 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Investigation of Particle Breakdown in the Production of Composite Magnesium Chloride and Zeolite Based Thermochemical Energy Storage Materials
Heriot Watt University, Institute of Mechanical, Process and Energy Engineering, Edinburgh, UK
* Corresponding Author: Louis F. Marie. Email:
Energy Engineering 2023, 120(10), 2193-2209. https://doi.org/10.32604/ee.2023.043075
Received 20 June 2023; Accepted 17 August 2023; Issue published 28 September 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Composite thermochemical energy storage (TCES) represents an exciting field of thermal energy storage which could address the issue of seasonal variance in renewable energy supply. However, there are open questions about their performance and the root cause of some observed phenomena. Some researchers have observed the breakdown of particles in their production phase, and in their use. This study seeks to investigate the underlying cause of this breakdown. SEM and EDX analysis have been conducted on MgCl2 impregnated 13X zeolite composites of differing diameters, as well as LiX zeolite. This was done in order to study the level of impregnation of salt into the zeolite matrix, as well as the effect this impregnation process has on the morphology of the zeolite. Analysis was conducted using ImageJ software to study the effect of the impregnation process on the diameter of the particles. It has been found that a by weight impregnation concentration of magnesium chloride of 11.90% for the LiX zeolite, and 7.59% and 5.26% for the large diameter 13X zeolite and the small diameter 13X zeolite respectively has been achieved. It has been found that the impregnation process significantly affects the morphology of 13X zeolite particles, causing large fissures to form, and eventually resulting in the previously found breakdown of these particles. It has been verified that a primary factor influencing the breakdown of the 13X zeolite particles is the efflorescence and sub-fluorescence phenomena, which leads to a build-up of crystals in the zeolite pores. It has also been found that prolonged impregnation times and the use of high concentration salt solutions in the soaking process can induce significant crystal growth which also leads to the breakdown of these particles. Results demonstrate that LiX zeolite is the optimum host matrix choice in these conditions. These results will allow for the design of more resilient composite TCES particles.Graphic Abstract
Keywords
Nomenclature
| σcra | Fracture Stress |
| Kp | Fracture Toughness |
| r | Radius |
Thermal energy storage is needed to increase the usage of renewable energy technologies. Improved energy storage could allow for renewable sources of energy to contribute more to the energy mix. This can be done by making up for the key deficiency of many renewable energy technologies. This is the issue that renewable sources of energy are generally intermittent. In the case of solar energy, this intermittency occurs in two separate cycles: diurnally, there is only solar energy during the day; and seasonally, there is significantly more solar energy available during summer months. This seasonal solar cycle leads to an energy deficit in the winter months [1]. This is especially pronounced in climates further from the equator, where the seasonal deficit in supply is greater and the seasonal increase in demand is also higher resulting from fewer daylight hours and colder ambient temperatures. Currently, the diurnal nature of solar thermal energy can be tackled with either: effective sensible thermal energy storage, or phase change material (PCM) based thermal energy storage. However, neither of these methods is likely to prove effective at making up seasonal deficits due to their rate of energy loss and their relatively low energy density.
A field, which has recently seen a significant increase in interest, is the storage of energy utilising thermochemical energy storage (TCES) pairs. This is a very broad field with TCES pairs that cover temperature ranges suitable for domestic heat, industrial waste heat recovery (low-temperature industrial heat recovery), and the storage of energy at very high temperatures produced by concentrated solar power (CSP). A typical TCES system is capable of providing an energy density of 250 kWh/m3, when comparing this with a water-based thermal energy storage system (50 kWh/m3) there is a clear benefit to TCES [2]. In the case of industrial waste heat recovery, TCES provides a lower economic cost to transport heat, especially when compared to district heating, and phase change materials [3,4]. This is attributed to the high energy density achieved through TCES, which has also allowed for TCES to be used as an effective method of upgrading waste heat [5]. However, it has been shown that this is only economically viable when a significantly large waste heat source is present. Flegkas et al. [6] suggested that in their generic system utilising the reaction of MgO/Mg (OH)2 within a fluidised bed reactor, a waste heat source of less than 5 MW was not economically viable to attempt heat recovery.
Broadly, TCES pairs are divided into two types; adsorption (physisorption)-based TCES, where the primary reaction taking place is the physical reaction of the TCES pair, dominated by Van der Waals forces, and reaction (chemisorption)-based TCES, where the primary reaction taking place is the chemical reaction of the TCES pair, dominated by covalent forces. Reaction-based TCES is particularly suited to CSP due to its temperature range (300°C–1000°C), which is useful in power generation, and when the energy supply is at a high temperature [7]. However, this application has faced significant issues in the degradation of the chemicals used resulting from these high temperatures [8,9]. The use of fluidised bed reactors (FBR) to replace packed beds is a possible approach, which would have the benefit of reducing hot spots due to the isothermal fluid-like properties of an FBR. This reduction in hot spots can prevent excessive degradation of the TCES material originating from sintering and melting at extreme temperatures, however, this comes at the expense of the 2nd law efficiency of the system as the thermal stratification present in a traditional packed bed is eliminated [10].
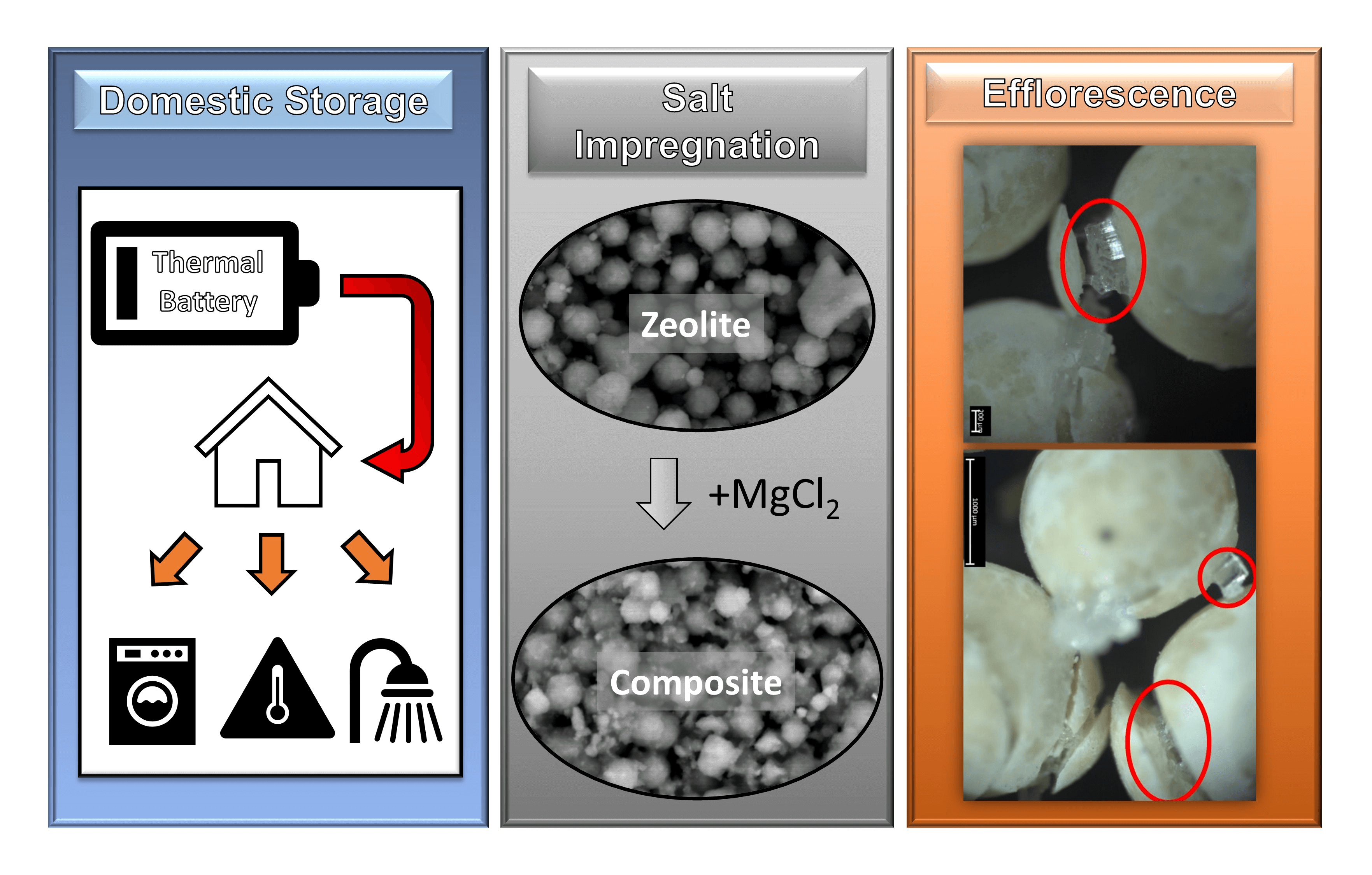
Sorption-based chemicals are suited to lower-temperature TCES pairs. These have been suggested for domestic hot water and domestic space heating due to their high energy density (when compared with sensible and latent thermal energy storage), and their low charging temperature, as well as their high cycle stability [11,12]. Initially, research in this field focused on the use of pure sorption materials (i.e., molecular sieves) such as zeolites and silica gels and more recently, metal-organic frameworks. Research demonstrated that these materials can be applied in district and domestic seasonal thermal energy storage [13–15]. These groups of materials have encountered issues with low energy density (<200 kWh/m3), meaning that any seasonal energy store would have to be very large to meet the seasonal deficit between energy demand and energy production [13]. Hence, efforts have been made to develop a class of materials which can meet the domestic demand to charge and discharge at a relatively low-temperature range (50°C−200°C), have a high cycle stability (50+ cycles), and have a high energy density (>400 kWh/m3). Composite TCES materials, which combine both sorption and reaction storage methods, have shown a potential to fit these requirements. The basic principle of composite TCES is to take a molecular sieve matrix and impregnate it with anhydrous salt [16,17] as indicated in Fig. 1. These composite TCES materials can significantly improve the sorption capacity through the combination of the physisorption of the molecular sieve, and the chemisorption of the salt hydrate impregnated into the sieve [18,19]. Composites also have the benefit of reducing agglomeration which leads to poor mass and heat transfer, a problem exhibited in pure anhydrous salt TCES. The impregnation of the salt into the molecular sieve takes advantage of their low propensity for agglomeration and cohesion, while maintaining higher energy density than standard molecular sieves [20].

Figure 1: Working principel of composite TCES [17]
Several potential molecular sieves have been proposed, as well as several anhydrous salts resulting in hundreds of possible composite TCES materials. However, the composite TCES must balance energy density with reaction rate, and cycle stability if a suitable composite TCES is to meet the needs of seasonal thermal energy storage. Early studies have seen success in composites utilizing zeolite (of various types) [20], vermiculite [21], and silica gel [11] as a host matrix. Hongois et al. [22] achieved an energy density of 166 kWh/m3 using a composite of magnesium sulphate impregnated into 13X zeolite, which represents a significant increase in the energy density of pure 13X zeolite in this setting (131 kWh/m3). Casey et al. [23] highlighted two vermiculite-based composites, the first impregnated with lithium bromide and the second with calcium chloride. These composites exhibited excellent sorption times but had low energy densities: 49.51 and 46.31 kWh/m3, respectively. Xu et al. [16] produced a composite of magnesium chloride impregnated into 13X zeolite and achieved an energy density of 308 kWh/m3. This increase in energy density is partially achieved by making use of both the absorption and deliquescence reactions of the magnesium chloride.
Despite very promising results obtained for many composites, there is potential to induce degradation of the sorption properties in the case of composite TCES [24]. In the case of Xu et al. [16], the cycling of these particles led to a very fast break-down after only a few cycles. The cracking observed can be seen in Fig. 2. Some authors have also found that in particular conditions TCES composites can degrade during their use, however, it has been reported that potassium carbonate-based composites do not degrade chemically [21,25].
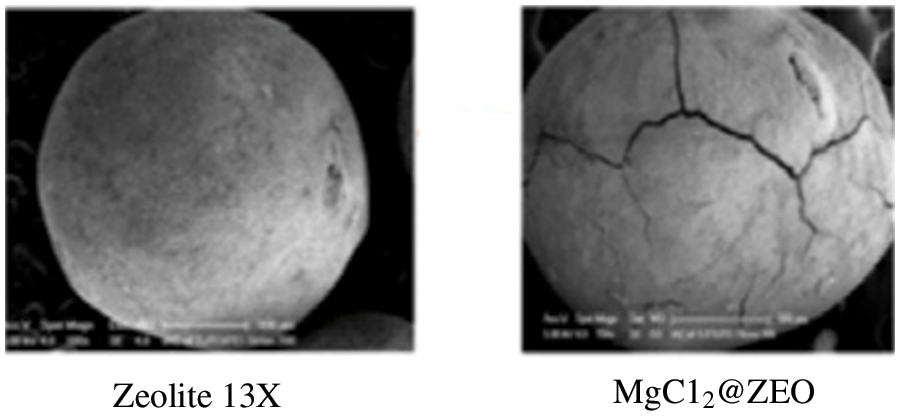
Figure 2: Cracking observed by Xu et al. [16]
One aspect that still lacks understanding, and has not been previously investigated, is the level of degradation of the host matrix that can be introduced during the production process of these TCES composites. If this could be better understood, and hence mitigated, then this could lead to increased cycle stability and extended time before TCES composites begin to degrade. Thus, the objective of this study is to investigate the level of degradation incurred and the factors influencing this within composite TCES materials. The mode by which these particles break down, and at which stage in the production process the particles experience breakdown, will be investigated.
Due to its increased heat release performance over other similar composites, a composite of 13X zeolite and magnesium chloride was selected (as in Xu et al. [16]) [26]. This was done for two reasons, the first being the very high energy density of the composite in this study, but also due to the visually observed, and remarked on cracking and breaking of the particle after several cycles.
2.2 Characteristics of Zeolites
In order to assess the effect of particle diameter on cracking and breaking, three composites have been prepared. One with 13X zeolite particles of Mesh 8–12 (1.68–2.38 mm), one with 13X zeolite particles Mesh 18–35 (0.5–1.0 mm), and one with LiX particles of Mesh 18–35 (0.5–1.0 mm). This has been done to look at whether 13X zeolite particles themselves are simply more susceptible to cracking and breaking, or whether the diameter of the particle is the variable influencing this. In order to verify the size of these particles, optically magnified images were taken of a sample of the particles; these images were processed using specialized image processing software (NIH ImageJ), processed images can be seen in Fig. 3.

Figure 3: ImageJ analysis image of small diameter particles (left); and large diameter particles (right)
In the analysis of the smaller particles, 1705 measurements were taken. These measurements resulted in a measured mean diameter of 0.636 mm (±0.075 mm, SD = 1), which is in keeping with the reported Mesh 18–35 (0.5–1.0 mm). It was noted that approximately 4% of the particles measured were below the 0.5 mm lower limit and none were above the 1.0 mm upper limit in diameter. This could possibly be due to some of the particles breaking apart in transit. In the analysis of the larger particles, 712 measurements were taken. These measurements resulted in a mean diameter of 2.028 mm (±0.285 mm, SD = 1), which is again in keeping with the reported Mesh 8–12 (1.68–2.38 mm). Again, it is noted that there are some outliers measured: 2.5% of particles measured had a diameter of less than 1.68 mm and 4.1% of particles measured had a diameter of greater than 2.38 mm. This is attributed to small errors in the production of the particles, and from breakages in the particles (in the case of the smaller threshold).
The particles were impregnated and prepared as follows: A 10% w/w solution of MgCl2 in deionized water was prepared at 80°C. Xu et al. [16] investigated much higher salt concentrations (up to 40% w/w), however as seen by Touloumet et al. [27], such high concentrations can significantly reduce pore volume. Xu et al. [16], also investigated 10% w/w solution and observed fracturing so this concentration is representative of similar concentrations found in the literature. This salt concentration was selected to balance the effects of the amount of impregnated salt and the corresponding reduction in observed pore volume. The zeolite particles were then immersed into this solution for a period of 72 h to allow for total impregnation of the salt into the zeolite matrix. A control was prepared in the same way but using deionised water instead of the salt solution. After soaking, samples were taken for analyses while the remaining materials were placed in a laboratory furnace and kept at 200°C for 3 h to activate the composite. This process is shown in Fig. 4. Further, the samples were sealed in ambient conditions and analysed using energy dispersive X-ray (EDX) to ascertain the level of salt impregnation and using scanning electron microscopy to investigate the morphology of the samples, as well as investigate the cracking phenomenon. To summarise, the analyses were conducted for three sample sets: pre-soaking, post-soaking and post-activation for both processes—with salt solution and with the deionized water.

Figure 4: Methodology of particle production
The previously described ImageJ analysis was then conducted on the particles to investigate whether the soaking process resulted in an increase in the diameter of the particles, as any increase in the particle diameter could result in a strain increase leading to the previously observed cracking and breaking.
2.4 Instrumentation and Equipment
Equipment and instrumentation used were as follows: The salt solution was produced using a Fisherbrand Isotemp hotplate stirrer combination, which is able to reach and maintain sufficient temperatures to assist dissolving of the salt. The furnace used was a Carbolite Gero PF laboratory oven with capabilities up to 300°C. The zeolite imaging was conducted with a Sony IMX703 optical sensor as well as a Zeiss Stemi 508 Greenough stereo microscope and processed using ImageJ software on a standard HP personal computer. SEM and EDX analysis were conducted with a Quanta 650FEG EDM scanning electron microscope.
EDX analysis of the large and small diameter zeolites demonstrates two interesting differences in the chemical composition of the zeolites. The amount of aluminium is comparable between the small diameter LiX zeolite and the large diameter 13X zeolite but is lower in the small diameter 13X zeolite. However, very little sodium (0.45 Mass %) is found in the LiX zeolite, compared to in the 13X zeolites. This is as expected given that 13X zeolite is comprised of aluminosilicate and sodium. There are also carbon atoms present in the small-diameter zeolites (7.92 Mass %), as presented in Table 1.
In all cases of both the small and large diameters and the LiX and 13X zeolites composites, EDX analysis shows that there is a clear increase in magnesium and chloride content. This indicates the impregnation process was successful. However, the degree of this success differs between the three samples. In the case of the small diameter LiX zeolite, a degree of 11.90% by weight impregnation of magnesium chloride was achieved from an initially measured 0%. In the case of the large diameter 13X zeolite, a degree of 7.59% by weight impregnation of magnesium chloride was achieved from an initially measured 1.11 %. Finally, in the case of the small diameter 13X zeolite, a degree of 5.26% by weight impregnation of magnesium chloride form an initially measured 0.71%. This is presented in Table 2.

The ratio of elements for each combination was analysed before and after the impregnation process to determine the salt content. This was done in order to directly compare values with Xu et al. [16]. The ratio between silicon and aluminium (Si:Al), sodium and aluminium (Na:Al), and magnesium and aluminium (Mg:Al) for each set of the samples are shown in Table 3. It can be seen that for the LiX zeolite, the ratio between silicon and aluminium remains similar between the pure zeolite and the composite material. This is also observed for the small diameter 13X zeolite. Such a small change (1.418 to 1.365 g/g) in the ratio between silicon and aluminium can also be seen in the large diameter 13X zeolite, although there is a slightly larger change. In all samples, a decrease in the ratio of sodium to aluminium can also be observed between the pure zeolite and the composite. What is more, for all samples, an increase in the ratio of magnesium to aluminium can be observed between pure zeolite and the composite. This data indicates that some sodium cations (Na+) have been exchanged for magnesium cations (Mg2+). The salt solution of 10% produced a ratio of Mg:Al in the large diameter 13X zeolite composite of 0.44, and a ratio of 0.325 in the small diameter 13X zeolite composite. Xu et al. [16] observed ratios of 0.388–0.525 between solution concentrations of 9% to 13%. These results show that the method reported by Xu et al. [16] is replicable (however, the mesh size of the particles investigated in Xu et al. is unknown). In the case of the LiX zeolite, the likely source of the ion exchange that has allowed impregnation is through the exchange between Li2+ and Mg2+ cations as observed by Guan et al. [30]. The EDX analysis is not able to detect the lithium present in the LiX zeolite. Lithium is too light for detection by standard EDX and can only be detected when the EDX detector is optimized for extremely low-energy X-rays (<0.1 keV) [31].
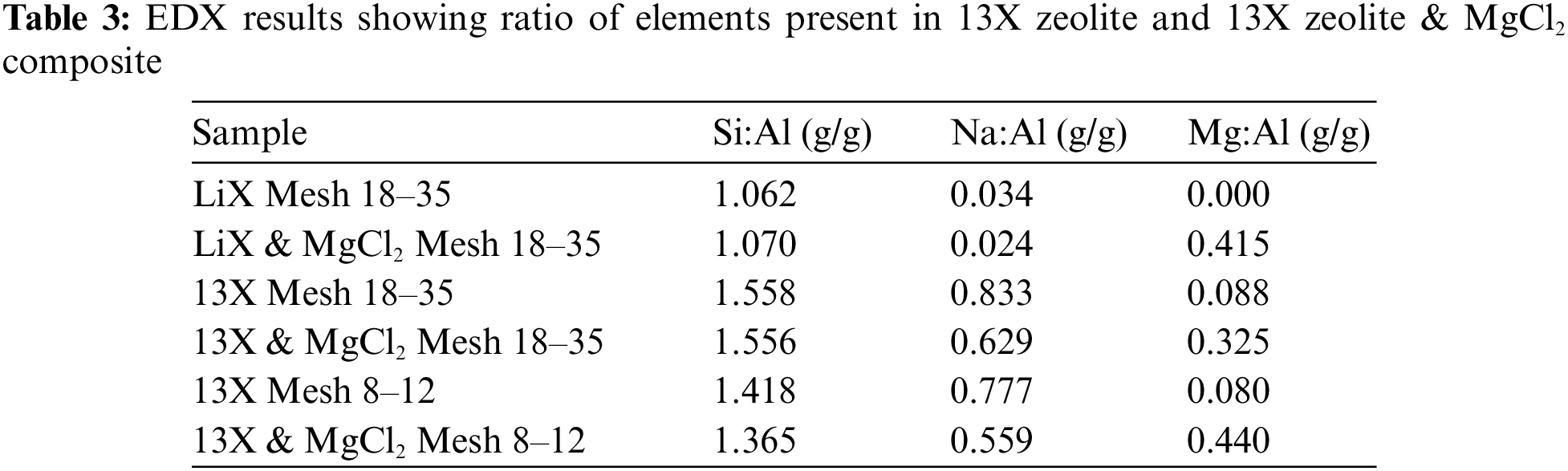
Given the small diameter LiX zeolite has a magnesium and chloride content of 11.90% by mass, the small diameter 13X zeolite has 5.26%, and the large diameter 13X zeolite has 8.70%, the theoretical energy density of this composite can be calculated if the energy density of the host zeolite is known. Janchen et al. [32] calorimetrically measured the heat of adsorption of LiX zeolite and 13X zeolite at 343 K, and measured heat of 59.8 kJ/mol (of water adsorbed) and 51.3 kJ/mol (of water adsorbed). This corresponds to a volumetric energy density of 105.60 kWh/m3 for the LiX zeolite and a volumetric energy density of 75.90 kWh/m3 for the 13X zeolite. As the heat of the reaction from anhydrous magnesium chloride to magnesium chloride hexahydrate is 164.3 kJ/mol, the theoretical energy density of each material can be calculated. These values are presented in Fig. 5. It should be noted that the actual energy density may differ as a result of inhomogeneous impregnation, and that these values are not universal. As the primary study of this work is to investigate material breakdown, the accurate measurement of the actual energy density of the materials produced is out of the scope of the study.
Figure 5: Theoretical energy storage density of composite materials
Scanning electron microscope (SEM) images of the small diameter LiX and 13X zeolites visually demonstrate the successful impregnation of MgCl2 into the zeolite. Fig. 6 shows the zeolites before and after impregnation. MgCl2 can be observed within the micropores of the zeolite after impregnation, in a similar fashion to the diagram in Fig. 1. On these small diameter zeolites, no cracking was observed in any of the scans taken of the zeolite prior to impregnation and after impregnation. SEM images taken of the large diameter 13X zeolite show little difference between the micropores of the zeolite prior to impregnation and post-impregnation (Fig. 6). Significantly however, large cracks in the surface of the zeolite are observed in the post impregnation composite material. These cracks were not visible in images taken of the pre-impregnation zeolite. It is likely that the previously studied efflorescence [33] of the salt has exacerbated this fracturing phenomenon in the large diameter 13X zeolite. Clearly, some supersaturation has occurred at the surface of the zeolite, resulting in this damage to the zeolite morphology, however, this damage is not visible when viewing the macro-morphology of the zeolite. This efflorescence has not been observed in the smaller diameter 13X zeolite, so it is likely that the pore radius is in fact smaller in this zeolite, leading to insufficient supersaturation within the zeolite pores.

Figure 6: SEM image of LiX zeolite, Mesh 18–35 (top left); SEM image of LiX zeolite impregnated with MgCl2, Mesh 18–35 (top right); SEM image of 13X zeolite, Mesh 18–35 (middle left); SEM image of 13X zeolite impregnated with MgCl2, Mesh 18–35 (middle right); SEM image of 13X zeolite, Mesh 8–12 (bottom left); SEM image of 13X zeolite impregnated with MgCl2, Mesh 8–12 (bottom right)
In order to verify that no cracking or other significant effects were produced in the morphology of the small-diameter zeolites during the impregnation process, further analysis was conducted on the material at each stage of the process. In all cases, no change in the morphology of the material was observed. In the case of the control, this is to be expected as the zeolite TCES has been shown to be stable over more than 60 cycles (particle diameter = 1.5 mm) [34]. As this process of soaking (discharging) and activating (charging) is comparable to a TCES cycle it demonstrates that, as expected, this zeolite does not incur any visible damage after a cycle. This is also the case for the impregnated 13X zeolite (small diameter) at each stage of the impregnation process. This can be compared to the larger diameter 13X zeolite which clearly incurs significant damage to the morphology, which is likely to lead to the breakdown of the particles as described by Xu et al. [16].
In order to investigate the cracking phenomenon further and to verify if cracking leads to the breakdown of the particles, magnified images of several hundred particles were analysed as described in the methodology. This analysis measured the cross-sectional area of each individual particle. A distribution of the particle diameters of pure 13X zeolite is presented in Fig. 7. As indicated in the methodology, these measurements resulted in a mean diameter of 2.028 mm (±0.285 mm, SD = 1). Nine samples measured are significantly outside the distribution, the smaller ones can be attributed to some very small diameter pieces of zeolite which have already broken off due to flaws in the manufacture and transport process. Likewise, a few large diameter zeolite beads have been measured to be larger than the maximum diameter reported (Mesh 8–12); these can be attributed to errors in the manufacturing process. These large particles can be clearly seen in Fig. 3. Despite this, the vast majority of the particles analysed are clearly seen to fall within the manufacturer’s specified diameter range.
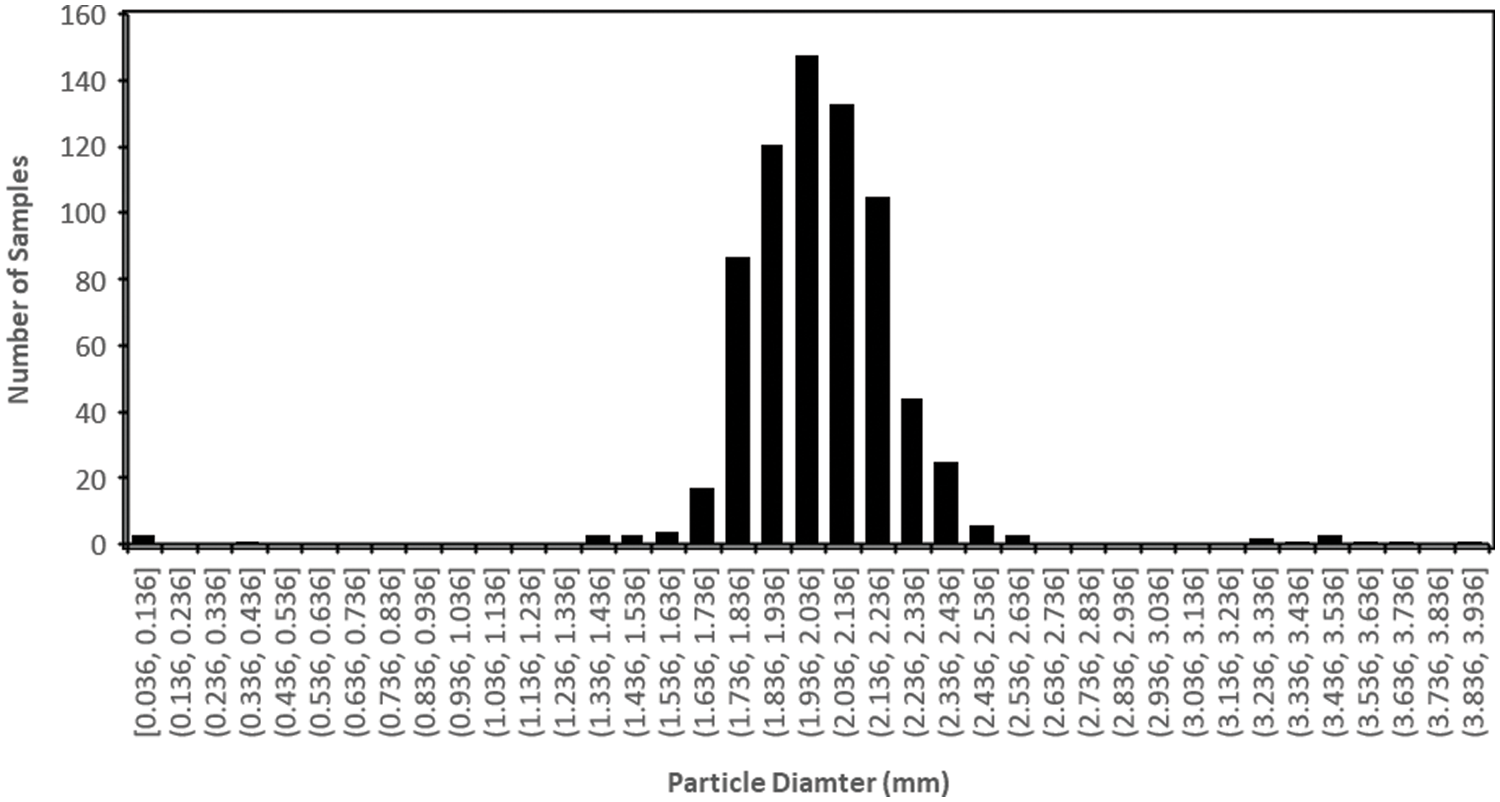
Figure 7: Histogram showing large diameter 13X zeolite particle size prior to impregnation process
The large-diameter 13X zeolite (Mesh 8–12) which was soaked in 10% magnesium chloride solution was also analysed after impregnation. The results are presented in Fig. 8. It is clear that the distribution is centred around a lower diameter (1.883 mm). The proportion of particles that are well outside the expected range appears to have remained the same. Formation of a new, small diameter distribution, and the skewing of the left side of the central distribution is apparent in Fig. 8. This evidences that particles are fracturing. The skewing of the central distribution on the left side suggests that larger particles are being broken into multiple pieces and large pieces are being broken off some particles. The new small diameter distribution originates from very small pieces of particles which have broken off.

Figure 8: Histogram showing large diameter 13X zeolite particle size after soaking in MgCl2 solution
It should be noted that the smallest diameter bucket/bin in Fig. 8 represents the lower limit to the diameter that can be measured by this method, but not the smallest particle than can be detected. Hence, many of the particles placed in this bin a likely to have smaller diameters than those that can be measured. Xu et al. [16] and de Gennaro et al. [34] suggested that cracking and breakdown of the particles were initiated by the expansion of the zeolite matrix during the soaking process. Results presented in Fig. 8 are in agreement with Xu et al. [16] and de Gennaro et al. [34] as it is clear there is some damage to the particles immediately following hydration. However, there is little difference in the mean diameter between the particles pre-soaking and post-soaking in the salt solution. Of the particles after soaking, a mean diameter of central distribution of 2.037 mm (±0.378 mm, SD = 1) was measured; this result will also be influenced by the skew of the central distribution toward the lower diameter. This is only marginally larger (0.44% change, the imaging has a precision of ±0.003 mm) than the measured diameter of 2.028 mm (±0.285 mm, SD = 1) prior to soaking. As expected, the overall measured mean diameter has reduced to a value of 1.883 mm (±0.630 mm, SD = 1) after the overall impregnation process is completed (i.e., after soak and activation); this is due to the increase in small diameter particles as a result of the cracking and breaking.
Evidentiality, the 13X particles differ in their resistance to cracking during the impregnation (and by extension charge/hydration) process. The large diameter 13X zeolite exhibited micro-fractures on its surface as seen under SEM imaging, whereas for the small diameter 13X and LiX zeolites such changes were not observed. The large diameter 13X zeolite also experienced a reduction in average diameter after the impregnation process. This was driven by the fracturing of the larger particles in the range into smaller particles of diameters approximately half the original diameter. There has also been a large increase in the number of particles of less than 180 µm in diameter after the impregnation process of the Mesh 8–12 (large) 13X zeolite. A particle will crack and break if the stress in the particle is higher than the Griffith criterion; the equation for which is given in Eq. (1) [35]. Where
As such, the particle sizes would be expected to crack at the same fracture stress. The surface strain resulting from an increase in diameter will be the same for both particles (calculated to be 0.0089).
While it is clear from the SEM images (Fig. 6) that salt impregnation into 13X zeolite can cause fissures and cracks to appear in the particle, optical imaging of these particles after a prolonged impregnation process demonstrates the effect that salt crystals can have on the zeolite particle on a macro scale (Fig. 9). Evidently, these regions of crystal growth have resulted in cracking and breakdown of the 13X zeolite particle on a macroscale. This phenomenon is as a result of the sub-florescence and the previously discussed efflorescence [36]. The pore-diameter in the large diameter zeolite is large enough that the supersaturation of salt in the pores has resulted in extensive crystal growth and fracturing. The prolonged impregnation time (168 h) here, as compared to the 72 h utilised in the initial methodology, has resulted in deeper penetration of the salt and extensive sub-florescence. It was not observed when exposing LiX zeolite to a prolonged period of crystal growth, which can be seen in Fig. 10. However, during the prolonged impregnation of the large diameter 13X zeolite, much of the water present at the beginning of the impregnation process was not present at the end, which suggests that the water evaporated or was absorbed. The reduction in water content will have resulted in an increase in the concentration of the solution remaining to a degree resulting in a supersaturated solution, which has triggered the excessive growth of crystals. This is evidenced by the lack of crystal growth in Fig. 10, where there is clearly still solution present on the surface of, and between the particles. From this, it is clear that the control of solution and concentration during the impregnation process is vital in order to avoid fracturing driven by florescence.
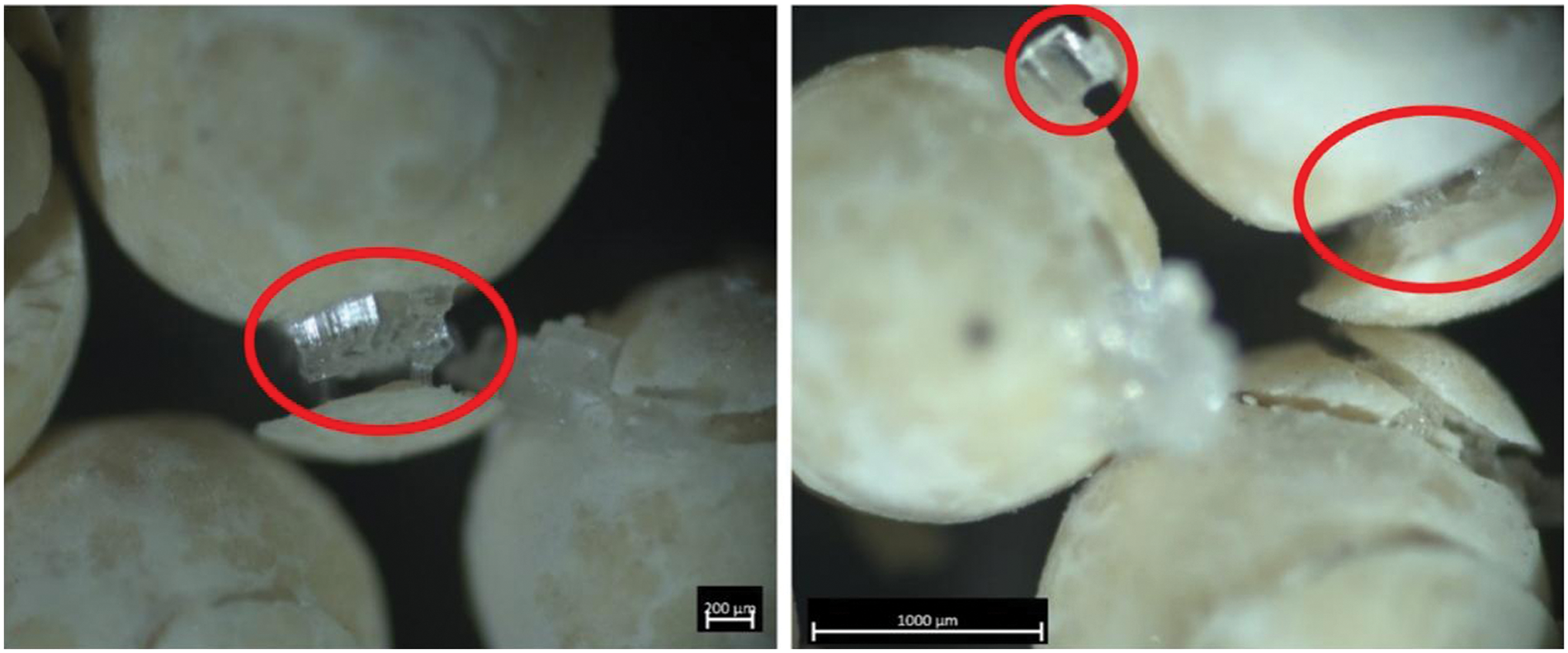
Figure 9: Optical microscopy image of 13X zeolite (Mesh 8–12) impregnated with MgCl2 following a prolonged impregnation time of approximately 168 h. Areas circled in red show regions of significant crystal growth

Figure 10: Optical microscopy image of LiX zeolite (Mesh 18–35) impregnated with MgCl2 following a prolonged impregnation time of approximately 168 h
Hence, care should be taken to avoid high-concentration solutions and long impregnation times when producing composite zeolites, as the high concentration can encourage crystal growth. This crystal growth can induce significant breakdown of the zeolite particles.
The breakdown of the particles into smaller diameter particles will have a great effect on the mass and heat transfer properties of the particle resulting from the change in surface area. The particles will also experience a large increase in cohesiveness, which can present problems such as limiting the mass transfer between the particles and any of their sorbate. The breakdown of the particles could also affect the application of these particles in novel thermochemical reactor types, which are beginning to be investigated in the field of thermochemical energy storage [37,38]. Particle beds of disparate diameters will experience non-ideal mixing, and particles of smaller diameters but the similar density will be in different Geldart classes, and thus fluidise differently [39,40]. Moreover, the effect on the performance of a fluidised bed in this setting is not well understood. It should also be investigated whether the breakdown of particles causes any loss to the salt impregnated into the pores. The sorption capacity of the composite over several repeated thermochemical cycles has been suggested to remain almost the same, but the issue of loss of material could potentially exhibit itself after many cycles [16].
It is clear that this method of impregnation can successfully produce composite thermochemical energy storage materials. In the case of impregnation, similar results to those already available in the literature were attained. A TCES material based on LiX zeolite has a high theoretical energy storage density, and also did not exhibit any cracking at any stage during the production of the composite. A TCES material based on 13X zeolite has also been produced, with a high theoretical energy storage density. However, the degree of impregnation resulting from the same method used to impregnate the LiX zeolite is significantly lower (by weight impregnation of magnesium chloride: 11.90% for the LiX zeolite, and 7.59% and 5.26% for the large diameter 13X zeolite and the small diameter 13X zeolite, respectively). The theoretical volumetric energy density of 143.25, 104.66, and 95.54 kWh/m3 for the LiX composite, the large diameter 13X composite, and the small diameter 13X composite, respectively. This demonstrates that LiX zeolite is able to be impregnated to a significantly higher degree (at the same solution concentration), allowing for a higher theoretical density to be achieved.
The LiX zeolite did not crack during discharging or charging. However, as previously observed, 13X zeolite of a large diameter impregnated with magnesium chloride cracks and breaks down [16]. This finding has been verified, and it has been demonstrated that the impregnation and activation process introduced cracks into the morphology of the large-diameter 13X zeolite composite. It has also been demonstrated that these particles begin to break apart even prior to their activation, and that the hydration step of the production generates many small pieces of composite broken off from the whole. The supersaturation of solution in the pores of the zeolite has resulted in efflorescence and sub-florescence-driven fracturing. This is something that has been shown to be effectively mitigated in smaller diameter particles through smaller pore diameters, and better concentration control, in order to avoid supersaturation.
The final interesting observation in the study is the propensity for crystal growth to induce the breakdown of particles in cases where these crystals are allowed to grow. If particles are left soaking in the solution for long periods, crystal growth has been observed breaking down these particles. However, this is likely due to the water loss resulting in a significant increase in the concentration of the soaking solution. High concentration will have allowed crystals to begin to form, and over the long soaking time grow into the matrix of the zeolite, until the sub-florescence results in fracturing. These results could possibly be applicable to other salts such as CaCl2 and MgSO4, provided the pore size of the zeolite is correctly sized to avoid particle breakdown.
The utilisation of alternative zeolite matrices such as LiX zeolite has the benefit of producing a composite of slightly higher theoretical energy storage density and is more easily impregnated with magnesium chloride. If these composites are to be used in moving and fluidised beds where particle size is especially important, then robust particles which do not breakdown through production should be considered as an important element in the selection of particles. As such, LiX zeolite is the preferential host matrix in these conditions.
Acknowledgement: The authors would like to acknowledge Ms. Saioa Ilincheta (Heriot-Watt University, Edinburgh) for her invaluable technical support of the work conducted.
Funding Statement: This research is funded by the UKRI DTP Scholarship: EP/R513040/1.
Author Contributions: Study conception and design: Louis F. Marie and Tadhg S. O’Donovan. Interpretation of results: Louis F. Marie and Karina Sałek. Manuscript draft preparation, data collection and data analysis: Louis F. Marie. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: Data available on request.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Böhm, H., Lindorfer, J. (2019). Techno-economic assessment of seasonal heat storage in district heating with thermochemical materials. Energy, 179, 1246–1264. [Google Scholar]
2. Kerskes, H. (2016). Thermochemical energy storage. Storing energy: With special reference to renewable energy sources. Netherlands: Elsevier. https://doi.org/10.1016/B978-0-12-803440-8.00017-8 [Google Scholar] [CrossRef]
3. Jarimi, H., Aydin, D., Yanan, Z., Ozankaya, G., Chen, X. et al. (2019). Review on the recent progress of thermochemical materials and processes for solar thermal energy storage and industrial waste heat recovery. International Journal of Low-Carbon Technologies, 14(1), 44–69. [Google Scholar]
4. Fernández, A. I., Barreneche, C., Miró, L., Brückner, S., Cabeza, L. F. (2015). Thermal energy storage (TES) systems using heat from waste. Advances in thermal energy storage systems: Methods and applications. UK: Woodhead Publishing Limited. https://doi.org/10.1533/9781782420965.4.479 [Google Scholar] [CrossRef]
5. Li, T. X., Wang, R. Z., Yan, T., Ishugah, T. F. (2014). Integrated energy storage and energy upgrade, combined cooling and heating supply, and waste heat recovery with solid-gas thermochemical sorption heat transformer. International Journal of Heat and Mass Transfer, 76, 237–246. [Google Scholar]
6. Flegkas, S., Birkelbach, F., Winter, F., Groenewold, H., Werner, A. (2019). Profitability analysis and capital cost estimation of a thermochemical energy storage system utilizing fluidized bed reactors and the reaction system MgO/Mg(OH)2. Energies, 12(24), 4788. https://doi.org/10.3390/en12244788 [Google Scholar] [CrossRef]
7. Pardo, P., Deydier, A., Anxionnaz-Minvielle, Z., Rougé, S., Cabassud, M. et al. (2014). A review on high temperature thermochemical heat energy storage. Renewable and Sustainable Energy Reviews, 32, 591–610. https://doi.org/10.1016/j.rser.2013.12.014 [Google Scholar] [CrossRef]
8. Kato, Y., Yamada, M., Kanie, T., Yoshizawa, Y. (2001). Calcium oxide/carbon dioxide reactivity in a packed bed reactor of a chemical heat pump for high-temperature gas reactors. Nuclear Engineering and Design, 210(1), 1–8. https://doi.org/10.1016/S0029-5493(01)00421-6 [Google Scholar] [CrossRef]
9. Tregambi, C., Padula, S., Galbusieri, M., Coppola, G., Montagnaro, F. et al. (2020). Directly irradiated fluidized bed reactor for thermochemical energy storage and solar fuels production. Powder Technology, 366, 460–469. [Google Scholar]
10. Almendros-Ibáñez, J. A., Fernández-Torrijos, M., Díaz-Heras, M., Belmonte, J. F., Sobrino, C. (2019). A review of solar thermal energy storage in beds of particles: Packed and fluidized beds. Solar Energy, 192, 193–237. https://doi.org/10.1016/j.solener.2018.05.047 [Google Scholar] [CrossRef]
11. Ousaleh, H. A., Said, S., Zaki, A., Faik, A., El Bouari, A. (2019). Silica gel/inorganic salts composites for thermochemical heat storage: Improvement of energy storage density and assessment of cycling stability. Materials Today: Proceedings, 30, 937–941. [Google Scholar]
12. Marie, L. F., Landini, S., Bae, D., Francia, V., O’Donovan, T. S. (2022). Advances in thermochemical energy storage and fluidised beds for domestic heat. Journal of Energy Storage, 53, 105242. [Google Scholar]
13. van Alebeek, R., Scapino, L., Beving, M. A. J. M., Gaeini, M., Rindt, C. C. M. et al. (2018). Investigation of a household-scale open sorption energy storage system based on the zeolite 13X/water reacting pair. Applied Thermal Engineering, 139, 325–333. https://doi.org/10.1016/j.applthermaleng.2018.04.092 [Google Scholar] [CrossRef]
14. Aydin, D., Casey, S. P., Riffat, S. (2015). The latest advancements on thermochemical heat storage systems. Renewable and Sustainable Energy Reviews, 41, 356–367. https://doi.org/10.1016/j.rser.2014.08.054 [Google Scholar] [CrossRef]
15. Bales, C. (2005). Thermal properties of materials for thermo-chemical storage of solar heat. IEA SHC—Task 32—Advenced Storage Concepts for Solar and Low Energy Buildings, 32. https://doi.org/10.1016/j.jviromet.2011.07.011 [Google Scholar] [CrossRef]
16. Xu, J. X., Li, T. X., Chao, J. W., Yan, T. S., Wang, R. Z. (2019). High energy-density multi-form thermochemical energy storage based on multi-step sorption processes. Energy, 185, 1131–1142. https://doi.org/10.1016/j.energy.2019.07.076 [Google Scholar] [CrossRef]
17. Aristov, Y. I. (2007). New family of solid sorbents for adsorptive cooling: Material scientist approach. Journal of Engineering Thermophysics, 16, 63–72. https://doi.org/10.1134/S1810232807020026 [Google Scholar] [CrossRef]
18. Yu, N., Wang, R. Z., Lu, Z. S., Wang, L. W., Ishugah, T. F. (2014). Evaluation of a three-phase sorption cycle for thermal energy storage. Energy, 67, 468–478. [Google Scholar]
19. Lu, Z., Wang, R., Gordeeva, L. (2017). Novel multi-step sorption-reaction energy storage cycles for air conditioning and temperature upgrading. Energy, 118, 464–472. [Google Scholar]
20. Ur Rehman, A., Maosheng, Z., Hayat, A. (2020). Water sorption studies on ZnSO4-zeolite composite as potential thermochemical heat storage materials. International Journal of Energy Research, 44(1), 269–281. https://doi.org/10.1002/er.4910 [Google Scholar] [CrossRef]
21. Shkatulov, A. I., Houben, J., Fischer, H., Huinink, H. P. (2020). Stabilization of K2CO3 in vermiculite for thermochemical energy storage. Renewable Energy, 150, 990–1000. [Google Scholar]
22. Hongois, S., Kuznik, F., Stevens, P., Roux, J. J. (2011). Development and characterisation of a new MgSO4-zeolite composite for long-term thermal energy storage. Solar Energy Materials and Solar Cells, 95(7), 1831–1837. https://doi.org/10.1016/j.solmat.2011.01.050 [Google Scholar] [CrossRef]
23. Casey, S. P., Elvins, J., Riffat, S., Robinson, A. (2014). Salt impregnated desiccant matrices for ‘open’ thermochemical energy storage—Selection, synthesis and characterisation of candidate materials. Energy and Buildings, 84, 412–425. https://doi.org/10.1016/j.enbuild.2014.08.028 [Google Scholar] [CrossRef]
24. Yu, N., Wang, R. Z., Wang, L. W. (2013). Sorption thermal storage for solar energy. Progress in Energy and Combustion Science, 39(5), 489–514. https://doi.org/10.1016/j.pecs.2013.05.004 [Google Scholar] [CrossRef]
25. Xu, S. Z., Lemington, L., Wang, R. Z., Wang, L. W., Zhu, J. (2018). A zeolite 13X/magnesium sulfate-water sorption thermal energy storage device for domestic heating. Energy Conversion and Management, 171, 98–109. [Google Scholar]
26. Whiting, G. T., Grondin, D., Stosic, D., Bennici, S., Auroux, A. (2014). Zeolite-MgCl2 composites as potential long-term heat storage materials: Influence of zeolite properties on heats of water sorption. Solar Energy Materials and Solar Cells, 128, 289–295. [Google Scholar]
27. Touloumet, Q., Postole, G., Massin, L., Lorentz, C., Auroux, A. (2022). Investigation of the impact of zeolite shaping and salt deposition on the characteristics and performance of composite thermochemical heat storage systems. Journal of Material Chemistry A, 11, 2737–2753. https://doi.org/10.1039/d2ta07615b [Google Scholar] [CrossRef]
28. Wang, M., Han, S., Chao, Z., Li, S., Tan, B. et al. (2020). Celgard-supported LiX zeolite membrane as ion-permselective separator in lithium sulfur battery. Journal of Membrane Science, 611(1), 1183–1186. https://doi.org/10.1016/j.memsci.2020.118386 [Google Scholar] [CrossRef]
29. Delmelle, R., Duarte, R. B., Franken, T., Burnat, D., Holzer, L. et al. (2016). Development of improved nickel catalysts for sorption enhanced CO2 methanation. International Journal of Hydrogen Energy, 41, 20185–20191. https://doi.org/10.1016/j.ijhydene.2016.09.045 [Google Scholar] [CrossRef]
30. Guan, L. L., Duan, L. Y., Xie, Y. C. (2005). Effects of Mn+ ion exchange on the adsorptive separation property of LiX with N2 and Ar. Wuji Cailiao Xuebao/Journal of Inorganic Materials, 20(2), 503–507. [Google Scholar]
31. Hovington, P., Timoshevskii, V., Burgess, S., Demers, H., Statham, P. et al. (2016). Can we detect Li K X-ray in lithium compounds using energy dispersive spectroscopy? Scanning, 38(6), 571–578. [Google Scholar] [PubMed]
32. Jänchen, J., Ackermann, D., Stach, H., Brösicke, W. (2004). Studies of the water adsorption on zeolites and modified mesoporous materials for seasonal storage of solar heat. Solar Energy, 76(1), 339–344. https://doi.org/10.1016/j.solener.2003.07.036 [Google Scholar] [CrossRef]
33. Roy, R., Weibel, J. A., Garimella, S. V. (2022). Modeling the formation of efflorescence and subflorescence caused by salt solution evaporation from porous media. International Journal of Heat and Mass Transfer, 189, 122645. https://doi.org/10.1016/j.ijheatmasstransfer.2022.122645 [Google Scholar] [CrossRef]
34. de Gennaro, B., Cappi, A., de Gennaro, M., Bianco, N., Langella, A. et al. (2022). Use of zeolites in the capture and storage of thermal energy by water desorption—Adsorption cycles. Materials, 15(16), 5574. https://doi.org/10.3390/ma15165574 [Google Scholar] [PubMed] [CrossRef]
35. Nan, C. W., Clarke, D. R. (1996). The influence of particle size and particle fracture on the elastic/plastic deformation of metal matrix composites. Acta Mater, 44, 3801–3811. [Google Scholar]
36. Fairhurst, D. (2015). Chapter 20—Droplets of ionic solutions. In: Droplet wetting and evaporation. USA: Academic Press. https://doi.org/10.1016/B978-0-12-800722-8.00020-5 [Google Scholar] [CrossRef]
37. Marie, L. F., O’Donovan, T. S. (2023). Fluidisation of thermochemical energy storage materials: Degradation assessment. Energy Sources, Part A: Recovery, Utilization, and Environmental Effects, 45(4), 10034–10050. https://doi.org/10.1080/15567036.2023.2242810 [Google Scholar] [CrossRef]
38. Bardy, D. A., Cruickshank, C. A., Tezel, F. H., Carrier, Y. H., Wong, B. (2020). An experimental investigation of fixed and fluidized beds as adsorbers in compact thermal energy storage systems. Journal of Energy Storage, 31, 101648. https://doi.org/10.1016/j.est.2020.101648 [Google Scholar] [CrossRef]
39. Khang, D. Y., Lee, H. H. (1997). Particle size distribution in fluidized beds for catalytic polymerization. Chemical Engineering Science, 52(3), 421–431. https://doi.org/10.1016/S0009-2509(97)86701-2 [Google Scholar] [CrossRef]
40. Kunii, D., Levenspiel, O. (1969). Introduction. In: Fluidization engineering. USA: John Wiley and Sons Inc. [Google Scholar]
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools