| Energy Engineering |
DOI: 10.32604/EE.2022.015477
ARTICLE
Experimental Research on Mercury Catalytic Oxidation over Ce Modified SCR Catalyst
Key Laboratory for Thermal Science and Power Engineering of the Ministry of Education, Department of Energy and Power Engineering, Tsinghua University, Beijing, 100084, China
*Corresponding Author: Yuqun Zhuo. Email: zhuoyq@mail.tsinghua.edu.cn
Received: 24 December 2020; Accepted: 29 March 2021
Abstract: In order to improve the ability of SCR catalyst to catalyze the oxidation of gaseous elemental mercury, a series of novel Ce modified SCR (Selection Catalytic Reduction, V2O5–WO3/TiO2) catalysts were prepared via two-step ultrasonic impregnation method. The performance of Ce/SCR catalysts on Hg0 oxidation and NO reduction as well as the catalytic mechanism on Hg0 oxidation was also studied. The XRD, BET measurements and XPS were used to characterize the catalysts. The results showed that the pore volume and pore size of catalyst was reduced by Ce doping, and the specific surface area decreased with the increase of Ce content in catalyst. The performance on Hg0 oxidation was promoted by the introduction of CeO2.Ce1/SCR (1%Ce, wt.%) catalyst exhibited the best Hg0 oxidation activity of 21.2% higher than that of SCR catalyst at 350°C, of which the NO conversion efficiency was also higher at 200–400°C. Furthermore, Ce1/SCR showed a better H2O resistance but a slightly weaker SO2 resistance than SCR catalyst. The chemisorbed oxygen and weak absorbed oxygen on the surface of catalyst were increased by the addition of CeO2. The chemisorbed oxygen and weak absorbed oxygen on the surface of catalyst were increased by the addition of CeO2. The Ce1/SCR possed better redox ability compared with SCR catalyst. HCl was the most effective gas responsible for the Hg0 oxidation, and the redox cycle (V4+ + Ce4+ ↔ V5+ + Ce3+) played an important role in promoting Hg0 oxidation.
Keywords: Mercury catalytic oxidation; SCR catalyst; Ce doping; reaction mechanism
Mercury is considered an important environmental pollutant due to its toxic, bioaccumulative and long-distance transport properties. The global mercury emissions reached 6,500 ton/year in 2014, and it will reach 8,600 tons per year in 2035 according to the ECHMERIT model [1]. In 2013, more than 80 countries and regions, including China, signed the Minamata Convention, which sets the binding standards for mercury-containing products and mercury emissions to atmosphere. Data show that over 3,000 tons of mercury from burning coal are emitted per year globally, and more than 90% of mercury from coal-fired power plants is released into the atmosphere [2]. After coal combustion, mercury mainly exists in coal cinder, fly ash and flue gas, and the content is about 2%, 23.1%∼26.9% and 56.3%∼69.7%, respectively [3]. The average distribution of mercury in flue gas was 56% elemental mercury (Hg0), 34% bivalent mercury (Hg2+) and 10% particulate mercury (HgP) [4]. Hg2+ is soluble and easy to react with limestone, more than 80% of which can be removed by wet limestone-gypsum FGD device [5]. About 90% of HgP can be captured by electrostatic precipitator, bag filter and other dust removal equipment [6]. However, the air pollution control device (APCDs) has little effect on Hg0 removal [7]. After long-term screening, SCR catalyst with the component of V2O5–WO3–TiO2 is the most widely used in coal-fired power plants.It is found that SCR catalyst can catalyze the conversion of Hg0 to Hg2+, which can be removed by subsequent dedusting and FGD equipment [8]. However, researches showed that Hg0 oxidation over SCR catalyst was limited, highly dependent on the concentration of HCl in flue gas with poor water resistance [9].
Doping metal oxides into SCR catalysts to improve the oxidation effect of mercury without decreasing the denitrification efficiency has become the focus of current research. CeO2 could provide significant oxygen storage capability through the redox shift between the two oxidation states (Ce3+ and Ce4+), which is helpful to enhance catalytic activity [10]. It was found that the catalytic activity of Ce/SCR catalyst was higher than that of Fe/SCR, Mn/SCR, Cu/SCR and Co/SCR catalysts [11]. The research of Ce/SCR catalyst is still at the initial stage, the optimal temperature for catalytic performance of Hg0 oxidation over Ce/SCR catalyst is low due to the large Ce doping amount and the catalyst performs poorly at the normal operating temperature (280°C–420°C) in power plant [12]. Within the operating temperature range of SCR catalyst in power plant, the optimum amount of Ce doping have not been determined at present [11–14]. Some research such as the effect of flue gas and the mechanism on Hg0 oxidation over Ce modified SCR catalysts also need to be studied.
In this study, a series of CeO2 modified support on V2O5–WO3/TiO2 catalysts were made by ultrasonic-assisted impregnation method. The Hg0 and NO oxidation efficiencies of the catalysts were tested under simulated flue gas. Besides, the effects of individual flue gas components on Hg0 oxidation were also evaluated. Furthermore, X-ray diffraction (XRD), Brunner−Emmet−Teller (BET) and X-ray photoelectron spectroscopy (XPS) were explored to characterize the catalysts, and the reaction mechanism was discussed based on the experimental and characterization results.
Cerium nitrate which was the precursor of CeOx will react with ammonium metavanadate precursor or ammonium tungstate precursor to produce cerium vanadate or cerium tungstate precipitation, affecting the uniformity of V2O5 and CeO2 on the surface of TiO2. Therefore, modified catalysts were prepared by multi-step ultrasonic impregnation method based on the common commercial SCR catalysts (containing 1%V2O5, 9%WO3 and 90%TiO2). TiO2 was added into mixed solutions of ammonium metavanadate, ammonium metatungstate and oxalic acid. The slurry was exposed to an ultrasonic bath for 4 h, dried at 105°C for 4 h and calcined at 500°C for 4 h in air to obtain SCR catalyst. Ce(NO3)3 was dissolved in deionized water at different concentrations. The powder of SCR catalysts was impregnated in Ce(NO3)3 solution for 4 h, dried at 105°C for 4 h and calcined at 500°C for 4 h. Finally, CeO2 doped catalyst V2O5–WO3/TiO2 was obtained and abbreviated as Cex/SCR. X represents the mass fraction of Ce in the sample, x = 0.5%, 1%, 3%, 5%. 80–100 mesh particles were selected by stainless steel sieve.
The schematic of the fixed-bed reactor system used for Hg0 oxidation was showed in Fig. 1. The reactor system consists of four parts: mercury generation, simulated flue gas mixing, temperature control and mercury analysis.The elemental mercury was generated by a PSA Cavkit 10.534 mercury generator. The water vapor was generated by an IAS Hovocal gas generator. The composition and concentration of simulated flue gas were controlled by mass flow controllers (MFCs). The fixed-bed reactor was made of a quartz tube heated in a vertical tubular furnace. The catalyst sample was placed between the quartz cotton and glass fiber filter membrane, which were loaded in the quartz reactor. The concentrations of Hg0 and Hg2+ in simulated flue gas were online monitored by Thermo CEMS. To avoid Hg contamination, all of the tubes, joints, and valves that Hg passed through were made of Teflon. To avoid condensation of water vapor, all pipelines with H2O (g)-containing gas passing through were heated up to 120°C. The tail gas was treated by an activated carbon trap before being released to the air.
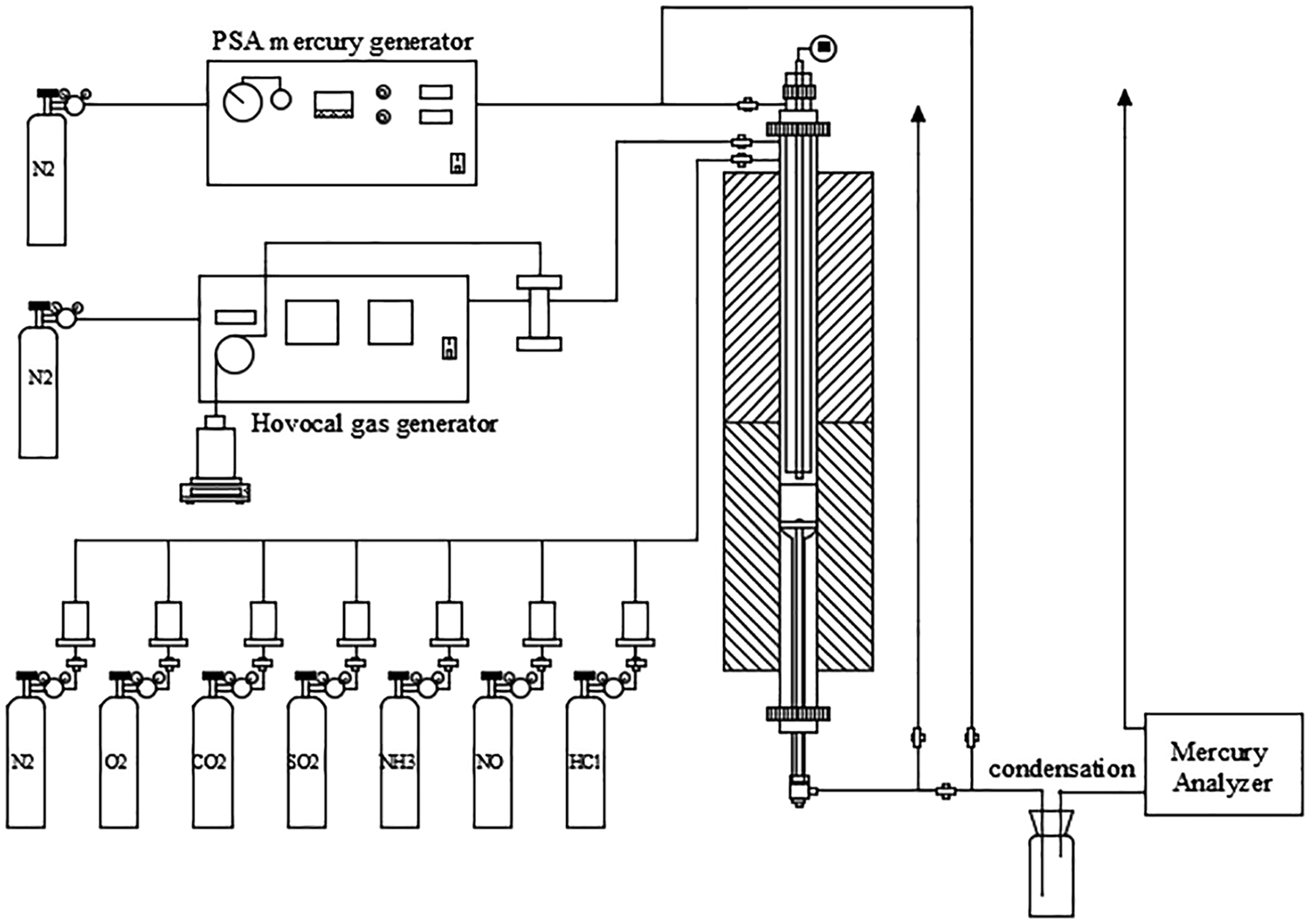
Figure 1: Schematic diagram of experimental system for mercury oxidation
The inlet Hg0 concentration was set at 30 μg/Nm3 and the total flow rate of simulated flue gas was controlled at 2 L/min. The concentration of Hg0 and Hg2+ at the outlet was recorded after adsorption balance, which was defined as the fluctuation of concentration less than 3%. The time for the balance was more than 2 h. The total Hg0 oxidation rate (Oxi) within a certain time was defined as follows:
where
XRD measurements were carried out on polycrystalline X-ray diffractometer (Smartlab, Rigaku, Japan) to examine the crystallinity and dispersivity of crystal pieces using Cu Ka radiation (9 KW) in the range of 3–90°. The specific surface area, pore volume and pore diameter of catalysts were tested on automatic nitrogen adsorption analyzer (Micrometritics ASAP2010). The surface area and pore size was calculated by Brunauer−Emmett−Teller (BET) method and Barrett−Joyner−Halenda (BJH) method, respectively. To investigate the elemental states, XPS was carried out by X-ray photoelectron spectrometer (PHI QuANTRO SXM, ULVAC-PHI, Japan). The observed spectra were calibrated with the C1s binding energy (BE) value of 284.6 eV.
3.1 Hg0 Oxidation Activity over Various Catalysts
Fig. 2 showed the Hg0 catalytic oxidation efficiencies over various catalysts with a space velocity of 60000 h−1 in the temperature range of 150–400°C. The simulated flue gas consisted of 10 ppm HCl, 12% CO2, 5% H2, 300 ppm NO, 400 ppm SO2 and N2 as balance gas. The results indicated that Ce doping did not change the optimum Hg0 catalytic oxidation temperature over SCR catalyst, which was still 350°C. It was clearly found that the addition of CeO2 resulted in enhancement of Hg0 oxidation activity in varying degrees. With the increase of Ce doping, the Hg0 oxidation efficiency over Cex/SCR catalyst first increased and then decreased at 350°C. Ce1/SCR performed the best mercury oxidation and approximately 84.24% mercury oxidation efficiency was obtained. It was worth noting that the Hg0 oxidation rate of Ce5/SCR decreased remarkably, even less than the Hg0 oxidation rate of SCR when the temperature exceeded 350°C. It was presumed that CeO2–TiO2 performed optimal Hg0 catalytic oxidation at about 150°C [15], and excessive Ce doping lead to poor Hg0 oxidation over Ce5/SCR at high temperature.
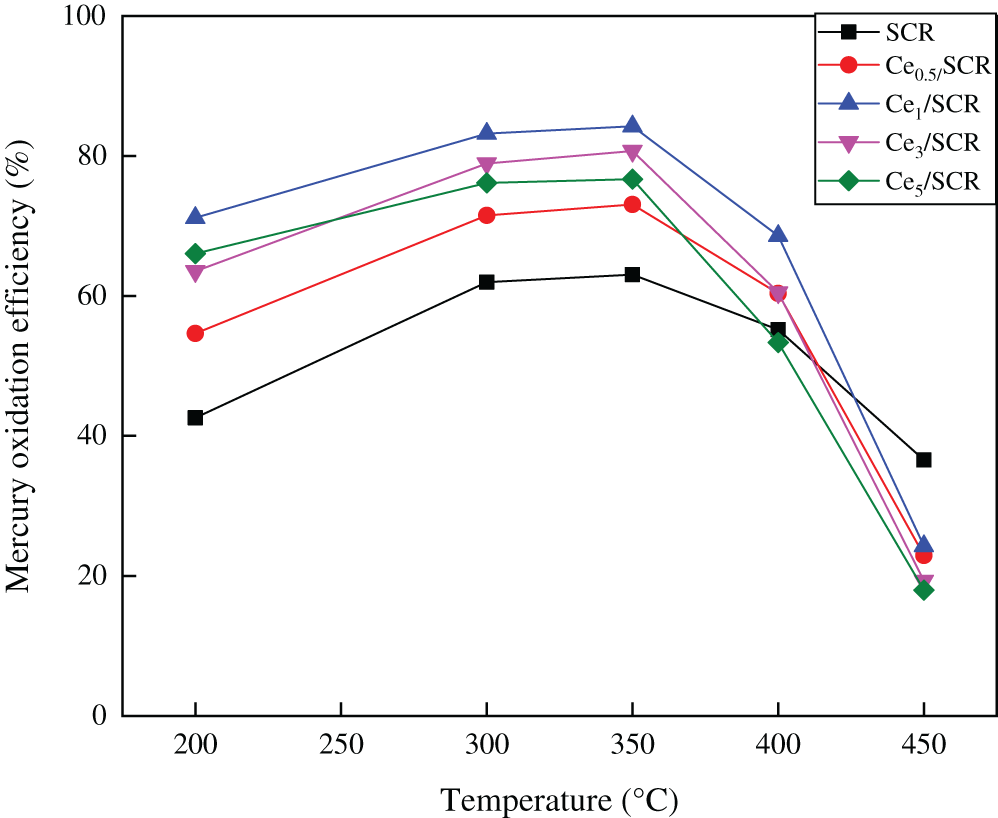
Figure 2: Comparison of catalytic activity for Hg0 oxidation over Cex/SCR catalysts
3.2 Effect of Flue Gas Constituents on Hg0 Oxidation
The comparison of effect of SO2 on Hg0 oxidation efficiency over Ce1/SCR and SCR catalyst was measured in Fig. 3. The results indicated that SO2 had a slightly promotional effect on Hg0 oxidation over SCR catalyst. SO2 was oxidized by chemisorbed oxygen to form SO3, which constituted new chemisorption sites for Hg0 and reacted with Hg0 to produce HgSO4 [15]. However, SO2 exhibited a weak inhibiting effect on Ce1/SCR catalytic activity. This could be due to that SO2 reacted with CexOy to form Ce(SO4)2 and Ce2(SO4)3, which reduced the participation of CexOy in Hg0 oxidation process and covered the surface of Ce1/SCR catalyst, reducing the contact area between the active components and the reaction gas. Because Ce content in Ce1/SCR catalyst was only 1%, SO2 had no obvious inhibition on Hg0 oxidation over Ce1/SCR catalyst.
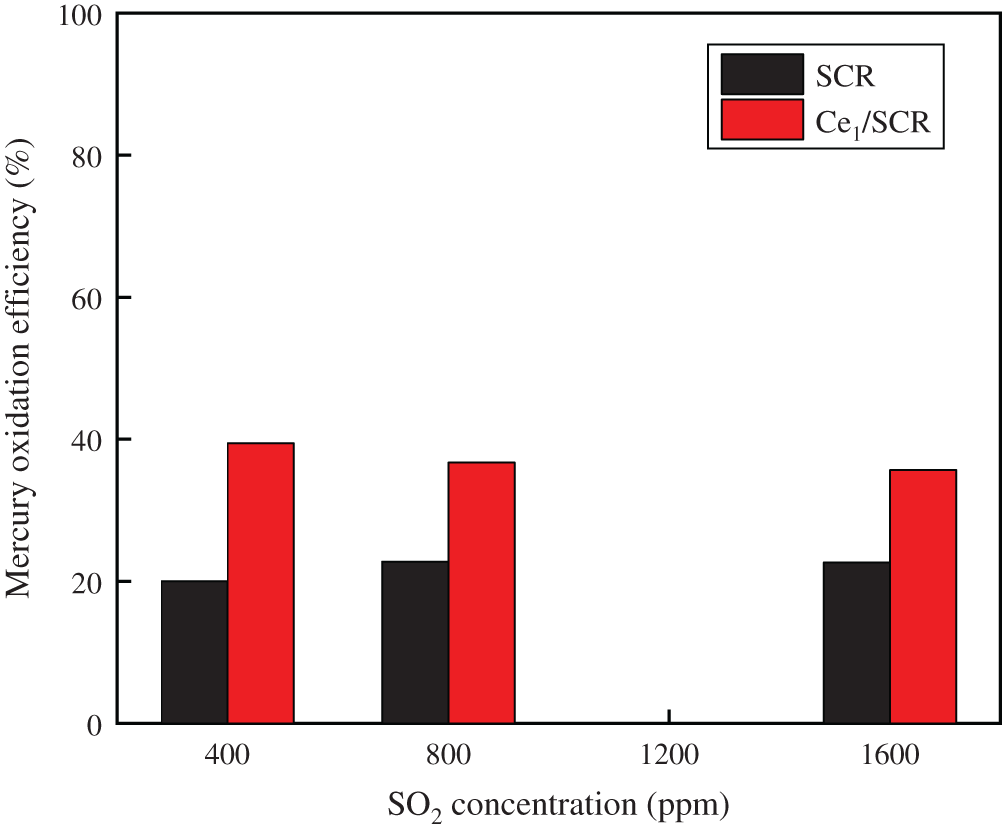
Figure 3: Effect of SO2 on Hg0 oxidation over SCR and Ce1/SCR catalyst at 350°C. Reaction conditions: 0.6 ppm HCl, 5% O2, N2 as balance gas
The results that the effect of H2O on Hg0 oxidation efficiency over Ce1/SCR and SCR catalyst was shown in Fig. 4. H2O inhibited Hg0 oxidation due to the competitive adsorption between H2O and Hg0 [13]. However, Ce1/SCR catalyst had a superior performance of H2O resistance than SCR catalyst. It was speculated that the active sites on the surface increased or the adsorption of water vapor decreased by doping Ce into SCR, or the transition between Ce4+/Ce3+ was likely to offset part of the inhibitory effect of H2O.
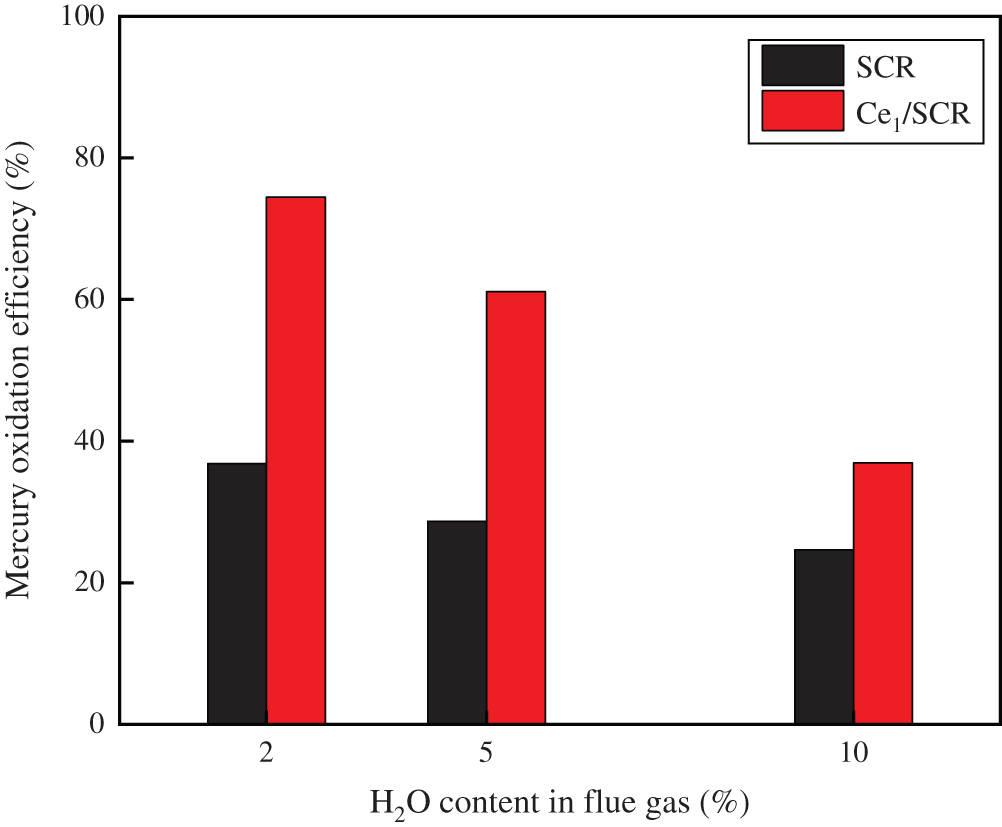
Figure 4: Effect of H2O on Hg0 oxidation over SCR and Ce1/SCR catalyst at 350°C. Reaction conditions: 30 ppm HCl, 5% O2, N2 as balance gas, GHSV = 600000 h−1
Fig. 5 showed the effect of NO on Hg0 oxidation over Ce1/SCR catalyst. It indicated that NO had a slight enhancing effect on Hg0 oxidation. This probably because NO reacted with chemisorbed oxygen to form NOx species, which could enhance Hg0 oxidation.
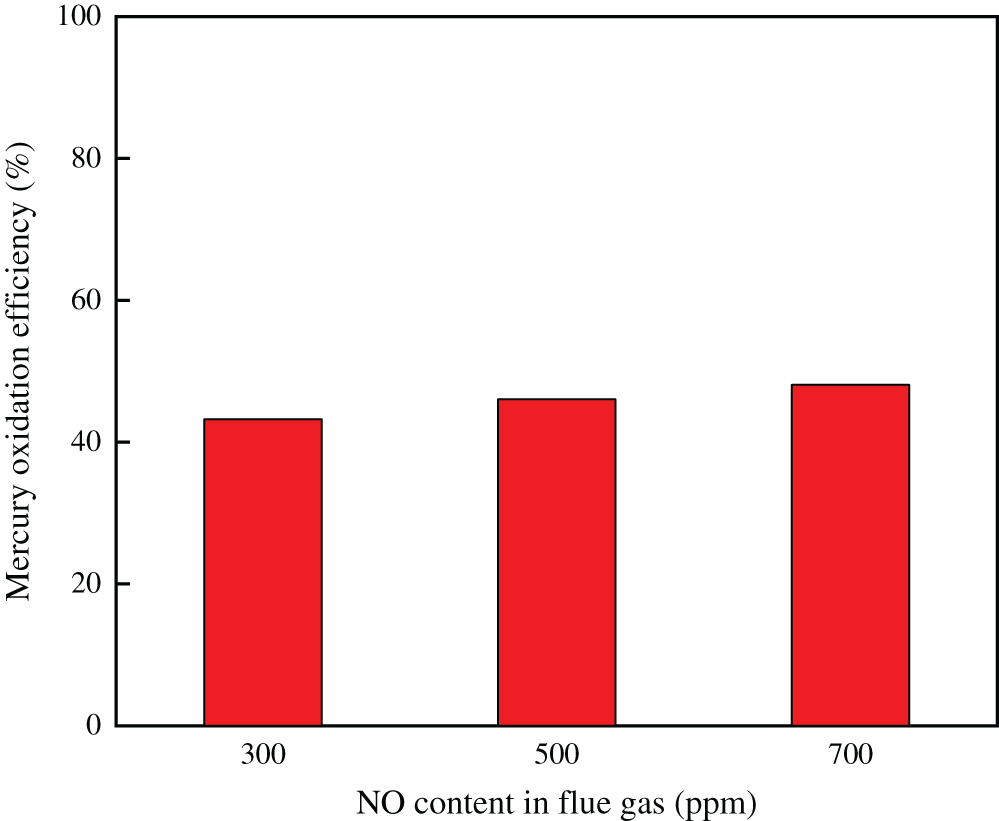
Figure 5: Effect of NO on Hg0 oxidation over Ce1/SCR catalyst at 350°C. Reaction conditions: 0.6 ppm HCl, 5% O2, N2 as balance gas
3.3 NO Reduction Activity over Various Catalysts
Catalytic activity evaluation was carried out using a flow-through powder reactor system referred in Pang et al. [16]. NO concentration was tested for 30 min at each steady state. The NO conversion was calculated using the equation below:
where NOin represents the inlet NO concentration maintained at 500 ppm and NOout represents the outlet average NO concentration.
The NO conversion over SCR and Ce1/SCR catalyst at various temperature were showed in Fig. 6. The NO conversion over SCR catalyst was noticeably enhanced by Ce doping. SCR catalyst performed best NO conversion at 400°C while Ce1/SCR catalyst performed best at 350°C. It was speculated that Ce had a good effect on catalytic oxidation at low temperature, which decreased the optimal denitrification temperature. At operating temperature (300–400°C) of catalyst in power plant, NO conversion over Ce1/SCR catalyst was greater than 90%, which had a small difference from the optimal denitrification efficiency. Therefore, Ce1/SCR can be well applied in power plant, which will have a better simultaneous removal performance of NO and Hg0 than SCR.
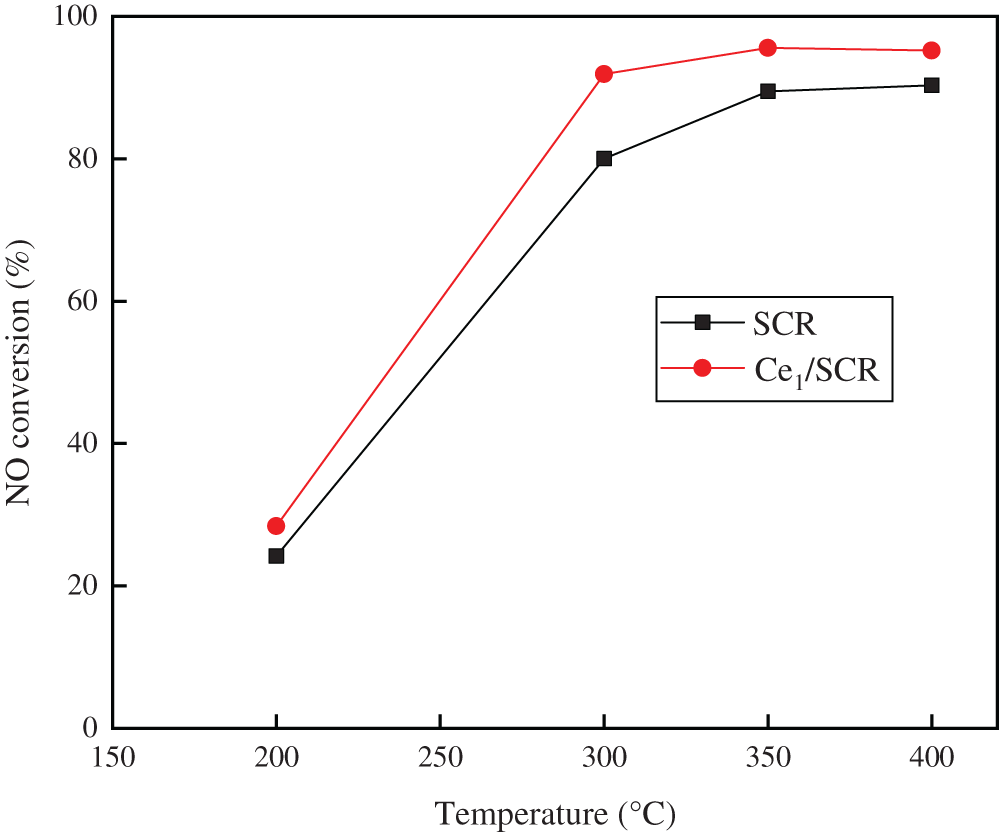
Figure 6: NO conversion over SCR and Ce1/SCR catalyst. Reaction conditions: NO 500 ppm, NH3 500 ppm, O2 3%, and SO2 400 ppm, balance N2, GHSV = 20000 h−1
The XRD patterns of the raw and modified SCR catalysts were displayed in Fig. 7. TiO2 phase other than rutile TiO2 phase was detected in all samples. The diffraction line of all samples were narrow and sharp, which indicated the high crystallinity. The characteristic peaks of V2O5 and WO3 were hardly detected, which were due to widely dispersion and poorer crystalline on the surface. CeO2 was not observed when the loading of Ce was lower than 3% (wt.%). There were weak characteristic CeO2 peaks at 28.38° when the loading of Ce was 5%, indicating a well-dispersed cluster with a small population of CeO2 may exist.
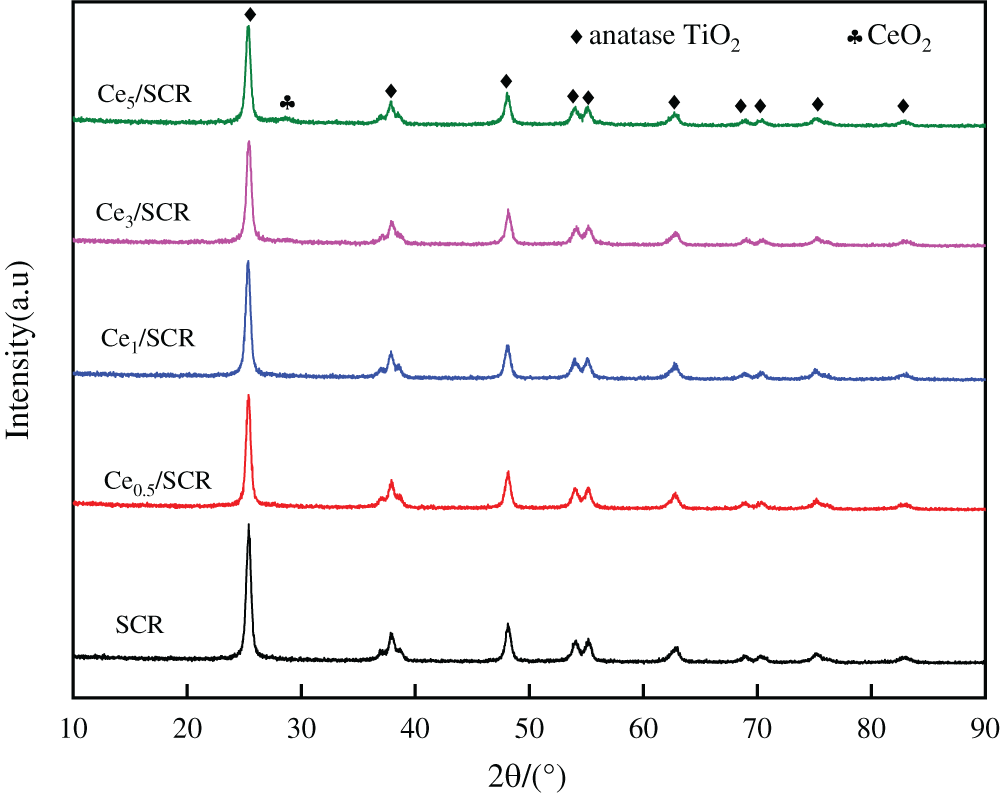
Figure 7: XRD profiles of Cex/SCR catalysts with different Ce contents
The pore structure parameters of different samples were summarized in Table 1. As can be seen from the result, the BET surface area, BJH pore volume and average pore volume of Cex/SCR catalyst were lower than those of SCR. The BET surface area of Cex/SCR decreased with the increase of Ce doping amount. The pore volume and pore diameter of Cex/SCR catalyst did not show obvious relationship with Ce doping amount. The Nitrogen adsorption-desorption isotherms of Cex/SCR were the IV adsorption-desorption isotherm specified by IUPAC. H1 hysteresis loop indicated mesoporous materials, which might facilitate mass transfer in the catalytic reaction (Fig. 8a). As displayed in Fig. 8b, Ce doping had no significant effect on the pore size distribution of catalysts. There were much mesoporous of 2∼50 nm in Cex/SCR catalysts, indicating strong interactions between catalyst surface and adsorbate.
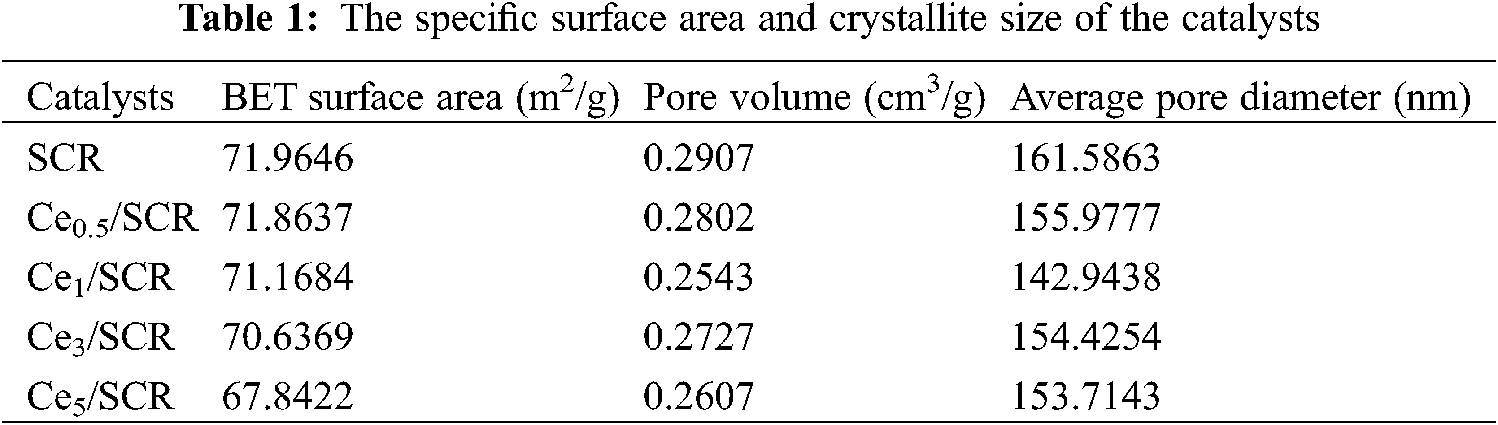
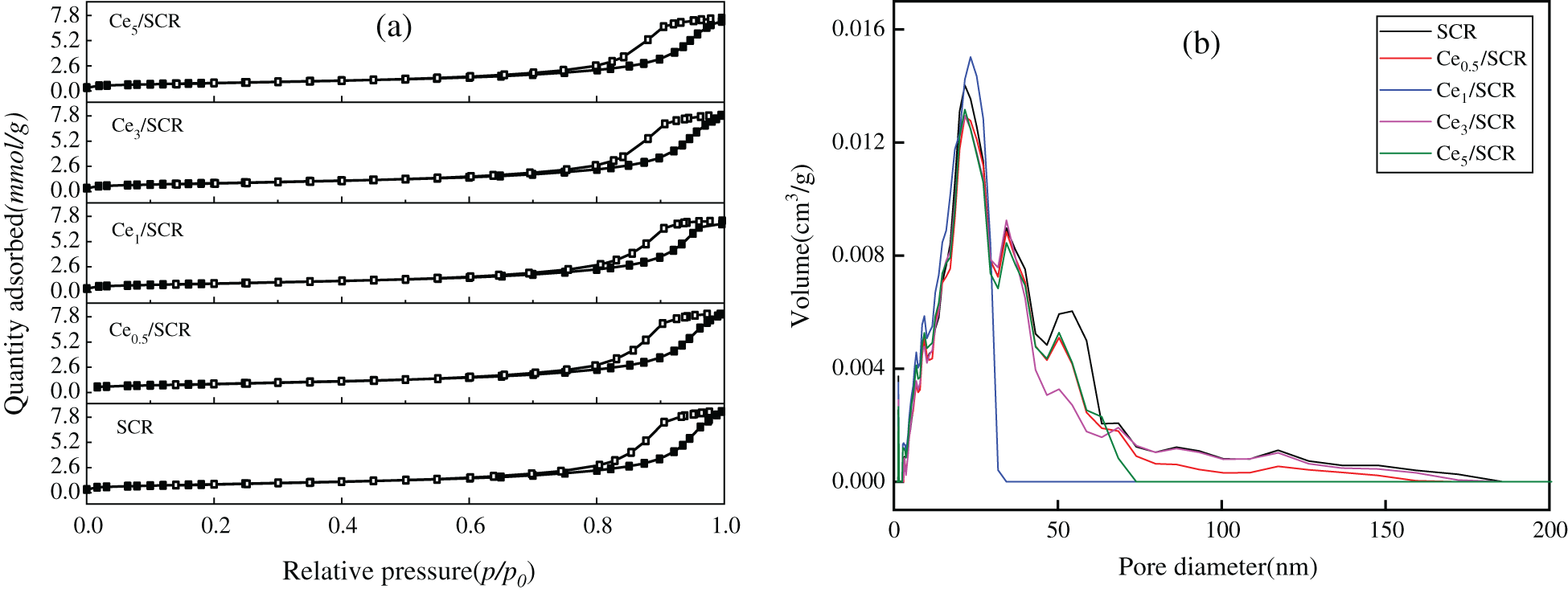
Figure 8: Physical properties of different catalysts. (a) N2 adsorption and desorption isotherms; (b) Particle size distribution
The effects of HCl and O2 on mercury oxidation over Ce1/SCR catalyst were shown in Fig. 9. Ce1/SCR catalyst showed less than 5% mercury oxidation efficiency whether 5% O2 was introduced into gas stream or not. Considering the measurement error, O2 had little effect on Hg0 oxidation. That was probably because the level of reaction between O2 and Hg0 was low, generating unstable Hg2+ which coule convert to Hg0 easily.
0.1 ppm HCl balanced in N2 resulted in Hg0 oxidation efficiency of 13.02%, which is higher than the 2.73% Hg0 oxidation efficiency under pure N2 condition. Hg0 oxidation efficiency of 64.68% was observed when HCl concentration further increased to 1 ppm. HCl exhibited a decisive effect on Hg0 oxidation over Ce1/SCR catalyst. 0.1 ppm HCl resulted in Hg0 oxidation efficiency of 50.42% with the aid of 5% O2, indicating more HCl can be oxidized to form active chlorine species in the presence of O2. That might because O2 replenished the consumed chemisorbed oxygen, regenerated the lattice oxygen and hence maintained the high surface oxygen concentration [15]. The reaction between HCl and Ce1/SCR catalysts may occur through
where Cl denoted an active chlorine species for oxidizing Hg0, and O represented chemisorbed or lattice oxygen on the surface of Ce1/SCR catalysts.
The entire reaction can be written as follows:
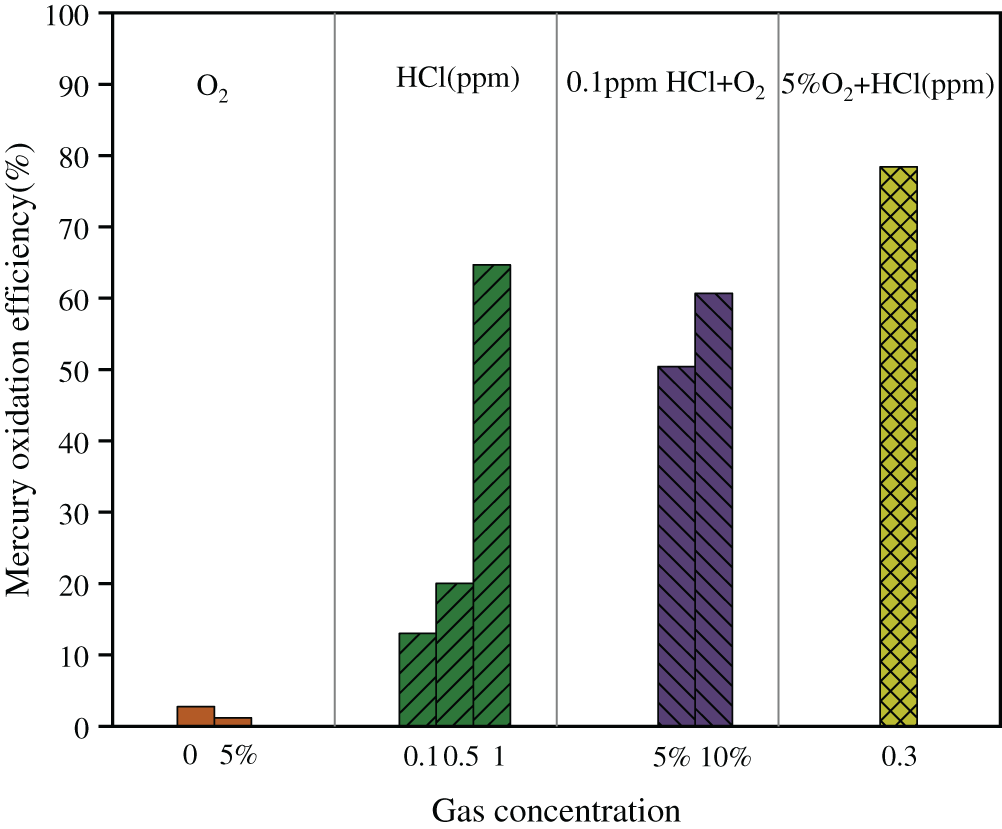
Figure 9: Effects of HCl and O2 on Hg0 oxidation of Ce1/SCR catalyst at 350°C. Reaction conditions: balance N2, GHSV = 20000 h−1
To determine the oxidation states of the element in these catalysts and to further explain the mechanism of Hg0 oxidation over Ce1/SCR catalyst, the catalysts were investigated by XPS technique. Ce1/SCR catalyst was pretreated in the gas composed of 50 μg/m3, 10 ppm HCl and 5% O2 at 350°C for 4 h to get sCe1/SCR catalyst. Fig. 10 showed the O1s XPS spectra for SCR, Ce1/SCR and sCe1/SCR catalyst. The peak appeared at low binding energy (529–530.5 eV) could be ascribed to be the lattice oxygen (denoted as Ob) [17], while the binding energy of 531.0–532.9 eV was ascribed to the chemisorbed oxygen and weakly bonded oxygen species (denoted as Oa) [18]. Oa has been thought to be the most active oxygen and played an important role in oxidation reaction [18–19]. The surface atomic concentrations of catalysts are given in Table 2. It could be found that Oa relative concentration of Ce1/SCR calculated by Oa/(Oa + Ob) was higher than that of SCR. The reason was that Ce doping created labile oxygen vacancies, which may conduce to the improvement of chemisorbed oxygen and was helpful for mercury oxidation. The Ce3d spectra of sCe1/SCR and Ce1/SCR catalysts were presented in Fig. 11. The bands labeled u1 and v1 represent the characteristic peaks of Ce3+ with 3d104f1 initial electronic state, whereas the peaks labeled u, u2, u3, v, v2, and v3 represent Ce4+ with 3d104f0 electronic state [20].
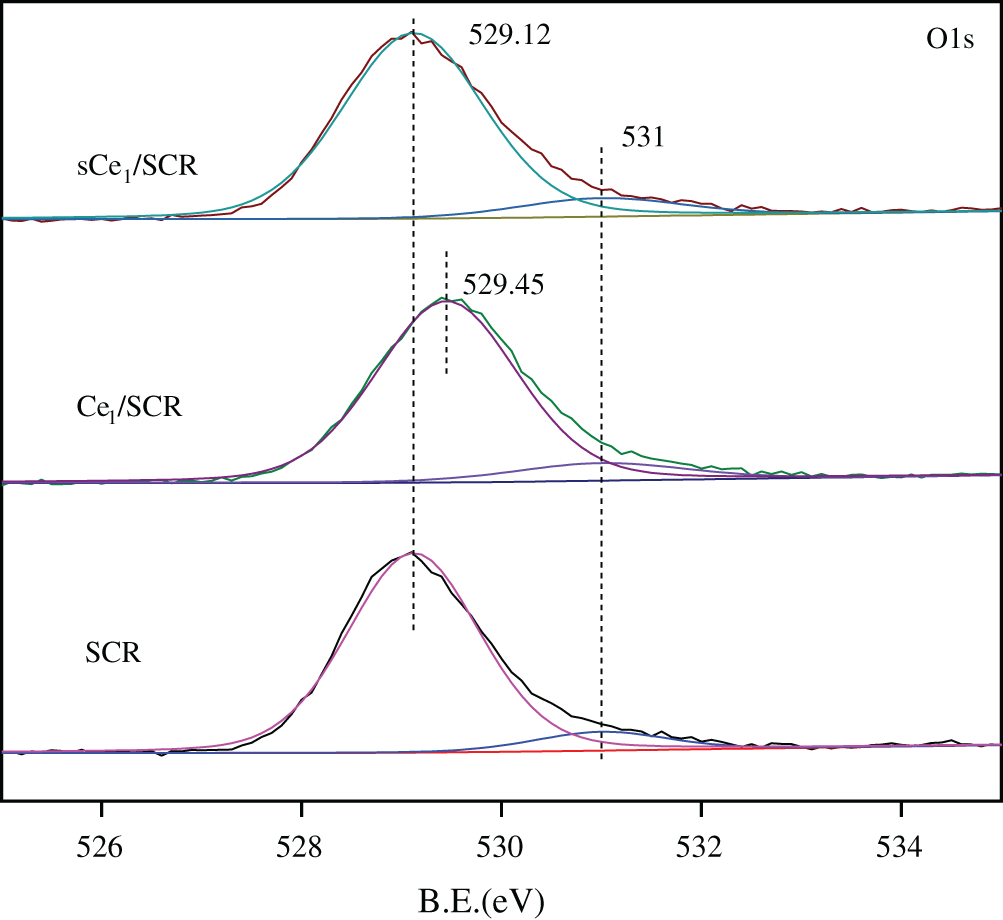
Figure 10: O1s XPS spectra for SCR,Ce1/SCR and Ce1/SCR
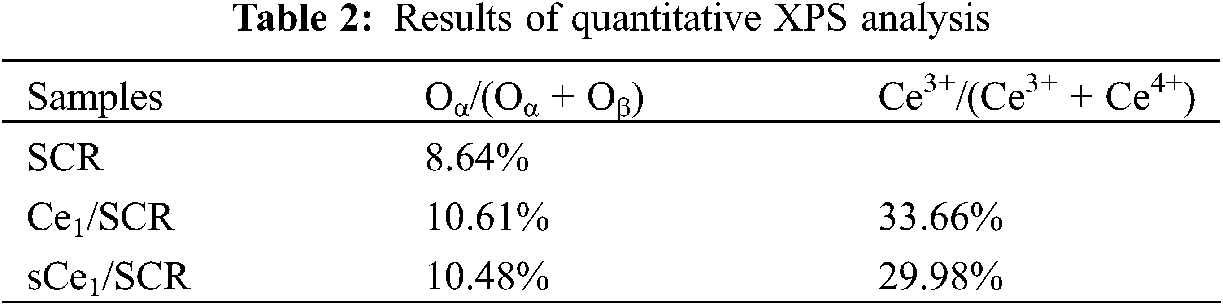
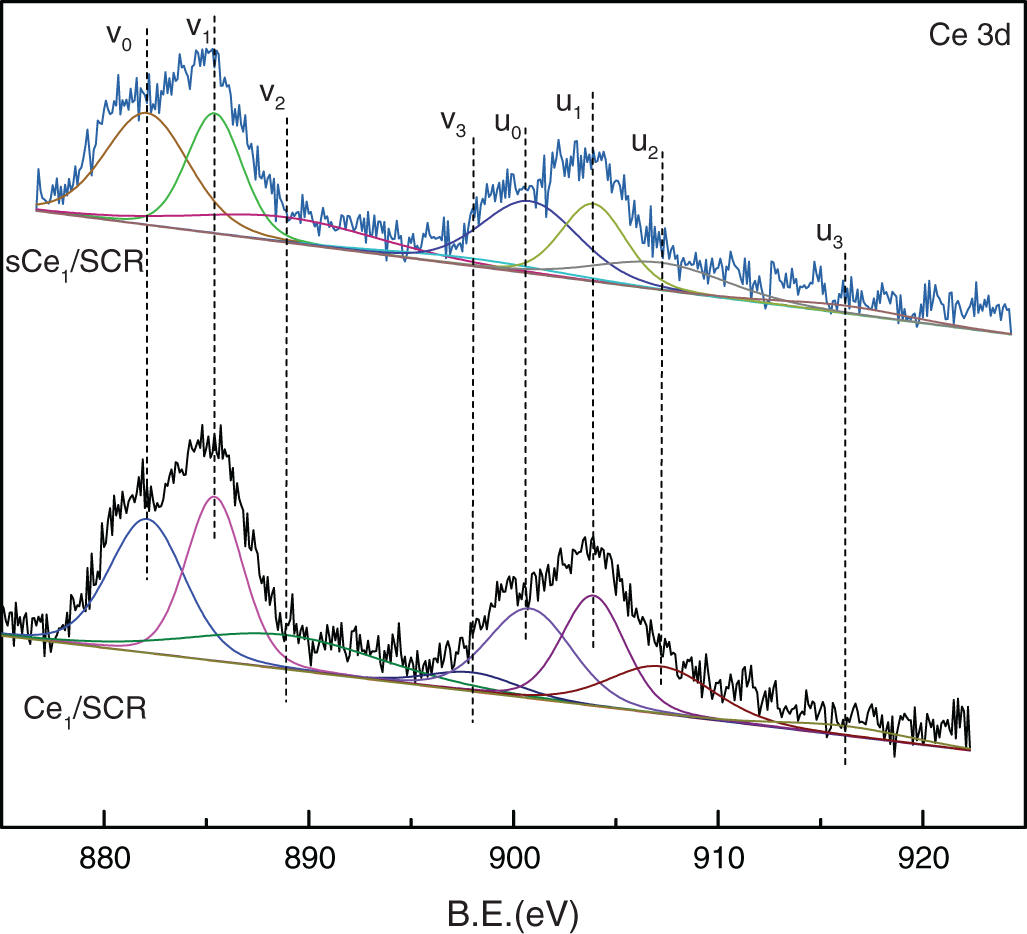
Figure 11: Ce3d XPS spectra for Ce1/SCR and Ce1/SCR
Table 2 showed a slight change in the Ce valence state ratios after the tests under a flue gas flow. This implied that the redox shift between Ce3+ and Ce4+ happened during the Hg0 catalytic oxidation process. In comparison with Ce1/SCR, the relative concentration of Ce3+ species decreased in sCe1/SCR, which might be due to sufficient O2 in flue gas to oxidize Ce3+ to Ce4+.
The Hg4f XPS profile spectrum of sCe1/SCR catalyst was presented in Fig. 12. The binding energies at 101.5 and 110.5 eV was corresponded to HgCl2 and HgO, respectively. No Hg0 was observed on the surface of sCe1/SCR catalyst, which probably because HCl accelerated Hg0 oxidation.
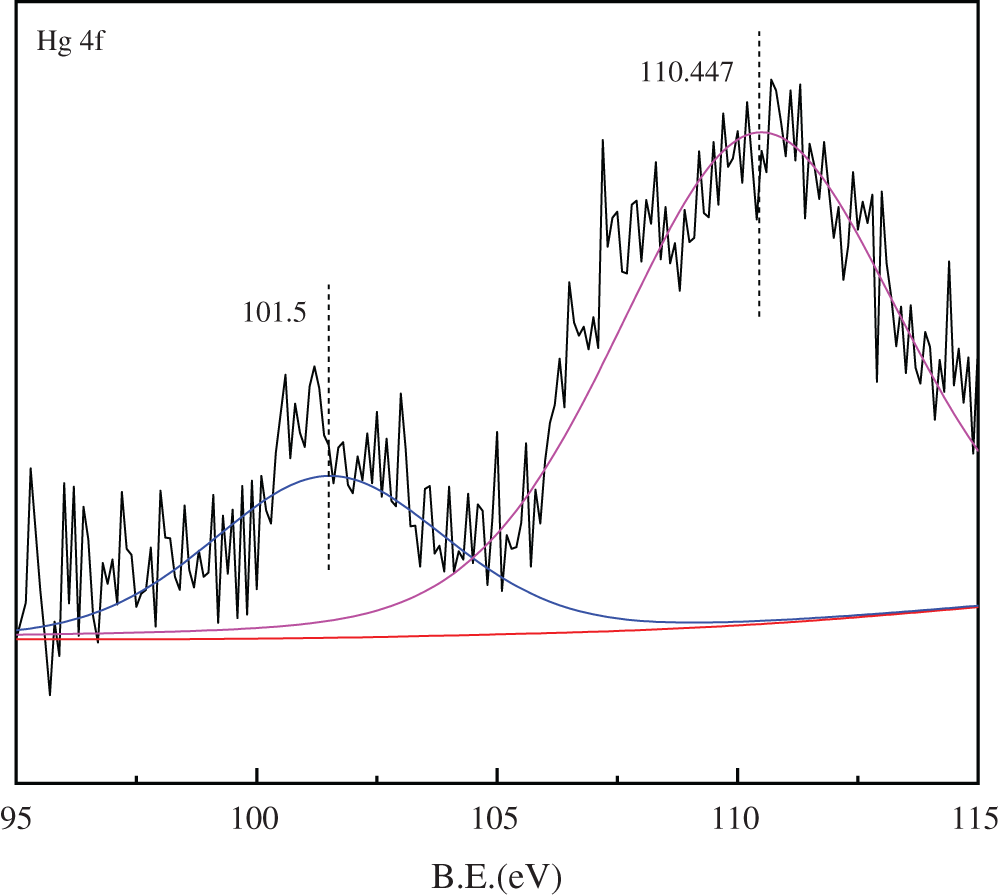
Figure 12: Hg4f XPS spectra for sCe1/SCR
Previous studies have shown that the interconversion of V4+/V5+ participates in the reaction process of Hg0 catalytic oxidation over SCR. Based on the results above, a possible mechanism for Hg0 oxidation can be proposed as follows:
HCl was adsorbed onto the active sites of catalyst and reacted with chemisorbed oxygen from V2O5 to form active surface chlorine species, which then would react with gas-phase or weakly bonded Hg0 to produce HgCl2. The redox process of V4+/V5+ was contributed by the shift of CeO2/Ce2O3. The missing lattice oxygen of CeO2 would be replaced by O2. The redox cycle (V4+ + Ce4+ ↔ V5+ + Ce3+) played an important role in promoting Hg0 oxidation.
Ce doping can significantly improve the catalytic activity for Hg0 oxidation of SCR catalyst Ce1/SCR (1%Ce, wt.%) displayed the best performance with an Hg0 oxidation efficiency of 21.2% higher than that of SCR catalyst in simulated coal-fired flue gas at 350°C. Ce1/SCR also enhanced NO conversion efficiency at 200–400°C. H2O in flue gas had an inhibitive effect on Hg0 oxidation. However, Ce1/SCR showed a better H2O resistance but a slightly weaker SO2 resistance than SCR catalyst. NO had a slight promotion effect on the Hg0 oxidation over Ce1/SCR. The XPS results indicated that Ce1/SCR possed more chemisorbed oxygen and better redox ability compared with SCR catalyst. The catalytic mechanism was that adsorbed HCl was oxidized by chemisorbed oxygen to form active chlorine which would react with gas-phase or weakly bonded Hg0 to form HgCl2. HCl played an essential role in Hg0 oxidation, and the redox cycle (
Funding Statement: This work was supported by the National Key Research and Development Program of China (No. 2016YFB0600603).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Pacyna, J. M., Travnikov, O., de Simone, F., Hedgecock, I. M., Sundseth, K. et al. (2016). Current and future levels of mercury atmospheric pollution on a global scale. Atmospheric Chemistry and Physics, 16(19), 12495–12511. DOI 10.5194/acp-16-12495-2016. [Google Scholar] [CrossRef]
2. Vidic, R. D., McLaughlin, J. B. (1996). Uptake of elemental mercury vapors by activated carbons. Journal of the Air & Waste Management Association, 46(3), 241–250. DOI 10.1080/10473289.1996.10467458. [Google Scholar] [CrossRef]
3. Ren, J., Zhou, J., Luo, Z., Cen, K. (2002). Study of mercury emission during coal combustion. Acta Scientiae Circumstantiae, 22(3), 289–293. DOI 10.13671/j.hjkxxb.2002.03.004. [Google Scholar] [CrossRef]
4. Zhang, L., Wang, S., Wu, Q., Wang, F., Lin, C. et al. (2016). Mercury transformation and speciation in flue gases from anthropogenic emission sources: A critical review. Atmospheric Chemistry and Physics, 16(4), 2417–2433. DOI 10.5194/acp-16-2417-2016. [Google Scholar] [CrossRef]
5. Wang, Y., Duan, Y., Yang, L., Meng, S., Wang, Q. (2008). Experimental study on mercury removed by combined wet-method flue gas desulphurization devices in combination with electrostatic precipitators. Proceedings of China Electric Machinery Engineering, 28(29), 64–69. DOI 10.13334/j.0258-8013.pcsee.2008.29.003. [Google Scholar] [CrossRef]
6. Yin, L., Zhuo, Y., Xu, Q., Zhu, Z., Du, W. et al. (2013). Mercury emission from coal-fired power plants in China. Proceedings of the CSEE, 33(29), 1–10. DOI 10.13334/j.0258-8013.pcsee.2013.29.004. [Google Scholar] [CrossRef]
7. Xu, W., Wang, J., Wang, W. (2010). Exploratory study of mercury removal effectiveness in various morphology and states during the dust removal and desulphurization treatment of coalfired flue gases. East China Electric Power, 38(1), 47–50. DOI CNKI:SUN:HDDL.0.2010-01-016. [Google Scholar]
8. Yan, R., Liang, D. T., Tay, J. H. (2003). Control of mercury vapor emissions from combustion flue gas. Environmental Science and Pollution Research, 10(6), 399–407. DOI 10.1065/espr2003.04.149. [Google Scholar] [CrossRef]
9. Chen, J., Yuan, D., Li, Q., Zheng, J., Zhu, Y. et al.(2008). Effect of flue-gas cleaning devices on mercury emission from coal-fired boiler. Proceedings of the CSEE, 28(2), 72–76. DOI 10.13334/j.0258-8013.pcsee.2008.02.012. [Google Scholar] [CrossRef]
10. Reddy, B. M., Khan, A., Yamada, Y., Kobayashi, T., Loridant, S. et al. (2003). Structural characterization of CeO2-TiO2 and V2O5/CeO2-TiO2 catalysts by Raman and XPS techniques. Journal of Physical Chemistry B, 107(22), 5162–5167. DOI 10.1021/jp0344601. [Google Scholar] [CrossRef]
11. Chi, G., Shen, B., Zhu, S., Chuan, H. (2016). Oxidation of elemental mercury over modified SCR catalysts. Journal of Fuel Chemistry and Technology, 44(6), 763–768. DOI 10.3969/j.issn.0253-2409.2016.06.018. [Google Scholar] [CrossRef]
12. Zhao, L., Li, C., Zhang, J., Zhang, X., Zhan, F. et al.(2015). Promotional effect of CeO2 modified support on V2O5-WO3/TiO2 catalyst for elemental mercury oxidation in simulated coal-fired flue gas. Fuel, 153, 361–369. DOI 10.1016/j.fuel.2015.03.001. [Google Scholar] [CrossRef]
13. Chi, G., Shen, B., Yu, R., He, C., Zhang, X. (2017). Simultaneous removal of NO and Hg0 over Ce-cu modified V2O5/TiO2 based commercial SCR catalysts. Journal of Hazardous Materials, 330, 83–92. DOI 10.1016/j.jhazmat.2017.02.013. [Google Scholar] [CrossRef]
14. Zhao, L., He, Q. S., Li, L., Lu, Q., Dong, C. Q. et al. (2015). Research on the catalytic oxidation of Hg0 by modified SCR catalysts. Journal of Fuel Chemistry and Technology, 43(5), 628–634. DOI 10.1016/S1872-5813(15)30018-9. [Google Scholar] [CrossRef]
15. Li, H., Wu, C., Li, Y., Zhang, J. (2011). CeO2-TiO2 catalysts for catalytic oxidation of elemental mercury in low-rank coal combustion flue Gas. Environmental Science & Technology, 45(17), 7394–7400. DOI 10.1021/es2007808. [Google Scholar] [CrossRef]
16. Pang, C., Zhuo, Y., Weng, Q. (2017). Mn/SAPO-34 as an efficient catalyst for the low-temperature selective catalytic reduction of NOx with NH3. RSC Advances, 7(51), 32146–32154. DOI 10.1039/C7RA05165D. [Google Scholar] [CrossRef]
17. Shan, W., Liu, F., He, H., Shi, X., Zhang, C. (2012). A superior Ce–W–Ti mixed oxide catalyst for the selective catalytic reduction of NOx with NH3. Applied Catalystis B: Environmental, 115, 100–6. DOI 10.1016/j.apcatb.2011.12.019. [Google Scholar] [CrossRef]
18. Fang, J., Bi, X., Si, D., Jiang, Z., Huang, W. (2007). Spectroscopic studies of interfacial structures of CeO2–TiO2 mixed oxides. Applied Surface Science, 253(22), 8952–61. DOI 10.1016/j.apsusc.2007.05.013. [Google Scholar] [CrossRef]
19. Wang, P., Su, S., Xiang, J., Cao, F., Sun, L. et al. (2013). Atalytic oxidation of Hg0 by CuO–MnO2–Fe2O3/--Al2O3 catalyst. Chemical Engineering Journal, 225, 68–75. DOI 10.1016/j.cej.2013.03.060. [Google Scholar] [CrossRef]
20. He, H., Dai, H. X., Au, C. T. (2004). Defective structure, oxygen mobility, oxygen storage capacity, and redox properties of RE-based (RE = Ce, Pr) solid solutions. Catalysis Today, 90(3–4), 245–254. DOI 10.1016/j.cattod.2004.04.033. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |