
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Deep Learning Algorithm for Detection of Protein Remote Homology
1 Kahramanmaras Sutcu Imam University, Kahramanmaras, 46100, Turkey
2 Adana Alparslan Turkes Science and Technology University, Adana, 01250, Turkey
3 Çukurova University, Adana, 01330, Turkey
* Corresponding Author: Fahriye Gemci. Email:
Computer Systems Science and Engineering 2023, 46(3), 3703-3713. https://doi.org/10.32604/csse.2023.032706
Received 26 May 2022; Accepted 12 July 2022; Issue published 03 April 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
The study aims to find a successful solution by using computer algorithms to detect remote homologous proteins, which is a significant problem in the bioinformatics field. In this experimental study, structural classification of proteins (SCOP) 1.53, SCOP benchmark, and the newly created SCOP protein database from the structural classification of proteins—extended (SCOPe) 2.07 were used to detect remote homolog proteins. N-gram method and then Term Frequency-Inverse Document Frequency (TF-IDF) weighting were performed to extract features of the protein sequences taken from these databases. Next, a smoothing process on the obtained features was performed to avoid misclassification. Finally, the proteins with balanced features were classified into remote homologs using the built deep learning architecture. As a result, remote homologous proteins have been detected with novel deep learning architecture using both negative and positive protein instances with a mean accuracy of 89.13% and a mean relative operating characteristic (ROC) score of 88.39%. This experiment demonstrated the following: 1) The successful outcome of this study in detecting remote homology is auspicious in discovering new proteins and thus in drug discovery in medicine. 2) Natural language processing (NLP) techniques were used successfully in bioinformatics, 3) the importance of choosing the correct n-value in the n-gram process, 4) the necessity of using not only positive but negative instances in a classification problem, and 5) how effective the processes, such as smoothing, are in the classification accuracy in an imbalanced dataset. 6) The deep learning architecture gives better results than the support vector machine (SVM) model on the smoothed data to detect proteins’ remote homology.Keywords
The detection of protein homology and protein remote homology in bioinformatics are two of the major problems that are useful in acquiring knowledge about proteins whose structure and function are unknown [1,2]. In addition, evolutionarily, the protein structure is more conserved than the protein sequence. Proteins containing similar structures and functions may akin low sequence similarity [3,4]. In homolog and remote homolog protein detection, protein sequence similarity can also be judged. Homolog proteins have a pairwise sequence identity of more than 40% similarity. Remote homology is defined as pairs of proteins with a pairwise sequence identity between 20% and 40%. Therefore, the determination of whether the proteins are remote homologs to each other can be decided by looking at the families and superfamilies of the two proteins. Any pair of proteins within the same family classification is considered a homology. Proteins from the same superfamily and different families are classified as remotely homologous to each other [1,2,5].
Many protein classification trials based on protein pairwise similarity have been conducted. Some have been based on positive protein instances, while others have been based on negative and positive protein instances [6]. Positive protein instances are instances within the same superfamily, while negative protein instances for the remote homology problem are instances outside the target superfamily. In this study, both negative and positive protein instances were used to discover remote homology. Most of the protein classification methods have depended on multiple sequence alignment. When there is a large amount of data, the multiple sequence alignments are expensive [7]. Due to the increasing number of protein sequences due to the rapid development of biotechnology, protein sequence alignment methods were not chosen in the study. Regarding homology detection, the literature comprises studies based chiefly on sequence similarity. Although the problems of protein remote homology and protein homology are similar, remote homology is a more difficult problem. Because it requires the discovery of very low sequence similarities [1,8–10].
Over recent years, it has been observed that the biological sequence is similar to NLP. Hence, the methods used in NLP have begun to be utilized in fields such as bioinformatics [2]. NLP methods, such as n-gram [2,3], Latent Semantic Analysis (LSA) [3], top-n-gram [11–13], and Latent Dirichlet Allocation (LDA) [6] have been used successfully to detect remote homolog proteins. Based on these studies, in this study, NLP techniques such as n-gram were used to extract remote homologous proteins. Machine learning algorithms are extensively utilized in dedicating purposeful information from big data in bioinformatics [14]. Deep learning is highly preferred for biological data since it provides high performance for many data solutions. The deep learning algorithm has been experimented on to extract information from many sequencing data, such as Deoxyribonucleic Acid (DNA), Ribonucleic acid (RNA), and protein sequences [8,14].
When are performed relation extraction using neural networks and log-linear models with supervised learning, a large amount of training data and much training time are needed [15]. Hence, new training representation methods are being developed to solve training time and big data problems. The technique named lexicalized dependency paths (LDPs) has been developed for this purpose in this article [15]. LDPs are a method developed by determining dependency paths between entities with the Australian Corpus of English (ACE) corpus [15].
One of the essential areas in bioinformatics is protein structure prediction studies that have been studied since the 1960s. Since it will be helpful to know protein homologs and remote homologs in estimating the structure of unknown proteins. In recent years, with machine learning and deep learning in complex problems, better predictions have been made by using these methods in protein structure prediction [16,17,18]. The strengths and difficulties associated with using deep learning methods in protein structure prediction [16,17] and protein local structural features [18] have been shown.
A new Convolutional Neural Network (CNN)-based method called ConvRes has been proposed for remote homolog protein detection to solve the problems faced by Long Short-Term Memory (LSTM) [19]. This method has been tested on the SCOP benchmark dataset, and time has been saved by training in a shorter time [19]. Furthermore, these methods inspire the use of deep learning techniques in remote homolog and other problems [15–19].
In the present study, it was determined whether or not the proteins obtained from the SCOP 1.53 protein database were remote homolog with each other by coding with the Keras and Biopython libraries and the Python programming language. The first step of the study was to obtain the protein data set from the SCOP database. After that, the second step was extracting the protein’s family and superfamily on the data set. In the third step, the remote homolog proteins were determined. Identifying the homolog and remote homolog proteins with each other can be conducted by looking at the similarity of the sequences of proteins. Proteins with more than 40% sequence similarity are defined as homologs.
Moreover, proteins with a pairwise sequence similarity between 20% and 40% were defined as remote homolog. Identifying whether the proteins were homolog or remote homolog to each other can be determined by recognizing the families and superfamily of the proteins. Next, the basic structure of the study was built on this knowledge.
SCOP database is known as a gold-standard protein database [20]. In the experimental study, three different data sets from the SCOP database were used to detect remote homolog proteins, 2 of which had previously been used to detect remote homolog proteins, SCOP 1.53, SCOP benchmark, and the newly created SCOP benchmark data set from SCOPe 2.07.
Families containing at least ten homolog proteins and at least five superfamilies outside of the superfamily of the target family were chosen from the SCOP 1.53 protein database as 54 families. Next, 4352 protein sequences were taken from the SCOP 1.53 protein database to detect remote homolog protein. The e-value of the pairwise alignments of these protein sequences taken from the Astral database was not higher than
The basic building blocks of the protein sequences were amino acids. A unique protein containing a unique genetic code was identified by linking various amino acids with varying numbers, from 9 to 700. The 20 major amino acids were in the protein production were as given in Q = {A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, Y} [13,14].
An n-gram is the n-length sequence of slices of an item, such as the number, digit, word, letter, etc. The n-gram is also called the multi-word unit. In n-gram, a unigram is a sequence with one length of an item; a bigram is a sequence with two lengths of an item, a trigram is a sequence with three lengths of an item, etc. In this study, the items were protein sequences composed of amino acids. Hence, the n-gram slices were composed of amino acids extracted from the proteins. Therefore, Bigrams, which are two amino acids, were used in the study.
The n-gram technique was used to obtain protein features to classify proteins in this study because studies in the field of bioinformatics have benefited from NLP studies over recent years. Character-level n-grams have been used to find morphological variations, such as misspelling and stemming [22]. As the maximum length of n-grams increases, the cost increases, even though the success of the classification increases. Conversely, for n-grams, if only get the maximum length of 2, it dramatically reduces the success of the classification. Generally, a maximum length of up to 3 or 4 is the most preferred for n-gram, even though it slightly increases the feature space and costs [22].
TF-IDF weighting is obtained from the TF-IDF matrix, created by multiplying the term frequency (TF) and inverse document frequency (IDF) values. Eq. (1) shows the common TF-IDF weighting formula, which comprises the TF in Eq. (2), which is an abbreviated term frequency and IDF in Eq. (3) is an abbreviated inverse document frequency.
where w(i,j) in Eq. (4) is the weight of n-gram i in protein sequence j, N is the number of protein sequences in the protein data set,
The imbalanced data problem appeared when the training samples were distributed at very different rates among the different classes. In other words, several samples in one class in the training data were much less or more than in the other classes. This is called the imbalanced data problem [23]. It is characteristic in an imbalanced data problem that the number of samples in the positive training data set is notably smaller than that of the negative training data set in the remote homology problem, which is a binary classification problem.
As Chawla described in 2002, the synthetic minority oversampling technique (SMOTE) is used to manufacture new samples from existing data to bring the data set into balance [19]. The basic working principle of the SMOTE algorithm is as follows:
I. It selects samples closest to the property space.
II. Among these selected samples, it draws a line in this property space.
III. It creates new samples by selecting new points on this line.
To gather the most accurate information from raw data for a specific purpose, deriving the best representative features is a difficult machine learning problem, especially in high-dimensional data. Therefore, it is a very important development to automatically learn and extract the most appropriate features of deep learning algorithms for learning representative from raw data [24,25]. In addition to this automated feature of learning and extraction, racing the computing capacity and power, the development of algorithms and remarkable effects in the big data area have made deep learning popular as a best-performing machine learning algorithm in recent years [25–36].
As a rule, a deep learning architecture comprises an input, hidden, and output layer. Publicly, various functions, such as activation and optimization functions on the layers, are used. The activation function choice is an essential factor in performance optimization and capacity of the deep learning architecture designed. The most preferred activation functions are rectified linear unit (ReLU), leaky ReLU, scaled exponential linear unit, softmax, and Hyperbolic Tangent (tanH). The softmax activation function is mainly used as an activation function in the output layer [27]. In this study, the softmax function was experimented with to classify proteins into remote homolog or non-remote homolog in the output layer.
In the deep learning model, the overfitting problem may be encountered when training with a limited number of learning samples the learnable parameter. Various functions, such as dropout, ReLU, initialization/momentum, denoising, and batch normalization, can be used to reduce the overfitting problem [34]. The dropout function was used in the recent research to reduce overfitting after the input layer and hidden layers.
There are many unique libraries, such as Caffe, Keras, Tensorflow, Torch, and Theano, for implementing deep learning [32]. Keras is an open-source Python deep learning library that has been widely used in many studies [37,38]. The current study was implemented using the Keras library backend Tensorflow.
The inputs for the study’s deep learning model are smoothed data sets of protein sequences obtained after TF-IDF weighting processes of n-grams. The smoothed protein sequences are classified with the deep learning architecture designed. The deep learning architecture of the experiment consisted of an input layer with 1200 neurons, three hidden layers with 600,300,100 neurons respectively, and an output layer. The dropout function with a 0.5 rate was used to reduce overfitting after the input layer and hidden layers. The model was trained using the Root Mean Squared Propagation (RMSprop) optimizer. The softmax function was used to classify the proteins into remote homolog or non-remote homolog in the output layer.
In this study, a new system was established using sequence similarity and family and superfamily similarity. Proteins belonging to the same superfamily but a different family was remote homolog proteins with each other. In contrast, proteins that belonged to the same superfamily and family were homolog proteins with each other. Hence, in this study, for coding the result, 1 was used as the remote homolog protein tag, and 0 was used for the non-remote homolog protein.
The system herein was created by labeling proteins from the same superfamily and different families as remote homolog proteins and proteins from a different fold as non-remote homolog proteins. Positive protein instances are taken from within the same superfamily, while negative protein instances are taken from outside the target superfamily. Negative instances are separated randomly into training and test sets. Positive test samples are taken within the same family as the target family. Positive training samples are taken from outside of the target family and within the same superfamily. The current experiment used negative and positive protein instances to detect remote homolog proteins. In the system herein, positive test instances were taken proteins from within the target family from the SCOP database. Positive train instances were taken from proteins inside the same protein superfamily with the target family and outside the target family from the SCOP database. Negative test and train instances were taken from outside of the superfamily. Three different data sets, SCOP 1.53, SCOP benchmark, and the newly created SCOP protein database, were used for remote homolog detection in this experimental study. The data sets of the experiment are detailed in Section 2.1. With this study, it has been observed that the SCOP database is useful in remote homologous protein detection studies.
Since remote homolog proteins look alike and have very small sequence similarities with one another, it is pretty challenging to determine remote homology with the entire and single amino acid sequence similarity of any protein. Hence, the next step was to extract n-grams from the protein sequences. The next step was to obtain the TF-IDF matrix using extracted n-grams from the protein sequences. After n-grams of protein sequences were taken and TF-IDF weighting calculation, the training data obtained was smoothed. Then, smoothing based on oversampling was applied to the data set reserved for the training process to prevent misclassification. With the smoothing process, new samples for the minority class were produced so that the number of samples in the minority class was equal to the number of samples in the majority class on the data set reserved for the training process. The smoothing process was performed to balance classes of the training protein data set using the synthetic minority oversampling technique (SMOTE). The next step was classifying the proteins into remote homolog and non-remote homolog. The deep learning architecture successfully classified the proteins into remote homolog or non-remote homolog The dropout function after the input and hidden layers was performed to avoid overfitting. This study used the softmax activation function to classify proteins as remote homologous or non-remote homologous in the output layer. The deep learning architecture of the experiment is detailed in Section 2.5.
Accuracy, ROC score, and confusion matrix results of the remote homology detection with smoothing and without smoothing process for between 1 and 5 grams of the target 1.4.1.1 family are shown in Table 1. As given in the confusion matrix in the remote homology detection without smoothing in Table 1, all 23 positive instances for the target 1.4.1.1 family were misclassified. Hence, remote homology detection without smoothing was unacceptable. Regarding accuracy, the confusion matrix, and ROC score, the best performance n-grams from 1 and 5 grams are seen in Table 1. In 1-gram and 4-gram trials, although the accuracy results seem to have decreased slightly after smoothing, the result of this study is not evaluated only by looking at the accuracy results.
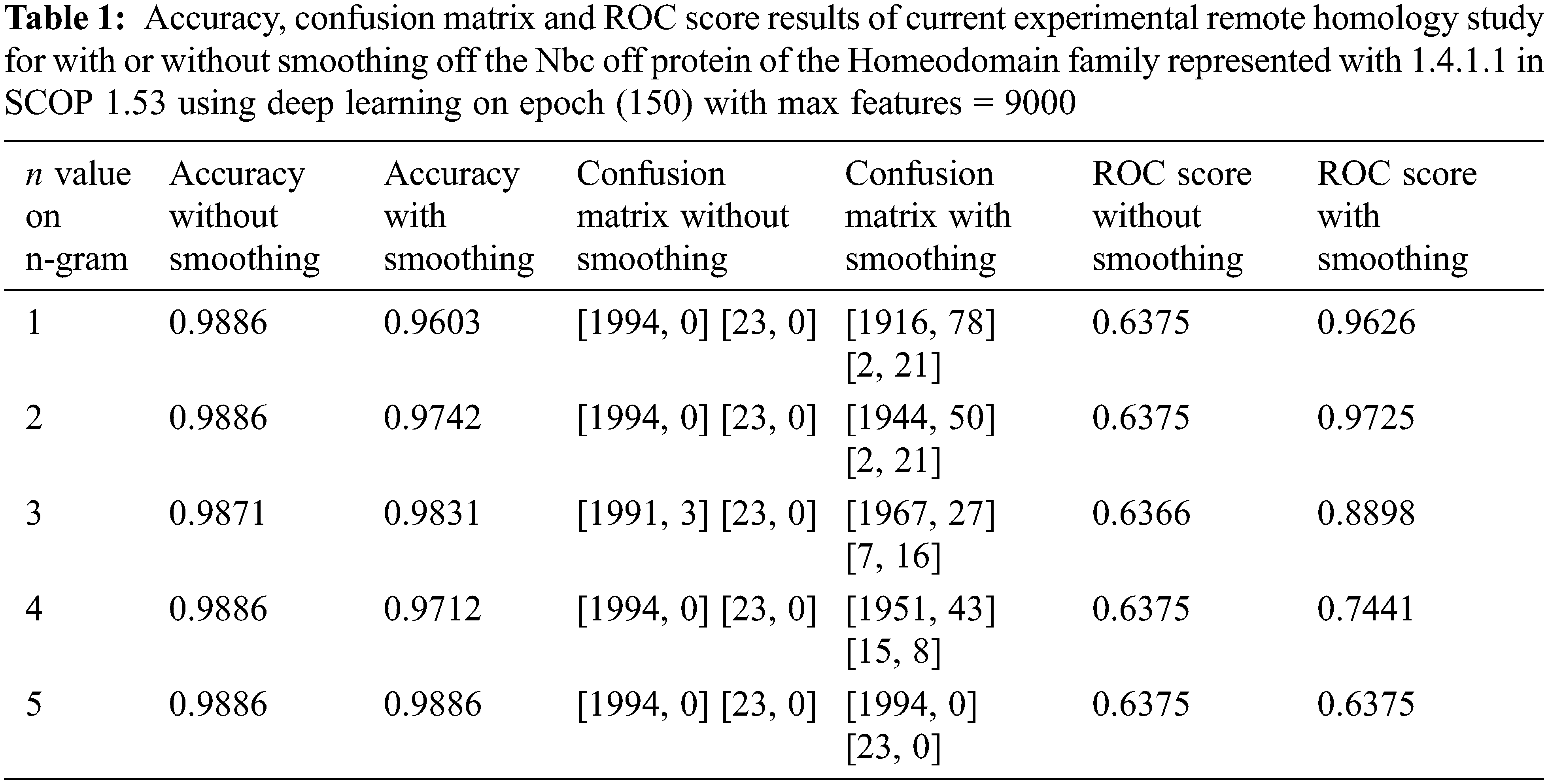
Table 1 shows a classification success should not be decided by looking only at the accuracy results. Only the accuracy results are considered; the classification seems successful; the confusion matrix results are considered too, and it was observed that there was a failure. This is because of the imbalanced data problem. In other words, although the accuracy of the trials without the smoothing process seems successful in this experiment, as it can be seen when looking at the confusion matrix, it finds the class of the samples belonging to the class with a small number of samples to be incorrect and makes a wrong classification. The smoothing process is carried out to prevent this situation.
After the smoothing process, there is a decrease in accuracy between 1 and 4 grams, while there is no change in the accuracy of remote homolog detection using 5 grams. However, the 5-grams trial cannot be considered successful, depending only on the accuracy results. The confusion matrix results in Tables. 1 and 2 show this study’s completely remote homolog results. While the “current experiment” in Table 3 is our remote homolog results of this study; references such as [3] in sources belong to previous studies. Table 1, the entire remote homolog class for 5-gram is misclassified after the smoothing process. As there are moved from 5-gram to 1-gram in Table 1, towards the smaller n-numbered grams, the success in more accurately labeling the remote homolog class in the confusion matrix increases. Hence, n-grams with small n numbers are more successful in smoothing numbers for remote homology detection. When it is desired to choose the most successful detection between 1 and 2 grams, it is observed that remote homolog protein detection using 2 grams in accuracy and ROC score values is more successful without using 1-gram. However, the confusion matrices are the same. Therefore, in this study, test results are given using 2-gram.


The experimental results of the detection using the deep learning of the remote homology of the proteins of the 54 preferred families are shown in Table 2. The average and highest ROC scores obtained by testing this newly designed method with three different protein data sets, SCOP 1.53, SCOP benchmark, and the new data set from SCOPe 2.07, are given in Table 2. Depending on Table 3, the Mean ROC score of this experimental remote homology detection study ranges between 86% and 90%; the best ROC score is over 99%.
Homology studies using NLP techniques such as n-gram have been conducted. The common aim in the current study and these studies in Table 3 was to perform an accurate remote homology detection without structural information about the proteins.
The bag-of-words (BoW) model has been used with the BoW feature model to extract useful features from protein data [3]. However, in the BoW model, while looking at how many times the word passes, the order in which the term is located is not kept. Loss of order information is a factor that reduces classification success. Therefore, the success of this study was increased by choosing TF-IDF vectorization instead of the BoW model.
Yeh et al. showed that n-gram and TF-IDF were used to eliminate noise and reveal biologically meaningful words [6]. As observed in the present experiment and other experiments, it was observed that the choice of n-numbers for n-gram processing was the vital factor influencing the success of the n-gram remote homology detection process.
In the study of Liu et al. [39], profile-based proteins (p1) or the kernel target alignment (KTA)-objective function was used to optimize the weight of each kernel method after the n-gram model, and it was seen to increase performance by between 3% and 13%. On the other hand, Yeh et al. [6] used both positive and negative samples to improve the accuracy of their study, and the lack of a balancing function, such as smoothing, was necessary because it was imbalanced data, which comprised the deficiency of their research. The current study balanced positive and negative train data by performing the smoothing procedure. On the other hand, Yeh et al. [6] owed its performance to using LDA.
The discovery of unknown protein structures and functions has an important impact on the discovery of new drugs in medicine today. In discovering protein structures, two important areas of bioinformatics, homologous protein, and remote homologous protein detection, are useful.
Although protein homology and remote protein homology are similar problems, it has been observed that remote homologous protein similarity is a more complex problem to solve. In this study, the focus was placed on detecting remote homolog proteins, a difficult and important problem in bioinformatics, to solve with low sequence similarity. Furthermore, due to the continuous increase of protein sequences, protein data storage and processing are other problems to be solved. In addition, in protein data, the small number of instances in remote homologous protein classes is a fundamental problem, as it causes unbalanced class distribution.
One of the crucial steps to detect remote homologous with the best performance is to obtain the properties of the proteins in the most useful way, while another is to classify these features in the best way. Experimental results of the study on one of the widely-used protein benchmark datasets, SCOP 1.53, showed that deep learning with smoothing protein features based on TF-IDF weighting and n-gram representative outperformed other related methods in terms of both the mean ROC score and accuracy.
Natural language processing can be used in bioinformatics problems involving text data like the n-gram algorithm used in this study. Likewise, image processing techniques are used in protein structure problems. Therefore, as seen in this study, it is inevitable to use computer algorithms such as natural language processing and image processing in medicine and bioinformatics problems.
Nowadays, new proteins are constantly being discovered whose structure and function are unknown. The discovery of new proteins, the need to store and process large amounts of protein data, and to extract information about these proteins bring to mind big data technology. Therefore, it will be beneficial to use big data technologies in bioinformatics problems with rapidly increasing data sets such as remote homologous protein. For this reason, it is planned to use big data technologies in the versions of the study. In conclusion, deep learning with TF-IDF weighting and n-gram representatives may be a useful tool for protein remote homology detection.
Funding Statement: This study is carried out by Cukurova University Scientific Research Projects (BAP) is supported with Project No: FDK-2019-11621.
Conflicts of Interest: The authors have no conflicts of interest to report regarding the present study.
References
1. G. Eason, B. Noble and I. N. Sneddon, “On certain integrals of Lipschitz-Hankel type involving products of bessel functions,” Philosophical Transactions of the Royal Society of London. Series A, Mathematical and Physical Sciences, vol. 247, no. 935, pp. 529–551, 1955. [Google Scholar]
2. A. Ben-Hur and D. Brutlag, “Remote homology detection: A motif based approach,” Bioinformatics, vol. 19, no. suppl_1, pp. i26–i33, 2003. [Google Scholar]
3. Q. W. Dong, X. L. Wang and L. Lin, “Application of latent semantic analysis to protein remote homology detection,” Bioinformatics, vol. 22, no. 3, pp. 285–290, 2006. [Google Scholar] [PubMed]
4. C. Pál, B. Papp and M. J. Lercher, “An integrated view of protein evolution,” Nature Reviews Genetics, vol. 7, no. 5, pp. 337–348, 2006. [Google Scholar]
5. A. Tripathy and S. K. Rath, “Classification of sentiment of reviews using supervised machine learning techniques,” International Journal of Rough Sets and Data Analysis (IJRSDA), vol. 4, no. 1, pp. 56– 74, 2017. [Google Scholar]
6. J. H. Yeh and C. H. Chen, “Protein remote homology detection based on latent topic vector model,” in IEEE Int. Conf. on Networking and Information Technology, Manila, Philippines, pp. 456–460, 2010. [Google Scholar]
7. A. Tomović, P. Janičić and V. Kešelj, “N-Gram-based classification and unsupervised hierarchical clustering of genome sequences,” Computer Methods and Programs in Biomedicine, vol. 81, no. 2, pp. 137–153, 2006. [Google Scholar] [PubMed]
8. F. Ghaffar, S. Khan and C. Yu-jhen, “Macromolecule classification based on the amino-acid sequence,” arXiv, arXiv:2001.01717, 2020. [Google Scholar]
9. H. Oğul and E. Ü. Mumcuoğlu, “A discriminative method for remote homology detection based on n-peptide compositions with reduced amino acid alphabets,” BioSystems, vol. 87, no. 1, pp. 75–81, 2007. [Google Scholar] [PubMed]
10. B. Liu, X. Wang, Q. Zou, Q. Dong and Q. Chen, “Protein remote homology detection by combining Chou’s pseudo amino acid composition and profile-based protein representation,” Molecular Informatics, vol. 32, no. 9–10, pp. 775–782, 2013. [Google Scholar] [PubMed]
11. B. Liu, J. Xu, Q. Zou, R. Xu, X. Wang et al., “Using distances between Top-n-gram and residue pairs for protein remote homology detection,” BioMed Central in BMC Bioinformatics, vol. 15, no. S2, pp. S3, 2014. [Google Scholar]
12. B. Liu, X. Wang, L. Lin, Q. Dong and X. Wang, “A discriminative method for protein remote homology detection and fold recognition combining top-n-grams and latent semantic analysis,” BMC Bioinformatics, vol. 9, no. 1, pp. 510, 2008. [Google Scholar] [PubMed]
13. B. Liu and Y. Zhu, “ProtDec-LTR3.0: Protein remote homology detection by incorporating profile-based features into Learning to Rank,” IEEE Access, vol. Jul 18, no. 7, pp. 102499–102507, 2009. [Google Scholar]
14. K. Lan, D. T. Wang, S. Fong, L. S. Liu, K. Wong et al., “A survey of data mining and deep learning in bioinformatics,” Journal of Medical Systems, vol. 42, no. 8, pp. 139, 2018. [Google Scholar] [PubMed]
15. H. Sun and R. Grishman, “Lexicalized dependency paths based supervised learning for relation extraction,” Computer Systems Science and Engineering, vol. 43, no. 3, pp. 861–870, 2022. [Google Scholar]
16. M. Torrisi, G. Pollastri and Q. Le, “Deep learning methods in protein structure prediction,” Computational and Structural Biotechnology Journal, vol. 18, pp. 1301–1310, 2020. [Google Scholar] [PubMed]
17. M. Torrisi, M. Kaleel and G. Pollastri, “Deeper profiles and cascaded recurrent and convolutional neural networks for state-of-the-art protein secondary structure prediction,” Scientific Reports, vol. 9, no. 1, pp. 1–12, 2019. [Google Scholar]
18. M. S. Klausen, M. C. Jespersen, H. Nielsen, K. K. Jensen, V. I. Jurtz et al., “NetSurfP-2.0: Improved prediction of protein structural features by integrated deep learning,” Proteins: Structure Function, and Bioinformatics, vol. 87, no. 6, pp. 520–527, 2019. [Google Scholar]
19. Y. Wang, J. Bao, F. Huang, J. Du and Y. Li, “Protein remote homology detection based on deep convolutional neural network,” Preprint (version 1) available at Research Square, 2019. [Online]. Available: https://www.researchsquare.com/article/rs-6054/v1 [Google Scholar]
20. A. Andreeva, D. Howorth, S. E. Brenner, T. J. Hubbard, C. Chothia et al., “SCOP database in 2004: Refinements integrate structure and sequence family data,” Nucleic Acids Research, vol. 32, no. suppl_1, pp. D226–D229, 2004. [Google Scholar] [PubMed]
21. S. E. Brenner, P. Koehl and M. Levitt, “The ASTRAL compendium for sequence and structure analysis,” Nucleic Acids Research, vol. 28, no. 1, pp. 254–256, 2000. [Google Scholar] [PubMed]
22. G. Ifrim, G. Bakir and G. Weikum, “Fast logistic regression for text categorization with variable-length n-grams,” in ACM Proc. of the 14th ACM SIGKDD Int. Conf. on Knowledge Discovery and Data Mining, Las Vegas, Nevada, USA, pp. 354–362, 2008. [Google Scholar]
23. Z. Zheng, X. Wu and R. Srihari, “Feature selection for text categorization on imbalanced data,” ACM Sigkdd Explorations Newsletter, vol. 6, no. 1, pp. 80–89, 2004. [Google Scholar]
24. N. V. Chawla, K. W. Bowyer, L. O. Hall and W. P. Kegelmeyer, “SMOTE: Synthetic minority over-sampling technique,” Journal of Artificial Intelligence Research, vol. 16, pp. 321–357, 2002. [Google Scholar]
25. C. Angermueller, T. Pärnamaa, L. Parts and O. Stegle, “Deep learning for computational biology,” Molecular Systems Biology, vol. 12, no. 7, pp. 878, 2006. [Google Scholar]
26. Y. LeCun, Y. Bengio and G. Hinton, “Deep learning,” Nature, vol. 521, no. 7553, pp. 436–444, 2015. [Google Scholar] [PubMed]
27. Y. Li, C. Huang, L. Ding, Z. Li, Y. Pan et al., “Deep learning in bioinformatics: Introduction, application, and perspective in the big data era,” Methods, vol. 166, pp. 4–21, 2019. [Google Scholar] [PubMed]
28. S. Ioffe and C. Szegedy, “Batch normalization: Accelerating deep network training by reducing internal covariate shift,” in Int. Conf. on Machine Learning, Lille, France, pp. 448–456, 2015. [Google Scholar]
29. A. Caliskan, M. E. Yuksel, H. Badem and A. Basturk, “Performance improvement of deep neural network classifiers by a simple training strategy,” Engineering Applications of Artificial Intelligence, vol. 67, pp. 14–23, 2018. [Google Scholar]
30. K. Lan, D. T. Wang, S. Fong, L. S. Liu, K. K. Wong et al., “A survey of data mining and deep learning in bioinformatics,” Journal of Medical Systems, vol. 42, no. 8, pp. 139, 2018. [Google Scholar] [PubMed]
31. J. de Dieu Uwisengeyimana and T. Ibrikci, “Diagnosing knee osteoarthritis using artificial neural networks and deep learning,” Biomedical Statistics and Informatics, vol. 2, no. 3, pp. 95, 2007. [Google Scholar]
32. E. M. Karabulut and T. Ibrikci, “Discriminative deep belief networks for microarray based cancer classification,” Biomedical Research, vol. 28, no. 3, pp. 0970–938X, 2017. [Google Scholar]
33. A. Caliskan, H. Badem, A. Basturk and M. E. Yuksel, “Diagnosis of the Parkinson disease by using deep neural network classifier,” Istanbul University-Journal of Electrical & Electronics Engineering, vol. 17, no. 2, pp. 3311–3318, 2017. [Google Scholar]
34. F. Gemci and T. Ibrikci, “Using deep learning algorithm to diagnose Parkinson disease with high accuracy,” Kahramanmaraş Sütçü İmam University Journal of Engineering Science, vol. 22, no. Special Issue, pp. 19–25, 2019. [Google Scholar]
35. D. Shen, G. Wu and H. I. Suk, “Deep learning in medical image analysis,” Annual Review of Biomedical Engineering, vol. 19, pp. 221–248, 2017. [Google Scholar] [PubMed]
36. S. Min, B. Lee and S. Yoon, “Deep learning in bioinformatics,” Briefings in Bioinformatics, vol. 18, no. 5, pp. 851–869, 2017. [Google Scholar] [PubMed]
37. Z. Wang, K. Liu, J. Li, Y. Zhu and Y. Zhang, “Various frameworks and libraries of machine learning and deep learning: A survey,” Archives of Computational Methods in Engineering, vol. 2019, pp. 1–24, 2019. [Google Scholar]
38. N. M. Kumar and R. Manjula, “Design of multi-layer perceptron for the diagnosis of diabetes mellitus using Keras in deep learning,” in Smart Intelligent Computing and Applications, Singapore: Springer, pp. 703–711, 2019. [Google Scholar]
39. B. Liu, D. Zhang, R. Xu, J. Xu, X. Wang et al., “Combining evolutionary information extracted from frequency profiles with sequence-based kernels for protein remote homology detection,” Bioinformatics, vol. 30, no. 4, pp. 472–479, 2014. [Google Scholar] [PubMed]
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools