
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
TECHNICAL REPORT

NJmat 2.0: User Instructions of Data-Driven Machine Learning Interface for Materials Science
1 Department of Internet Engineering, School of Software Engineering, Nanjing University of Information Science & Technology, Nanjing, 210044, China
2 Department of Materials Physics, School of Chemistry and Materials Science, Nanjing University of Information Science & Technology, Nanjing, 210044, China
* Corresponding Author: Lei Zhang. Email:
Computers, Materials & Continua 2025, 83(1), 1-11. https://doi.org/10.32604/cmc.2025.062666
Received 24 December 2024; Accepted 17 February 2025; Issue published 26 March 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
NJmat is a user-friendly, data-driven machine learning interface designed for materials design and analysis. The platform integrates advanced computational techniques, including natural language processing (NLP), large language models (LLM), machine learning potentials (MLP), and graph neural networks (GNN), to facilitate materials discovery. The platform has been applied in diverse materials research areas, including perovskite surface design, catalyst discovery, battery materials screening, structural alloy design, and molecular informatics. By automating feature selection, predictive modeling, and result interpretation, NJmat accelerates the development of high-performance materials across energy storage, conversion, and structural applications. Additionally, NJmat serves as an educational tool, allowing students and researchers to apply machine learning techniques in materials science with minimal coding expertise. Through automated feature extraction, genetic algorithms, and interpretable machine learning models, NJmat simplifies the workflow for materials informatics, bridging the gap between AI and experimental materials research. The latest version (available at https://figshare.com/articles/software/NJmatML/ 24607893 (accessed on 01 January 2025)) enhances its functionality by incorporating NJmatNLP, a module leveraging language models like MatBERT and those based on Word2Vec to support materials prediction tasks. By utilizing clustering and cosine similarity analysis with UMAP visualization, NJmat enables intuitive exploration of materials datasets. While NJmat primarily focuses on structure-property relationships and the discovery of novel chemistries, it can also assist in optimizing processing conditions when relevant parameters are included in the training data. By providing an accessible, integrated environment for machine learning-driven materials discovery, NJmat aligns with the objectives of the Materials Genome Initiative and promotes broader adoption of AI techniques in materials science.Keywords
Materials science has traditionally relied on empirical methods and domain-specific expertise to study and design new materials [1–3]. However, the increasing complexity and vastness of the research space in modern materials science—characterized by high-dimensional and multi-modal data—have rendered conventional approaches insufficient for addressing many of the field’s intricate challenges. Data-driven methods, which leverage large-scale data analysis and advanced algorithms, have revolutionized the way materials science is conducted [4,5]. Unlike traditional methods, which often depend on manual data analysis, data-driven approaches provide more efficient, accurate, and scalable solutions for discovering new materials and understanding their properties. This paradigm shift has laid the foundation for the Materials Genome, which aims to accelerate the development of new materials by fostering collaboration across experimental, computational, and database domains [6,7]. The materials genome initiative emphasizes the importance of open data sharing and collaboration to optimize the design, synthesis, and processing of materials. To achieve the objectives of materials genome engineering, there is an urgent need for advanced computational tools that can facilitate the discovery and innovation of materials.
In recent years, natural language processing (NLP) and large language models (LLMs) have gained significant traction in materials science [8,9]. These technologies enable the efficient extraction and analysis of information from vast amounts of unstructured data, such as scientific literature, patents, and research articles. LLMs, such as GPT-based models, possess the capability to understand and generate text, making them particularly valuable for extracting meaningful insights from materials-related documents. By embedding domain-specific knowledge, these models can identify relationships between materials properties and their corresponding terminologies. For instance, NLP techniques can be utilized to automatically extract key features, relationships, and patterns from scientific texts, which can subsequently inform material design and discovery related to the processing-structure-property-performance (PSPP) framework. As these models continue to evolve, their integration into materials science workflows will accelerate the discovery of novel materials and significantly enhance the overall efficiency of research in the field. Machine learning potentials (MLP) represent another essential aspect of modern materials science research [10,11]. MLP refers to machine learning models that predict material properties based on atomic structures. These models, trained on large datasets of known materials, offer a powerful tool for simulating and predicting material behavior without the need for costly and time-consuming experiments. MLP can be particularly effective in predicting key material properties, such as stability, conductivity, and reactivity, which are critical for the design of new materials with targeted characteristics. By leveraging MLP, researchers can explore a much broader space of materials, including those that have yet to be synthesized, thus enabling the rapid discovery of materials with tailored properties.
In addition, large language models, such as LLaMA3, further enhance this process by providing interactive data analysis, literature summarization, and hypothesis generation. Beyond textual data, structured numerical datasets require specialized machine learning approaches [12,13]. Crystal graph convolutional neural networks (CGCNNs) capture the intricate atomic and bonding relationships within crystalline materials, enabling accurate predictions of formation energy, electronic properties, and other key characteristics [14–16]. MLPs that are trained on high-precision quantum mechanical datasets offer an efficient alternative to traditional interatomic potentials, allowing for large-scale atomistic simulations with near first-principles accuracy [16,17].
While several powerful software packages for materials science have emerged in recent years, many require specialized knowledge of Linux and programming, making them challenging for experimentalists to use effectively. Although influential, the present material informatics tools often lack user-friendly interfaces, which limits their accessibility. To address this gap, NJmat 2.0 is developed as a data-driven software package designed with an intuitive interface that allows researchers to easily access advanced machine learning and natural language algorithms. NJmat enables experimentalists to analyze their data without the need for coding or complex programming expertise.
In this manuscript, we present the user instructions for NJmat, the machine learning interface designed specifically for materials science (Fig. 1). Featuring multiple user-friendly, click-to-use interactive buttons, NJmat integrates machine learning, deep learning, language models, natural language processing, large language model, machine learning potential and graph neural network to offer a robust solution for materials property prediction and analysis. It is a versatile tool that is particularly beneficial for materials, chemical, and physical scientists in the field of materials genomics and informatics. With no coding expertise required, it is accessible to a broad user base.
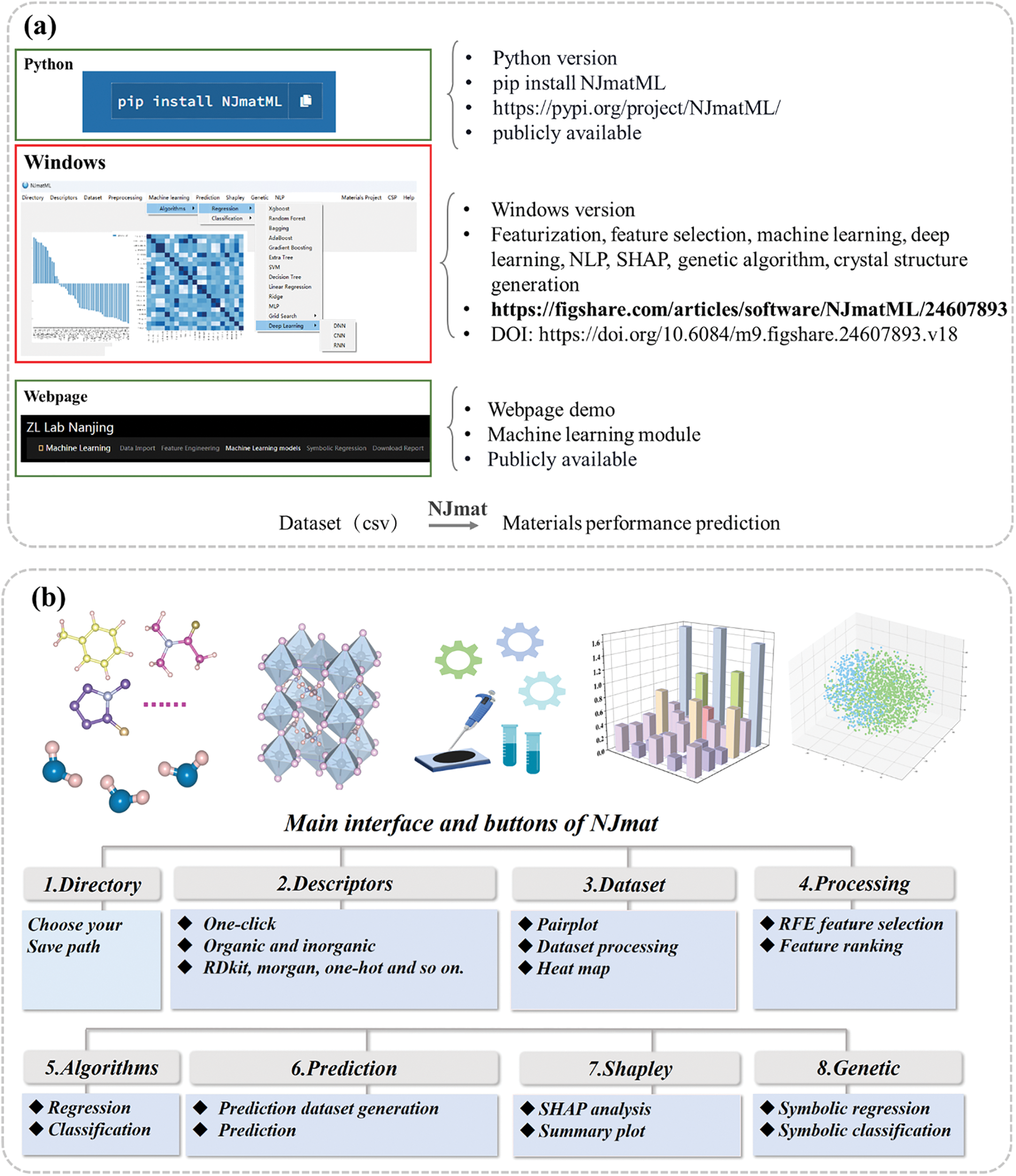
Figure 1: (a) Release of windows version of NJmat. The windows version is the principal version of NJmat, which is tailored for user-friendliness, providing a graphical user interface (GUI) that eliminates the need for extensive coding knowledge. (b) Representative buttons of NJmat, which encompasses a wide array of functionalities designed to address key challenges in materials science research
NJmat is a comprehensive button-based user-friendly software package developed to streamline materials science research through data-driven approaches. To accommodate diverse user needs and expertise levels, NJmat is available in three versions: Python, Windows, and Webpage. (1) The Python version NJmatML is designed for researchers who prefer programmatic access and the flexibility to integrate the software into custom workflows. It can be installed using the command: pip install NJmatML. This version is suitable for users with coding expertise and the need for advanced customization. (2) Windows Version. The Windows version is the principal version of NJmat, which is tailored for user-friendliness, providing a graphical user interface (GUI) that eliminates the need for extensive coding knowledge. This version is developed with the goal of reducing the learning curve and enabling efficient use of NJmat’s core functionalities. (3) Webpage-Based Version. To provide an alternative option for users without local installation requirements, a web-based demo of NJmat is available at this address https://patrick007.shinyapps.io/zllab_ofi_ml/ (accessed on 01 January 2025), with limited capabilities provided at the moment compared with the windows version. This webpage allows users to explore the software’s conventional capabilities such as feature selection and machine learning model construction interactively and serves as a convenient platform for preliminary evaluations with a varieties of machine learning algorithms.
NJmat encompasses a wide array of functionalities designed to address key challenges in materials science research [18]. These include featurization (automated extraction of material descriptors from datasets), feature selection (identification of critical features for improving predictive model performance), machine learning and deep learning (tools for developing predictive models and optimizing material properties), natural language processing (NLP) (capabilities for extracting insights from unstructured text data, such as scientific literature), SHAP analysis (tools for interpretable machine learning, enabling the explanation of model predictions), genetic algorithms (optimization strategies for designing materials with desired properties) and crystal structure generation (methods for randomly generating and visualizing material structures).
The NJmat platform (https://figshare.com/articles/software/NJmatML/24607893 (accessed on 01 January 2025)) provides a comprehensive suite of tools for materials science data analysis, encompassing both regression and classification tasks (Fig. 2a). The main menu includes several key components that facilitate various stages of the analysis pipeline, including file directory management, feature generation, machine learning model construction, and more. By supporting publicly available datasets in CSV format, NJmat further facilitates streamlined workflows for data-driven materials research. The “Directory” button allows users to define file paths for data import and export. The “Descriptors” button automatically generates molecular and material features based on chemical formulas and SMILES representations from the dataset. “Dataset” enables direct visualization of the data distribution, while “Preprocessing” provides feature selection, heatmap visualizations, and feature importance rankings. The “Machine Learning” section incorporates a wide range of algorithms for both regression and classification tasks. Regression algorithms include XGBoost, Random Forest, Bagging, AdaBoost, Gradient Boosting, Extra Tree, SVM, Decision Tree, Linear Regression, Ridge, MLP, Grid Search, and Deep Learning models (DNN, CNN, and RNN). Classification algorithms encompass Random Forest, Extra Tree, Gaussian Process, Decision Tree, SVM, AdaBoost, XGBoost, CatBoost, and Deep Learning (DNN, CNN, and RNN). The “Prediction” feature allows for the generation of virtual data outputs, which can then be used for further validation. The “Shapley” tool provides Shapley plots for the interpretation of model results, while the “Genetic” module offers genetic algorithms for both classification and regression tasks (Fig. 2c). These algorithms enable more interpretable machine learning models, which are represented using tree structures derived from mathematical operators (e.g., addition, subtraction, multiplication, division, and square roots) and terminals, such as NJmat descriptors. The “NLP” module represents the natural language processing component of NJmat, used for extracting chemical information from textual data. The “Visualizer” module enables the visualization of crystal structures and CIF file downloads. “CSP” facilitates random crystal generation based on material formulas, and “Help” provides access to useful documentation and resources.
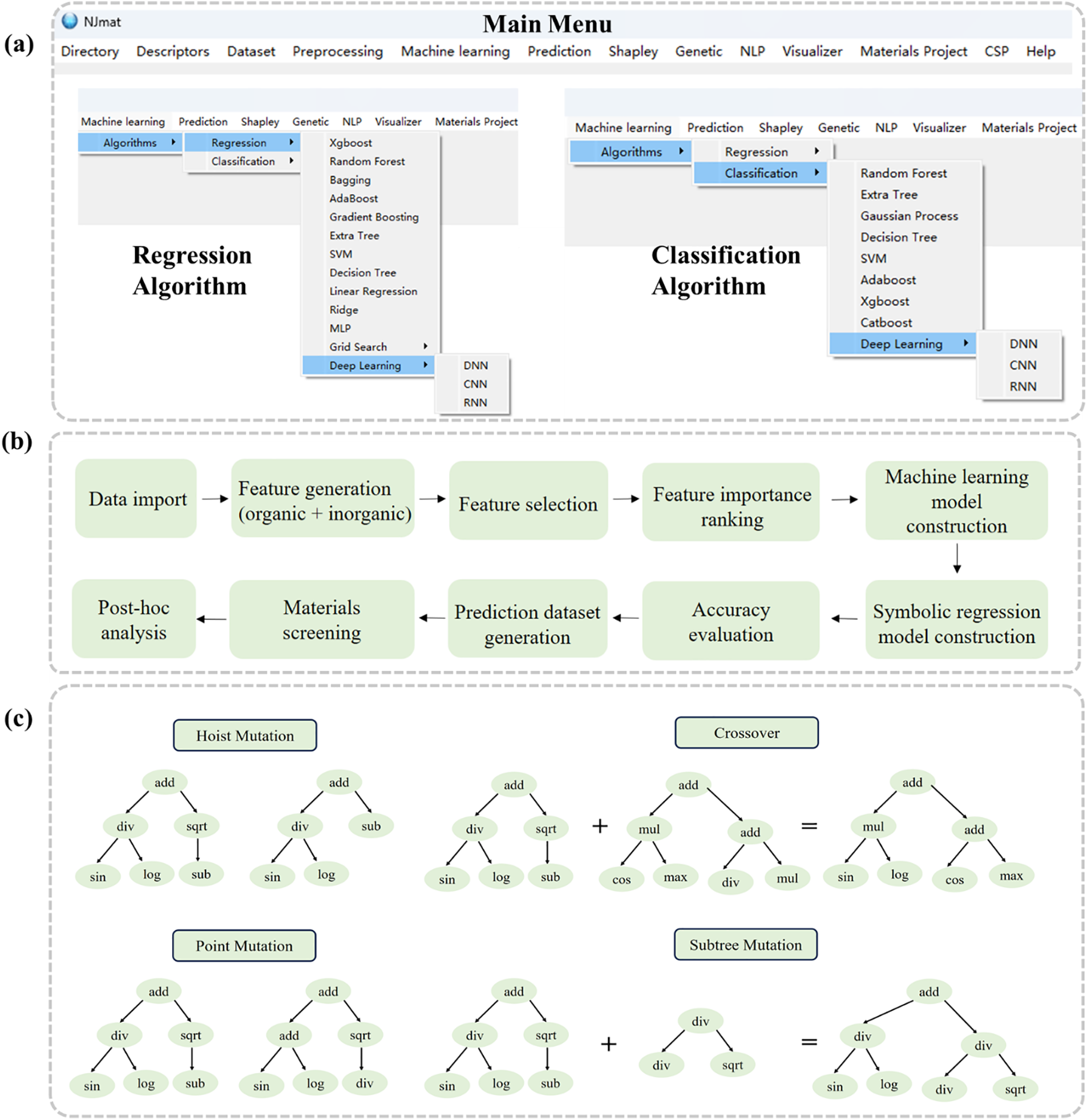
Figure 2: (a) Main menu buttons of NJmat. (b) Flowchart illustrating the use of NJmat for materials science: data import, feature generation (for both organic and inorganic materials), feature selection, feature importance ranking, and machine learning model construction, symbolic regression/classification model construction, accuracy evaluation, prediction dataset generation, materials screening, and post-hoc analysis. (c) Genetic algorithm operations, including hoist mutation, crossover, point mutation, and subtree mutation, are presented. Unlike traditional machine learning techniques (e.g., Random Forest, Support Vector Machine (SVM), and Multi-Layer Perceptron (MLP)), the genetic algorithm provides more interpretable models by using mathematical operators (such as addition, subtraction, multiplication, division, square roots, etc.) and terminals (which are different NJmat descriptors) to form tree-like representations
The current version of NJmat includes MatBERT [19], Word2Vec [20,21], and Crystal Hamiltonian Graph Neural Network (CHGNet) [22] as preliminary examples to represent NLP, MLP and GNN functionalities proposed in recent years. MatBERT is a deep learning model based on transformer architectures, fine-tuned with over 200 million materials science articles, enabling it to predict material properties by understanding contextual relationships. Word2Vec, a popular natural language processing method, transforms material-related terms into continuous vector representations, capturing semantic relationships between terms. For example, it can identify strong associations such as “lithium” and “battery”, while distinguishing terms like “carbon” and “steel” in different contexts. The Word2Vec models in NJmat are trained on over 1 million or 50,000 materials science articles, and users can also create custom language models using an NJmat submodule. CHGNet employs graph-based neural networks, representing atoms as nodes and bonds as edges. Through graph neural networks (GNNs), CHGNet learns atomic interactions and can efficiently predict material structures and properties. By integrating these models, NJmat leverages a hybrid approach that combines language processing, machine learning, and graph analysis to accelerate material discovery and property prediction.
A case study on machine learning model construction, specifically focusing on perovskite adsorption energy, is provided in https://github.com/cxxhub/expert-potato (accessed on 01 January 2025).
NJmat automatically facilitates the construction of machine learning models, supporting both regression and classification tasks based on various algorithms. The adsorption energy (in eV) fitting results for ionic adsorption on 2D Ruddlesden-Popper (RP) halide perovskites are analyzed using NJmat. The dataset, available at https://github.com/cxxhub/expert-potato (accessed on 01 January 2025), is used for training and evaluation. NJmat automatically generates heat maps and feature rankings based on Pearson correlation coefficients to assess feature importance (Fig. 3). The fitting results for various machine learning algorithms on both training and test datasets exhibit high accuracy, evaluated using metrics such as mean absolute error (MAE), mean squared error (MSE), and the coefficient of determination (R2). Algorithms tested include XGBoost, Random Forest, Bagging, AdaBoost, Gradient Boosting, Linear Regression, Decision Tree, Extra Tree, MLP, SVM, and Ridge Regression. For instance, the XGBoost model achieves an MAE of 0.386, an MSE of 0.783, and an R2 of 0.994, indicating decent predictive performance over a wide range of adsorption energies. Among the models, AdaBoost demonstrated the best MAE of 0.371, the smallest error among all tested algorithms for this particular case. Based on these results, the AdaBoost model is recommended for further predictions on virtual datasets, which can be directly executed using the prediction functionality available in NJmat. However, the choice of machine learning algorithm depends on the specific task and may vary across different use cases. This workflow enables efficient and accurate predictions of material properties, simplifying the analysis of large and complex datasets.
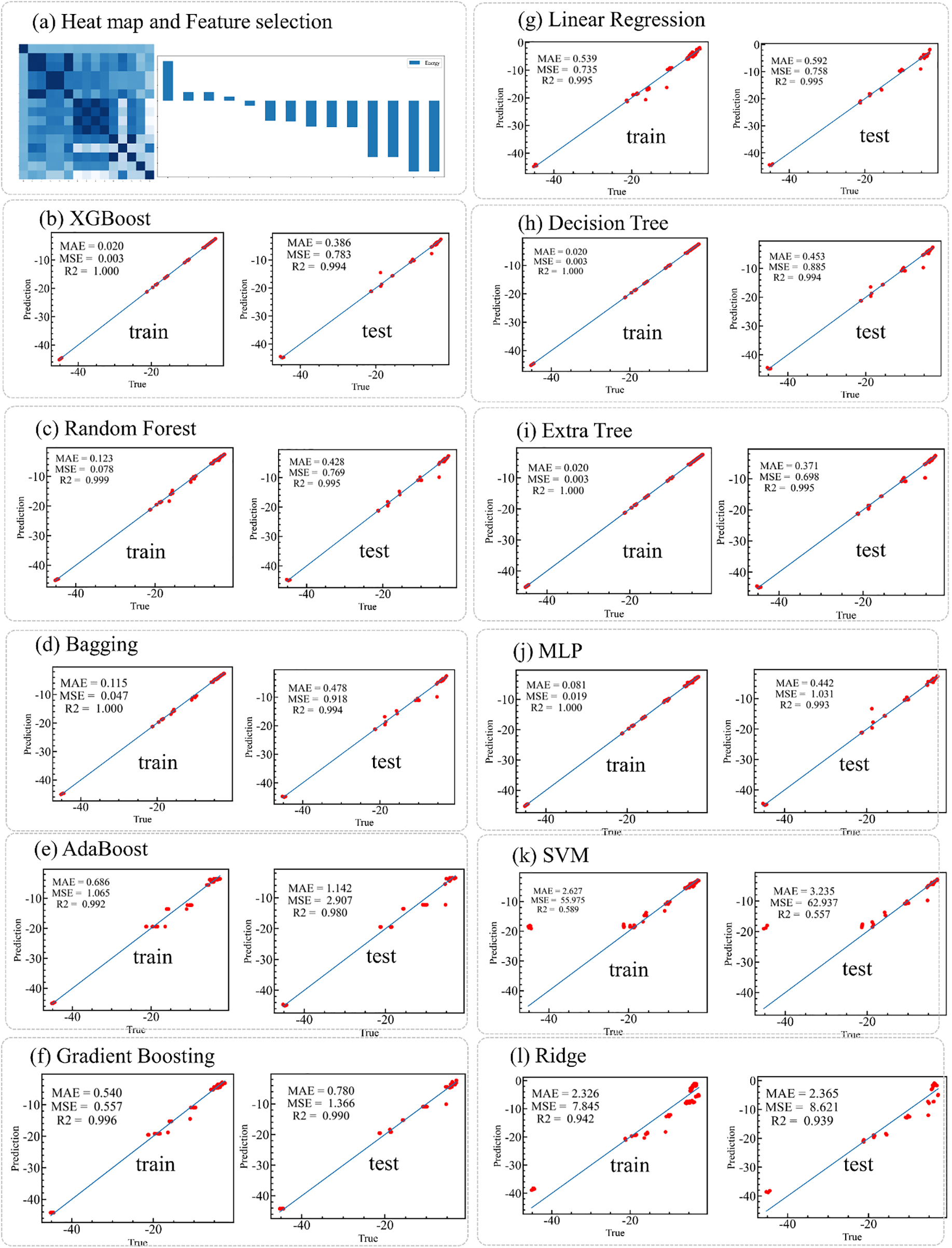
Figure 3: Fitting results using NJmat for a perovskite adsorption energy case. (a) Heat maps and Pearson coefficient-based rankings analyze feature importance. (b–l) Performance of machine learning algorithms, including XGBoost, Random Forest, Bagging, AdaBoost, Gradient Boosting, Linear Regression, Decision Tree, Extra Tree, MLP, SVM, and Ridge, for both training and test datasets. Results are evaluated based on MAE, MSE, and R2, demonstrating the effectiveness of NJmat for predicting material properties
The NJmat platform includes a natural language processing (NLP) module, NJmatNLP (Fig. 4a), designed to harness advanced language models for materials science applications. This module utilizes MatBERT (Fig. 4b) and Word2Vec models, trained on extensive datasets comprising millions of material science abstracts or subsets of 50,000 papers. NJmatNLP supports materials prediction tasks through cosine similarity and clustering techniques, with results visualized via UMAP for enhanced interpretability. The module includes NJmatNLPSetup, an installation file facilitating the materials prediction using the MatBERT language model integrated with UMAP visualization, and NJmatCDE, which enables the extraction of proper chemical formulas and material names from tabulated datasets, streamlining data preparation for downstream analysis. NJmatVIS, a dedicated visualizer submodule, provides an intuitive platform for visualizing and editing crystal structures from CIF files. The module allows users to add or delete atoms, view structures in detail, and download CIF files. By integrating directly with the Materials Project database, NJmatVIS facilitates access to structural crystallographic data, enhancing the efficiency of materials research workflows.
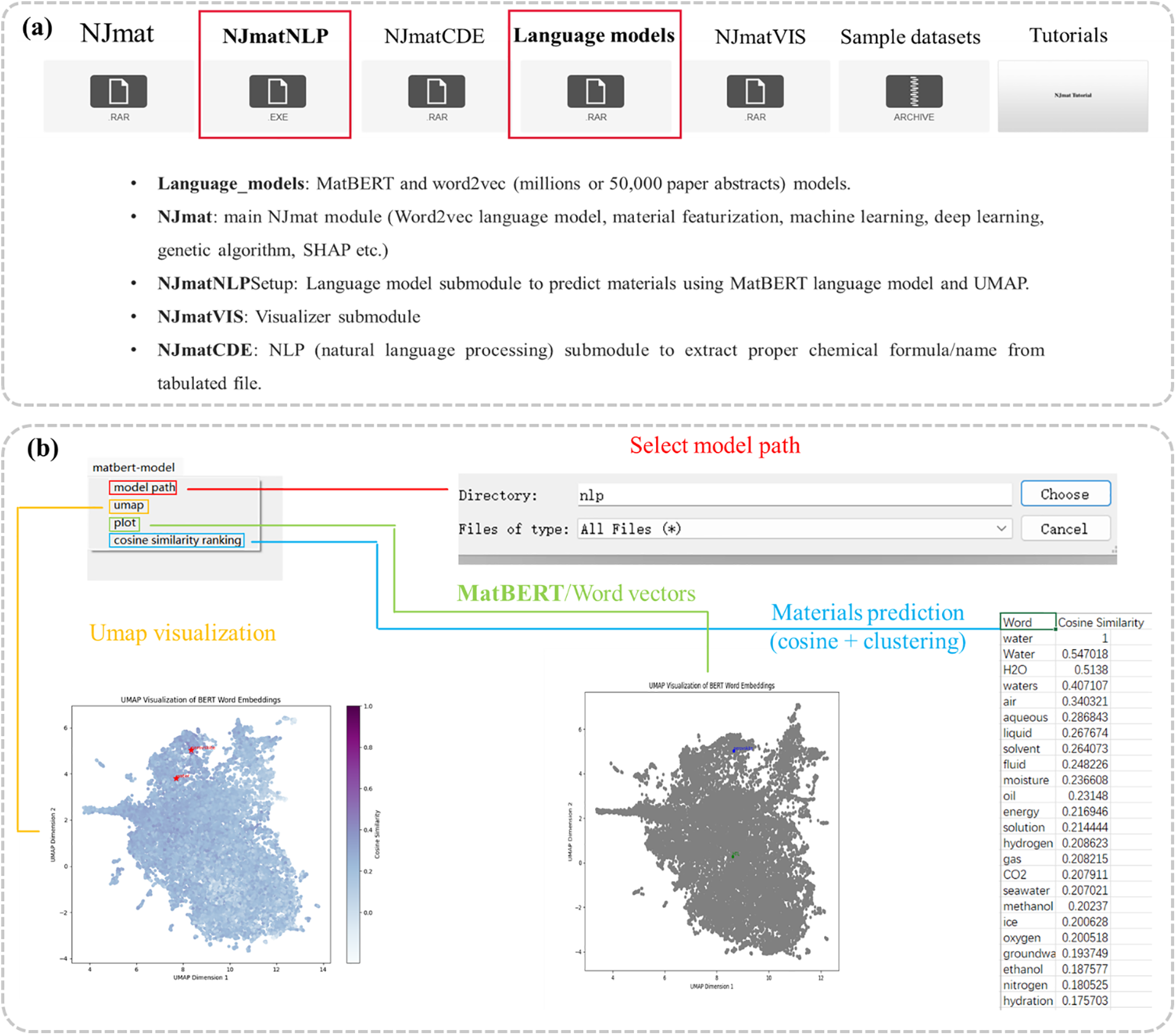
Figure 4: NJmatNLP: a submodule of NJmat for materials prediction using advanced language models. (a) Overview of the NLP modules in NJmat, including language models and the NJmatNLP framework. (b) Implementation of MatBERT within NJmatNLP, featuring model selection, UMAP visualization, plotting, and cosine similarity calculations
The NJmat platform incorporates CHGNet, a graph neural network designed for accurate simulations of material properties based on a crystal structure. It leverages a machine learning potential framework that encodes atomic interactions within crystal structures, allowing for precise predictions of energy, forces, and magnetic moments. Using the NJmatCHG submodule, users can import a CIF file and perform key computational tasks, including energy prediction, geometry optimization, and molecular dynamics simulations, with minimal user intervention (Fig. 5). The energy, forces, and magnetic moments of the input structure are automatically computed and displayed in the interface. For energy predictions, the module achieves a high level of accuracy due to its extensive training on diverse datasets, capturing the complex interatomic potentials of various materials. Geometrical optimization using the submodule rapidly converges to stable structures within seconds in the case of a perovskite structure while maintaining a balance between computational efficiency and accuracy. The optimization results align closely with DFT benchmarks, with structural parameters such as lattice constants deviating by less than 1%. This capability ensures that NJmat can provide robust predictions suitable for high-throughput materials screening. Furthermore, molecular dynamics simulations performed through NJmat capture dynamic behavior with a time resolution comparable to ab-initio methods. These features highlight NJmat’s utility as an interface to accelerate materials discovery with minimal coding requirement.
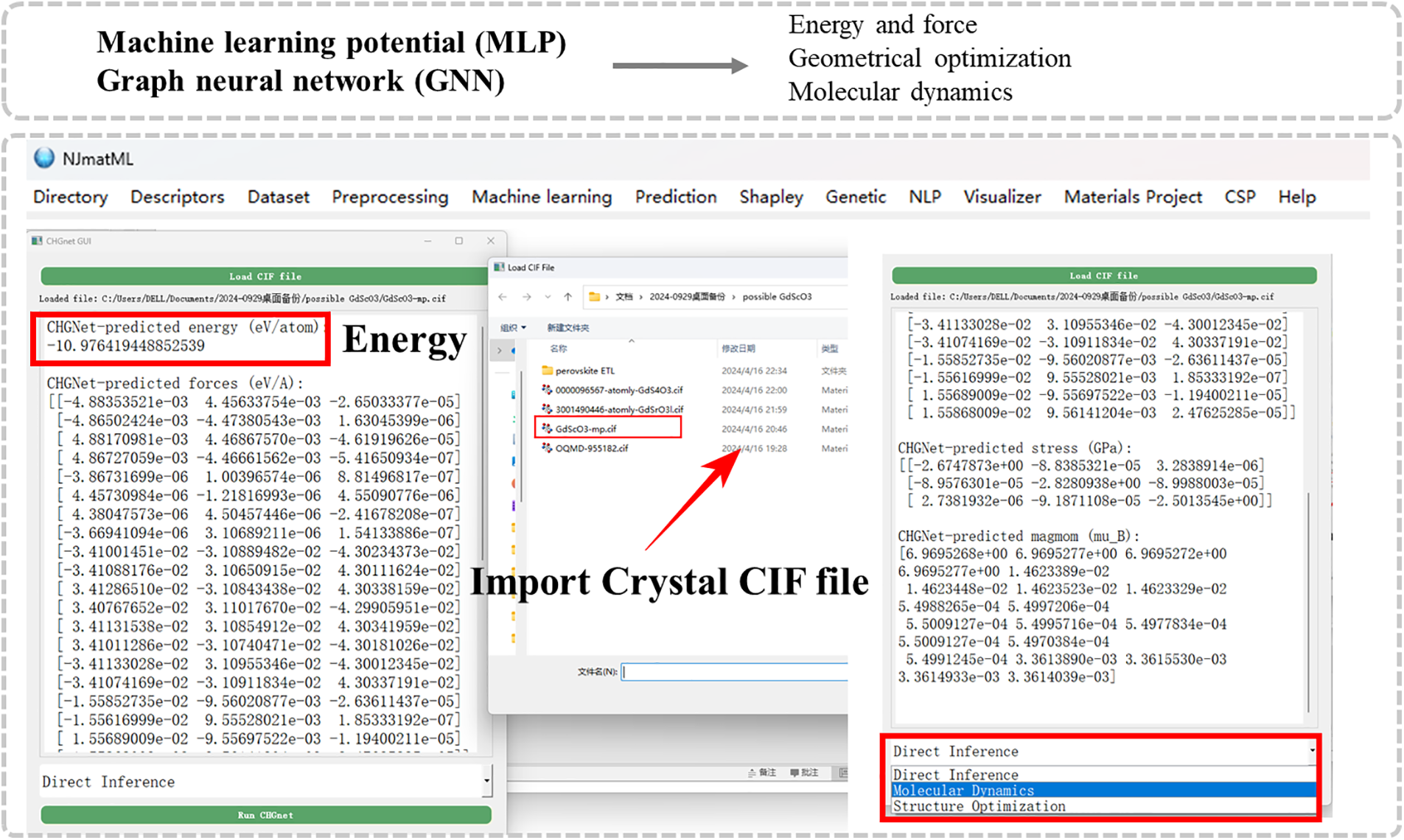
Figure 5: NJmat offers a user-friendly submodule for performing machine learning potential (MLP) and graph neural network (GNN) calculations, integrated with CHGNet. The interface supports three key tasks: (1) direct energy inference, (2) molecular dynamics simulations, and (3) structural optimization
The current machine learning interface does not comprehensively account for processing and microstructural factors. However, its primary focus is on structure-property relationships and the discovery of novel chemistries, rather than the optimization of existing materials. That said, the tool can assist in optimizing processing conditions, such as temperature and fabrication time, provided these parameters are included in the initial training and testing dataset. Several applications of the tool across different fields of materials research are summarized. (1) NJmat can be applied in perovskite surface design to predict stability and optoelectronic properties, aiding the development of next-generation solar cells and optoelectronic devices. (2) In catalyst discovery, it enables rapid screening by selecting and designing meaningful features (via the genetic algorithm module) and predicting catalytic activity, accelerating the search for efficient energy conversion and storage materials. (3) For battery materials, NJmat facilitates virtual screening of electrodes and electrolytes by predicting key electrochemical properties once the starting train-test dataset involving chemical formulas and their outputs is prepared, contributing to high-performance energy storage systems. (4) In structural alloy design, it assists in predicting mechanical properties and phase stability, supporting the development of advanced structural materials. (5) Additionally, in molecular informatics, the tool enables automatic featurization from SMILES strings, streamlining the exploration of novel molecules with tailored mechanical, thermal, or electronic properties. By integrating automated feature generation, Shapley plots, and interpretable machine learning models, NJmat enhances usability across diverse materials domains, promoting broader adoption of machine learning in materials science and aligning with the goals of the Materials Genome Initiative.
NJmat provides an educational capability that integrates machine learning techniques specifically designed for materials science and molecular science. The platform enables students to automatically apply machine learning models to predict material properties, optimize structures, and simulate molecular interactions. Features like automated feature extraction, feature selection, and the integration of genetic algorithms streamline the process, allowing students to focus on understanding the core principles behind these techniques. NJmat’s machine learning potentials help students explore the predictive capabilities of data-driven approaches, providing a hands-on learning experience in computational material science. By offering interpretable results, NJmat allows students to gain insights into how machine learning can be used to drive innovations in materials and molecular research.
In conclusion, NJmat is a user-friendly machine learning interface developed to support materials design and analysis, enabling integration of advanced data-driven methodologies. The platform combines a variety of functionalities, including data visualization, feature selection, feature analysis, automatic machine learning model construction, and virtual property prediction. Additionally, NJmat highlights recent computational techniques such as natural language processing (NLP), large language models (LLM), machine learning potentials (MLP), and graph neural networks (GNN), making it a comprehensive tool for materials property prediction and analysis. Tailored to meet the needs of real-world applications, NJmat is specifically crafted to support physical scientists, particularly experimentalists, by providing an intuitive, no-code interface that simplifies the application of machine learning techniques in materials research. By providing a robust and accessible platform for data-driven materials science, NJmat aims to bridge the gap between experimentalists and advanced computational methods, empowering researchers to make informed decisions and drive innovation in materials design.
Acknowledgement: The authors are thankful to researchers in Nanjing University of Information Science and Technology for the helpful discussion.
Funding Statement: This work was supported by the Jiangsu Provincial Science and Technology Project Basic Research Program (Natural Science Foundation of Jiangsu Province) (No. BK20211283).
Author Contributions: The authors confirm their contributions to the paper as follows: study conception and design: Lei Zhang; data collection: Lei Zhang; analysis and interpretation: Lei Zhang, Hangyuan Deng; draft manuscript preparation: Hangyuan Deng, Lei Zhang. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The data that support the findings of this study are available within the article.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest regarding the present study.
References
1. Venugopal V, Olivetti E. MatKG: an autonomously generated knowledge graph in material science. Sci Data. 2024;11(1):217. doi:10.1038/s41597-024-03039-z. [Google Scholar] [PubMed] [CrossRef]
2. Deb J, Saikia L, Dihingia KD, Sastry GN. ChatGPT in the material design: selected case studies to assess the potential of ChatGPT. J Chem Inf Model. 2024;64(3):799–811. doi:10.1021/acs.jcim.3c01702. [Google Scholar] [PubMed] [CrossRef]
3. Wang Z, Chen A, Tao K, Han Y, Li J. MatGPT: a vane of materials informatics from past, present, to future. Adv Mater. 2024;36(6):e2306733. doi:10.1002/adma.202306733. [Google Scholar] [PubMed] [CrossRef]
4. Yan Z, Liang H, Wang J, Zhang H, da Silva AK, Liang S, et al. PDGPT: a large language model for acquiring phase diagram information in magnesium alloys. Mater Genome Eng Adv. 2024;2(4):e77. doi:10.1002/mgea.77. [Google Scholar] [CrossRef]
5. Wang WY, Zhang S, Li G, Lu J, Ren Y, Wang X, et al. Artificial intelligence enabled smart design and manufacturing of advanced materials: the endless Frontier in AI+ era. Mater Genome Eng Adv. 2024;2(3):e56. doi:10.1002/mgea.56. [Google Scholar] [CrossRef]
6. Jain A, Ong SP, Hautier G, Chen W, Richards WD, Dacek S, et al. Commentary: the materials project: a materials genome approach to accelerating materials innovation. APL Mater. 2013;1(1):011002. doi:10.1063/1.4812323. [Google Scholar] [CrossRef]
7. Butler KT, Davies DW, Cartwright H, Isayev O, Walsh A. Machine learning for molecular and materials science. Nature. 2018;559(7715):547–55. doi:10.1038/s41586-018-0337-2. [Google Scholar] [PubMed] [CrossRef]
8. Zhang L, Zhou J, Chen X. Data-driven exploration and first-principles analysis of perovskite material. J Mater Inf. 2024;4(3):1–14. doi:10.20517/jmi.2024.20. [Google Scholar] [CrossRef]
9. Huang Y, Li S, Hu W, Shao S, Li Q, Zhang L. Language model-assisted machine learning, photoelectrochemical, and first-principles investigation of compatible solvents for a CH3NH3PbI3 film in water. ACS Appl Mater Interfaces. 2024;16(38):51595–607. doi:10.1021/acsami.4c06276. [Google Scholar] [PubMed] [CrossRef]
10. Wang J, Gao H, Han Y, Ding C, Pan S, Wang Y, et al. MAGUS: machine learning and graph theory assisted universal structure searcher. Natl Sci Rev. 2023;10(7):nwad128. doi:10.1093/nsr/nwad128. [Google Scholar] [PubMed] [CrossRef]
11. Ghalati MK, Zhang J, El-Fallah GMAM, Nenchev B, Dong H. Toward learning steelmaking—a review on machine learning for basic oxygen furnace process. Mater Genome Eng Adv. 2023;1(1):e6. doi:10.1002/mgea.6. [Google Scholar] [CrossRef]
12. Yuan Y, Sui Y, Li P, Quan M, Zhou H, Jiang A. Multi-model integration accelerates Al-Zn-Mg-Cu alloy screening. J Mater Inf. 2024;4(4):1–22. doi:10.20517/jmi.2024.34. [Google Scholar] [CrossRef]
13. Zhang S, Wang WY, Wang X, Li G, Ren Y, Gao X, et al. Large language models enabled intelligent microstructure optimization and defects classification of welded titanium alloys. J Mater Inf. 2024;4(34):1–28. doi:10.20517/jmi.2024.64. [Google Scholar] [CrossRef]
14. Xie T, Grossman JC. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys Rev Lett. 2018;120(14):145301. doi:10.1103/PhysRevLett.120.145301. [Google Scholar] [PubMed] [CrossRef]
15. Antunes LM, Butler KT, Grau-Crespo R. Crystal structure generation with autoregressive large language modeling. Nat Commun. 2024;15(1):10570. doi:10.1038/s41467-024-54639-7. [Google Scholar] [PubMed] [CrossRef]
16. Yang Y, Zhang S, Ranasinghe KD, Isayev O, Roitberg AE. Machine learning of reactive potentials. Annu Rev Phys Chem. 2024;75(1):371–95. doi:10.1146/annurev-physchem-062123-024417. [Google Scholar] [PubMed] [CrossRef]
17. Zhang H, Juraskova V, Duarte F. Modelling chemical processes in explicit solvents with machine learning potentials. Nat Commun. 2024;15(1):6114. doi:10.1038/s41467-024-50418-6. [Google Scholar] [PubMed] [CrossRef]
18. Huang Y, Zhang L, Deng H, Mao J. NJmat: data-driven machine learning interface to accelerate material design. J Chem Inf Model. 2024;64(16):6477–91. doi:10.1021/acs.jcim.4c00493. [Google Scholar] [PubMed] [CrossRef]
19. Trewartha A, Walker N, Huo H, Lee S, Cruse K, Dagdelen J, et al. Quantifying the advantage of domain-specific pre-training on named entity recognition tasks in materials science. Patterns. 2022;3(4):100488. doi:10.1016/j.patter.2022.100488. [Google Scholar] [PubMed] [CrossRef]
20. Zhang L, He M, Huang E, Ma X, You J, Jen AKY, et al. Overcoming language barrier for scientific studies via unsupervised literature learning: case study on solar cell materials prediction. Sol RRL. 2024;8(10):2301079. doi:10.1002/solr.202301079. [Google Scholar] [CrossRef]
21. Mikolov T, Chen K, Corrado G, Dean J. Efficient estimation of word representations in vector space. arXiv:1301.3781. 2013. [Google Scholar]
22. Deng B, Zhong P, Jun K, Riebesell J, Han K, Bartel CJ, et al. CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling. Nat Mach Intell. 2023;5(9):1031–41. doi:10.1038/s42256-023-00716-3. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools