
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Enhancing Cancer Classification through a Hybrid Bio-Inspired Evolutionary Algorithm for Biomarker Gene Selection
Department of Information Technology, College of Computer and Information Sciences, King Saud University, P.O. Box 145111, Riyadh, 4545, Saudi Arabia
* Corresponding Authors: Hala AlShamlan. Email: ; Halah AlMazrua. Email:
Computers, Materials & Continua 2024, 79(1), 675-694. https://doi.org/10.32604/cmc.2024.048146
Received 29 November 2023; Accepted 19 February 2024; Issue published 25 April 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
In this study, our aim is to address the problem of gene selection by proposing a hybrid bio-inspired evolutionary algorithm that combines Grey Wolf Optimization (GWO) with Harris Hawks Optimization (HHO) for feature selection. The motivation for utilizing GWO and HHO stems from their bio-inspired nature and their demonstrated success in optimization problems. We aim to leverage the strengths of these algorithms to enhance the effectiveness of feature selection in microarray-based cancer classification. We selected leave-one-out cross-validation (LOOCV) to evaluate the performance of both two widely used classifiers, k-nearest neighbors (KNN) and support vector machine (SVM), on high-dimensional cancer microarray data. The proposed method is extensively tested on six publicly available cancer microarray datasets, and a comprehensive comparison with recently published methods is conducted. Our hybrid algorithm demonstrates its effectiveness in improving classification performance, Surpassing alternative approaches in terms of precision. The outcomes confirm the capability of our method to substantially improve both the precision and efficiency of cancer classification, thereby advancing the development of more efficient treatment strategies. The proposed hybrid method offers a promising solution to the gene selection problem in microarray-based cancer classification. It improves the accuracy and efficiency of cancer diagnosis and treatment, and its superior performance compared to other methods highlights its potential applicability in real-world cancer classification tasks. By harnessing the complementary search mechanisms of GWO and HHO, we leverage their bio-inspired behavior to identify informative genes relevant to cancer diagnosis and treatment.Keywords
Authors are required to adhere to this Microsoft Word template in preparing their manuscripts for submission. It will speed up the review and typesetting process.
In 2020, cancer claimed the lives of almost 10 million people worldwide, as reported by the World Health Organization (WHO). Disturbingly, there are dire projections indicating a 50% surge, leading to approximately 15 million new cases. Consequently, there is an urgent requirement for effective cancer prevention and treatment strategies [1,2]. In cancer, cells within the organs or tissues of the human body exhibit uncontrolled growth, potentially spreading to adjacent regions or, in more advanced stages, to distant organs. Early detection is of paramount importance to enhance survival rates. Therefore, the identification of effective and predictive genes for cancer classification is critical. Achieving this objective entails the careful selection of an adequate number of features (genes) from DNA microarrays.
It can be difficult to extract meaningful information from gene expression data due to the vast number of genes involved [3]. The growing popularity of microarray data in cancer research classification has been attributed to the emergence of gene expression technologies, due mainly to the abundance of gene expression information (features/genes) available for detecting common patterns within a collection of samples. Cancer cells are identified with microarrays, which analyze DNA proteins to gain insight into their genes. Microarray data are organized according to a gene expression matrix, in which rows represent genes, and columns indicate experimental conditions [4].
Our understanding of disease-gene associations is greatly enhanced by the use of microarray technology. When dimensionality is present, irrelevant genes can complicate cancer classification and data analysis. In order to solve this problem and extract useful information, a feature selection method and classification algorithm are employed. Through the use of these algorithms, cancer-related genes can be removed from microarrays in order to facilitate cancer classification [5]. In the context of existing work, previous studies have focused on gene selection methods to improve the accuracy of cancer classification. However, the vast number of genes involved in gene expression data makes it difficult to extract meaningful information [3]. This issue has led to the emergence of feature selection techniques, such as filtering, wrapping, and embedding, to identify informative genes and reduce dimensionality [5]. Nevertheless, there is a need for more effective approaches that can overcome the limitations of existing methods. Recent publications indicate a growth in hybrid approaches to feature selection as part of the overall framework, as evidenced by the growing number of publications in recent years [6]. Gene selection is a process that identifies the most informative and valuable genes for the classification problem. Achieving this is possible by eliminating irrelevant genes and noise in the data, resulting in a more accurate classification of cancer [4]. By combining both filter and wrapper techniques, the hybrid method is able to provide the best of both worlds. The development of hybrid approaches has increased in recent years, including the combination of two wrappers and the merger of filter and wrapper methods. For accurate diagnosis, these approaches identify valuable genes. As a result of integrating the strengths of both techniques, hybrid methods achieve optimal results [4].
In recent years, there has been significant research and development in the field of meta-heuristic algorithms to tackle complex optimization problems. Notably, several novel algorithms have emerged, which have shown promising results in various domains. The liver cancer algorithm (LCA) [7] draws inspiration from the behavior of liver cells in response to tumor growth and has been successfully applied in tasks related to liver cancer, such as feature selection and classification. The slime mould algorithm (SMA) [8] replicates the foraging behavior of slime molds and has been used for optimization problems like routing optimization, image segmentation, and clustering. The moth search algorithm (MSA) [9] mimics moths’ attraction to light and has found applications in feature selection, image processing, and data clustering. The hunger games search (HGS) [10] algorithm incorporates survival of the fittest principles, and it has been employed in feature selection, scheduling, and parameter tuning. The Runge Kutta method (RUN) [10], originally a numerical integration technique for solving differential equations, has been adapted for optimization problems in engineering design, control systems, and economic modeling. The colony predation algorithm (CPA) [10] is inspired by predator-prey interactions and has been applied to data clustering, image segmentation, and function optimization. The weighted mean of vectors (INFO) [10] algorithm combines candidate solutions using weighted means and has been used in feature selection, image processing, and data mining tasks. These recently proposed meta-heuristic algorithms offer innovative approaches to optimization and have the potential to address complex real-world challenges.
In this study, we propose a novel hybrid feature selection method that addresses the challenges in cancer classification. Our approach combines two bio-inspired wrapper feature selection techniques and leverages the evolutionary and bio-inspired optimization algorithm GWO-HHO (Grey Wolf Optimizer-Harris Hawks Optimization), which integrates Grey Wolf Optimizer (GWO) and Harris Hawks Optimization (HHO). The main contributions of our research can be summarized as follows:
Development of a Hybrid Feature Selection Method: We introduce a novel approach that combines bio-inspired wrapper techniques and the GWO-HHO algorithm. This hybrid method aims to overcome the limitations of existing feature selection methods and enhance the accuracy of cancer classification.
Effective Selection of Relevant and Informative Genes: By utilizing our hybrid feature selection method, we aim to identify the most relevant and informative genes for cancer classification. This process helps to reduce dimensionality, eliminate irrelevant genes, and improve the accuracy of cancer diagnosis.
Comprehensive Evaluation and Comparison: We conduct a comprehensive evaluation of our proposed method by analyzing six diverse binary and multiclass gene expression microarray datasets. Through this evaluation, we compare the performance of our hybrid approach against recently published algorithms, showcasing its superior accuracy in cancer classification and gene selection.
Advancements in Cancer Research: By accurately classifying cancer and identifying informative genes, our approach contributes to advancing our understanding of the underlying biology of cancer. This knowledge can potentially lead to the development of more effective diagnostic and therapeutic strategies.
In summary, our study addresses the challenges in cancer classification through the introduction of a hybrid feature selection method. By effectively selecting relevant and informative genes, our approach overcomes the limitations of existing methods and achieves superior performance in accurately classifying cancer. The results of our research contribute to advancements in cancer research and hold promise for the development of improved diagnostic and therapeutic interventions.
In the subsequent sections of the paper, we have organized the content as follows: Section 2 provides the necessary background information related to the perspectives discussed in the related works section, which significantly influenced the development of our proposed method. In Section 3, we describe the approaches we propose for gene selection, outlining the methodologies and techniques employed. Section 4 presents the analysis of the data and experimental results, where we evaluate the performance of our method using diverse gene expression microarray datasets. Finally, in Section 5, we provide a comprehensive conclusion that summarizes the key findings of our research and discusses the implications, limitations, and potential future directions.
In recent years, several methods have been proposed for cancer classification and gene selection in bioinformatics research. In this section, we review some of the related works that have addressed similar problems using different optimization algorithms and machine learning techniques. One of the related works is the GWO-SVM method proposed by AlMazrua et al. [11]. They combined the Grey Wolf Optimizer (GWO) algorithm with Support Vector Machines (SVM) for feature selection in cancer classification. Their approach aimed to enhance the classification accuracy by selecting the most relevant genes for cancer diagnosis. Another related work is the GWO-KNN method, also proposed by AlMazrua et al. [11]. In this study, they employed the GWO algorithm with the k-Nearest Neighbors (KNN) classifier for cancer classification. The GWO algorithm was used to optimize the feature subset selection process, while KNN was utilized for the classification task.
The HHO-SVM method proposed by AlMazrua et al. [12] is another relevant work in this area. Here, they combined the Harris Hawks Optimization (HHO) algorithm with SVM for gene selection in cancer classification. Their approach aimed to improve the classification performance by identifying the most informative genes associated with cancer.
Similarly, the HHO-KNN method proposed by AlMazrua et al. [12] utilized the HHO algorithm with KNN for cancer biomarker gene detection. By applying the HHO algorithm, they aimed to identify the most discriminative genes that could serve as potential biomarkers for cancer diagnosis. The HS-GA method [13] is also worth mentioning, as it employed a Hybridization of Harmony Search (HS) and Genetic Algorithm (GA) for gene selection in cancer classification. The hybrid algorithm aimed to improve the search efficiency by combining the exploration capabilities of HS and the exploitation abilities of GA.
Additionally, the FF-SVM method proposed by Almugren et al. [14] utilized a Fuzzy Expert System (FF) along with SVM for classification of microarray data. Their approach aimed to improve the classification accuracy by incorporating fuzzy logic to handle the uncertainty in gene expression data. Lastly, the GBC method proposed by Alshamlan et al. [15] introduced the Genetic Bee Colony (GBC) algorithm for gene selection in microarray cancer classification. The GBC algorithm aimed to optimize the gene selection process by simulating the foraging behavior of bee colonies.
In the context of evolutionary algorithms and feature selection, several relevant studies have contributed to the advancement of this field. One study proposed a method for learning correlation information for multi-label feature selection [16]. Their approach aimed to enhance the discrimination capability of selected features by integrating correlation learning and feature selection. Another study presented a multi-label feature selection technique that considers label correlations and utilizes a local discriminant model [17]. Additionally, a study introduced a multi-label feature selection approach based on label correlations and feature redundancy [18]. Their method aimed to select a subset of features that maximized discriminative power while considering relationships among labels and features. Moreover, another study explored manifold learning with a structured subspace for multi-label feature selection [19]. They leveraged the inherent data structure to identify informative features for multi-label classification tasks. In addition to these studies, another investigation also focused on manifold learning with a structured subspace for multi-label feature selection [19]. Lastly, an approach was proposed that integrates a differential evolution algorithm with dynamic multiple populations based on weighted strategies [20], demonstrating improvements in feature selection performance.
These related works have contributed to the field of cancer classification and gene selection by employing various optimization algorithms and machine learning techniques. However, the reviewed algorithms have certain limitations regarding the number of selected genes, overfitting, and computational complexity. Firstly, they may not effectively reduce the number of selected genes, resulting in larger feature sets that can be challenging to interpret and may introduce noise into the classification process. This can hinder the identification of the most informative genes and potentially lead to reduced performance. Secondly, the algorithms may not explicitly address the issue of overfitting, which can occur when the model becomes overly complex relative to the available data, leading to poor generalization performance on unseen data. Overfitting can limit the practical utility of the models in real-world applications. Lastly, these methods may lack efficient strategies to handle computational complexity, particularly when dealing with large-scale datasets. The computational demands of these algorithms can become prohibitive, requiring substantial time and resources for analysis. These limitations highlight the need for a hybrid algorithm that combines the strengths of different optimization techniques to overcome these challenges and improve the accuracy, interpretability, and efficiency of gene selection in cancer classification. In the following sections, we will present our novel GWO-HHO hybrid algorithm, which aims to address these limitations and demonstrate its effectiveness in cancer analysis.
The primary objective of our study is to develop a novel hybrid bio-inspired evolutionary algorithm, termed GWO-HHO, which aims to identify predictive and relevant genes for achieving high classification accuracy in cancer classification tasks. The proposed model consists of three phases: The GWO phase, the HHO phase, and the classification phase.
In the GWO phase, we employ the Grey Wolf Optimizer algorithm to identify the most informative genes from a pool of candidate genes. This phase focuses on selecting the genes that exhibit significant discriminatory power for cancer classification. Subsequently, in the HHO phase, we utilize the Harris Hawks Optimization algorithm to further refine the selected genes and enhance their predictive power. This phase aims to optimize the selected genes by leveraging the search capabilities of the HHO algorithm.
Finally, in the classification phase, we employ a classification algorithm to accurately classify cancer samples based on the selected genes. The classification algorithm will be chosen based on its suitability for the dataset and its ability to handle the selected features effectively.
By integrating these three phases, our proposed hybrid algorithm aims to improve the accuracy of cancer classification by identifying and utilizing the most relevant genes. We believe that this approach has the potential to significantly contribute to the field of cancer research and facilitate the development of more effective diagnostic and treatment strategies.
For a visual representation, we have included Fig. 1, which illustrates the proposed model and the steps involved in the GWO-HHO algorithms. Additionally, Algorithm 1 presents the pseudo code for our proposed algorithm.
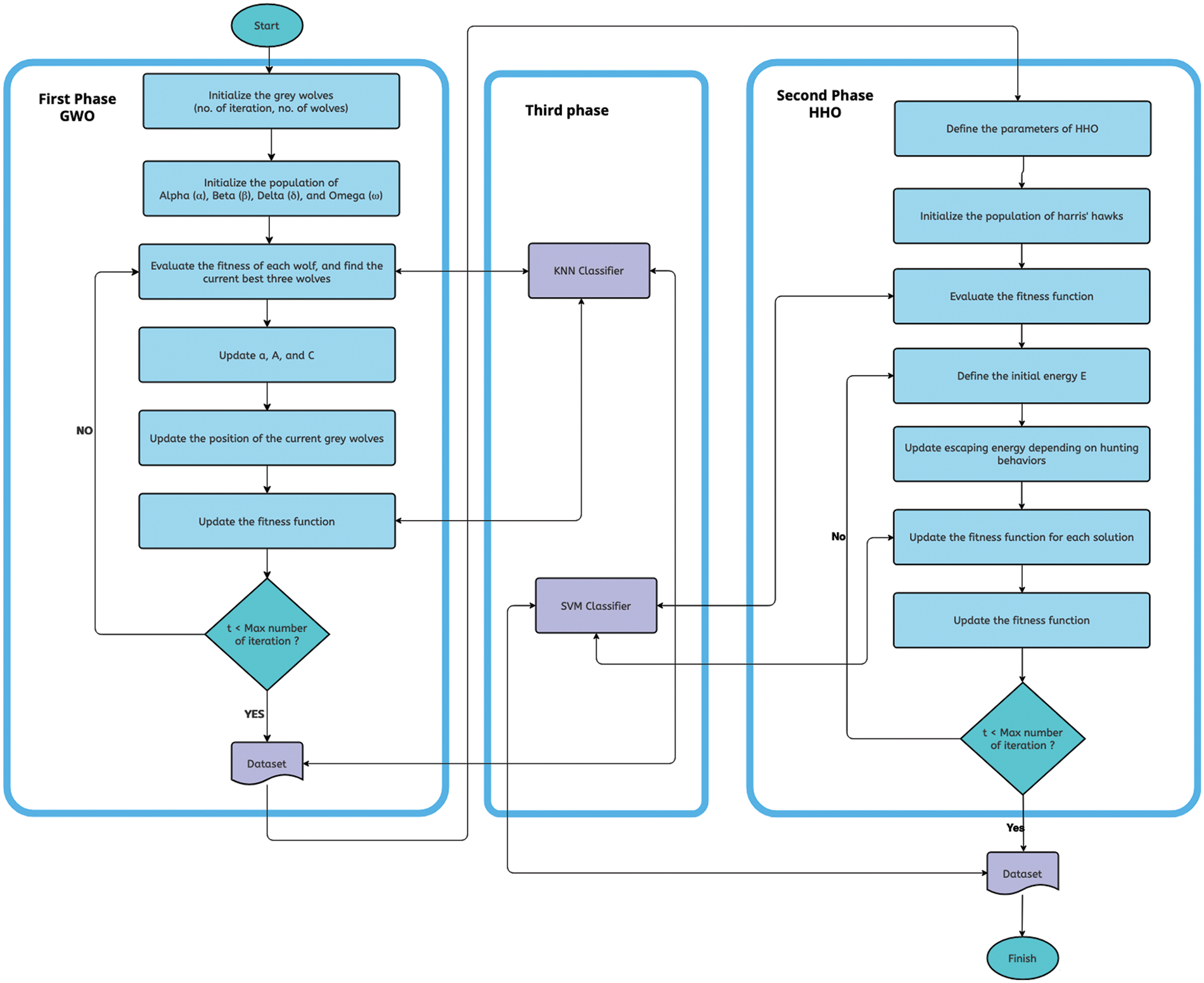
Figure 1: The GWO-HHO algorithm flowchart
3.1 Proposed GWO-HHO Algorithm
In this section, we will delve into the various phases of our proposed GWO-HHO algorithm.

3.1.1 First Phase: Pre-Processing Using Gray Wolf Optimization (GWO)
The Gray Wolf Optimization (GWO) algorithm represents an advanced swarm intelligence optimization technique, particularly well-suited for wrapper feature selection. It draws inspiration from the coordinated hunting behavior of gray wolves in their natural habitat, where each member of the wolf pack plays a distinct role in identifying and capturing prey. In a similar fashion, the GWO algorithm adopts a strategy reminiscent of gray wolf hunting behavior to discern and select the most informative features for classification tasks.
The GWO algorithm has exhibited promising outcomes in the realm of feature selection, proving its effectiveness across various applications such as image classification, prediction, and modeling. This algorithm was initially introduced by Mirjalili et al. [21] in 2014. The GWO approach leverages a nature-inspired optimization technique to craft optimal solutions for various problem domains. In the context of enhancing accuracy, GWO proves to be a highly effective feature selection method. Notably, it stands out by not requiring the utilization of a threshold parameter to weed out irrelevant features, a step often necessary in other methods.
As the number of variables increases, the task of selecting the most relevant subset of features becomes increasingly complex. However, GWO’s wrapper feature selection method is notably less time-consuming compared to alternative techniques in addressing this complexity.
GWO possesses a unique advantage in that it circumvents the challenge of getting trapped in local minima, a problem frequently encountered by other bio-inspired optimization methods. For more in-depth information regarding GWO and its efficacy in feature selection, interested readers can refer to a previously published paper [11]. This paper not only presents a mathematical model for gray wolves but also provides experimental results utilizing GWO as a standalone feature selection algorithm.
The GWO algorithm was selected as the first phase of the proposed model because of the high dimensionality of the dataset. Among other things, GWO’s demonstrated ability to deal with real-world problems, such as spring tension and compression, welded beams, and pressure vessels, contributed to the decision. This has resulted in GWO being widely recognized as an excellent tool for solving complex optimization problems. The dataset used in this study has a high dimensionality, so it is a great candidate to address it.
The GWO was designed based on a mathematical model of gray wolves’ social hierarchy in which selected the fittest solution as the Alpha(α). Due to this, the second and third best solutions are known as Beta(β) and Delta(δ), respectively. There are no more possible solutions, so Omega(ω) is assumed to be the only solution available. Alpha, beta, and delta are all used in the GWO algorithm in order to lead the search. In this case, the ω wolves are pursuing these three wolves.
There are usually three stages of predation by wolves: Hunting, encircling, and attacking.
• Encircling: In GWO, the hunting behavior of gray wolves is emulated, specifically the encircling behavior during a hunt. This behavior is mathematically represented by Eqs. (1)–(3).
where t is the number of iterations, X(t) is one gray wolf, X(t + 1) will be the next position it lands at, and Xp(t) corresponds to one of the following: α, β, δ. coefficient vectors A and C are represented as follows:
where r1 and r2 are random vectors in the range [0,1] and is a decreasing number in the range [0,2], with a = 2 − 2t/MaxIteration being the most prominent example.
• Hunting: In the context of gray wolf hunting behavior, it is noteworthy that these wolves can collectively locate and encircle their prey. Although beta or delta wolves may take part in the hunt at times, it is primarily the alpha wolf that leads the charge. However, when dealing with an abstract search space where the optimal solution (prey) is unknown, we make certain assumptions to mathematically mimic the hunting behavior of gray wolves. In this abstraction, we consider the alpha wolf to represent the best candidate solution, with beta and delta wolves also possessing superior knowledge regarding the probable location of potential prey. As a result, we keep track of the three best solutions that have been developed thus far. We then require other search agents to update their positions based on the information obtained from the positions of these best search agents. This concept is elucidated by Eqs. (5)–(7).
• Attacking: The gray wolves encircled the prey and were preparing to catch it (con-165 vergence and get results). Because of A ∈ [−2a, 2a], this operation was normally 166 performed by lowering a in Eq. (3). When |A| ≥ 1 occurs, the gray wolves stay 167 away from the prey to achieve global search; when |A| < 1 occurs, the gray wolf pack 168 approaches the prey and finally finishes it.
3.1.2 Second Phase: Gene Selection Using Harris Hawks Optimization (HHO)
The GWO algorithm operates on a continuous search space, which means that it is not capable of solving the feature selection problem on its own. in addition, as revealed by Niu et al. [22] who analyzed the GWO, and demonstrated that it has a severe structural defect. The algorithm performs best when the solution to an optimization problem is zero. Nevertheless, if the optimal solution is not zero, the same performance cannot be achieved. As a result, the further the function’s optimal solution is from zero, the lower the performance of GWO. To address this limitation, an additional feature selection method must be applied after GWO. To this end, HHO was used in the second phase after GWO. HHO was applied to the dataset produced by GWO to resolve the issue of the continuous search space. By combining these two techniques, the proposed hybrid feature selection algorithm can effectively address the dimensionality problem of the dataset and identify the most informative features for accurate cancer classification.
HHO is a swarm computation technique that was developed by Heidari et al. in 2019 [23]. It draws inspiration from the cooperative hunting and chasing behavior observed in Harris’s hawks, particularly their “surprise pounces” or “the seven kills”. During a cooperative attack, multiple hawks work together and attack a rabbit that has revealed itself. The HHO algorithm employs a similar strategy, where multiple solutions explore the search space and converge on the most effective solution. This cooperative approach enables the algorithm to effectively navigate complex search spaces and identify optimal solutions to problems. In another published paper, AlMazrua et al. [12] discussed the inspiration behind HHO, the mathematical model employed by the algorithm, and presented the results of using HHO as a standalone feature selection method. The authors explained how HHO drew inspiration from the cooperative hunting behavior of Harris’s hawks and elaborated on the mathematical model used to implement the algorithm. Additionally, they presented the results of their experiments, which demonstrated the effectiveness of HHO as a feature selection method. Overall, their study provides valuable insights into the capabilities of HHO and its potential applications in various domains.
Hawks are known to chase their prey by tracing, encircling, and eventually striking and killing.
The mathematical model, inspired by the hunting behaviors of Harris’s hawks, encompasses three distinct stages: Exploration, the transition between exploration and exploitation, and exploitation. Throughout each stage of the hunt, the Harris’s hawks represent the candidate solutions, while the targeted prey represents the best candidate solution, nearly approaching the optimal.
During their search for prey, Harris’s hawks employ two distinct exploration techniques. The candidate solutions aim to position themselves as close to the prey as possible, while the best candidate solution is the one intended to be the prey.
In the first technique, Harris’s hawks select a location by taking into account the positions of other hawks and their prey. In the second method, the hawks perch atop random tall trees. Eq. (8) enables the simulation of these two methods with equal probabilities denoted as q.
• Vector x(t) is the current hawk position, whereas vector x(t + 1) is the hawk’s position at the next iteration.
• The hawk xrandom(t) is selected at random from the population.
• The rabbit position is xrabbit(t).
• q, r1, r2, r3 and r4 are randomly generated numbers inside (0, 1).
• LB and UB are the upper and lower bounds of variables.
• xmean(t) is the average position of the current population of hawks, which is calculated as shown in Eq. (9).
• t is the total number of iterations.
• xi(t) is the position for each hawk in iteration t.
• The total number of hawks is represented by N.
The algorithm switches from exploration to exploitation (transition from exploration to exploitation) depending on the rabbit’s running or escaping energy, as shown in Eq. (10).
• E represents the prey’s escaping energy.
• The initial state of the energy is indicated by E0,which changes randomly inside (−1, 1) at each iteration.
When |E| ⩾ 1, hawks seek out more areas to investigate the rabbit’s whereabouts; alternatively, the exploitation stage begins. The algorithm formulates the rabbit’s escape success p ⩾ 0.5 or failure p < 0.5 with an equal chance p. The Hawks also will also carry out a soft |E| ⩾ 0.5 or hard siege |E| < 0.5, based on the rabbit’s energy. The soft siege is defined as in Eqs. (11)–(13).
• The difference between the hawk and rabbit positions is represented by ∆x(t).
• J is a random number used to generate the rabbit’s random jump force.
A hard siege, on the other hand, can be calculated as follows in Eq. (14):
A soft siege with repeated fast dives is attempted when |E| ⩾ 0.5 and p < 0.5, as the rabbit could successfully escape. The hawks have the option of selecting the best dive. Lévy flight is employed to imitate the prey’s hopping. The hawks’ next action is calculated as shown in Eq. (15) to determine whether the dive is successful or not.
The hawks will dive following Eq. (16), the Lévy flight L pattern, if the previous dive turns out to be ineffective.
• The problem dimension dim is the size of the RandomVector, and dim is the dimension of the problem.
Eq. (17) has been used to update the final soft-siege rapid dives.
Eqs. (8) and (9) are used to calculate k and z, respectively. A hard siege with progressive rapid dives occurs when |E| ⩾ 0.5 and p < 0.5 are not sufficient for the rabbit to flee, as it no longer possesses enough energy. The rabbit’s z is calculated via Eq. (16), while k is updated using Eq. (18).
3.1.3 Third Phase: Classification
In this study, two different classifiers, Support Vector Machine (SVM) and K-Nearest Neighbors (KNN), were used. This decision was based on the demonstrated performance of GWO with KNN and HHO with SVM. In the classification phase, the fitness function score was calculated using both classifiers, with the primary objective being to improve classification accuracy while minimizing the number of selected genes. To evaluate the robustness of the proposed approach, a cross-validation method was employed. Specifically, Leave-One-Out Cross-Validation (LOOCV), which is widely used in machine learning, was used to assess the performance of the proposed algorithm.
In the first phase of the proposed hybrid method, the initial microarray gene dataset is preprocessed and filtered using the GWO gene selection technique. Each gene is evaluated individually and sorted according to its importance. An analysis is then conducted to identify a subset of genes that yield the highest classification accuracy when analyzed using a KNN classifier. The KNN classifier is employed in place of the SVM classifier due to its superior classification accuracy. Through the GWO approach, a dataset is produced that contains fewer redundant and more relevant genes. By using GWO to filter out irrelevant and noisy genes, the computational load during the next phase of the algorithm, which utilizes the HHO technique and SVM classifier, is reduced. This approach allows for the identification of the most informative genes, resulting in improved classification accuracy and reduced computational cost.
In the second phase of the proposed hybrid method, a gene selection process utilizing HHO is employed to identify the most predictive and informative genes from the GWO dataset, which yield the highest accuracy for the KNN classifier. In the HHO search space, each solution is represented by one or more gene indices drawn from the GWO dataset. The fitness value associated with each solution is determined by the SVM classifier, which measures the classification accuracy of the solution to a gene selection problem. By using HHO, the proposed method can explore the search space more efficiently and identify the optimal subset of genes, resulting in improved classification accuracy. The use of SVM as the fitness function enables the algorithm to identify the most informative genes that yield the best classification results.
In the final phase of the proposed hybrid method, the informative and predictive genes identified by the GWO algorithm in the first phase are utilized to train a KNN classifier using LOOCV. Next, the HHO algorithm is employed to train an SVM classifier using LOOCV. This phase is designed to evaluate the efficiency of the pro-posed hybrid method. By utilizing both classifiers and LOOCV, the algorithm can identify the most informative genes and achieve the highest possible classification accuracy while minimizing the number of selected genes. The use of LOOCV allows for unbiased estimates of classification accuracy, which is important for assessing the effectiveness of the proposed approach. The aim of this phase is to identify the most significant genes for improving the performance of the SVM and KNN classifiers and determine which classifier performs best when utilized with GWO. GWO is employed as a pre-processing phase to reduce the dimensionality of the microarray data, eliminating duplicate and irrelevant genes. Statistically similar genes are then selected as inputs for the gene selection phase.
The fitness function, as shown in Eq. (19), is calculated for GWO, allowing the algorithm to exclude as many features as possible while maintaining high levels of accuracy. This approach enables the identification of the most informative genes, which are subsequently utilized to train both classifiers and evaluate their performance. By comparing the performance of the SVM and KNN classifiers, the proposed method aims to determine the most effective approach for cancer classification using the GWO algorithm.
In this study, two kind of publicly available microarray cancer datasets were utilized, consisting of binary and multiclass datasets. To evaluate the performance and effectiveness of the proposed algorithms, six benchmark microarray datasets were employed. By utilizing these datasets, the study aims to provide a comprehensive evaluation of the proposed approach and demonstrate its potential applicability in real-world cancer classification tasks. The use of benchmark datasets also allows for the comparison of the proposed approach against existing methods, providing valuable insights into its relative effectiveness. The study employed three binary datasets for colon tumors [24], lung cancer [25], and leukemia3 [26]. Additionally, three multiclass datasets were used, including leukemia2 [24], lymphoma [27] and SRBCT [27]. A detailed breakdown of the experimental datasets, including information on the number of samples and classes, can be found in Table 1. By utilizing a diverse range of datasets, the study aims to demonstrate the applicability and effectiveness of the proposed approach across various cancer types and classification scenarios.

The best solution was determined by using KNNs and SVMs. We determined the value of k using a trial-and-error approach. Across all datasets in the experiments, k = 7 produced the best results.
The number of iterations (Max_iter) and the size (dim) of a method play an important role in its practicality. Additional parameters to k, dim, up, and lp can be found in Table 2.

The proposed method will be evaluated based on two factors: Classification accuracy and the number of selected genes. The objective is to minimize the number of selected genes while maximizing the accuracy of classification. To obtain a reliable estimation of the accuracy of a classification algorithm on a small number of samples, LOOCV can be used. In contrast to LOOCV, k-fold cross validation incorporates randomness so that the mean accuracy associated with k-fold cross validation on a data set is not constant. Due to the low number of samples in our datasets, LOOCV is used in the evaluation process. As validation data, LOOCV uses one sample of the original data, whereas residual samples are used for training. The process is repeated in order to ensure that each sample in the data is only used once for the purposes of validation. Therefore, LOOCV is a special case of k-fold cross validation, where k equals the number of samples in the data, and every sample is evaluated exactly once for validity [28]. In addition, we applied k-fold cross validation at 5-fold, to compare the results between LOOCV and k-fold in order to determine if there are any overfitting issues. Moreover, LOOCV maximizes data utilization, provides an unbiased estimate of performance, minimizes variance, and ensures robust evaluation. LOOCV evaluates the proposed method on all instances, identifies overfitting, and reduces reliance on specific train-test splits, resulting in a comprehensive and reliable assessment of the method’s effectiveness on the given dataset.
Furthermore, LOOCV (Leave-One-Out Cross-Validation) was chosen in our study to address the challenges associated with high-dimensional gene expression data, limited sample sizes, and noise. LOOCV is suitable for limited sample sizes as it maximizes the use of available data by leaving out one sample at a time for validation [28]. Moreover, it is worth noting that LOOCV has been extensively utilized and demonstrated its effectiveness in handling high-dimensional datasets, as supported by multiple previous studies [11–15,28]. It helps mitigate the risk of overfitting in high-dimensional gene expression data by providing a more rigorous assessment of the classification algorithm’s performance. By employing LOOCV, we aimed to ensure a robust and reliable evaluation of our proposed hybrid algorithm’s performance in cancer classification tasks.
The classification accuracy will be determined using Eq. (20).
4 Experimental Results and Discussions
In this section, we will delve into the experimental results of our proposed hybrid method, the GWO-HHO algorithm. Subsequently, we will conduct a comparative analysis with state-of-the-art research to demonstrate the efficiency of our algorithm.
4.1 The Experiments Results of (GWO-HHO) Method
The primary objective of this study was to assess and compare the performance of the proposed hybrid feature selection method across a selection of six cancer microarray expression datasets. The aim was to determine its effectiveness in terms of classification accuracy. Additionally, we evaluated the proposed feature selection method using both Leave-One-Out Cross-Validation (LOOCV) and 5-fold Cross-Validation and compared its performance with that of other evolutionary algorithms. Tables 3 and 4 provide insights into how the proposed hybrid feature selection method performs with regard to accuracy and the number of selected features. Notably, LOOCV was employed in Table 3 for assessing classification accuracy, while Table 4 utilized the 5-fold Cross-Validation approach. The experimental results clearly indicate that overfitting does not adversely impact the accuracy of microarray data.

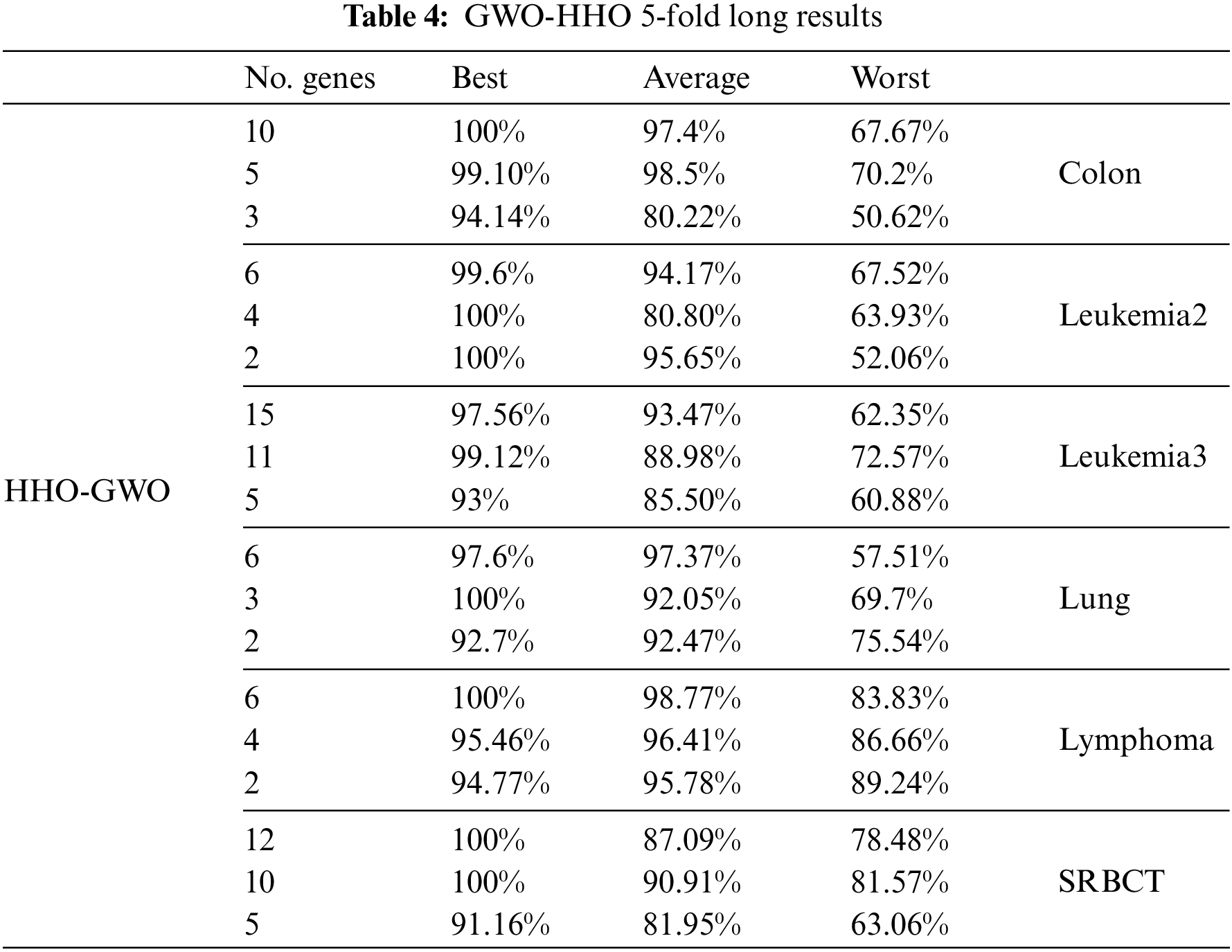
In the first phase of the proposed method, we applied GWO independently to each dataset and classifier to identify the optimal number of informative genes while achieving high accuracy. Next, HHO was utilized on the GWO-selected dataset to further decrease the number of selected genes and improve accuracy. Based on the experimental results, it was observed that GWO performed best with the KNN classifier while HHO performed best with the SVM classifier. As a result, each phase was associated with a specific classifier.
As shown in Tables 3 and 4, the results highlight the remarkable performance of the proposed hybrid method. It achieved an impressive 100% accuracy for most datasets, demonstrating its robustness in accurately classifying cancer samples. Notably, the algorithm achieved perfect classification accuracy for leukemia2, leukemia3, lung, and lymphoma datasets. For these datasets, the number of selected genes was found to be less than 5, indicating the algorithm’s ability to identify a small set of highly informative genes for accurate classification.
For the leukemia3, colon, and SRBCT datasets, the proposed method achieved 100% accuracy while selecting less than 13 genes. This demonstrates the algorithm’s effectiveness in accurately classifying cancer samples with a relatively small number of selected genes. The ability to minimize the number of genes is crucial in reducing the complexity and computational burden associated with cancer diagnosis and treatment.
Although the colon dataset did not achieve 100% accuracy, the proposed hybrid method still achieved notable results. The algorithm accurately classified the colon dataset with an accuracy above 78% while selecting a small number of genes. This suggests that the selected genes are highly relevant to the classification task, contributing to the algorithm’s performance.
Overall, the results from Tables 3 and 4 provide strong evidence for the effectiveness of the proposed hybrid method. The algorithm demonstrated high accuracy in most datasets, while simultaneously minimizing the number of selected genes. These findings have significant implications for cancer diagnosis and treatment, as they highlight the potential of the proposed method to accurately classify cancer samples using a small subset of informative genes.
In order to assess its effectiveness, we compared the proposed GWO-HHO algorithm with previous metaheuristic bio-inspired methods, focusing on classification accuracy and the number of selected genes as shown in Table 5.
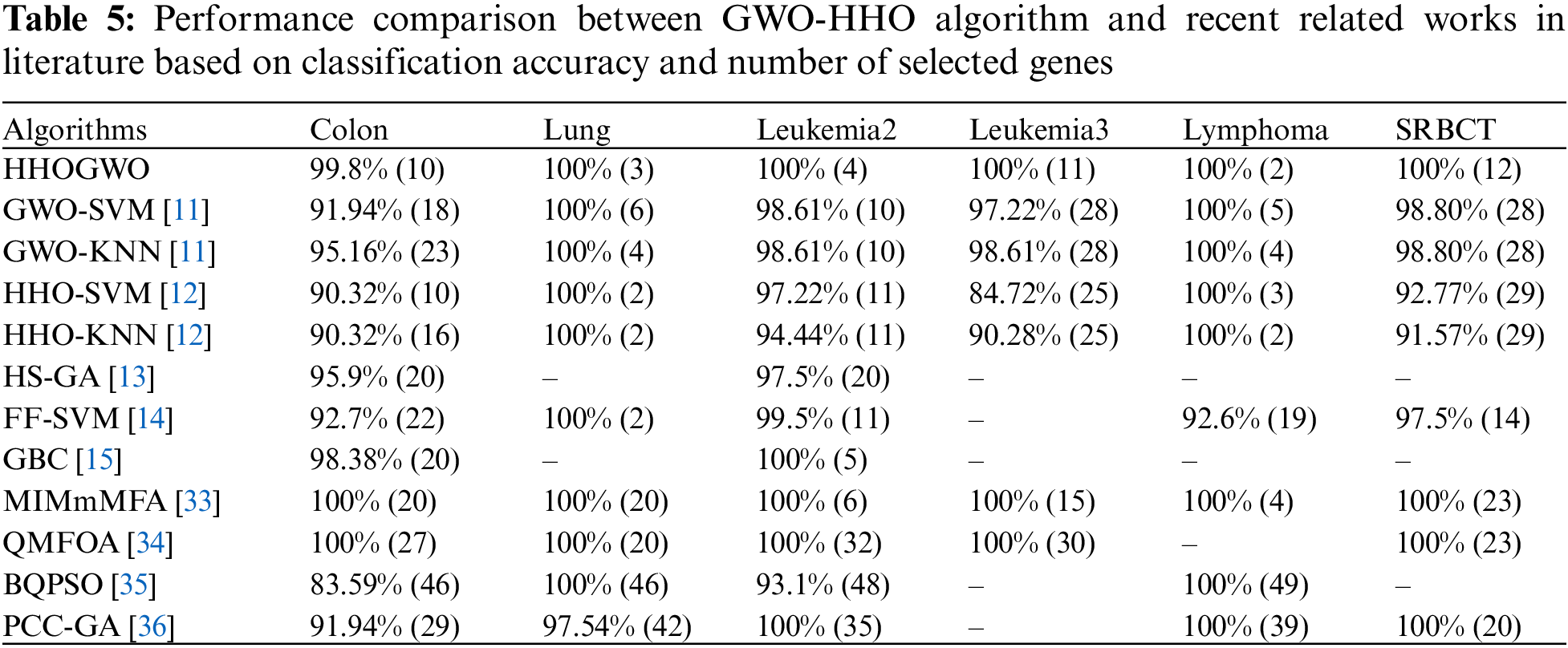
The results of this comparison revealed that GWO-HHO outperformed other bio-inspired gene selection methods in terms of accuracy. Remarkably, it achieved a remarkable 100% classification accuracy while selecting fewer genes. While it did not yield the smallest number of genes for the lung dataset, it still achieved a perfect accuracy rate of 100%. Additionally, GWO-HHO consistently outperformed its competitors across all datasets except for lung. This demonstrated the algorithm’s ability to accurately classify cancer datasets while keeping the gene selection to a minimum.
As shown in Table 5, the proposed approach can be applied to real-world cancer classification tasks compared with other state-of-the-art methods. A promising tool for cancer diagnosis and treatment, the GWO-HHO algorithm selects informative genes efficiently and accurately while minimizing computational costs.
We have integrated several studies to address relevant aspects of our research and identify research gaps in the related works. Al-Khafaji et al. [29] focused on the classification of breast cancer images using new transfer learning techniques, highlighting a research gap in innovative transfer learning techniques specific to breast cancer image classification. Li et al. [30] proposed a dual transfer learning approach for medical image analysis, emphasizing the need for improved methodologies and addressing challenges in transfer learning, which highlights the research gap in the application of this approach for breast cancer detection. Liang et al. [31] introduced an enhanced approach for breast cancer detection by incorporating improved multi-fractal dimension techniques and feature fusion methods, emphasizing the importance of advanced image analysis techniques, and highlighting the research gap in integrating these techniques for breast cancer detection. Additionally, Rahmah et al. [32] conducted a systematic review of computing approaches for breast cancer detection through computer-aided diagnosis, emphasizing the need for a comprehensive understanding of existing approaches and their limitations, which highlights the research gap in developing more effective and accurate computer-aided diagnosis systems for breast cancer detection. By incorporating these studies, we provide a clearer understanding of the research landscape and highlight the research gaps that our proposed research aims to address.
One of the limitations we identified is the reliance on microarray data. While microarray technology has been widely used in cancer research, it is important to acknowledge that the field has seen advancements in other high-throughput technologies such as RNA sequencing. Future studies could explore the effectiveness of the proposed hybrid method on RNA sequencing data to validate its performance across different data modalities.
This study tackled the challenge of dimensionality within microarray gene expression profiles by introducing a hybrid feature selection approach. This approach integrates the Gray Wolf Optimization (GWO) and Harris Hawks Optimization (HHO) algorithms with the support vector machines (SVM) and K-nearest neighbors (KNN) algorithms. The objective of our method was to enhance the precision of cancer gene selection and classification while simultaneously minimizing the number of genes selected.
The effectiveness of our hybrid technique was confirmed through the evaluation of multiple cancer microarray datasets. In comparison to other wrapper-based feature selection methods and the standalone GWO and HHO algorithms, our hybrid approach consistently exhibited superior performance in both accuracy and the reduction of selected genes. This underscores the substantial improvements our approach brings not only in terms of performance but also in functionality compared to existing methods. Furthermore, our hybrid approach outperformed recently published hybrid feature selection algorithms, further demonstrating its superiority.
The theoretical implications of our research lie in the development of a bio-inspired algorithm, the GWO-HHO hybrid, for solving gene selection problems. By combining the strengths of GWO and HHO algorithms, we achieved improved accuracy and efficiency in cancer classification tasks. This contributes to the field of bioinformatics and supports the advancement of gene selection methodologies.
Practically, our findings have several advantages that can benefit real-world cancer classification problems. The ability of our hybrid method to accurately classify cancer datasets while reducing the number of selected genes holds great potential for clinical applications. The identification of a smaller set of highly informative genes can aid in the development of more targeted and effective diagnostic and therapeutic strategies.
While our study presents promising results, it is important to acknowledge its limitations. Firstly, our research relied on microarray data, and future investigations should explore the performance of the proposed hybrid method on other data modalities, such as RNA sequencing data, to validate its effectiveness across different technologies. Additionally, our evaluation was based on cross-validation techniques, and further studies should incorporate independent validation on external datasets to assess the generalizability of our method.
Future research should focus on investigating the integration of the proposed hybrid method with complementary techniques like feature extraction or dimensionality reduction methods, enabling the development of a comprehensive framework for cancer analysis. Additionally, exploring the application of the GWO-HHO hybrid algorithm in other areas of bioinformatics, such as protein structure prediction or biomarker identification, would expand its potential beyond cancer classification. Furthermore, there is a need to consider the development of an interpretable framework that provides insights into the biological relevance of the selected genes, offering a deeper understanding of the underlying mechanisms driving cancer development and progression. Such efforts will contribute to advancements in cancer research and pave the way for improved diagnostic and therapeutic strategies.
Acknowledgement: The authors extend their appreciation to the Deputyship for Research and In-novation, “Ministry of Education” in Saudi Arabia for supporting this research (IFKSUOR3-014-3).
Funding Statement: The authors extend their appreciation to the Deputyship for Research and In-novation, “Ministry of Education” in Saudi Arabia for funding this research (IFKSUOR3-014-3).
Author Contributions: Hala AlShamlan, and Halah AlMazrua equally contributed to the manuscript and revised the codes. All authors approved the manuscript.
Availability of Data and Materials: The microarray dataset used in this study is available at the following website: Https://csse.szu.edu.cn/staff/zhuzx/datasets.html.
Conflicts of Interest: All authors declare that they have no competing interests.
References
1. World Health Organization, “Cancer,” Accessed: Oct. 21, 2023. [Online]. Available: https://www.who.int/news-room/fact-sheets/detail/cancer [Google Scholar]
2. “Global cancer rates could increase by 50% to 15 million by 2020,” Accessed: Jun. 03, 2022. [Online]. Available: https://www.who.int/news/item/03-04-2003-global-cancer-rates-could-increase-by-50-to-15-million-by-2020 [Google Scholar]
3. H. C. King and A. A. Sinha, “Gene expression profile analysis by DNA microarrays: Promises and pitfalls,” JAMA, vol. 286, no. 18, pp. 2280–2288, 2001. doi: 10.1001/jama.286.18.2280 [Google Scholar] [PubMed] [CrossRef]
4. S. Alagukumar and R. Lawrance, “Classification of microarray gene expression data using associative classification,” in 2016 Int. Conf. Comput. Technol. Intell. Data Eng. (ICCTIDE’16), Jan. 2016, pp. 1–8. doi: 10.1109/ICCTIDE.2016.7725362. [Google Scholar] [CrossRef]
5. I. Guyon, S. Gunn, M. Nikravesh, and L. Zadeh, “Feature extraction: Foundations and applications (Studies in Fuzziness and Soft Computing),” 2006. Accessed: Nov. 07, 2021. [Online]. Available: https://www.semanticscholar.org/paper/Feature-Extraction%3A-Foundations-and-Applications-in-Guyon-Gunn/9d62d9fa8172f2e65d2cd41ab76a51be977cbf1c [Google Scholar]
6. H. Almazrua and H. Alshamlan, “A comprehensive survey of recent hybrid feature selection methods in cancer microarray gene expression data,” IEEE Access, vol. 10, pp. 71427–71449, 2022. doi: 10.1109/ACCESS.2022.3185226. [Google Scholar] [CrossRef]
7. E. H. Houssein, D. Oliva, N. A. Samee, N. F. Mahmoud, and M. M. Emam, “Liver cancer algorithm: A novel bio-inspired optimizer,” Comput. Biol. Med., vol. 165, no. 24, pp. 107389, Oct. 2023. doi: 10.1016/j.compbiomed.2023.107389 [Google Scholar] [PubMed] [CrossRef]
8. S. Li, H. Chen, M. Wang, A. A. Heidari, and S. Mirjalili, “Slime mould algorithm: A new method for stochastic optimization,” Future Gener. Comput. Syst., vol. 111, pp. 300–323, Oct. 2020. doi: 10.1016/j.future.2020.03.055. [Google Scholar] [CrossRef]
9. J. Li, Y. H. Yang, Q. An, H. Lei, Q. Deng and G. G. Wang, “Moth search: Variants, hybrids, and applications,” Math., vol. 10, no. 21, pp. 4162, Jan. 2022. doi: 10.3390/math10214162. [Google Scholar] [CrossRef]
10. Z. Chen et al., “Dispersed differential hunger games search for high dimensional gene data feature selection,” Comput. Biol. Med., vol. 163, pp. 107197, Sep. 2023. doi: 10.1016/j.compbiomed.2023.107197 [Google Scholar] [PubMed] [CrossRef]
11. H. AlMazrua and H. AlShamlan, “A new approach for selecting features in cancer classification using grey wolf optimizer,” in Int. Conf. Deep Learn., Big Data and Blockchain (DBB 2022), Springer, 2022, pp. 53–64. [Google Scholar]
12. H. AlMazrua and H. AlShamlan, “A new algorithm for cancer biomarker gene detection using harris hawks optimization,” Sens., vol. 22, no. 19, pp. 7273, 2022. doi: 10.3390/s22197273 [Google Scholar] [PubMed] [CrossRef]
13. S. A. Vijay and P. Ganeshkumar, “Fuzzy expert system based on a novel hybrid stem cell (HSC) algorithm for classification of micro array data,” J. Med. Syst., vol. 42, no. 4, pp. 1–12, Apr. 2018. doi: 10.1007/s10916-018-0910-0 [Google Scholar] [PubMed] [CrossRef]
14. N. Almugren and H. M. Alshamlan, “New bio-marker gene discovery algorithms for cancer gene expression profile,” IEEE Access, vol. 7, pp. 136907–136913, 2019. doi: 10.1109/ACCESS.2019.2942413. [Google Scholar] [CrossRef]
15. H. M. Alshamlan, G. H. Badr, and Y. A. Alohali, “Genetic bee colony (GBC) algorithm: A new gene selection method for microarray cancer classification,” Comput. Biol. Chem., vol. 56, no. 6769, pp. 49–60, Jun. 2015. doi: 10.1016/j.compbiolchem.2015.03.001 [Google Scholar] [PubMed] [CrossRef]
16. Y. Fan, J. Liu, J. Tang, P. Liu, Y. Lin and Y. Du, “Learning correlation information for multi-label feature selection,” Pattern Recognit., vol. 145, no. 8, pp. 109899, Jan. 2024. doi: 10.1016/j.patcog.2023.109899. [Google Scholar] [CrossRef]
17. Y. Fan, J. Liu, W. Weng, B. Chen, Y. Chen and S. Wu, “Multi-label feature selection with local discriminant model and label correlations,” Neurocomput., vol. 442, pp. 98–115, Jun. 2021. doi: 10.1016/j.neucom.2021.02.005. [Google Scholar] [CrossRef]
18. Y. Fan, B. Chen, W. Huang, J. Liu, W. Weng and W. Lan, “Multi-label feature selection based on label correlations and feature redundancy,” Knowl.-Based Syst., vol. 241, no. 11, pp. 108256, Apr. 2022. doi: 10.1016/j.knosys.2022.108256. [Google Scholar] [CrossRef]
19. Y. Fan, J. Liu, P. Liu, Y. Du, W. Lan and S. Wu, “Manifold learning with structured subspace for multi-label feature selection,” Pattern Recognit., vol. 120, no. 2, pp. 108169, Dec. 2021. doi: 10.1016/j.patcog.2021.108169. [Google Scholar] [CrossRef]
20. X. Li, L. Wang, Q. Jiang, and N. Li, “Differential evolution algorithm with multi-population cooperation and multi-strategy integration,” Neurocomput., vol. 421, no. 1, pp. 285–302, Jan. 2021. doi: 10.1016/j.neucom.2020.09.007. [Google Scholar] [CrossRef]
21. S. Mirjalili, S. M. Mirjalili, and A. Lewis, “Grey wolf optimizer,” Advances in Engineering Software, vol. 69, pp. 46–61, Mar. 2014. doi: 10.1016/j.advengsoft.2013.12.007. [Google Scholar] [CrossRef]
22. P. Niu, S. Niu, L. Chang, and N. liu, “The defect of the grey wolf optimization algorithm and its verification method,” Knowl.-Based Syst., vol. 171, pp. 37–43, 2019. doi: 10.1016/j.knosys.2019.01.018. [Google Scholar] [CrossRef]
23. A. A. Heidari, S. Mirjalili, H. Faris, I. Aljarah, M. Mafarja and H. Chen, “Harris hawks optimization: Algorithm and applications,” Future Gener. Comput. Syst., vol. 97, pp. 849–872, Aug. 2019. doi: 10.1016/j.future.2019.02.028. [Google Scholar] [CrossRef]
24. T. R. Golub et al., “Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring,” Sci., vol. 286, no. 5439, pp. 531–537, Oct. 1999. doi: 10.1126/science.286.5439.531 [Google Scholar] [PubMed] [CrossRef]
25. D. G. Beer et al., “Gene-expression profiles predict survival of patients with lung adenocarcinoma,” Nat. Med., vol. 8, no. 8, pp. 816–824, Aug. 2002. doi: 10.1038/nm733 [Google Scholar] [PubMed] [CrossRef]
26. S. A. Armstrong et al., “MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia,” Nat. Genet., vol. 30, no. 1, pp. 41–47, Jan. 2002. doi: 10.1038/ng765 [Google Scholar] [PubMed] [CrossRef]
27. J. Khan et al., “Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks,” Nat. Med., vol. 7, no. 6, pp. 673–679, Jun. 2001. doi: 10.1038/89044 [Google Scholar] [PubMed] [CrossRef]
28. T. T. Wong, “Performance evaluation of classification algorithms by k-fold and leave-one-out cross validation,” Pattern Recognit., vol. 48, no. 9, pp. 2839–2846, 2015. doi: 10.1016/j.patcog.2015.03.009. [Google Scholar] [CrossRef]
29. A. A. Alhussan et al., “Classification of breast cancer using transfer learning and advanced Al-biruni earth radius optimization,” Biomimetics, vol. 8, no. 3, pp. 270, Jul. 2023. doi: 10.3390/biomimetics8030270 [Google Scholar] [PubMed] [CrossRef]
30. A. A. Mukhlif, B. Al-Khateeb, and M. A. Mohammed, “Incorporating a novel dual transfer learning approach for medical images,” Sens., vol. 23, no. 2, pp. 570, Jan. 2023. doi: 10.3390/s23020570 [Google Scholar] [PubMed] [CrossRef]
31. D. A. Zebari et al., “Breast cancer detection using mammogram images with improved multi-fractal dimension approach and feature fusion,” Appl. Sci., vol. 11, no. 24, pp. 12122, 2021. doi: 10.3390/app112412122. [Google Scholar] [CrossRef]
32. D. A. Zebari et al., “Systematic review of computing approaches for breast cancer detection based computer aided diagnosis using mammogram images,” Appl. Artif. Intell., vol. 35, no. 15, pp. 2157–2203, Dec. 2021. doi: 10.1080/08839514.2021.2001177. [Google Scholar] [CrossRef]
33. A. Dabba, A. Tari, S. Meftali, and R. Mokhtari, “Gene selection and classification of microarray data method based on mutual information and moth flame algorithm,” Expert Syst. Appl., vol. 166, pp. 114012, Mar. 2021. doi: 10.1016/j.eswa.2020.114012. [Google Scholar] [CrossRef]
34. A. Dabba, A. Tari, and S. Meftali, “Hybridization of moth flame optimization algorithm and quantum computing for gene selection in microarray data,” J. Ambient Intell. Humaniz. Comput., vol. 12, no. 2, pp. 2731–2750, Feb. 2021. doi: 10.1007/s12652-020-02434-9. [Google Scholar] [CrossRef]
35. M. Xi, J. Sun, L. Liu, F. Fan, and X. Wu, “Cancer feature selection and classification using a binary quantum-behaved particle swarm optimization and support vector machine,” Comput. Math. Methods Med., vol. 2016, pp. 3572705, 2016. doi: 10.1155/2016/3572705 [Google Scholar] [PubMed] [CrossRef]
36. S. S. Hameed, F. F. Muhammad, R. Hassan, and F. Saeed, “Gene selection and classification in microarray datasets using a hybrid approach of PCC-BPSO/GA with multi classifiers,” J. Comput. Sci., vol. 14, no. 6, pp. 868–880, Jun. 2018. doi: 10.3844/jcssp.2018.868.880. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools