
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Acidic Magnetic Biocarbon-Enabled Upgrading of Biomass-Based Hexanedione into Pyrroles
1
School of Chemistry and Materials Science, Guizhou Normal University, Guiyang, 550001, China
2
National Key Laboratory of Green Pesticide, Key Laboratory of Green Pesticide and Agricultural Bioengineering, Ministry of
Education, State-Local Joint Laboratory for Comprehensive Utilization of Biomass, Center for R&D of Fine Chemicals of Guizhou
University, Guiyang, 550025, China
* Corresponding Authors: Haoli Qin. Email: ; Hu Li. Email:
(This article belongs to the Special Issue: Biochar Based Materials for a Green Future)
Journal of Renewable Materials 2023, 11(11), 3847-3865. https://doi.org/10.32604/jrm.2023.030122
Received 23 March 2023; Accepted 19 April 2023; Issue published 31 October 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Sustainable acquisition of bioactive compounds from biomass-based platform molecules is a green alternative for existing CO2-emitting fossil-fuel technologies. Herein, a core–shell magnetic biocarbon catalyst functionalized with sulfonic acid (Fe3O4@SiO2@chitosan-SO3H, MBC-SO3H) was prepared to be efficient for the synthesis of various N-substituted pyrroles (up to 99% yield) from bio-based hexanedione and amines under mild conditions. The abundance of Brønsted acid sites in the MBC-SO3H ensured smooth condensation of 2,5-hexanedione with a variety of amines to produce N-substituted pyrroles. The reaction was illustrated to follow the conventional PallKnorr coupling pathway, which includes three cascade reaction steps: amination, loop closure and dehydration. The prepared MBC-SO3H catalyst could effectively activate 2,5-hexanedione, thus weakening the dependence of the overall conversion process on the amine nucleophilicity. The influence of different factors (e.g., reaction temperature, time, amount of catalyst, molar ratio of substrates, and solvent type) on the reaction activity and selectivity were investigated comprehensively. Moreover, the MBC-SO3H possessed excellent thermochemical stability, reusability, and easy separation due to the presence of magnetic core-shell structures. Notably, there was no activity attenuation after 5 consecutive catalytic experiments. This work demonstrates a wide range of potential applications of developing functionalized core-shell magnetic materials to construct bioactive backbones from biomass-based platform molecules.Graphic Abstract
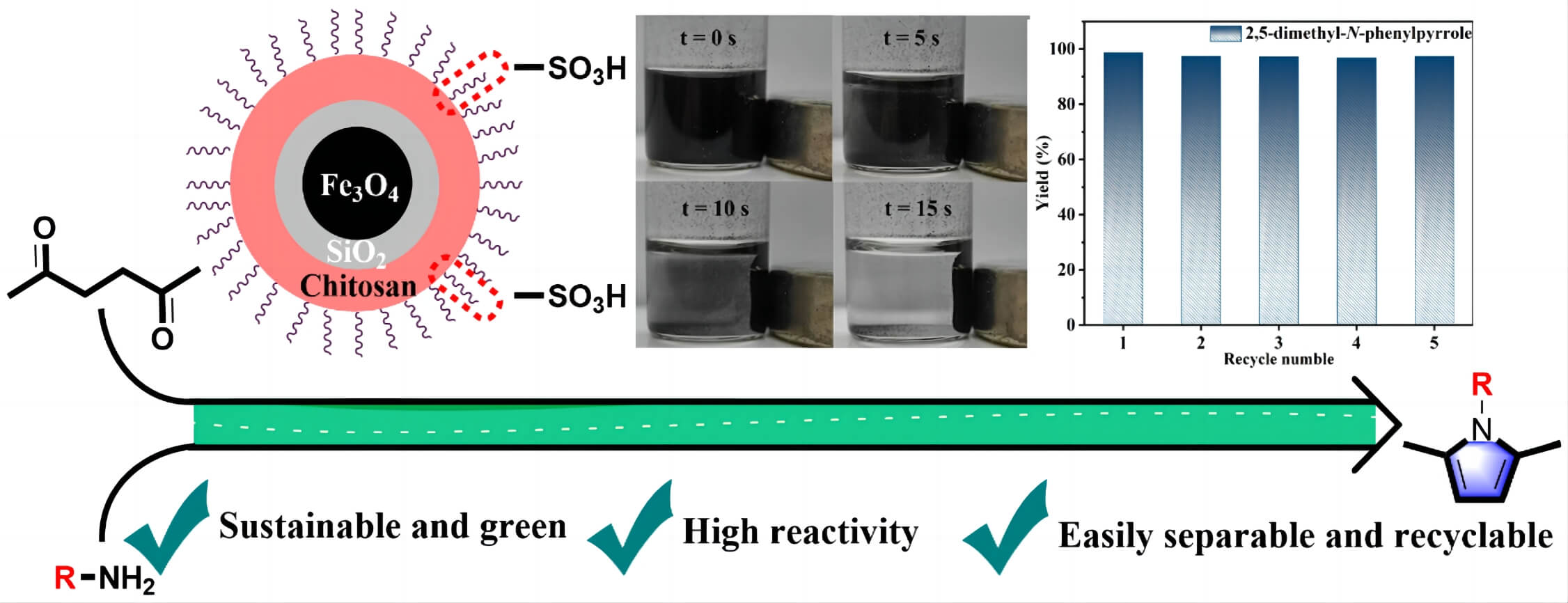
Keywords
Reducing reliance on fossil energy is the key to achieving the “double carbon” target and addressing global climate change. In recent years, renewable and sustainable biomass energy has received much attention [1]. The construction of nitrogen-containing heterocycles based on biomass-derived platform molecules is encouraging. Among them, pyrrole heterocycles, especially N-substituted pyrroles are widely used in the manufacture of pesticides, pharmaceuticals, materials, perfumes, and other organic compounds [2]. It is worth noting that N-substituted pyrroles are functional modules in many pharmaceuticals and active formulations with various pharmacological effects such as anti-inflammatory, anticancer, and antifungal activities [3–5]. Currently, the annual production of just pyrrole is up to 10,000 tons to meet the demand of chemical manufacturing industries [6]. Notably, the need for pyrrole is expected to continue to grow with the outbreak of novel coronaviruses [7]. With decreasing fossil resources and increasing demand for pyrroles and derivatives, sustainable green synthetic strategies to produce N-substituted pyrroles are highly desired.
In general, N-substituted pyrroles are synthesized via cross-coupling of pyrroles and arylboronic acids, coupling of pyrroles with aromatic amines, Hantzsch reactions, etc. [8]. The synthesized precursors mainly originate from CO2-emitting chemical manufacturing industries by refining crude oil. Serious environmental problems related to the overuse of fossil energy have driven people to seek green and sustainable alternatives. Recently, the tailoring of efficient conversion routes for upgrading biomass-derived molecules (e.g., furan, polyols, and catechol) into nitrogen-containing heterocyclic compounds have painted a rosy blueprint for reducing the carbon footprint of the chemical industry and meeting the green economy [9]. Oxygen-rich biomass can be easily converted to various oxygenated hydrocarbons and further to aliphatics, aromatics, lactones, and olefins by hydrodeoxygenation, which means that oxygen-rich biomass-derived compounds offer unlimited possibilities for the introduction of heteroatoms to build nitrogen-containing heterocycles [10]. 2,5-hexanedione is an important biomass-derived platform molecule [10], in which the carbonyl carbon exhibits electrophilic properties allowing nucleophilic attack of heteroatoms containing lone pairs electrons to replace the oxygen atom, motivating researchers to complete the transformation of 2,5-hexanedione to N-substituted pyrroles.
A “one-pot” strategy was developed for the synthesis of pyrrole and its derivatives using 2,5-hexanedione and nitro compounds as substrates via Pd-, Pt-, and Ru-based noble metal catalysts at temperatures of higher than 150°C [11,12]. Completing this domino reaction required noble metals for the hydrogenation of nitro to amino, while 2,5-hexanedione needed to be adsorbed and activated, thus resulting in relatively low yields. Up to 90% yields of N-substituted pyrroles could be achieved through a noble metal indium-mediated Paal-Knorr coupling reaction using aromatic amines as nitrogen sources at 150°C [13]. Although N-substituted pyrroles could be obtained under autocatalysis, the reactivity was strongly dependent on the basicity of the substrate to complete the amination, and the ring-closing and dehydration processes, thus as long as 24 h of reaction time was required to achieve high yields [14]. Until now, most of the catalysts employed are precious metals-based or homogeneous catalysts, which makes the catalyst recovery process complex. The efficient synthesis of N-substituted pyrroles using low-cost catalysts under mild conditions remains a great challenge.
In this connection, inexpensive magnetic materials, such as Fe3O4, Fe, and Co, have emerged as one of the most promising alternatives for homogeneous catalysis. The functionalization of magnetic materials can not only preserve their magnetic properties but also allow them to be quickly and easily separated from the reaction system by an external magnetic field, exhibiting excellent reusability [15]. Further, a magnetic core-shell structure can be designed to improve the accessibility and lifetime of the catalysts, especially using a high porosity shell layer (e.g., mesoporous silica) [16]. In addition, for obtaining N-substituted pyrroles via the Pall-Knorr process, acid functionalization of the magnetic core-shell structure was essential. Hence, a rational integration of new material with magnetic and reactive sites can be developed for the construction of catalytic systems with excellent activity, stability, and separability. In this work, a magnetic core-shell Fe3O4@SiO2@chitosan-SO3H catalyst (MBC-SO3H) was designed and prepared for the efficient synthesis of N-substituted pyrroles using 2,5-hexanedione and amines as feedstock under mild conditions. It is expected that magnetic acid catalysts can provide a new perspective for the green synthesis of N-substituted pyrroles and guide future research in the field of catalytic biomass upgrading.
FeCl3⋅6H2O (99%), FeSO4⋅7H2O (>99%), NH3⋅H2O (25 wt.%), tetraethylorthosilicate (TEOS, 99%), chitosan (95%), p-toluenesulfonic acid (98%), glutaraldehyde (50 wt.% in H2O), Amberlyst-15, HCOOH (>96%), AlCl3 (99%), MgO (98%), toluene (>99.5%), dichloromethane (99.5%), ethyl acetate (99.5%), tetrahydrofuran (99%), acetone (>99.5%), ethanol (>99.5%), methanol (99.9%), acetonitrile (99.9%), aniline (99%), benzylamine (99%), o-toluidine (98%), 2,5-dimethylaniline (97%), 2-methoxyaniline (>98%), 4-ethoxyaniline (98%), 3-aminophenol (99%), 4-chloroaniline (98%), 4-bromoaniline (99%), 4-(trifluoromethyl)aniline (98%), 2-fluoro-5-nitroaniline (98%), n-propylamine (98%), and n-pentylamine (98%) were purchased from Shanghai Aladdin Reagent Co., China, and used without further purification.
2.2.1 Preparation of Magnetic Fe3O4
In a typical procedure for the preparation of Fe3O4, FeCl3⋅6H2O (5.40 g) and FeSO4⋅7H2O (2.78 g) were added to deionized water (80 mL). Then, NH3⋅H2O (25 wt.%) was used to create an alkaline environment (pH = 10). The metal precursor solution was stirred at 80°C for 1.5 h. Finally, the precipitation could be effectively separated by a magnet and dried at 80°C for 6 h to give Fe3O4.
2.2.2 Preparation of Magnetic Fe3O4@SiO2
As-prepared Fe3O4 (1.0 g) was uniformly dispersed in a mixed solvent containing ethanol (80 mL), distilled water (20 mL), and NH3⋅H2O (25%, 1 mL). To the above mixture, tetraethyl orthosilicate (TEOS, 1 mL) was added and reacted for 6 h at room temperature. The solid obtained by magnet separation was washed with ethanol and distilled water, respectively, and dried at 80°C for 6 h. The final solid is named Fe3O4@SiO2.
The as-prepared Fe3O4@SiO2 (1.0 g) was dispersed evenly in CH3COOH (1 wt.%, 50 mL) homogeneous solution containing chitosan (0.5 g). Then, p-toluenesulfonic acid (5.7 g) was added to the solution under continuous stirring. Subsequently, the cross-linking reaction was carried out under the action of glutaraldehyde (1 mL) for 3 h. The obtained gelatinous solid was dried for 6 h at 80°C and ground to obtain MBC-SO3H. For comparison, the Fe3O4@SiO2-SO3H catalyst was prepared in the absence of chitosan.
The morphology of the prepared catalyst was observed by scanning electron microscopy (SEM, JSM-6700F). Transmission electron microscopy (TEM) and HR-TEM were conducted on TALOS F200C. Functional group information is collected by Fourier transforms infrared (FT-IR, Thermo Fisher Scientific Nicolet iS50) under ambient conditions in KBr disks, and OMNIC software was used to analyze the FT-IR spectrum. Powder X-ray diffraction (PXRD) patterns were collected using a Tongda TD-3500 X-ray diffractometer with a Cu Kα radiation (λ = 0.154056 nm). The patterns were recorded in the 2θ range of 5° to 80°. X-ray photoelectron spectroscopy (XPS) was performed on Thermo ESCALAB 250 equipment. Thermogravimetric analysis (TGA) was conducted by Mettler-TGA/DSC1 at a temperature of 25°C–800°C thermal analysis at a temperature of 10°C/min. Brunner-Emmet-Teller (BET) measurements were performed on ASAP 2460 equipment (Micromeritics). The acid amount of the catalyst was studied by temperature-programmed desorption (TPD, TP5080) of the ammonia technique. The hysteresis lines were tested by a superconducting quantum interferometer (PMS-XL-7). Acid–base titration was used to determine the H+ ion concentration before and after using catalysts. A NaOH solution (10 mL, 0.1 M) was added to the catalyst (50 mg) in an Erlenmeyer flask and stirred for 60 min. The addition of an HCl solution (0.1 M) neutralized the excess amount of base to the equivalence point of the titration.
All reactions were carried out in a pressure-resistant tube, a glass reaction tube equipped with a thread cap, ensuring a sealed reaction environment. In a general process, the catalyst (MBC-SO3H), 2,5-hexanedione (1 mmol), amine (1.2 mmol), internal standard (naphthalene, 20 mg), and solvent (methanol, 2 mL) were added sequentially to 10 mL of the pressure-resistant tube. After tightening the threaded cap of the pressure-resistant tube, the reactor was fixed in an oil pot equipped with a magnetic stirring device at 50°C for 1 h. Upon completion, the reaction solution was diluted with methanol (5 mL). 1 mL of the diluted reaction solution was taken for further analysis by gas chromatography (GC, Agilent GC6890, HP-5 column (30 m × 0.320 mm × 25 μm)).
3.1 Catalyst Preparation and Characterization
The core-shell Fe3O4@SiO2@Chitosan (MBC) catalyst was facilely prepared based on Fe3O4 cores and renewable biocarbon, as depicted in Fig. 1a. Subsequently, the sulfonic acid group was introduced by interacting with the abundant amino and hydroxyl groups on the surface of chitosan. The SEM image of MBC-SO3H shows a block-stacked porous structure (Fig. 1b). Noteworthily, the core-shell structure of MBC-SO3H was observed in Fig. 1c (red dotted line area). The HR-TEM also proved the existence of the core-shell structure (Fig. 1d).
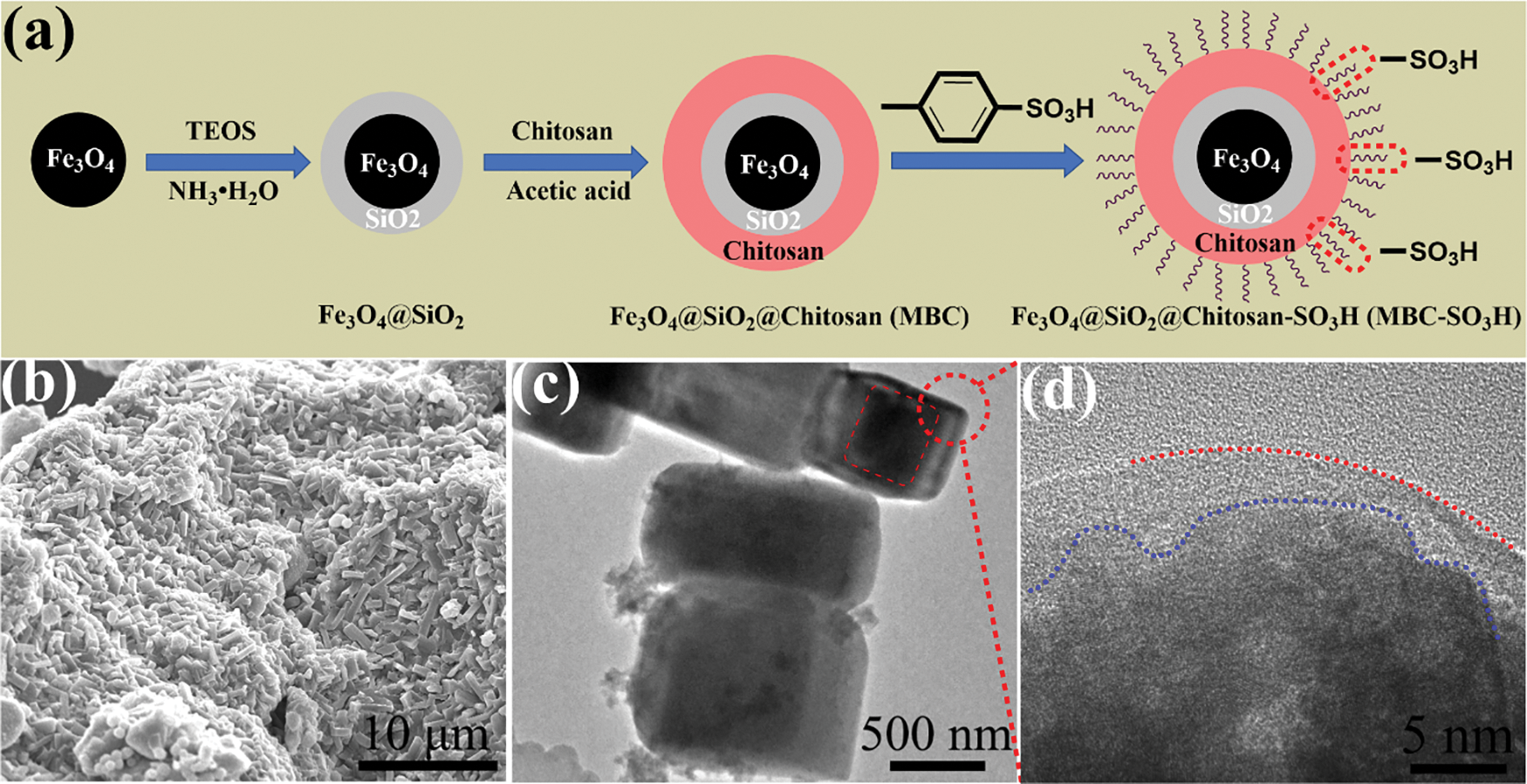
Figure 1: (a) Schematic diagram for preparation of MBC-SO3H. (b) SEM, (c) TEM, and (d) HR-TEM images of MBC-SO3H
The crystal phase of the MBC-SO3H catalyst was tested by XRD patterns and analyzed in the 2θ range of up to 70° [17–19]. The major diffraction peaks of the catalyst were located at 18.5°, 30.1°, 35.5°, 37.1°, 43.0°, 53.5°, 57.2°, and 62.7°, which coincided with the (111), (220), (311), (222), (400), (422), (511), and (440) crystal plane of Fe3O4 (PDF #: 00-001-1053) (Fig. 2a) [20,21], indicating that the catalyst magnetic core was successfully constructed. The Fe2O3 (PDF #: 00-001-1111) was in the form of an impurity phase [22,23].
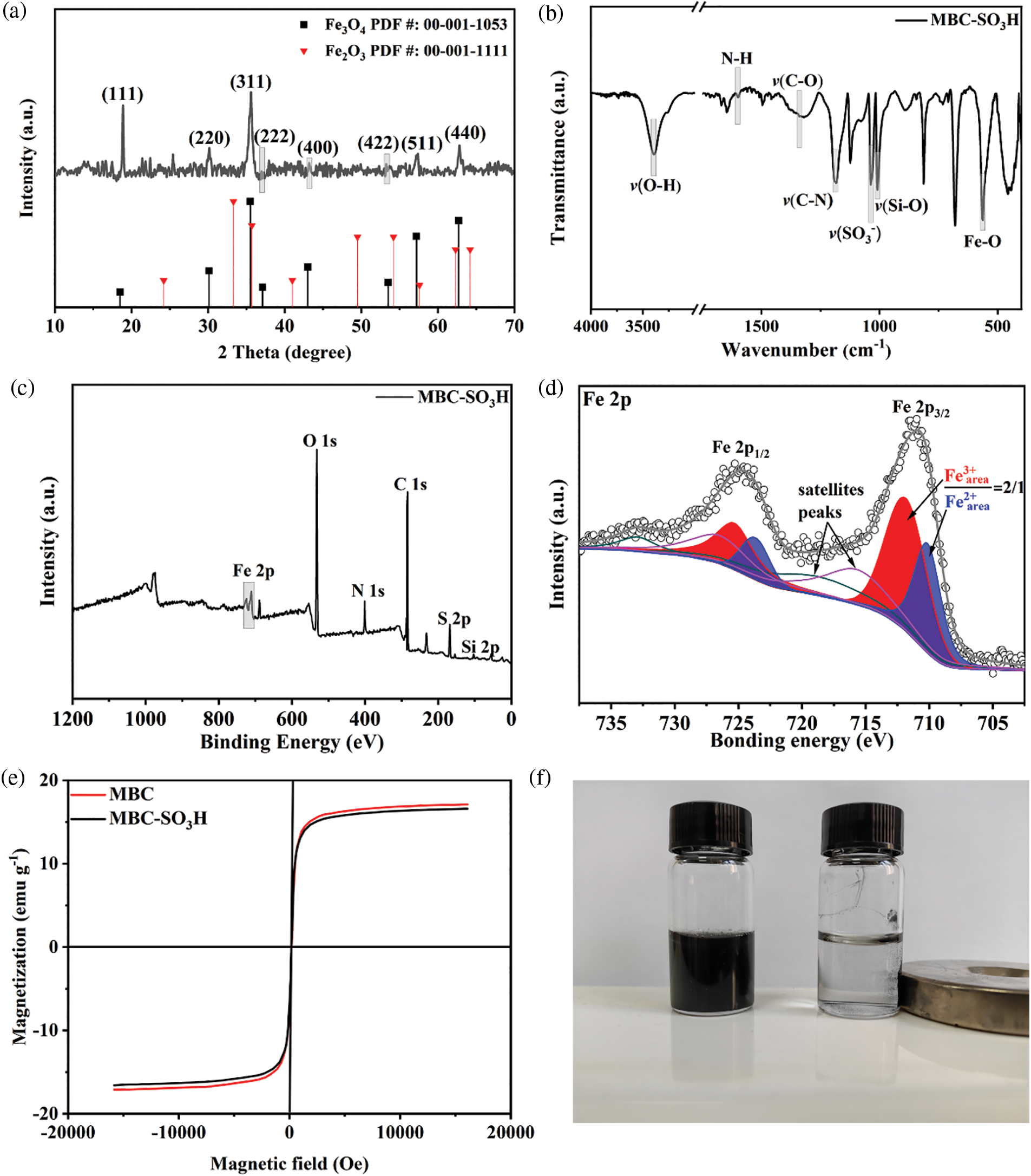
Figure 2: (a) XRD pattern, (b) FT-IR spectrum, (c) XPS survey spectrum, (d) Fe 2p XPS spectrum of MBC-SO3H, (e) the magnetic hysteresis loops of MBC-SO3H and MBC, and (f) magnetic separation image of MBC-SO3H
Functional group information of the MBC-SO3H catalyst was investigated by FT-IR (Fig. 2b). The characteristic adsorption band of the Fe−O bond was observed at 565 cm−1 [24], again confirming the presence of the magnetic Fe3O4 core. The stretching vibration of the Si−O bond was observed at 1010 cm−1 [25]. Meanwhile, the presence of O−H (3396 cm−1), N−H (1600 cm−1), C−O (1382 cm−1), and C−N (1182 cm−1) bonds indicated the successful coating of chitosan. The core-shell MBC-SO3H was confirmed by the layered coating, which agrees with the findings of TEM and HRTEM. The symmetric stretching vibration band of SO3− was observed at 1058 cm−1 (Fig. 2b), showing the catalyst functionalization with sulfonic acid groups [26]. The elements of C, N, O, Si, S, and Fe were detected by the XPS survey (Fig. 2c), further proving the successful preparation of MBC-SO3H. To verify that the core in the MBC-SO3H was Fe3O4 instead of Fe2O3, the fitting of the Fe 2p orbit was carried out (Fig. 2c). The Fe 2p orbital was split into 2p1/2 and 2p3/2 owing to spin-orbit coupling. The Fe 2p3/2 spectrum was fitted to two peaks with binding energies of 711 and 709.6 eV, which is attributed to Fe3+ and Fe2+, respectively (Fig. 2d). While the ratio of the peak area of Fe3+ (red zone) and Fe2+ (blue zone) was 2:1 [27], which was a typical characteristic of Fe3O4. Two satellite peaks were also identified at 716 and 718.5 eV, which are caused by vibrational or charge transfer processes [28]. The above results adequately demonstrated that sulfonic acid-functionalized magnetic biocarbon (MBC-SO3H) was successfully prepared. A vibrating sample magnetometer (VSM) was applied to study the magnetic properties of MBC-SO3H and acidic carrier MBC (Fig. 2e). The strong magnetic field response of both catalysts could be observed, demonstrating that the catalysts possess excellent ferromagnetism. The magnetic properties of MBC-SO3H could be visually observed in Fig. 2f, once again confirming that the catalyst is highly magnetic and easily recovered by magnetic separation methods.
The dates of the specific surface area (SBET = 92.8 m2 g−1) and pore volume (VTotal = 0.22 cm3 g−1) of the MBC-SO3H were presented in Fig. 3a and Table S1. According to the IUPAC classification system, the obtained Brunauer-Emmett-Teller (BET) curve is type-IV, indicating the typical microporous structure of the MBC-SO3H (Fig. 3a). The mean pore size was found to be 13.0 nm by the BJH method (Fig. 3b), which was conducive to the adsorption of substrate molecules and the mass transfer process of the reaction. The acid strength distribution of the MBC-SO3H was examined by NH3-TPD (Fig. 3c). The results showed the presence of weak (50°C–200°C), moderate (300°C–500°C), and strong (500°C–650°C) acid sites. Meanwhile, the acid amount of MBC-SO3H was calculated to be 4.17 and 3.98 mmol g−1 (Table S1) using the NH3-TPD measurement and acid-based titration, respectively. The weight loss of the MBC-SO3H can be roughly divided into three stages at 50°C–700°C in thermal stability testing (Fig. 3d). At the temperature range of 50°C–250°C, the mass loss of the catalyst was 3.2%, which was due to the volatilization of the residual solvent in the catalyst. The most significant mass loss (32.3% weight loss) at 250°C–550°C was due to the carbonation of chitosan. Finally, relatively higher temperatures of 550°C–700°C led to partial decomposition of Fe3O4 (weight loss of 2.4%). The MBC-SO3H is expected to exhibit good thermal stability since the temperature of the reaction system was not higher than 125°C
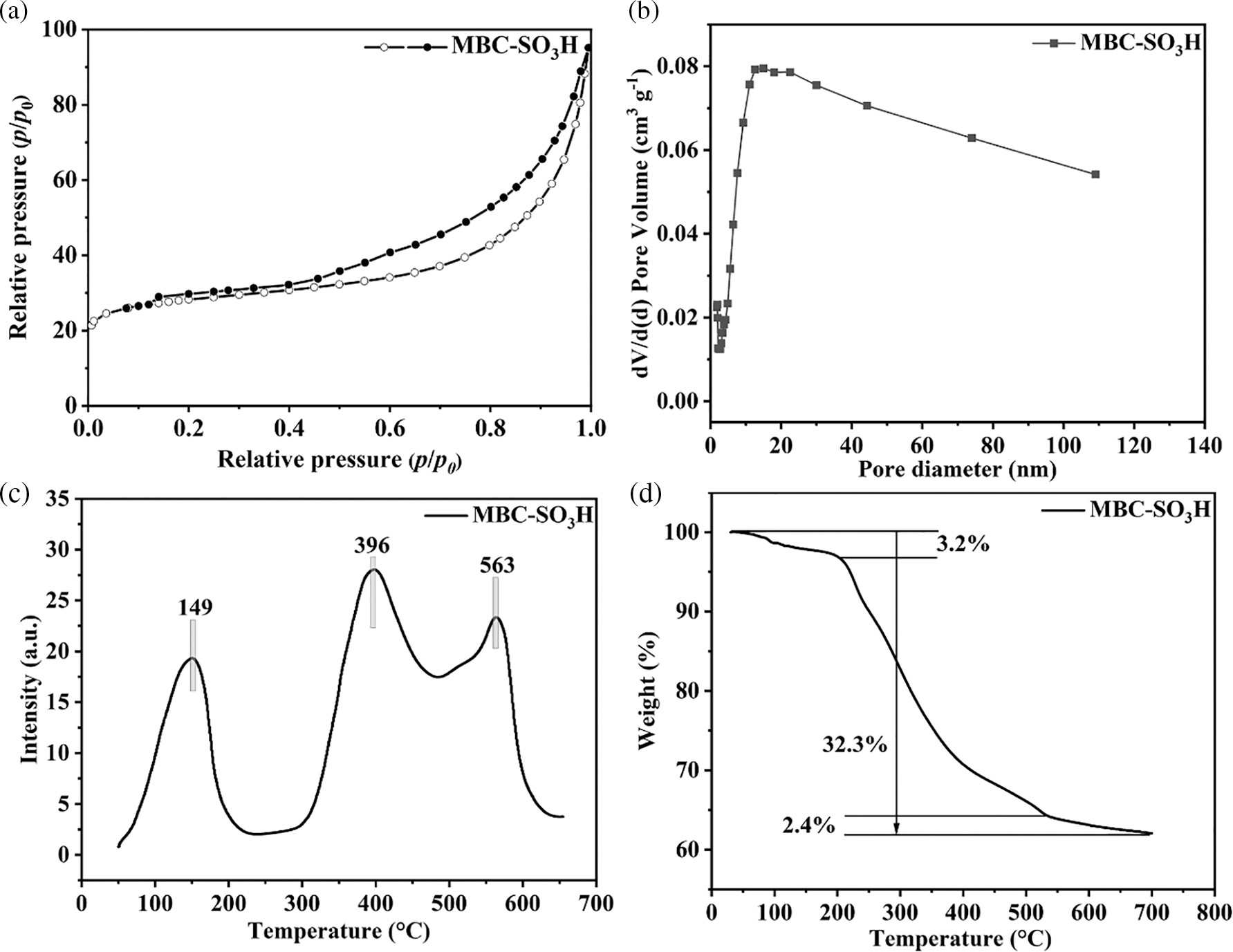
Figure 3: (a) N2 adsorption-desorption isotherms, (b) pore distribution, (c) NH3-TPD profile, and (d) TGA curve of MBC-SO3H
3.2 Optimization of Reaction Conditions
The synthesis of 2,5-dimethyl-N-phenylpyrrole (1c) from biomass-based 2,5-hexanedione (1a) and aniline (1b) was carried out to screen efficient catalysts containing different acid sites, and the conversion of 1a and yield of 1c was summarized in Table 1. It was found that the catalytic activity of Lewis acid catalysts (Entries 1, 8, and 9) was significantly lower than that of Brønsted acid catalysts (Entries 5, 6, and 7). Compared with HCOOH and Amberlyst-15, MBC-SO3H exhibited the highest catalyst activity (yield: 57.1%, 60.5% vs. 84.4%), indicating that catalyst activity was proportional to acid strength of Brønsted acid (MBC-SO3H > Amberlyst-15 > HCOOH). In addition, the catalyst activity could be further enhanced by the encapsulation of SiO2 and chitosan in a series of magnetic catalysts (Entries 1–5). As a result, the pore structure of the functionalized catalysts facilitated the enrichment of substrate molecules and thus enhanced the overall reaction activity. Importantly, compared to using SiO2 as the acid carrier, relatively higher catalytic activity was obtained using renewable chitosan as the acid carrier in the presence of glutaraldehyde as a crosslinker (Entry 5). Two reasons can explain the high activity: (i) chitosan can effectively anchor acid species due to rich amino and hydroxyl functional groups, resulting in a high amount of acid; and (ii) the simultaneous wrapping of SiO2 and chitosan can more effectively avoid the reaction of acid species with Fe3O4.
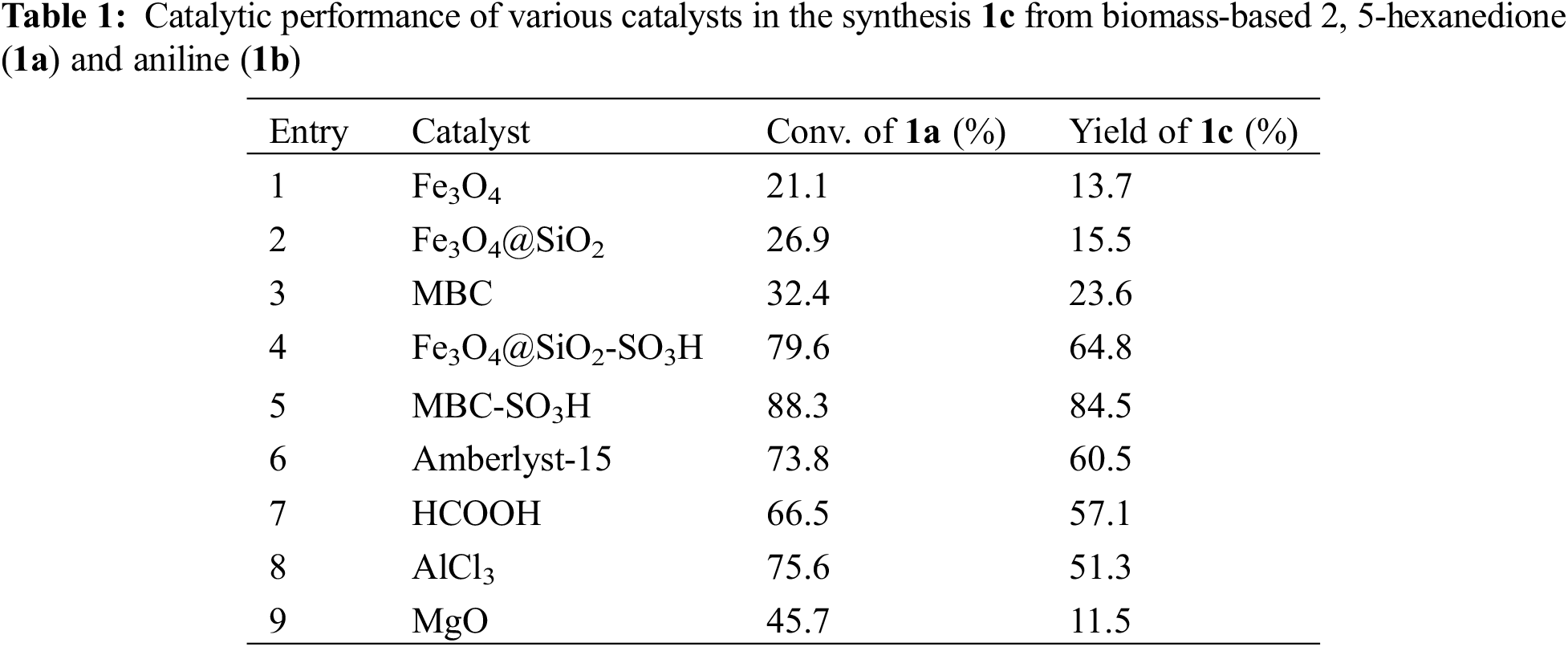
Based on the experimental conditions of catalyst screening, the whole reaction system was firstly optimized using temperature as a single variable, and 5 time periods were set at 25°C intervals, as summarized in Fig. 4a. The yield of 1c showed a volcano-shaped distribution with increasing temperature and reached up to 84.5% at 50°C, which was twice as high as that at 25°C (42.2% yield). The significant variation of the yield well demonstrated the sensitivity of the reaction process of synthesis 1c to temperature. It is speculated that the increase in temperature facilitates multi-step reactions such as amination, ring closure, and dehydration. Unexpectedly, the yield decreased by 12.9% as the temperature was sequentially elevated to 125°C. Therefore, the product analysis was performed by GC-MS to fully understand the sensitivity of the reaction to temperature (Fig. S1). In addition to 1c (Fig. S2), some byproducts like N-(5-(phenylimino)hexan-2-yl)aniline (1c’, Fig. S3), and N2,N5-diphenylhexane-2,5-diimine (1c’’, Fig. S4) were detected at 75°C and the amount increased with increasing temperature (Fig. S1). These results suggest that temperature can effectively promote the amination and dehydration process, but too high a temperature will lead to excessive amination. Therefore, more factors affecting the reaction should be considered to obtain a high yield of the target product 1c at 50°C.
Figure 4: Optimization of reaction conditions: (a) temperature, (b) time, (c) amount of catalyst, and (d) molar ratio of 1b to 1a
Then, the synthesis of 1c in 2 mL of anhydrous methanol using 1 mmol 1b and 1 mmol 1a as substrate in the presence of 5 wt.% MBC-SO3H at 50°C was investigated by varying reaction times. As shown in Fig. 4b, it can be found that the yield of 1c increased from 76.2% to 90.4% when the time was extended from 20 to 60 min. Notably, the yield (83.6%) of 1c decreased slightly as the reaction time continued to prolong to 100 min. It may be because pyrrole derivatives are unstable in an acidic environment and easily decompose into other nitrogen-containing compounds. Thus, 60 min was selected for the subsequent study considering a superior yield of 1c.
The yield of 1c was only 2.3% without any catalyst (Fig. 4c), which could be up to 93.3% when the amount of catalyst was 5 wt.% relative to 1a. However, the yield decreased to 88.0% and 79.8% with increasing the catalyst dosage to 7.5 wt.% or 10 wt.%. This result implied that a moderate increase in the catalyst amount could ensure that the acidic sites are not weakened and thus accelerate the reaction. However, an excessive number of acidic sites may lead to further conversion and decomposition of the target product. Hence, the catalyst amount of 5 wt.% was used for further experiments.
The basicity of the amine is an important parameter to accelerate the amination in the 1c synthesis. Moderately increasing the proportion of 1b was expected to accelerate the reaction. As shown in Fig. 4d, the yield of 1c increased from 93.3% to 98.7% when the molar ratio of 1b to 1a was increased from 1:1 to 1.2:1. However, When the proportion of 1b increased, the yield of 1c decreased accordingly. For example, when the ratio was 1.4:1, 1.6:1, and 1.8:1, the corresponding yield of 1c was 96.5%, 94.4%, and 91.7%, respectively. From the kinetic point of view, an excess of 1b can drive the reaction equilibrium toward the target product and thus accelerate the reaction rate. However, too high a concentration of amine may deplete some of the acidic sites in the catalyst or lead to over-amination, thus discouraging the reaction. Therefore, the appropriate molar ratio of 1b to 1a was considered to be 1.2:1.
In summary, the yield of 1c could reach 98.7% under the reaction temperature of 50°C, the reaction time of 60 min, the catalyst amount of 5 wt.%, and the molar ratio of 1b to 1a of 1.2:1.
The solvent effect on the synthesis of 1c was investigated under the above-optimized reaction conditions (Table 2). Low to moderate yields (Entries 1–5, 8) were obtained in toluene (25.7%), dichloromethane (40.3%), ethyl acetate (44.4%), tetrahydrofuran (51.3%), acetone (56.5%), and acetonitrile (60.1%). It was not difficult to find that the yields increased with the increasing polarity of the solvent. However, the yields in ethanol and methanol were higher than those of the more polar acetonitrile (Entries 6–8). This result indicated that in addition to solvent polarity, protonic solvents were also an important factor in accelerating the reaction. Protonic solvents were more conducive to the protonation of 1a and thus facilitated the amination process of the reaction. Meanwhile, the highest yield (98.7%) was achieved in the more polar methanol rather than in ethanol. It can be speculated that the strong polar proton solvent was more favorable for the protonation of carbonyl and hydroxyl groups, which significantly facilitates the amination and dehydration processes. Consequently, methanol was considered the optimal solvent.
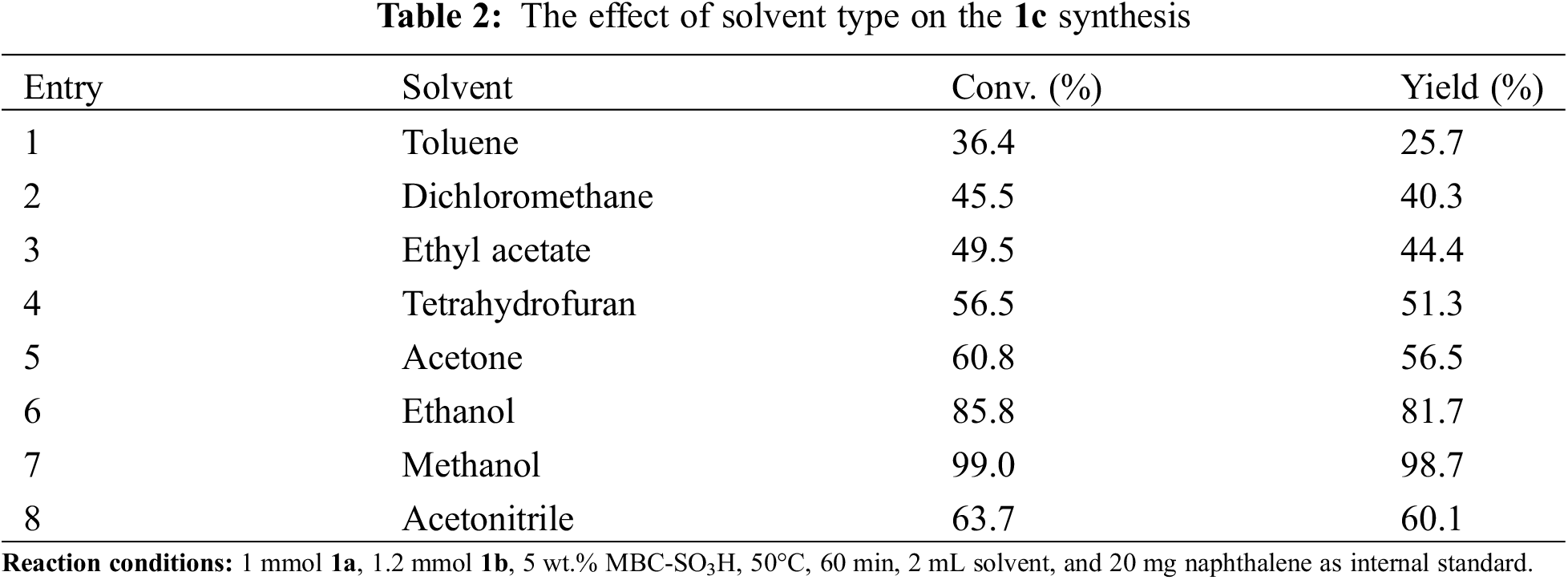
The range and universality of MBC-SO3H for the synthesis of N-substituted pyrroles were investigated under optimized conditions. Moderate to high yields were obtained by coupling various amines with 1a to produce N-substituted pyrroles in the presence of MBC-SO3H (Table 3). Initially, pyrroles could be synthesized by the Paal-Knorr reaction using aromatic amines. The 1c was obtained with excellent yield (98.8%) using 1b as a nitrogen source. When benzylamine was used as feedstock, the yield of 1-benzyl-2,5-dimethylpyrrole (2c) was acceptable with an appropriately prolonged reaction time, despite the low nucleophilicity and basicity of benzylamine. Further, the substrate range was extended to aromatic amines with electron-withdrawing and electron-donating groups to investigate the substituent effect. The reaction completion time of anilines containing the electron-withdrawing group (m-OH, p-Cl, p-Br, p-CF3, o-F, and m-NO2) was slightly longer than that of amines attached to the electron-donating group (o-CH3, o-CH3, m-CH3, o-OCH3, and p-OCH3). But it is far shorter than the required reaction time of 24 h reported in the past [25]. The aliphatic amines were also used as nitrogen sources after the successful experiments with aromatic amines. Reaction completion time of propylamine and n-amylamine with 1a to produce the corresponding N-substituted pyrrole (1-propyl-2,5-dimethylpyrrole 12c, and 1-n-amyl-2,5-dimethylpyrrole 13c) was shorter than that of aromatic amines, while the obtained yields were higher. The above analysis shows that two main factors can reduce the reactivity: (i) The anilines containing the electron-withdrawing group reduce the electron density on N, resulting in poor reactivity; however, the MBC-SO3H greatly reduces the dependence of reactivity on substrate nucleophile and basicity. (ii) The benzene ring is directly attached to the -NH2 group, which hinders the attack on N in space and thus reduces the reactivity.
The recyclability of the MBC-SO3H catalyst is critical, which will affect the economic viability of large-scale use of the catalyst in the production of N-substituted pyrrole derivatives.
The reusability of the MBC-SO3H was measured for 10 consecutive reaction cycles under optimized experimental conditions. Notably, MBC-SO3H could be quickly separated (15 s) by a magnet at the end of the reaction (Fig. 5a). This simple separation method has strong industry competitiveness in subsequent mass production. More importantly, the yield of 1c remained almost constant after 7 consecutive experiments (Fig. 5b), showing that the catalyst possesses excellent reusability. Further cycling found that the yield decreased by 8.5% after 10 cycles compared to the original. The N2 adsorption-desorption analysis showed that there was almost no change in the structure of the MBC-SO3H after 10 cycles (Fig. 5c, Table S1). The slight decrease in specific surface area and pore volume may be owing to the adsorption of the organic species (e.g., CH3OH). The average pore size of the catalyst was almost unchanged after 10 consecutive experiments (Fig. 5d, Table S1). It was speculated that the activity attenuation may result from the shedding of sulfonic acid groups (-SO3H). The reduced absorption intensity of the S element of MBC-SO3H in the XPS survey tentatively demonstrated this speculation (Figs. 5e and 5f). The results of the acid-base titration showed that the acid amount of the MBC-SO3H decreased by 0.53 mmol g−1 after 10 cycles (Table S1), fully confirming that leaching of -SO3H would directly lead to the decreased catalyst activity.
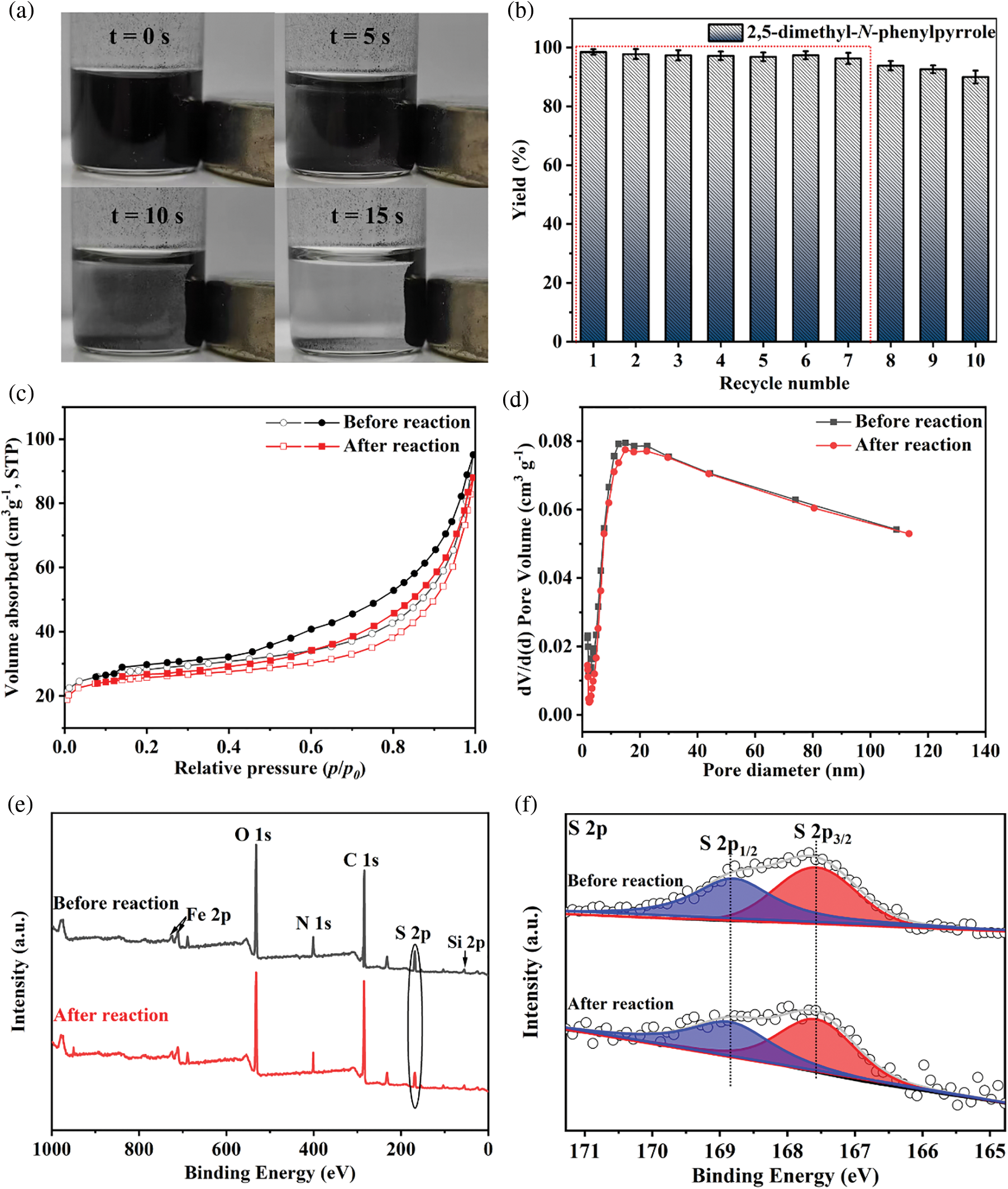
Figure 5: (a) Separation of MBC-SO3H from reaction systems. (b) The yields of 1c during 10 consecutive experiments. (c) N2 adsorption-desorption isotherms, (d) pore distribution, (e) XPS survey, and (f) S 2p XPS profiles of MBC-SO3H before and after the reaction
The Paal-Knorr reaction is a classic reaction whose mechanism has been extensively studied [29–31]. A reasonable catalytic mechanism can be proposed for the current catalytic system to explain the experimental phenomena, as summarized in Fig. 6. Firstly, the carbonyl group of 1a is activated by proton to form intermediate I in the presence of MBC-SO3H. In the subsequent amination process, the activated intermediate I is more likely to be nucleophile attacked by a nitrogen atom containing the lone pair electron in the primary amine to produce intermediate II. The intermediate III is offered after undergoing deprotonation and protonation. The formation of the pyrrole ring (intermediate IV) is also a nucleophilic N attack on the activated carbonyl carbon. Finally, the corresponding N-substituted pyrrole (VIII) is obtained by two-step dehydration under the action of acid catalysis. The reaction process goes through three processes including amination, ring-closure, and dehydration. The catalyst can effectively accelerate the amination and dehydration process by activating the carbonyl and hydroxyl groups. Notably, the nucleophilic attack is the key to the amination and closing ring, which fully demonstrates the dependence of the reaction on the substrate nucleophilicity. Thus, when anilines are used as the substrates, the electron-absorbing properties of phenyl will reduce the electron density of N and the availability of the lone pair of nitrogen. In this case, amination and cyclization require higher temperatures to compensate for the activation energy required for the reaction. From the perspective of the reaction mechanism, the existence of over-amination in product distribution is a complex process determined by many factors [22].
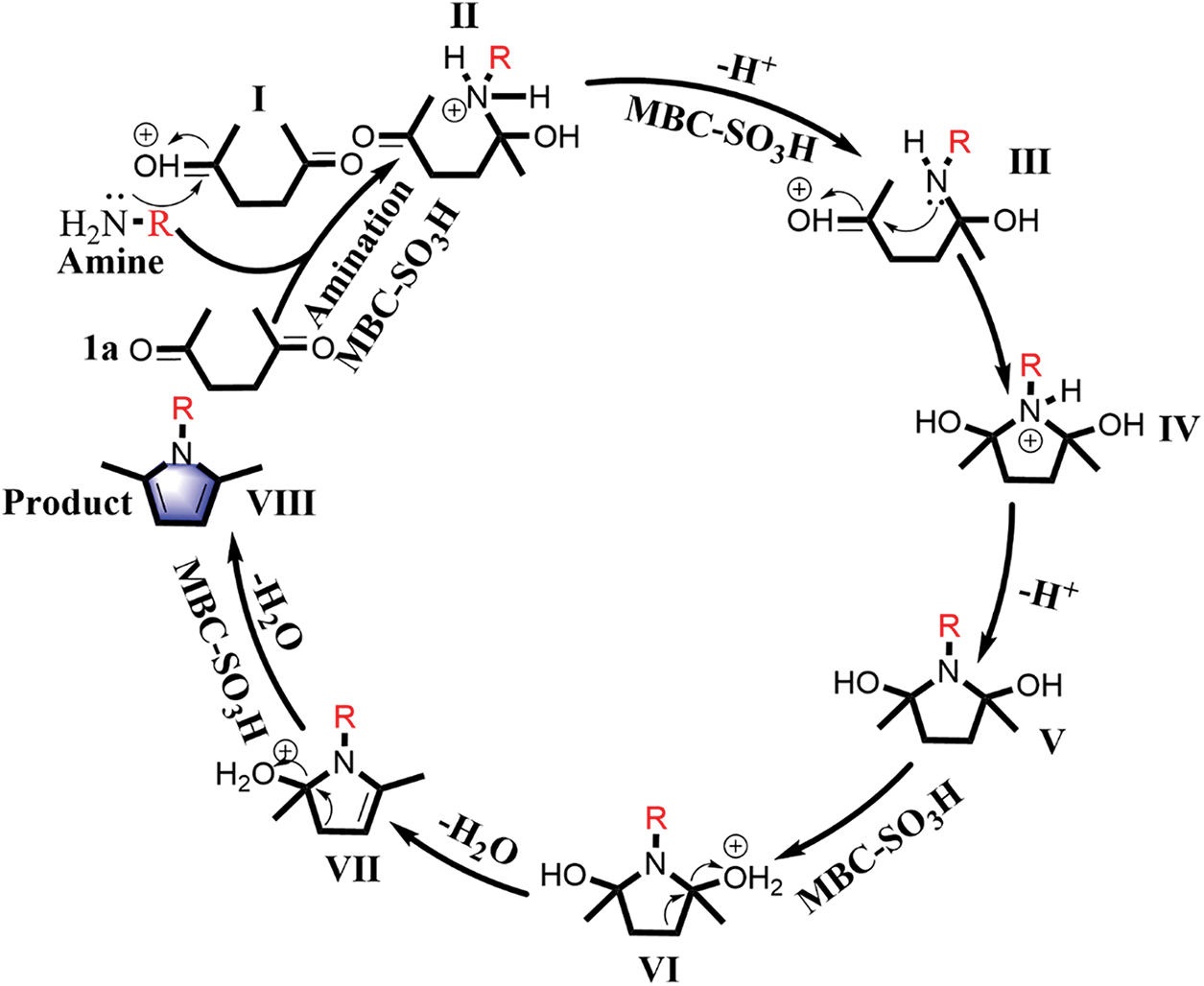
Figure 6: Plausible reaction mechanism for the synthesis of N-substituted pyrrole VIII from bio-based 1a and amine catalyzed by MBC-SO3H
In summary, a magnetic acid catalyst with core-shell structure (MBC-SO3H) was prepared and used as a heterogeneous catalyst for the efficient synthesis of N-substituted pyrroles with yields up to 99% from biomass-based 2,5-hexadione and amines under mild conditions. The moderate specific surface and mesoporous structure in MBC-SO3H were conducive to the substrate adsorption and mass transfer process. Importantly, abound Brønsted acid sites could effectively activate carbonyl and hydroxyl groups and thus accelerate amination and dehydration during the conversion process. The MBC-SO3H catalyst weakened the dependence of the reaction on the nucleophilicity of the amine so that the reaction could be completed quickly. MBC-SO3H exhibited good thermochemical stability with no activity decay after 5 cycles and possessed the great advantage of efficient and rapid separation from the reaction mixture. This study shows that magnetic core-shell materials can establish efficient conversion pathways from biomass to value-added N-substituted pyrroles.
Acknowledgement: The authors would like to thank all the editors and anonymous reviewers for their comments and suggestions.
Funding Statement: The study was funded by the Guizhou Provincial S&T Project (ZK[2022]011), Guizhou Natural Science Foundation (20201Y182), and College Students’ Innovation and Entrepreneurship Training Program (S202110657036).
Author Contributions: Study conception and design: Hu Li, Haoli Qin; Data collection: Zhimei Li; Analysis and interpretation of results: Zhimei Li, Kuan Tian, Keping Wang, and Zhengyi Li; Draft manuscript preparation: Zhimei Li, Kuan Tian. All authors reviewed the results and approved the final version of the manuscript.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Li, H., Riisager, A., Saravanamurugan, S., Pandey, A., Sangwan, R. S. et al. (2018). Carbon-increasing catalytic strategies for upgrading biomass into energy-intensive fuels and chemicals. ACS Catalysis, 8(1), 148–187. [Google Scholar]
2. Chen, X., Song, S., Li, H., Gözaydın, G. K., Yan, N. (2021). Expanding the boundary of biorefinery: Organonitrogen chemicals from biomass. Accounts of Chemical Research, 54(7), 1711–1722. [Google Scholar] [PubMed]
3. Fürstner, A. (2003). Chemistry and biology of roseophilin and the prodigiosin alkaloids: A survey of the last 2500 years. Angewandte Chemie International Edition, 42(31), 3582–3603. [Google Scholar]
4. Lee, D., Swager, T. M. (2003). Defining space around conducting polymers: Reversible protonic doping of a canopied polypyrrole. Journal of the American Chemical Society, 125(23), 6870–6871. [Google Scholar] [PubMed]
5. Pei, M., Wang, A., Xie, X., Hu, X., Liu, Y. (2022). Gold-catalyzed cyclization of ynones involving cis-hydrofunctionalizations: Rapid assembly of C-, O-, or S-functionalized pyrroles by a single methodology. Organic Letters, 24(7), 1541–1545. [Google Scholar] [PubMed]
6. Manal, A. K., Srivastava, R. (2023). Zr-KIT-6 catalyzed renewable synthesis of N-aryl pyrroles for producing bioactive synthetic compounds. Applied Catalysis A: General, 650, 119018. [Google Scholar]
7. Pyrrole Market Share, Size (2022). Consumption analysis by applications, future demand, top leading players, competitive situation, emerging trends and forecast to 2026. [Google Scholar]
8. Trautwein, A. W., Süßmuth, R. D., Jung, G. (1998). Hantzsch pyrrole synthesis on solid support. Bioorganic & Medicinal Chemistry Letters, 8(17), 2381–2384. [Google Scholar]
9. Wu, H., Li, H., Fang, Z. (2021). Hydrothermal amination of biomass to nitrogenous chemicals. Green Chemistry, 23(18), 6675–6697. [Google Scholar]
10. Gallezot, P. (2012). Conversion of biomass to selected chemical products. Chemical Society Reviews, 41(4), 1538–1558. [Google Scholar] [PubMed]
11. Hua, M., Song, J., Huang, X., Hou, M., Fan, H. et al. (2021). Support effect of Ru catalysts for efficient conversion of biomass-derived 2,5-hexanedione to different products. ACS Catalysis, 11(13), 7685–7693. [Google Scholar]
12. Cirujano, F. G., Leyva-Pérez, A., Corma, A., Llabres i Xamena, F. X. (2013). MOFs as multifunctional catalysts: Synthesis of secondary arylamines, quinolines, pyrroles, and arylpyrrolidines over bifunctional MIL-101. ChemCatChem, 5(2), 538–549. [Google Scholar]
13. Kim, B. H., Bae, S., Go, A., Lee, H., Gong, C. et al. (2016). Synthesis of two distinct pyrrole moiety-containing arenes from nitroanilines using Paal–Knorr followed by an indium-mediated reaction. Organic & Biomolecular Chemistry, 14(1), 265–276. [Google Scholar]
14. Cho, H., Madden, R., Nisanci, B., Török, B. (2015). The Paal–Knorr reaction revisited. A catalyst and solvent-free synthesis of underivatized and N-substituted pyrroles. Green Chemistry, 17(2), 1088–1099. [Google Scholar]
15. Wang, A., Sudarsanam, P., Xu, Y., Zhang, H., Li, H. et al. (2020). Functionalized magnetic nanosized materials for efficient biodiesel synthesis via acid–base/enzyme catalysis. Green Chemistry, 22(10), 2977–3012. [Google Scholar]
16. Chiang, Y. D., Dutta, S., Chen, C. T., Huang, Y. T., Lin, K. S. et al. (2015). Functionalized Fe3O4@silica core–shell nanoparticles as microalgae harvester and catalyst for biodiesel production. ChemSusChem, 8(5), 789–794. [Google Scholar] [PubMed]
17. Shao, D., Xu, K., Song, X., Hu, J., Yang, W. et al. (2009). Effective adsorption and separation of lysozyme with PAA-modified Fe3O4@silica core/shell microspheres. Journal of Colloid and Interface Science, 336(2), 526–532. [Google Scholar] [PubMed]
18. Hu, H., Wang, Z., Pan, L. (2010). Synthesis of monodisperse Fe3O4@silica core–shell microspheres and their application for removal of heavy metal ions from water. Journal of Alloys and Compounds, 492(1–2), 656–661. [Google Scholar]
19. Salehi, S., Hosseinifard, M., Mohammadi Meyabadi, A. (2023). Comparison of core–shell and hollow Fe3O4/silica/chitosan magnetic nanoparticles in vanadium removal: Experimental design and optimization analysis. Cellulose, 30, 2969–2996. [Google Scholar]
20. Sun, S., Zeng, H., Robinson, D. B., Raoux, S., Rice, P. M. et al. (2004). Monodisperse MFe2O4 (M = Fe, Co, Mn) nanoparticles. Journal of the American Chemical Society, 126(1), 273–279. [Google Scholar] [PubMed]
21. Pileni, M. P. (2003). The role of soft colloidal templates in controlling the size and shape of inorganic nanocrystals. Nature Materials, 2(3), 145–150. [Google Scholar] [PubMed]
22. Hyeon, T., Lee, S. S., Park, J., Chung, Y., Na, H. B. (2001). Synthesis of highly crystalline and monodisperse maghemite nanocrystallites without a size-selection process. Journal of the American Chemical Society, 123(51), 12798–12801. [Google Scholar] [PubMed]
23. Rockenberger, J., Scher, E. C., Alivisatos, A. P. (1999). A new nonhydrolytic single-precursor approach to surfactant-capped nanocrystals of transition metal oxides. Journal of the American Chemical Society, 121(49), 11595–11596. [Google Scholar]
24. Park, J., An, K., Hwang, Y., Park, J. G., Noh, H. J. et al. (2004). Ultra-large-scale syntheses of monodisperse nanocrystals. Nature Materials, 3(12), 891–895. [Google Scholar] [PubMed]
25. Elsayed, I., Mashaly, M., Eltaweel, F., Jackson, M. A. (2018). Dehydration of glucose to 5-hydroxymethylfurfural by a core-shell Fe3O4@SiO2-SO3H magnetic nanoparticle catalyst. Fuel, 221, 407–416. [Google Scholar]
26. Alemi-Tameh, F., Safaei-Ghomi, J., Mahmoudi-Hashemi, M., Teymuri, R. (2016). A comparative study on the catalytic activity of Fe3O4@SiO2-SO3H and Fe3O4@SiO2-NH2 nanoparticles for the synthesis of spiro [chromeno [2, 3-c] pyrazole-4, 3′-indoline]-diones under mild conditions. Research on Chemical Intermediates, 42, 6391–6406. [Google Scholar]
27. Ke, Q., Tang, C., Liu, Y., Liu, H., Wang, J. (2014). Intercalating graphene with clusters of Fe3O4 nanocrystals for electrochemical supercapacitors. Materials Research Express, 1(2), 025015. [Google Scholar]
28. Radu, T., Petran, A., Olteanu, D., Baldea, I., Potara, M. et al. (2020). Evaluation of physico-chemical properties and biocompatibility of new surface functionalized Fe3O4 clusters of nanoparticles. Applied Surface Science, 501, 144267. [Google Scholar]
29. Khaghaninejad, S., Heravi, M. M. (2014). Paal–Knorr reaction in the synthesis of heterocyclic compounds. Advances in Heterocyclic Chemistry, 95–146. [Google Scholar]
30. Minetto, G., Raveglia, L. F., Taddei, M. (2004). Microwave-assisted Paal−Knorr reaction. A rapid approach to substituted pyrroles and furans. Organic Letters, 6(3), 389–392. [Google Scholar] [PubMed]
31. Banik, B. K., Banik, I., Renteria, M., Dasgupta, S. K. (2005). A straightforward highly efficient Paal–Knorr synthesis of pyrroles. Tetrahedron Letters, 46(15), 2643–2645. [Google Scholar]

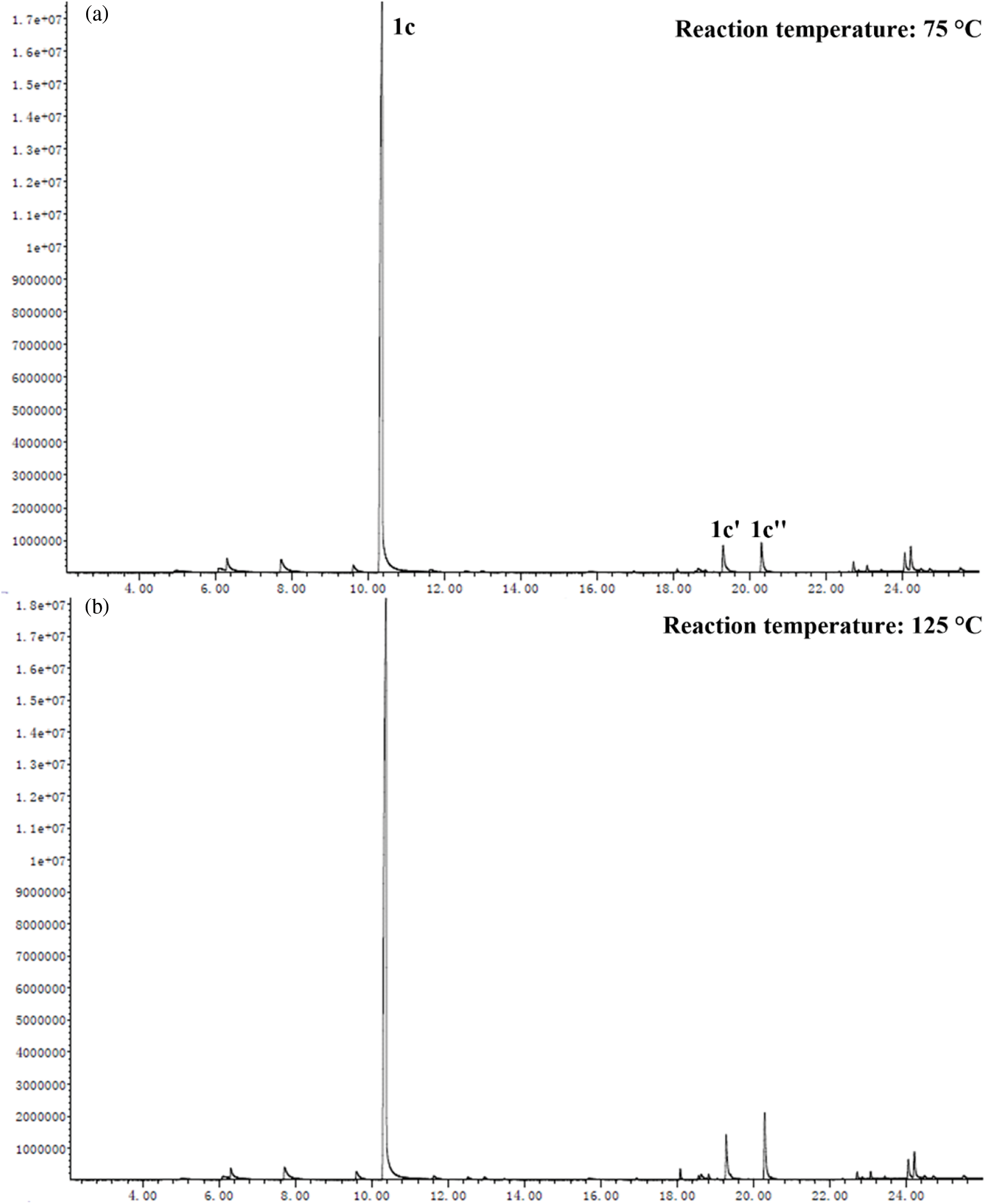
Figure S1: GC spectra of the liquid solution after reaction at (a) 75°C, and (b) 125°C
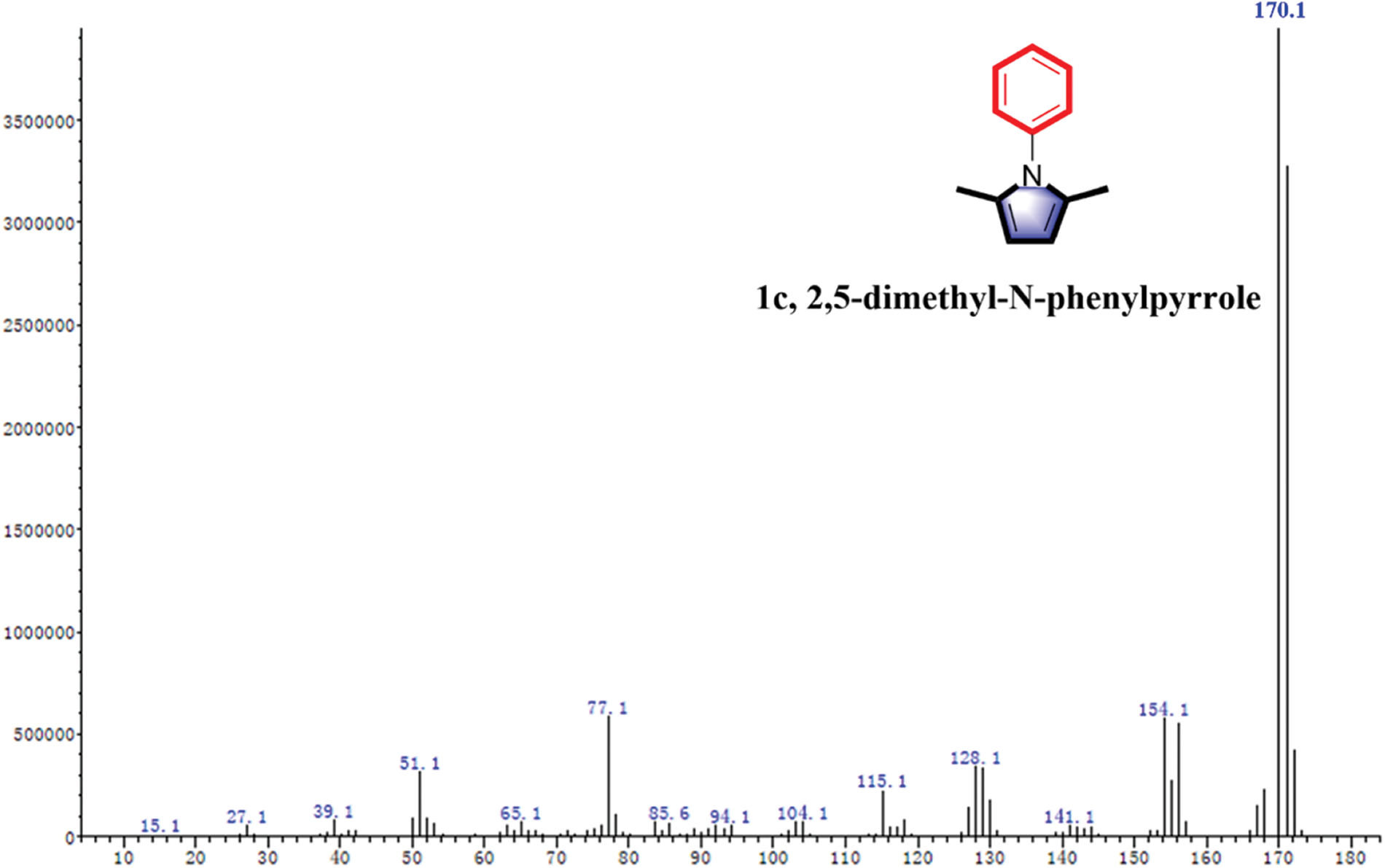
Figure S2: The MS spectrum of 1c
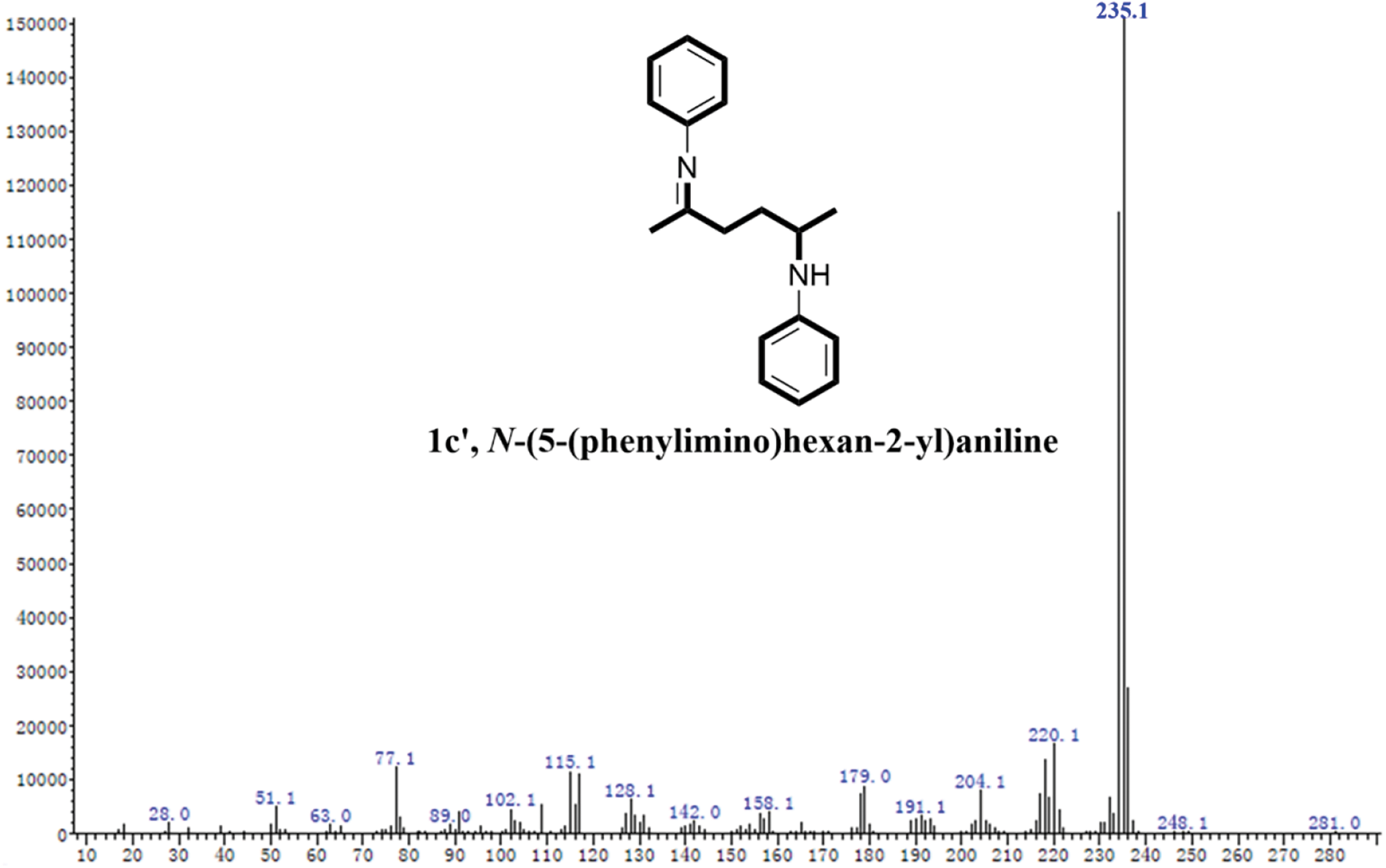
Figure S3: The MS spectrum of 1c’
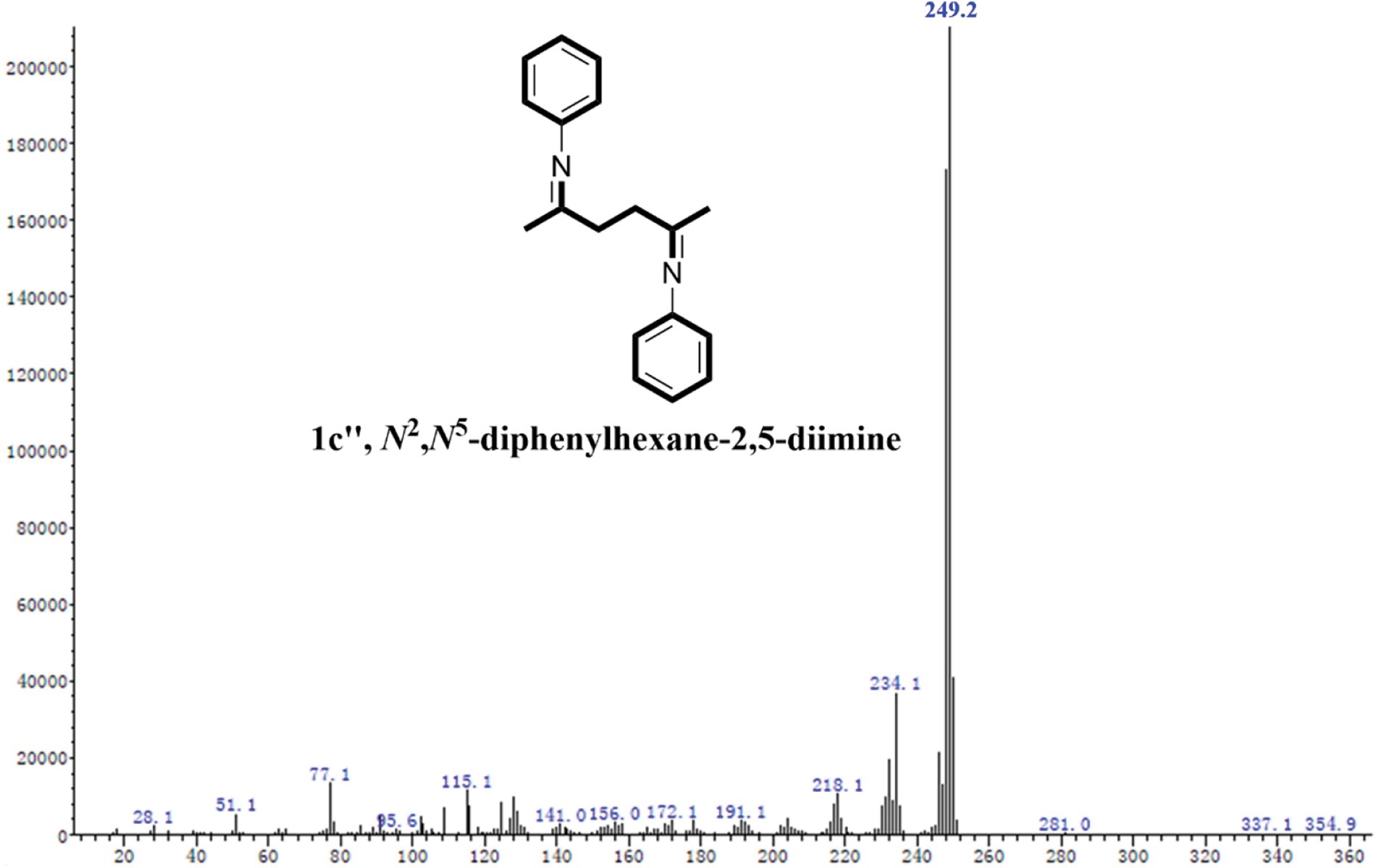
Figure S4: The MS spectrum of 1c”
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools