1710 cm−1 corresponds to C=O symmetric stretching (υ CO sym) of the carbonyl group of starch furoate and BM rings. It is chosen as the standard peak [34,35] since it does not significantly change during the cross-linking reaction [26]. The normalized intensity ratios of the peak at absorption bands of 1174–1178 cm−1 (I1176/I1707) for the cross-linked products of different DS and their annealed products are given in Table 3.
It is plausible that the intensity ratio of the two peaks (I1176/I1710) changes at different annealing temperatures (Table 3), where the maximum intensity value was observed at the temperature of 70°C for the cross-linked product DS of 0.32 (Table 3, data numbers 4–6). The results show that the intensity of the cycloadduct (C-O-C) peak increases with temperature from 50°C to 70°C, and the intensity of the cycloadduct decreases at the temperature of 150°C. The decrease in the intensity implies that within the temperature range of 50°C–150°C, the cross-linked starch product undergoes reversibility, where an increase in the DA cross-linking reaction rate at a temperature of 70°C followed by an increase in de-cross-linking reaction (retro DA) rates at the highest temperature (T = 150°C) was observed.
Therefore, based on the changes in the intensity of the cycloadduct peak (Table 3), one may expect that the maximum degree of cross-linking is obtained at a temperature of 70°C. Indeed, in all cases, the degree of cross-linking reaches the maximum at 70°C, and the value decreases at a temperature of 150°C as qualitatively determined from the solubility measurement (see Figs. 6 and 7).
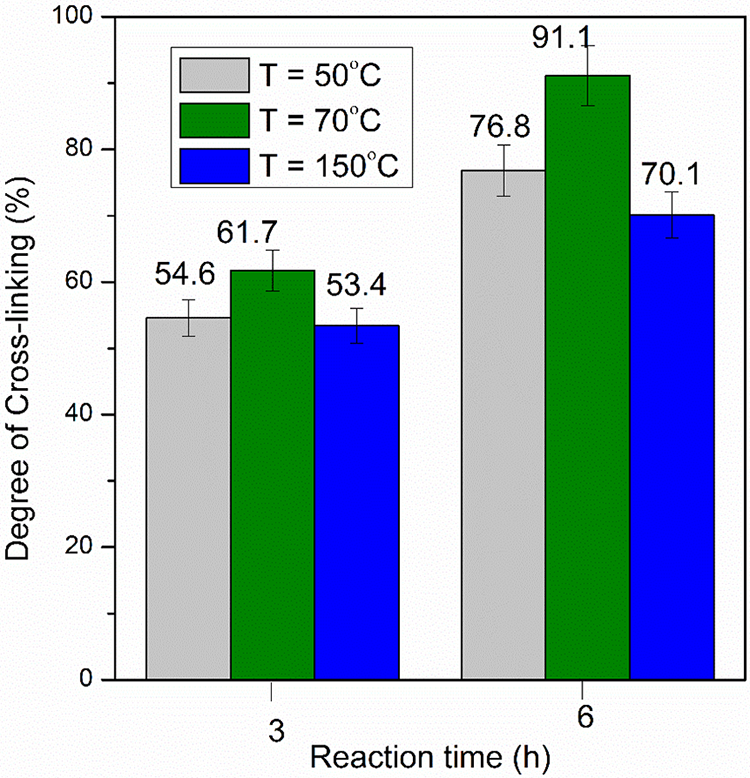
Figure 6: Degree of cross-linking values at different reaction temperatures and times. The data presented in this figure were obtained from experiments using starch furoate with DS of 0.32, BM intake of 2 mol/mol AGU, reaction temperature of 50°C, and annealed for 24 h

Figure 7: Degree of cross-linking values at different annealing temperatures and BM intake. The data presented in this figure were obtained from experiments using starch furoate with DS of 0.61, DA reaction time of three hours, and annealed for 24 h
Furthermore, as expected, our finding on the temperature ranges for both DA (50°C–70°C) and retro DA (150°C) reactions agrees with the ones reported in the literature not only for starch [15] but also for various polymer [17,18,21,22], wherein the DA cross-linking reaction involving furan pendant and BM, the DA reaction is dominant within the temperature range of 50°C–80°C while the retro DA (de-cross-linking) takes place at the temperature range higher than 120°C (T > 120°C).
Despite the maximum intensity ratio that can be reached at the temperature of 70°C for the cross-linked products at DS of 0.32, a different trend was observed for the cross-linked product with a DS of 0.21 (Table 3, data numbers 1–3) and 0.61 (Table 3, data numbers 7–9) where the intensity ratio values increase even at the temperature of 150°C. One may argue that, in this case, the maximum degree of cross-linking should be obtained at 150°C. However, this is not the case, as shown in Fig. 7; the degree of cross-linking from the product with DS of 0.61 at 150°C is lower than that of 70°C (see above). A similar trend was also observed for the cross-linking degree of the product with a DS of 0.21 (data not shown for brevity). This result suggests that although an increase in the intensity of the cycloadduct at the temperature of 150°C was observed, it may be possible that due to the occurrence of the retro DA reaction at 150°C, one of the two DA adducts between furan and BM are eliminated (de-cross-linked). The remaining cycloadduct (as detected by FTIR) is available only on one side of the BM. Since it requires the presence of two cycloadducts to form a cross-linking network between furan and BM (Scheme 1), no cross-linking occurs if only one adduct is formed between furan and BM. The incomplete formation of cross-linking adduct eventually leads to a lower degree of cross-linking of the final product, in line with the result shown in Fig. 7.
Thus far, the observation provided by FTIR and solubility measurement imply that the DA cross-linking and retro DA de-cross-linking occur within the experimental windows; however, it remains uncertain whether the product is thermally reversible when subjected to a change in temperature with repetitive heating and cooling cycles. Therefore, thermal reversibility was evaluated using DSC analysis, as shown in Fig. 8 [17,22], where three cycles of temperature (heating and cooling) were employed.

Figure 8: Thermal behavior of starch furoate cross-linked with BM (DS = 0.18) in the heating and cooling cycles
As expected, the changes in equilibrium between DA (cross-linking) and retro DA (de-cross-linking) reactions can be observed from the appearance of exothermic transitions in the cooling cycles and endothermic transitions in the heating cycles during the successive thermal cycles, respectively [22]. As shown in the heating cycles (Fig. 8), two transitions were observed at the temperature range of 50°C–70°C and at the temperature range of 125°C–150°C, where both transitions are absent in the thermogram of native sago starch and starch furoate. The presence of initial transitions (50°C–70°C) and second endothermic transitions (range 125°C–150°C, in the third cycle) in the cross-linked starch [36] may be related to the glass transition of the amorphous portion and retro DA reaction [22,33,37], respectively Furthermore, as described in the literature [38], the glass transition temperature (Tg) of BM is expected to show at temperatures greater than 400°C (T > 400°C), which is beyond of the measurement scope in our work.
Similar observations can be made with the cooling cycles where broad exothermic transitions ascribed to the DA reaction step occur at 50°C–100°C (Fig. 8). These transitions are not present in both native sago starch and starch furoate thermograms (Fig. 8), suggesting the occurrence of DA reaction between starch furoate and bismaleimide. The findings from DSC analysis imply that the cross-linked starch furoate with BM is indeed thermally reversible and confirms the previous explanations provided by FTIR and the degree of cross-linking experiments results (see above).
3.2.2 The Effect of Degree of Substitution (DS), Cross-Linking Reaction Time, and BM Intake on the Degree of Cross-Linking
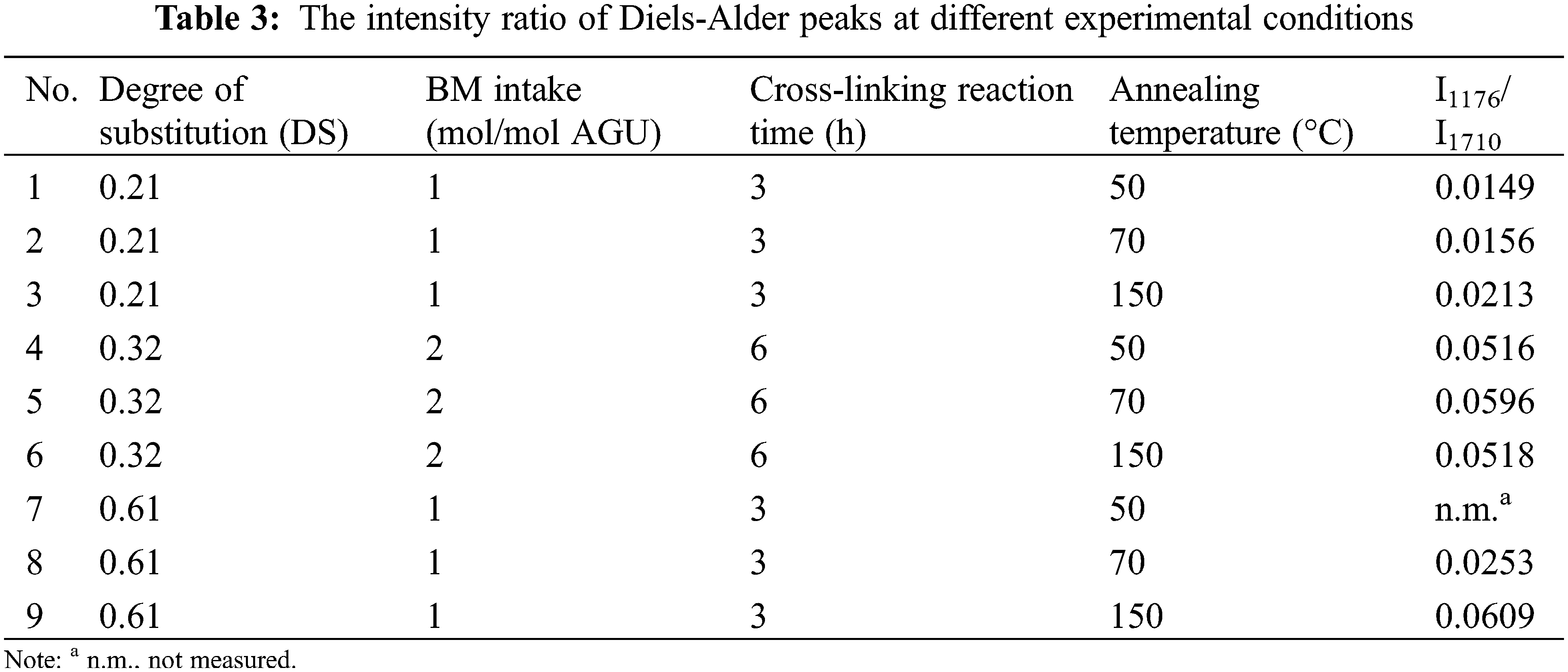
The effect of ester linkage (DS value) on the reactivity of the DA cross-linking reaction step is presented in Table 2, where the different DS of starch ester products were varied, resulting in different cross-linking values. By comparing the results obtained when starch esters with DS values of 0.21 (Table 2, data points 1–3) and 0.61 (Table 2, data points 13–15) were applied in the DA cross-linking reaction step with the same BM intake (1 mol/mol AGU) and reaction time (3 h), it is evident that higher amounts of furan pendant group attached in the starch ester (higher DS) increase the reactivity of the Diels-Alder reaction with BM, which in turn leads to the higher amount of cross-linking degree of the starch ester with a DS of 0.61 (maximum of 90% at an annealing temperature of 70°C) compared with the DS of 0.21 (maximum of 32% at an annealing temperature of 70°C, Table 2) [14].
Figs. 6 and 7 show that the DC values depend on reaction time and BM intake changes, respectively. As shown in Fig. 6, the DC values increase at longer reaction times for all annealing temperatures. The DC value at a reaction time of 6 h is significantly higher than its value at a faster reaction time (three hours). The result may be related to the fact that more DA reaction between furan and BM takes place at a longer reaction time, leading to higher cross-linking after annealing.
In addition, the positive effect of BM intake on the DC values was observed (Fig. 7). As expected, in all cases, a higher BM intake enhances the DA reaction rate [39] and increases the DC of the final product; for instance, at an annealing temperature of 70°C the DC values increase from 70% to 90% when the BM intake increases from 0.5 to 1 mol/mol AGU. A similar trend to the annealing temperature of 70°C can also be observed for the other annealing temperature where an increase of DC values from 30% to 55% and from 59% to 80% was achieved at temperatures of 50°C and 150°C, respectively.
3.3.1 Changes in Crystallinity and Morphology of Modified Starch Products
Despite the changes in chemical structure, the crystallinity and morphology of the modified starch product are altered after transesterification with methyl 2-furoate and cross-linking with BM. The diffraction patterns of native starch, starch ester, and cross-linked starch products are shown in Fig. 9. It is obvious that native starch show diffraction peaks at 2θ of 5.6°, 13.7°, 17.1°, 18.1°, and 21.8°, which is the typical range of diffraction pattern for type C crystallinity of starch [40,41]. The crystallinity of native sago starch decreases, and amorphous material is formed after transesterification, as shown by the broad region in the range of 2θ between 5° and 40°.

Figure 9: X-ray diffraction patterns of native sago starch (a), sago starch furoate with DS of 0.21 (b), and starch cross-linked with BM (c). The data presented in this figure were obtained from experiments using BM intake of 2 mol/mol AGU, the annealing temperature of 50°C, and annealed for 24 h
Surprisingly, the diffraction pattern of the final product changes and shows several distinct peaks at 7.9°, 12.9°, 13.8°, 15.8°, 19.9°, 22°, 26.1°, 27.6°, 31.1°, and 33.9° in combination with a broad region under the peaks at 12.9°–26.1° suggesting the formation of semi-crystalline polymer structure after cross-linking with BM. Moreover, the peaks in the diffraction region of 12.9°–33.9° are the typical peaks in the highly crystalline BM diffraction pattern [42]. The latter suggests that the starch ester structure does not solely determine the crystallinity of the cross-linked starch product but is also strongly influenced by the presence of BM as the cross-linker.
The changes in the morphology of starch after modification are clearly shown in Fig. 10. The SEM images from the native sago starch granule (Figs. 10a and 10b) show an oval and smooth surface in line with the one reported in the literature [41]. The loss of the starch granules after transesterification, as shown in Fig. 10c, may be related to the loss of crystallinity and the formation of amorphous starch furoate products after transesterification with MF in DMSO as in agreement with the observation made with the XRD analysis (see above). By comparing with Fig. 10c, it is evident that more solid granules are formed as expected due to higher networking in the starch backbone between furan and BM after cross-linking (Fig. 10d). The higher networking in starch matrices eventually leads to higher crystallinity of the end products compared with the intermediate starch furoate products, which is in line with the findings in XRD analysis (see above).

Figure 10: SEM images of native sago starch with the magnification of 400 (a) and 5000 (b) times, starch furoate with DS of 0.61 (c), and starch cross-linked with BM and annealed at a temperature of 70°C for 24 h (d). Both (c) and (d) images were acquired with a magnification of 5000 times
3.3.2 Thermal Stability of the Modified Starch Product
Thermal gravimetry analysis was used to measure the thermal stability of the modified products. By comparing with the thermogram of native starch, it is clear that the intermediate starch furoate (DS = 0.32, Fig. 11) has a lower thermal degradation. The degradation temperature range (Tonset and Toffset) were determined from the first derivatization of each thermograms as given in Supplementary Materials Fig. S4. As shown in Fig. 11, the degradation of native starch occurs at the temperature range of 206.4°C–452.9°C with the maximum at 293.8°C (65% wt/wt loss) while the starch furoate product (Fig. 11) starts to degrade at an even lower temperature (T = 187.3°C–415.9°C, 60% wt/wt loss). The lower degradation temperature of the starch ester may be related to the loss of crystallinity of the starch-modified product after the transesterification reaction.

Figure 11: Thermal degradation of native sago starch, starch furoate with DS 0.32, and starch cross-linked annealed at 70°C for 24 h
In addition, a significant improvement in the thermal stability of the cross-linked product is shown in Fig. 11. The cross-linked product has a higher first thermal degradation (Temperature range of = 212.8°C–354.3°C, with a maximum temperature of 274.1°C) than both native and intermediate starch ester products. This result implies that the cross-linking network formed between the furan pendant and BM moieties enhances the thermal stability of the final product [15]. The cross-linked product also shows a second thermal degradation at the temperature range of 380.3°C–608.1°C (Fig. 11), which may be corresponded to the degradation or evaporation temperature of BM monomer after retro DA reaction [19,43]. Based on the corresponding mass loss of the DA cross-linked products (Fig. 11), it is plausible that the amount of char formed in the cross-linking starch products (43% wt/wt) is higher compared with the ones of natives (17.7% wt/wt) and the starch ester product (15.6% wt/wt). The high amount of char from the cross-linked starch that formed in the TGA analysis may be due to the presence of aromatics in the starch backbone, which comes from the furan pendant group, bismaleimide, and also from the aromatization of cycloadduct at high temperature (T > 150°C) [17,26,44]. These aromatics may contribute to the high char residue of the cross-linked product as measured by the TGA analysis [44].
Several significant findings are reported in this piece of research. The findings include the successful application of the new synthetic route to synthesize thermo-reversible starch cross-linked products via two reaction steps which are the transesterification of starch with methyl 2-furoate as reagent and K2CO3 as the catalyst, followed by the Diels-Alder reaction of starch furoate with 1,1′-(methylenedi-4,1-phenylene) bismaleimide (BM). The FTIR analyses and 1H-NMR analyses acquired the proof of principle of both reaction steps.
The influence of several important process parameters on transesterification and DA crosslinking reaction steps was evaluated from the changes in the degree of substitution (DS) and degree of crosslinking (DC) values, respectively. Within the experimental windows, the DS values increase with MF intake and temperatures while the changes in catalyst intakes results with a maximum DS value at K2CO3 of 0.3 mol/mol AGU. Moreover, the DA cross-linking and retro DA de-cross-linking reaction may be tuned at different annealing temperatures. The thermal reversibility of the final products is also confirmed by the presence of exothermic DA (temperature range of 50°C–70°C) and the endothermic retro DA transitions (temperature range of 125°C–150°C) in the DSC thermograms.
This work shows that not only has the tunable cross-linking properties with temperature, but the final product has higher thermal stability compared with the native starch and even with the intermediate starch furoate product. This work opens an opportunity to apply the novel synthetic pathways in the synthesis of starch cross-linking, resulting in different product performances (thermo-reversible and thermal stability), which gives a new perspective on the potential application of the novel product as bioplastics.
Acknowledgement: The authors would like to thank Prof. Francesco Picchioni (University of Groningen) for his valuable and stimulating discussion. We acknowledge the facilities, scientific and technical support from Advanced Characterization Laboratories Bandung (National Research and Innovation Agency), the Research Center for Nanosciences and Nanotechnology (Bandung Institute of Technology), and GreenLabs Bandung.
Funding Statement: This work was funded by the Indonesia Toray Science Foundation (No.: 001/I/ITSF/SEK/2019).
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Henky Muljana; data collection: Ivana Hasjem, Merianawati Sinatra, Dicky Joshua Pesireron, Michael Wilbert Puradisastra, Kevin Yovan Hermanto, Ryan Hartono; analysis and interpretation of results: Henky Muljana, Tony Handoko; draft manuscript preparation: Henky Muljana. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: Data available within the article or its supplementary materials.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Gandini, A., Lacerda, T. M., Carvalho, A. J. F., Trovatti, E. (2016). Progress of polymers from renewable resources: Furans, vegetable oils, and polysaccharides. Chemical Reviews, 116(3), 1637–1669. https://doi.org/10.1021/acs.chemrev.5b00264 [Google Scholar] [PubMed] [CrossRef]
2. Elschner, T., Obst, F., Heinze, T. (2018). Furfuryl- and maleimido polysaccharides: Synthetic strategies toward functional biomaterials. Macromolecular Bioscience, 18(5), 1800258. https://doi.org/10.1002/mabi.201800258 [Google Scholar] [PubMed] [CrossRef]
3. Muljana, H., Irene, C., Saptaputri, V., Arbita, E., Sugih, A. K. et al. (2018). Synthesis of sago starch laurate in densified carbon dioxide. Polymer Engineering & Science, 58(3), 291–299. https://doi.org/10.1002/pen.24569 [Google Scholar] [CrossRef]
4. Dastidar, T. G., Netravali, A. N. (2012). ‘Green’ crosslinking of native starches with malonic acid and their properties. Carbohydrate Polymers, 90(4), 1620–1628. https://doi.org/10.1016/j.carbpol.2012.07.041 [Google Scholar] [PubMed] [CrossRef]
5. Falua, K. J., Pokharel, A., Babaei-Ghazvini, A., Ai, Y., Acharya, B. (2022). Valorization of starch to biobased materials: A review. Polymers, 14(11), 2215. https://doi.org/10.3390/polym14112215 [Google Scholar] [PubMed] [CrossRef]
6. Abe, M. M., Martins, J. R., Sanvezzo, P. B., Macedo, J. V., Branciforti, M. C. et al. (2021). Advantages and disadvantages of bioplastics production from starch and lignocellulosic components. Polymers, 13(15), 2484. https://doi.org/10.3390/polym13152484 [Google Scholar] [PubMed] [CrossRef]
7. Chakraborty, I., Pooja, N., Banik, S., Govindaraju, I., Das, K. et al. (2022). Synthesis and detailed characterization of sustainable starch-based bioplastic. Journal of Applied Polymer Science, 139(39), e52924. https://doi.org/10.1002/app.52924 [Google Scholar] [CrossRef]
8. Tsakona, M., Baker, E., Rucevska, I., Maes, T., Rosendahl Appelquist, L. et al. (2021). Drowning in plastics: Marine litter and plastic waste vital graphics. https://www.unep.org/resources/report/drowning-plastics-marine-litter-and-plastic-waste-vital-graphics [Google Scholar]
9. Gutierrez, T. J. (2018). Active and intelligent films made from starchy sources/blackberry pulp. Journal of Polymers and the Environment, 26(6), 2374–2391. https://doi.org/10.1007/s10924-017-1134-y [Google Scholar] [CrossRef]
10. Sagnelli, D., Hooshmand, K., Kemmer, G. C., Kirkensgaard, J. J. K., Mortensen, K. et al. (2017). Cross-linked amylose bio-plastic: A transgenic-based compostable plastic alternative. International Journal of Molecular Science, 18(10), 2075. https://doi.org/10.3390/ijms18102075 [Google Scholar] [PubMed] [CrossRef]
11. Amaraweera, S. M., Gunathilake, C., Gunawardene, O. H. P., Fernando, N. M. L., Wanninayaka, D. B. et al. (2021). Development of starch-based materials using current modification techniques and their applications: A review. Molecules, 26(22), 6880. https://doi.org/10.3390/molecules26226880 [Google Scholar] [PubMed] [CrossRef]
12. Dang, X., Yu, Z., Yang, M., Woo, M. W., Song, Y. et al. (2022). Sustainable electrochemical synthesis of natural starch-based biomass adsorbent with ultrahigh adsorption capacity for Cr(VI) and dyes removal. Separation and Purification Technology, 288(38), 120668. https://doi.org/10.1016/j.seppur.2022.120668 [Google Scholar] [CrossRef]
13. Dang, X., Du, Y., Wang, X., Liu, X., Yu, Z. (2023). New indoleacetic acid-functionalized soluble oxidized starch-based nonionic biopolymers as natural antibacterial materials. International Journal of Biological Macromolecules, 242(3), 125071. https://doi.org/10.1016/j.ijbiomac.2023.125071 [Google Scholar] [PubMed] [CrossRef]
14. Antunes, B. F., Ferreira, A. G., Amaral, A. C., Carvalho, A. J. F., Gandini, A. et al. (2022). Crosslinking starch with diels-alder reaction: Water-soluble materials and water-mediated processes. Polymer International, 71(11), 1340–1346. https://doi.org/10.1002/pi.6438 [Google Scholar] [CrossRef]
15. Nossa, T. S., Belgacem, N. M., Gandini, A., Carvalho, A. J. F. (2015). Thermoreversible crosslinked thermoplastic starch. Polymer International, 64(10), 1366–1372. https://doi.org/10.1002/pi.4925 [Google Scholar] [CrossRef]
16. Briou, B., Améduri, B., Boutevin, B. (2021). Trends in the diels-alder reaction in polymer chemistry. Chemical Society Reviews, 50(19), 11055–11097. https://doi.org/10.1039/D0CS01382J [Google Scholar] [PubMed] [CrossRef]
17. Gandini, A. (2013). The furan/maleimide diels-alder reaction: A versatile click-unclick tool in macromolecular synthesis. Progress in Polymer Science, 38(1), 1–29. https://doi.org/10.1016/j.progpolymsci.2012.04.002 [Google Scholar] [CrossRef]
18. Bose, R. K., Koetteritzsch, J., Garcia, S. J., Hager, M. D., Schubert, U. S. et al. (2014). A rheological and spectroscopic study on the kinetics of self-healing in a single-component diels-alder copolymer and its underlying chemical reaction. Journal of Polymer Science Part A Polymer Chemistry, 52(12), 1669–1675. https://doi.org/10.1002/pola.27164 [Google Scholar] [CrossRef]
19. Fujisawa, N., Takanohashi, M., Chen, L., Uto, K., Matsumoto, Y. et al. (2021). A diels-alder polymer platform for thermally enhanced drug release toward efficient local cancer chemotherapy. Science and Technology of Advanced Materials, 22(1), 522–531. https://doi.org/10.1080/14686996.2021.1939152 [Google Scholar] [PubMed] [CrossRef]
20. Gevrek, T. N., Sanyal, A. (2021). Furan-containing polymeric materials: Harnessing the Diels-Alder chemistry for biomedical applications. European Polymer Journal, 153(22), 110514. https://doi.org/10.1016/j.eurpolymj.2021.110514 [Google Scholar] [CrossRef]
21. Polgar, L. M., van Duin, M., Broekhuis, A. A., Picchioni, F. (2015). Use of diels-alder chemistry for thermoreversible cross-linking of rubbers: The next step toward recycling of rubber products? Macromolecules, 48(19), 7096–7105. https://doi.org/10.1021/acs.macromol.5b01422 [Google Scholar] [CrossRef]
22. Zhang, Y., Broekhuis, A. A., Picchioni, F. (2009). Thermally self-healing polymeric materials: The next step to recycling thermoset polymers? Macromolecules, 42(6), 1906–1912. https://doi.org/10.1021/ma8027672 [Google Scholar] [CrossRef]
23. Ningrum, R. S., Rosalina, R., Sondari, D., Fazriyah, L., Putri, R. et al. (2023). Physicochemical properties of sago starch fractionated by butanol. Starch, 75(3–4), 2200157. https://doi.org/10.1002/star.202200157 [Google Scholar] [CrossRef]
24. Xu, Y., Miladinov, V., Hanna, M. (2004). Synthesis and characterization of starch acetates with high substitution. Cereal Chemistry, 81(6), 735–740. https://doi.org/10.1094/CCHEM.2004.81.6.735 [Google Scholar] [CrossRef]
25. Chen, W. C., Syed Mohd Judah, S. N. M., Ghazali, S. K., Munthoub, D. I., Alias, H. et al. (2021). The effects of citric acid on thermal and mechanical properties of crosslinked starch film. Chemical Engineering Transactions, 83, 199–204. https://doi.org/10.3303/CET2183034 [Google Scholar] [CrossRef]
26. Muljana, H., Arends, S., Remerie, K., Boven, G., Picchioni, F. et al. (2022). Cross-linking of polypropylene via the diels-alder reaction. Polymers, 14(6), 1176. https://doi.org/10.3390/polym14061176 [Google Scholar] [PubMed] [CrossRef]
27. Köhler, S., Heinze, T. (2007). Efficient synthesis of cellulose furoates in 1-N-butyl-3-methylimidazolium chloride. Cellulose, 14(5), 489–495. https://doi.org/10.1007/s10570-007-9138-8 [Google Scholar] [CrossRef]
28. Marefat Seyedlar, R., Imani, M., Mirabedini, S. M. (2021). Bio-based furan coatings: Adhesion, mechanical and thermal properties. Polymer Bulletin, 78(2), 577–599. https://doi.org/10.1007/s00289-020-03124-4 [Google Scholar] [CrossRef]
29. Zhang, K., Cheng, F., Zhang, K., Hu, J., Xu, C. et al. (2019). Synthesis of long-chain fatty acid starch esters in aqueous medium and its characterization. European Polymer Journal, 119(3), 136–147. https://doi.org/10.1016/j.eurpolymj.2019.07.021 [Google Scholar] [CrossRef]
30. Winkler, H., Vorwerg, W., Wetzel, H. (2013). Synthesis and properties of fatty acid starch esters. Carbohydrate Polymers, 98(1), 208–216. https://doi.org/10.1016/j.carbpol.2013.05.086 [Google Scholar] [PubMed] [CrossRef]
31. Muljana, H., van der Knoop, S., Keijzer, D., Picchioni, F., Janssen, L. P. B. M. et al. (2010). Synthesis of fatty acid starch esters in supercritical carbon dioxide. Carbohydate Polymers, 82(2), 346–354. https://doi.org/10.1016/j.carbpol.2010.04.067 [Google Scholar] [CrossRef]
32. Escobar, A., Sathicq, Á., Pizzio, L., Blanco, M., Romanelli, G. (2015). Biomass valorization derivatives: Clean esterification of 2-furoic acid using tungstophosphoric acid/zirconia composites as recyclable catalyst. Process Safety and Environmental Protection, 98(37), 176–186. https://doi.org/10.1016/j.psep.2015.07.008 [Google Scholar] [CrossRef]
33. Araya-Hermosilla, E., Giannetti, A., Lima, G. M. R., Orozco, F., Picchioni, F. et al. (2021). Thermally switchable electrically conductive thermoset rGO/PK self-healing composites. Polymers, 13(3), 339. https://doi.org/10.3390/polym13030339 [Google Scholar] [PubMed] [CrossRef]
34. Sclavons, M., Franquinet, P., Carlier, V., Verfaillie, G., Fallais, I. et al. (2000). Quantification of the maleic anhydride grafted onto polypropylene by chemical and viscosimetric titrations, and FTIR spectroscopy. Polymer, 41(6), 1989–1999. https://doi.org/10.1016/S0032-3861(99)00377-8 [Google Scholar] [CrossRef]
35. Coates, J. (2006). Interpretation of infrared spectra, a practical approach. In: Meyers, R. A. (Ed.Encyclopedia of analytical chemistry, pp. 1–23. Hoboken New Jersey: John Wiley & Sons. https://doi.org/10.1002/9780470027318.a5606 [Google Scholar] [CrossRef]
36. Chung, H., Woo, K., Lim, S. (2004). Glass transition and enthalpy relaxation of cross-linked corn starches. Carbohydrate Polymers, 55(1), 9–15. https://doi.org/10.1016/j.carbpol.2003.04.002 [Google Scholar] [CrossRef]
37. Araya-Hermosilla, E., Gabbani, A., Mazzotta, A., Ruggeri, M., Orozco, F. et al. (2022). Rapid self-healing in IR responsive plasmonic indium tin oxide/polyketone nanocomposites. Journal of Materials Chemistry A, 10(24), 12957–12967. https://doi.org/10.1039/D2TA01286C [Google Scholar] [CrossRef]
38. Ren, Z., Hao, S., Xing, Y., Yang, C., Dai, S. (2019). Asymmetric bismaleimide-based high-performance resins with improved processability and high Tg over 400°C. High Performance Polymers, 31(9–10), 1132–1139. https://doi.org/10.1177/0954008319826368 [Google Scholar] [CrossRef]
39. Beljaars, M., Heeres, H. J., Broekhuis, A. A., Picchioni, F. (2022). Bio-based aromatic polyesters reversibly crosslinked via the Diels-Alder reaction. Applied Sciences, 12(5), 2461. https://doi.org/10.3390/app12052461 [Google Scholar] [CrossRef]
40. Okazaki, M. (2018). The structure and characteristics of sago starch. In: Ehara, H., Toyoda, Y., Johnson, D. V. (Eds.Sago palm: Multiple contributions to food security and sustainable livelihoods, pp. 247–259. Singapore: Springer. https://doi.org/10.1007/978-981-10-5269-9_18 [Google Scholar] [CrossRef]
41. Du, C., Jiang, F., Jiang, W., Ge, W., Du, S. (2020). Physicochemical and structural properties of sago starch. International Journal of Biological Macromolecules, 164(3), 1785–1793. https://doi.org/10.1016/j.ijbiomac.2020.07.310 [Google Scholar] [PubMed] [CrossRef]
42. Satheesh Chandran, M., Krishna, M., Salini, K., Rai, K. S. (2010). Preparation and characterization of chain-extended bismaleimide/carbon fibre composites. International Journal of Polymer Science, 2010, 987357. https://doi.org/10.1155/2010/987357 [Google Scholar] [CrossRef]
43. Feng, J. L., Yue, C. Y., Chian, K. S. (2006). Development and characterization of bismaleimides containing aliphatic chain for microelectronics application. E-Polymers, 6(1), 49. https://doi.org/10.1515/epoly.2006.6.1.633 [Google Scholar] [CrossRef]
44. Factor, A. (1990). Char formation in aromatic engineering polymers. In: Nelson, G. L. (Ed.Fire and polymers: Hazards identification and prevention, pp. 274–287. Washington: American Chemical Society. [Google Scholar]
Supplementary Materials

Scheme S1: The transesterification scheme between starch and MF using an alkaline salt base (K2CO3) as a catalyst

Figure S1: FTIR spectra of native sago starch, methyl 2-furoate and starch furoate with DS of 0.21

Figure S2: FTIR spectra of starch furate with DS of 0.21, bismaleimide and starch cross-linked

Figure S3: FTIR spectra of cross-linked starch with DS of 0.32 annealed at different temperatures: T = 50°C, T = 70°C, and T = 150°C. All samples were annealed for 24 h

Figure S4: First derrivative of thermogram for native sago starch, starch furoate with DS 0.32, and starch cross-linked annealed at 70°C for 24 h
Note: To = Tonset, Toff = Toffset, Tp = Tpeak.
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools