DOI:10.32604/cmc.2022.021787
| Computers, Materials & Continua DOI:10.32604/cmc.2022.021787 | |
| Article |
An Optimized Neural Network with Bat Algorithm for DNA Sequence Classification
1Faculty of Computing and Information Technology, Sohar University, Sohar, 311, Sultanate of Oman
2Soft Computing & Data Mining Centre (SMC), Faculty of Computer Science & Information Technology, Universiti Tun Hussein Onn Malaysia (UTHM), Batu Pahat, 86400, Malaysia
3School of Electronics, Computing and Mathematics, University of Derby, Derby, DE22 1GB, United Kingdom
4Institute of Computer Sciences and Information Technology, The University of Agriculture, 25120, Peshawar, Pakistan
5College of Computing and Informatics, Saudi Electronic University, Riyadh, Saudi Arabia
*Corresponding Author: Muhammad Aamir. Email: m.aamir@derby.ac.uk
Received: 14 July 2021; Accepted: 30 September 2021
Abstract: Recently, many researchers have used nature inspired metaheuristic algorithms due to their ability to perform optimally on complex problems. To solve problems in a simple way, in the recent era bat algorithm has become famous due to its high tendency towards convergence to the global optimum most of the time. But, still the standard bat with random walk has a problem of getting stuck in local minima. In order to solve this problem, this research proposed bat algorithm with levy flight random walk. Then, the proposed Bat with Levy flight algorithm is further hybridized with three different variants of ANN. The proposed BatLFBP is applied to the problem of insulin DNA sequence classification of healthy homosapien. For classification performance, the proposed models such as Bat levy flight Artificial Neural Network (BatLFANN) and Bat levy Flight Back Propagation (BatLFBP) are compared with the other state-of-the-art algorithms like Bat Artificial Neural Network (BatANN), Bat back propagation (BatBP), Bat Gaussian distribution Artificial Neural Network (BatGDANN). And Bat Gaussian distribution back propagation (BatGDBP), in-terms of means squared error (MSE) and accuracy. From the perspective of simulations results, it is show that the proposed BatLFANN achieved 99.88153% accuracy with MSE of 0.001185, and BatLFBP achieved 99.834185 accuracy with MSE of 0.001658 on WL5. While on WL10 the proposed BatLFANN achieved 99.89899% accuracy with MSE of 0.00101, and BatLFBP achieved 99.84473% accuracy with MSE of 0.004553. Similarly, on WL15 the proposed BatLFANN achieved 99.82853% accuracy with MSE of 0.001715, and BatLFBP achieved 99.3262% accuracy with MSE of 0.006738 which achieve better accuracy as compared to the other hybrid models.
Keywords: DNA sequence classification; bat algorithm; levy flight; back propagation neural network; hybrid artificial neural networks (HANN)
In the field of biological sciences, it very important to know the important aspect of DNA. Because all the genetic information of an organism related to the functioning and reproduction is contained in the DNA. It carries all the information in encrypted form from cell to another cell as well as from parents to the offspring. Recently, the DNA sequences can be easily read with the development of sequencing technologies [1]. It is like a repository of methods that holds all the instructions for making of protein in the human body [2]. Normally, a DNA sequence consists of four types of similar chemicals such as Adenine (A), Guanine (G), Thiamine (T), and Cytosine (C), all these types of chemicals are repeated billions of times in the genome, called nucleotides or base pairs of the DNA sequence. In the DNA sequences, all the four base pair such as Adenine is bonded to Thiamine and Guanine is bonded to cytosine [2]. For the understanding and to decrypt the information related to biological field, a new area of interest known as bioinformatics has evolved [3]. It is a new emerging research domain of the 21st century, it combines many areas like biology, Mathematics, statistics, and computer science etc. It is an important interdisciplinary field which uses information technology for successfully solving the biological problems [4]. The speed of the data generation and growth is exponential in the area of bioinformatics. But it is very complex to generate any useful information from analyzing DNA sequences [3]. Currently, GenBank is a well-known DNA sequence database, which consists of more than 2 million nucleotides or base pairs [1]. To extract information from the massive quantity of the biological data, many cutting-edge computer technologies, algorithms and tools are required [4]. The vital issue in the domain of bioinformatics is the prediction of secondary structure of protein, multiple sequence alignment, inferencing for the construction of phylogenetic trees. These problems are non-deterministic and non-polynomial in nature [3]. Previously, several conventional statistical models and computer science techniques are used [4]. The statistical techniques such as Hidden Markov model (HMM), and Distance based classification are practiced for the aim of the classification of DNA Sequences [5]. More recently, data mining techniques like rule learning (RL), Naïve Bayes(NB), and nonlinear integral classifier (NIC) are found to be more useful and practiced for the classification of DNA sequences [6]. Furthermore, the decision tree algorithm is practically utilized for the DNA sequence classification [7]. The use of numerous traditional techniques for classification of DNA sequence having limitations with respect to accuracy and the time complexity.
For overriding the glitch of having low accuracy, progressive techniques like hybrid machine learning algorithms are used with an intention for DNA sequence classification accurately [6]. The computational models called hybrid Artificial Neural Network (HANN) are primarily inspired from the biological neural systems called neurons [8,9]. ANN mimics the working functionality of the human brain [10]. Many researchers are working in the field of hybrid neural networks for tackling the problems of DNA and Proteins. Recently, Eickholt studied boosting and neural networks for the prediction of disorder in proteins [11]. Countless nature inspired optimization methods are also trained on bioinformatics to improve convergence during search and for the alignment of multiple DNA sequences; as it is the major and core problem in this bioinformatics field [12]. This research work proposed a hybrid method for the classification of Insulin DNA sequence of a healthy human. Many researchers in the past used numerous traditional statistical and data mining techniques for the above-mentioned problem but all the methods and techniques having flaws with respect to accuracy. Furthermore, this research work hybrid the proposed model with Bat a nature inspired optimization algorithm, Levy Flight with artificial neural network and back propagation neural network. With the hybridization approach the performance of the neural network and back propagation algorithm is increased up to the mark.
The main contributions of this paper are given below;
• This research paper proposed hybrid metaheuristic methods combined with a Bat algorithm for the aim to classify the healthy human insulin DNA sequences.
• During the preprocessing, the alignment of DNA sequences is done with the help of omega cluster tool in the first phase. In the second phase the sequence of healthy human is converted into binary for achieving machine readability.
• The methods proposed in this research work is a nature inspired optimization technique called Bat algorithm combined with Leavy flight and simple artificial neural networks and back propagation neural network for the aim to enhance the accuracy in DNA sequence classification.
The structure of the paper is organized as follows: Section 2 sheds some light on the literature review and the algorithms used for the classification of the DNA sequence. Furthermore, Section 3 explains the proposed BatLFBP algorithm, and the methodology for DNA sequence classification is discussed in the Section 4. The simulation results are discussed in the Section 5 and finally, the paper is concluded in the Section 6.
Classification is the basic problem in the field of supervised learning in artificial intelligence. Until now, numerous methods and techniques are used for the purpose of classification. Various methods and models are used for classification, such as Hidden Markov Model (HMM), sequence-sequence classification technique and regression etc. are practiced to solve and classify the biological sequences. In these models the highest alignment score is based in the target classification. Furthermore, another class is used known as featured based selection. In this procedure the biological sequences are changed into a sequence of features and attributed vectors and then classification methods are applied to classify them into the required classes. Sometimes by converting the sequences into the features, the data loses its true nature or form [5].
Another technique employed for classification of DNA sequence is distance-based classification. Numerous distance calculation functions are used to calculate the similarity between the sequences and clearly displays the quality of the classification. Various methods of data mining and machine learning like KNN, SVM with local alignment are used for this type of classification, but these methods offer non-polynomial time and mostly are slow in learning [5–13]. Many other Machine learning is practiced for the aim of the classification of DNA sequences. Despite providing ample results, all of these methods have limitations. But with rapid advancements in the machine learning field, hybrid metaheuristic approaches try to attain efficient outcomes within the minimum ratio of time. More recently, decision tree variant ID3 has been used to classify the DNA sequences. During the implementation process of the decision tree algorithm different statistical parameters are used for the evaluation purposes. The results obtained by ID3 algorithm were accurate up to 88 percent, thus showing that ID3 is efficient and accurate [7]. Similarly, Kassim et al. [6] presented a technique to classify the sequences of DNA with the help of convolutional neural networks (CNN). The CNN comprised of one input layer, followed by numerous hidden layers and has the capability to encounter np-hard problems like DNA sequences. This research article utilized CNN to quantify the efficiency of various data mining techniques. They were able to obtain accuracies of 67.4, 68.2 and 71.6 percent on SVM, decision tree, and rule-based learning respectively. Concurrently, the CNN attained an accuracy rate of 90% for the classification of DNA sequences.
In addition to the current issue, [13] suggested another technique for DNA sequence classification via wavelet neural network (WNN). The research work uses numerous techniques such like Least trimmed squares (LTS) and Genetic Algorithm (GA) combined with WNN for solving the problem of convergence. WNN is estimating the function f(x) of the signal of the DNA sequence. The technique comprises of the arrangements and processing of the spectrum of the DNA sequence signals. K-mean classification technique is used to combine the same DNA sequences related to the criteria. The researcher used Pearson correlation is used for evaluating the relationship between two vectors of DNA sequences. The result outcomes with the help of WNN of training is 98% and for testing is 92%. Their technique was much better than the BPNN which attained an accuracy of 83% and 85% for training and testing respectively. From the literature discussed in this paper, it is found that the DNA sequence classification is highly enhanced with the help of hybrid metaheuristics rather than simple ANNs.
Shadab et al. in [14] identify DNA-Binding proteins (DBPs) by using deep learning methods. In this research the author proposed two different deep learning based methods for identifying DBPs: DeepDBP-ANN and DeepDBP-CNN. The DeepDBP-ANN was used for generated set of features trained on traditional neural network. And DeepDBP-CNN was pre-learned embedding and Convolutional Neural Network. Both proposed methods were tested on standard benchmark datasets. DeepDBP-ANN had achieved test accuracy of 82.80%. While DeepDBP-CNN achieved 84.31% accuracy. But still need to improve the accuracy of the used model in this paper. Further (Gunasekaran et al., 2021) [15] employed convolutional neural network (CNN), convolutional neural network long short term memory (CNN-LSTM), and CNN-Bidirectional LSTM architectures using Label and K-mer encoding for DNA sequence classification. The models are evaluated on different classification metrics. From the experimental results, the CNN and CNN-Bidirectional LSTM with K-mer encoding offers high accuracy with 93.16% and 93.13%, respectively, on testing data.
Therefore, this study utilize a novel metaheuristic bat algorithm to classify DNA sequences in ANN. The proposed methodology is discussed in the next section.
3.1 Implementation of the Bat Algorithm
Metaheuristic algorithms are used for various optimization problems. Among all these algorithms bat algorithms is one of the algorithm which is developed by Yang in 2010 [16], inspired from the natural searching behavior of the bat [17]. Through echolocation, bat finds the place of the food. Bats communicates efficiently and detect very rapidly the optimal solution with continuously changes occur in the emission and loudness using random walk. The bat algorithm uses three rules which are given below;
• Through echolocation, all bats find the distance and also acknowledge the difference between food/prey and the background obstacle in some magical way.
• Bat flies with a random velocity of (
• There are numerous options to adjust the loudness. For simplicity: the loudness is assumed to be varied from a positive large A0 to a minimum constant value, which is represented by Amin.
The initial position
With the interval of [0,1],
where,
where
Benoit Mandelbrot used the Levy Flight for the very first time for the distribution of a specific pitch size. Later, instead of using Levy’s continuous flight named after a French mathematician Paul Levy. The researchers’ employees’ random strolls in levy flight on a discrete grid. This is randomly selected walk with a large tail of probability distribution [20]. Levy motion, commonly known as a levy flight, is a type of non-Gaussian random process in which the random walks are drawn from the levy stable distribution. This distribution is a simple, easy formula of law of power where 0 < 2 is a clue.
3.3 The Proposed BatLFBP Algorithm
The Bat with levy flight back propagation (Bat LFBP) algorithm was proposed in this paper and is used to classify a human insulin DNA sequence. The Bat population is first initialized in the suggested Bat LFBP models. After that, the BP system structure is built. Next, the entry value is then used to create the BP network. The initial weights and bias values are initialized using the Bat levy flight algorithm and then those weights are passed into the BP. Each weight is calculated and compared to the others. In the coming pass, Bat will update the weights until to reach the best possible solution is found, and then continue to search for optimal weights until the network’s last cycle or epoch of the network is reached or the MSE is achieved.
The following equations are used by the BPNN algorithm to calculate weight and biases.
where as
From BPNN algorithm it is easy to calculate MSE for every weight matrix in
The total output of m unit for the output layer is given in Eq. (11);
where,
The index of the hybrid network can be calculated form the following Eq. (13) as given below.
whereas, the average performance of
when each epoch ends the average of average MSE for the ith epoch can be computed from the given Eq. (15).
The MSE is replicating by the bat algorithm and is found when all the inputs are processed for each population of the bat so the bat prey
New solution
The movement of the bats
The bats can move from
where,

The research methodology of this research consists of two phases, the first phase is the pre-processing and the second phase is the post- model training phase. In the pre-processing phase, the data is convert to a standard form on removing any unwanted materials from the data. The dataset is taken from the NCBI, the world's largest on-line database of biological data. The DNA sequences of healthy (human) homosapien insulin are taken, in FASTA format. FASTA is a DNA sequence text format. The DNA sequences in FASTA are a sequential line of congestive characters with no spacing as shown in the Fig. 1.
Figure 1: Normal view of the DNA sequence
The DNA sequence consist the combination of different pair A (Adenine), T (Thymine), C (Cytosine), G (Guanine). All the pair in the sequences are different combination of these base pair. The DNA sequences of this dataset have 495 base pairs after alignment using standard shape alignment. The dataset are divided in two group such as 30% and 70%. 70% of the data is used for the training of the algorithms and 30% of the data is used for testing purpose. Fig. 2 shows the nucleotide density and the relationship of A with T and C with G in the DNA sequence. Tab. 1 shows the description of the dataset.
Figure 2: The pairs between nucleotides of the DNA

In order to reduce the number of mismatches in the preprocessing steps and increase the number of matches in the DNA sequence, initial alignment of the DNA sequences is performed with the omega cluster tool. A sequence will be considered the best sequence which have a larger number of correspondences. The DNA sequences are in characters, and it will be converted to the binary form to readable for the machine. The DNA sequence Binary schema used for binarization of is given as; A = 0 0 1 is Adenine, T = 0 1 0 is Thymine, C = 0 1 1 is Cytosine, G = 1 0 0 is Guanine, N = 1 0 1 is Gap.
So, according to the above rule, this study converts the DNA Sequence into Binary. Nucleotides of the DNA sequence ATCGN. Fig. 3 shows the binary form of the DNA sequence according to the above-mentioned schema.

Figure 3: DNA sequence binarization
In the post processing step, first the hybrid BatLFBP model discussed in Section (3.3) is design and then to train the proposed model with the clean data is fed to the model. Bat starts random search and picks the best value from the specified location by the levy flight and then fed these best values to the artificial neural networks and finally generates the output results. In the Fig. 4, the proposed BatLFBP algorithm for DNA sequence classification is given.

Figure 4: The proposed BatLFBP algorithm for DNA sequence classification
This section includes the classification of DNA sequence outputs for the three templates using multiple window lengths (WL); WL 5, WL 10 and WL 15. The data set is taken from NCBI of a multicellular organism. The total number of DNA base pairs in this dataset are 469, and after alignments the number of base pair sequence reach to 495 by adding 26 gap to equate with other DNA sequences. With the help of “mRNA Nm_000207” number Homo sapiens insulin (INS), transcript variant 1, is access. The sequence can be traced easily with the help of this number, in the large database like NCBI. The basic theme of the research work is the classification of the insulin DNA sequence of human using metaheuristic optimization techniques hybrid with neural network models. The highlighted BatLFBP and BatLFANN are the proposed algorithms and the four other hybrid algorithms are used for comparison and given in the Tabs. 2–4.

In this portion of the research work, the machines used for the aim of simulations are equipped with an Intel core i7 turbo processor having the strength of 2.7 and 2.9 GHz, seventh generation and having 8GB RAM. The tool used for the intent of implementation of the proposed algorithms is MATLAB R2014b with Windows 10 operating system. The proposed Bat Algorithm with Levy flight back propagation neural network is tested on the benchmark DNA sequences dataset taken from the world famous and largest biological National Center for Biotechnology Information (NCBI) data repository. The proposed algorithms used in this paper are given below.
1. Bat with Levy Flight Artificial Neural Network algorithm (BATLFANN).
2. Bat with levy Flight Backpropagation algorithm (BATLFBP).
The performance of the proposed algorithms used in this research work are equated with the following four neural network algorithms merged with optimization algorithms on two datasets with two different variants and three separate window sizes.
1. Bat with Artificial Neural Network algorithm (BatANN) [10].
2. Bat with Back Propagation Neural Network algorithm (BatBP) [19].
3. BAT with Gaussian distribution Artificial Neural Network algorithm (BatGDNN) [21].
4. BAT with Gaussian Distribution Back Propagation algorithm (BatGDBP) [21].
While performing the experiments, performance parameters used are accuracy and mean square error (MSE). The evaluation of the proposed algorithms is done on two dataset variants of insulin DNA sequences having different windows length sizes like five, ten and fifteen. The maximum number of the epochs for this experimental work were set to 1000.
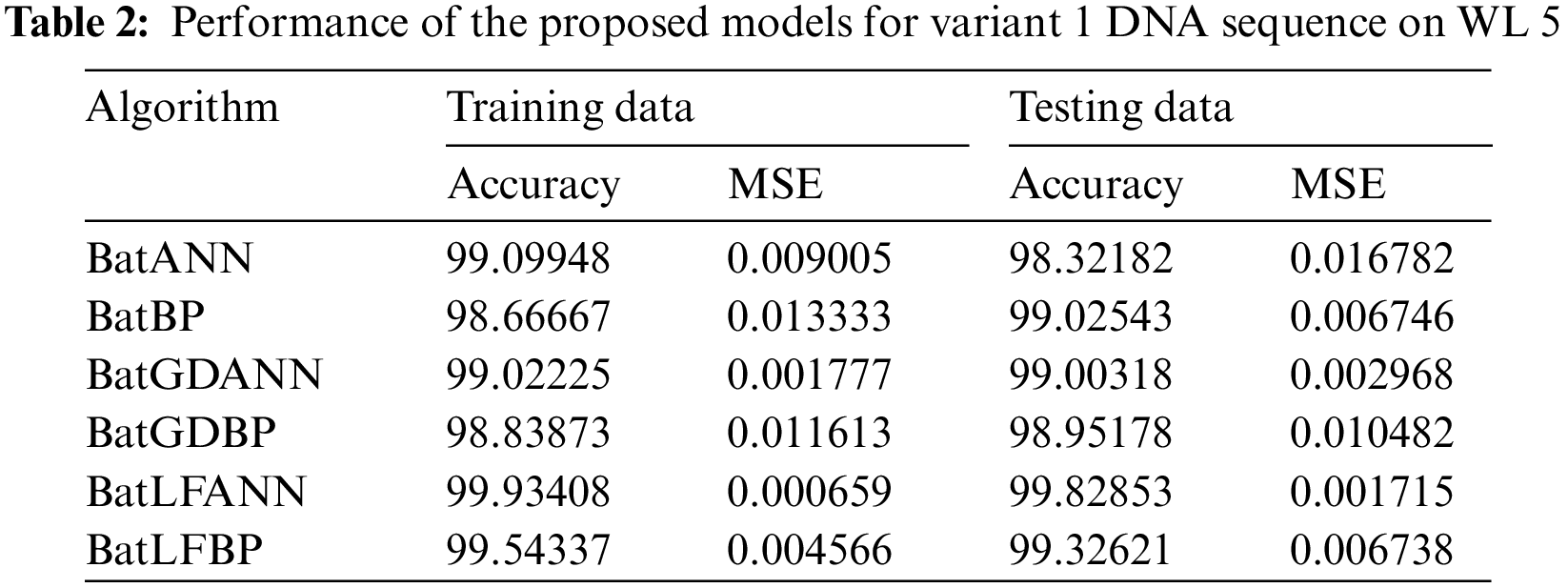
5.2 Results Performance for WL 5
Tab. 2 illustrates the performance of the proposed hybrid algorithms against other comparison algorithms which are applied to the DNA sequence of Insulin Variant 1 (V1) data set with a window length of five. The values in the Tab. 2 demonstrate the accuracy and MSE of the proposed hybrid models and other conventional hybrid models. Here in this research, the most efficient results are given by the proposed BatLFANN and BatLFBP with the accuracy rate of 99.93408%, 99.54337%, and an MSE of 0.000659 and 0.004566 on 70% training datasets. Furthermore, BatGDBP is left behind the BatLFANN for accuracy and MSE. BatLFBP provides 99.54337% accuracy with 0.004566 of MSE for 70% of workout datasets. Likewise, BatGDANN achieves 99.02225% precision with an MSE of 0.001777 for the 70% of drive data after BatGDANN, BatANN delivers 99.09948% precision and MSE of 0.009005. Finally, BatBP converges with an accuracy of 98.6667% and an MSE of 0.013333.
Whereas for the test data sets, the proposed hybrid algorithms BatLFANN and BatLFBP achieved 99.82853%, 99.32621%, with an MSE of 0.001715, and 0.006738 respectively. The rest of the algorithms like BatANN, BatBP, BatGDANN, and BATGNBP achieved accuracies of 98.32182%, 99.02543%, 99.00318%, 98.95178% and MSE’s of 0.016782, 0.006746, 0.002968, 0.010482 respectively. The above results concluded that BatLFANN and BatLFBP gave best optimal solutions 99.93408% and 99.54337% at 70% of training datasets which are almost near to actual results. Also, BatLFANN and BatLFBP gave promising outcomes of 99.82853% and 99.32621% on the 30% testing dataset with MSE’s of 0.001715 and 0.006738 respectively. Additionally, Fig. 5 shows a graphical representation of the convergence performance of the MSE to test the data set of each hybrid algorithm.
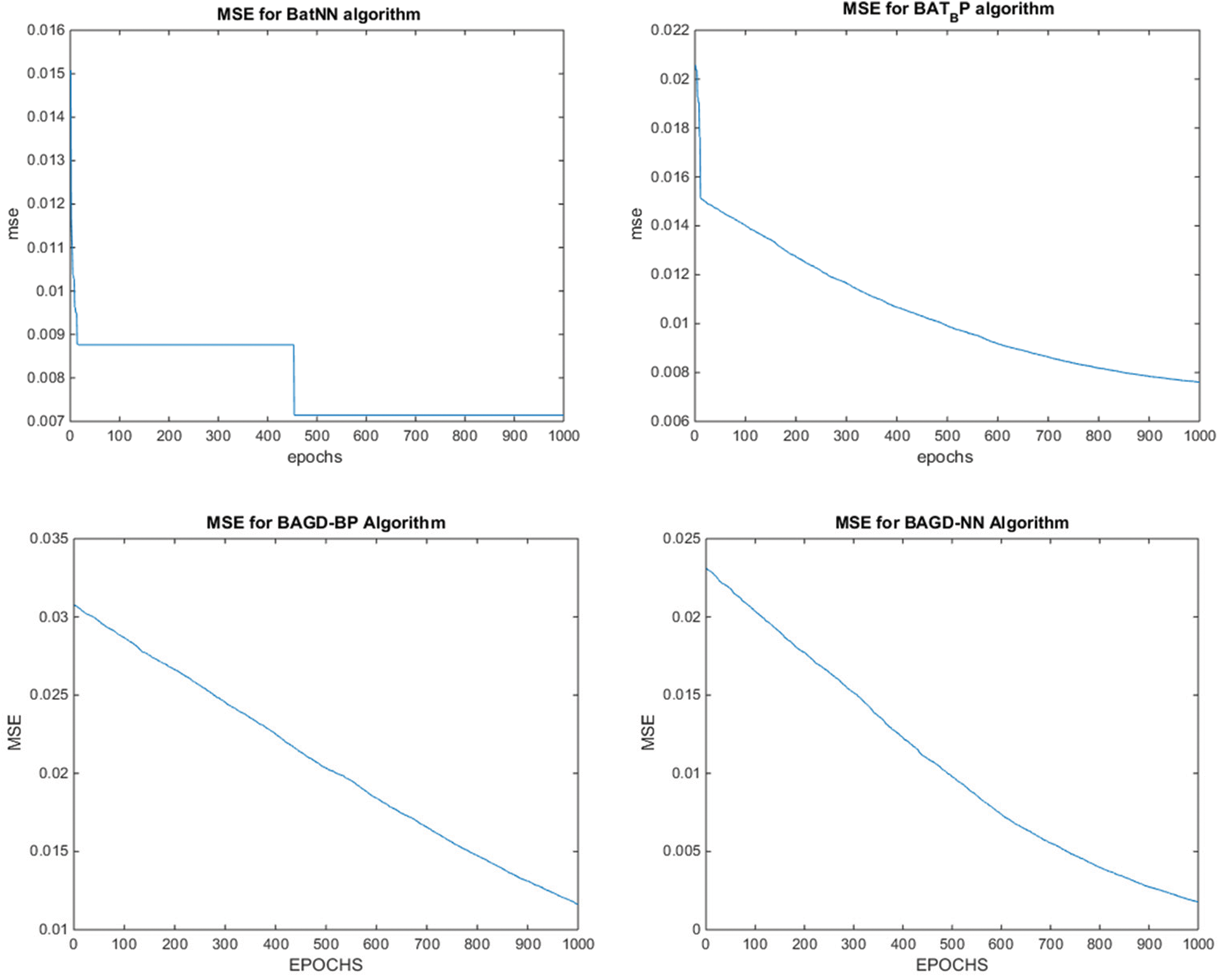

Figure 5: MSE convergence of the proposed BatLFBP with other hybrid algorithms for WL 5 on variant1 (V1) dataset
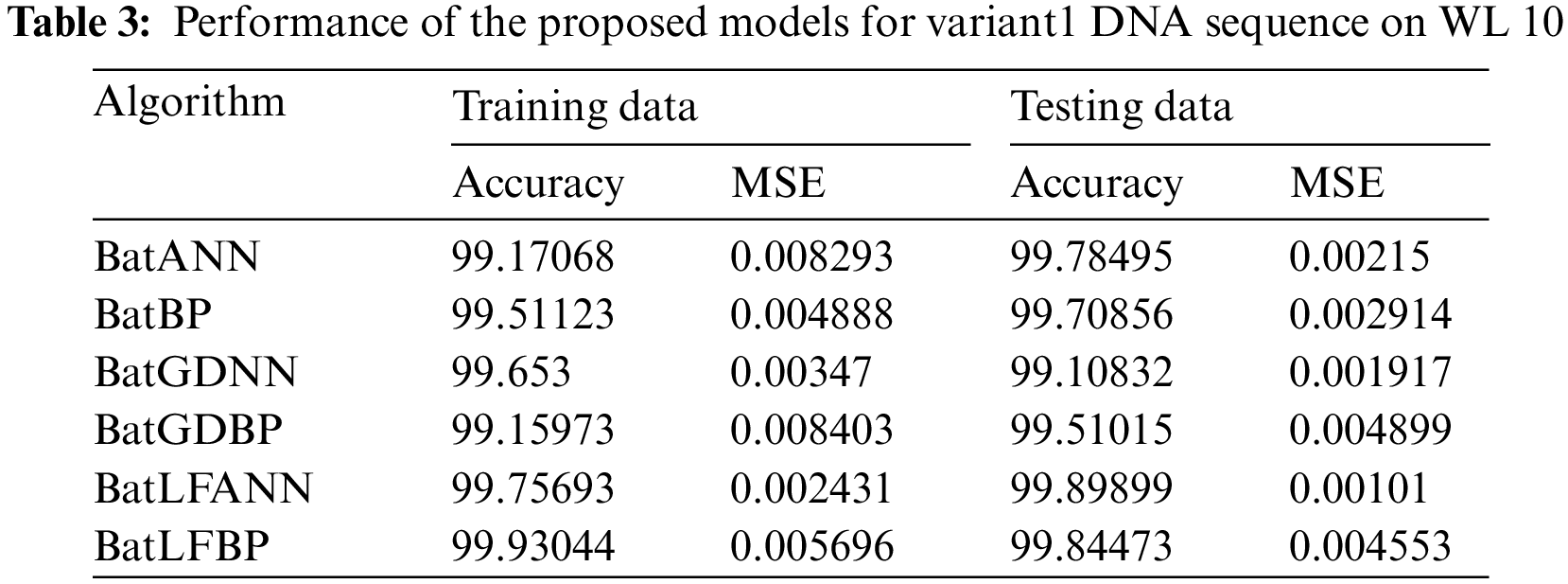
5.3 Results Performance for WL 10
Tab. 3 presents various records that are based on six different hybrid models applied to the dataset of a Variant1 (V1) insulin DNA sequence. That offers performance in terms of precision and average squared error. In this research, the most efficient result is delivered by of the proposed BatLFANN and BatLFBP with accuracies of 99.75693%, 99.93044 and MSE’s 0.002431, 0.005696 on 70% of the training datasets. Parallel to the proposed models, the performance of the other hybrid BatANN achieves an accuracy rate of 99.17068% and an MSE of 0.008293 on 70% of the training data. Apart from these, BatBP’s accuracy rate on 70% of training set is 99.51123 with an MSE of 0,004888. Moreover, BatGDANN achieved performance in-terms of accuracy is 99.653% with an MSE of 0.00347. Finally, BatGDBP was 99.1597% accurate with an MSE of 0.0084 ono 70% of the training sets. It is concluded from the above results, the proposed hybrid models BatLFANN and BatLFBP gave promising results on training datasets.
Similarly, on 30% of testing datasets BatLFANN and BatLFBP achieved accuracies of 99.89899%, 99.54473% with MSE of 0.00101, 0.004553 respectively. The remaining hybrid algorithms like simple BatANN, BatBP, BatGDNN and BatGDBP delivered accuracies on 30% of the testing sets are 99.78495%, 99.70856%, 99.10832%, and 99.51015% with MSE’s of 0.00215, 0.002914, 0.001917, and 0.004899 respectively. Based on the results, it is concluded that the proposed BatLFANN and BatLFBP algorithms perform better than the rest of the comparable hybrid algorithms. The graphical representation of the performance of each hybrid algorithm is presented in Fig. 6.

Figure 6: MSE convergence of the proposed BatLFBP with other hybrid algorithms for WL 10 on variant1 (V1) dataset
5.4 Results Performance for WL 15
Tab. 4 Comprises of number of results which are obtained from different hybrid models which have been applied to the DNA sequence of Insulin Variant 1 (V1) dataset with fifteen window length size. In the Tab. 4, the proposed hybrid models are BatLFANN and BatLFBP which delivered accuracies of 70% on the training datasets i.e., 99.82023% and 99.61878% with MSE’s of 0.001798 and 0.003812 respectively. Beside these proposed hybrid algorithms, BatANN, BatBP, BatGDANN, and BatGDBP attained accuracies on the 70% of the training set of 99.03144%, 98.08247%, 99.00282%, and 99.49946% and MSE’s of 0.000686, 0.011175, 0.001172, and 0.005005 respectively.
Additionally, after completing the dataset training, testing is done on the 30% of the data. During the test phase, the proposed BatLFANN and BatLFBP provided 99.88153% and 99.83418% accuracies with MSE’s of 0.001185 and 0.001658 respectively. The comparing algorithms like simple BatANN, and BatBP achieved accuracies of 99.3449% and 99.33194%. Whereas, BatGDNN and BatGDBP gave accuracies of 99.73115%, and 99.32988% with MSE’s 0.002688, and 0.006701 respectively. It is summarized from the above Tab. 4, the results of all the hybrid models such as BatLFANN and BatLFBP achieved better accuracies for the training and the testing datasets. The graphical representation of the performance of the hybrid algorithms is provided below in Fig. 7.
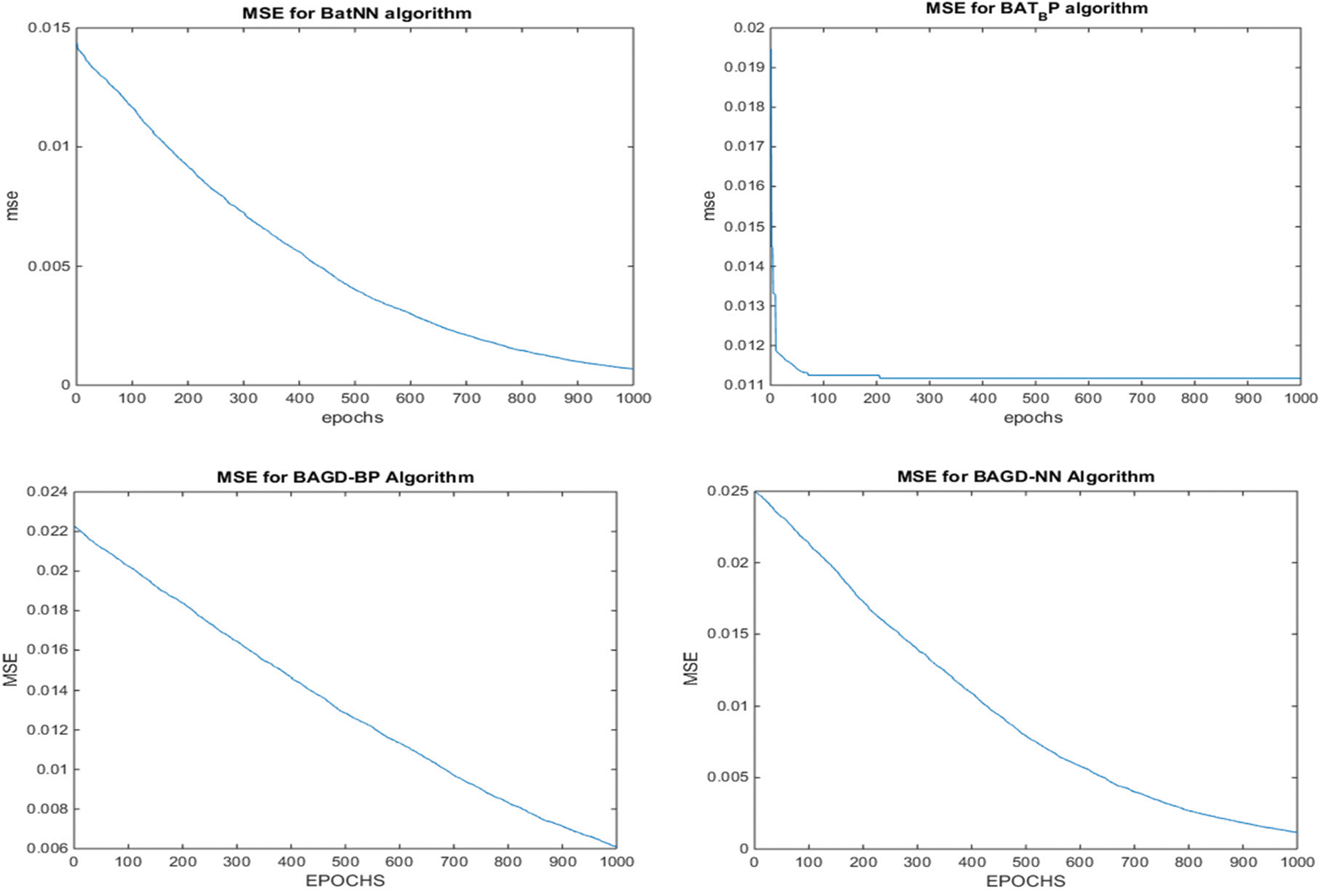

Figure 7: MSE convergence of the proposed BatLFBP with other hybrid algorithms for WL 15 on variant1 (V1) dataset

This research work targeted the hybridization of the metaheuristic optimization techniques with Artificial Neural Network (ANN) and Backpropagation Neural Network (BPNN) to achieve a high level of results and accuracy for the task of classification. In this research work, the Bat algorithm is a hybrid of ANN and BPNN. A total of six hybrid algorithms are used for the classification of DNA sequences, two of which are proposed hybrid algorithms and the remaining four are comparable hybrid algorithms. Datasets on insulin DNA sequences are used for assessments with three different window sizes: window length 5, window length 10 and window length 15. For classification performance, the proposed models such as BatLFANN and BatLFBP are compared with the other state-of-the-art algorithms like BatANN, BatBP, BatGDANN and BatGDBP in-terms of MSE and accuracy. From the perspective of simulations results, it is show that the proposed BatLFANN and BatLFBP achieved better accuracies as compared to the other hybrid models. After the favorable outcomes from the practical evaluations, it is concluded that Bat optimization techniques with ANN and BP gave accuracies of almost 99 percent within 1000 epochs on DNA sequence classification.
In the Future, this research will be enhanced with some of the most recent metaheuristic algorithms like Sine-Cosine to achieve better classification on some other biological DNA sequences. It has the ability to find out the infected and healthy DNA sequence for diseases like COVID19. Please refer to Tab. 5 for all the terminologies and notations used in this paper.
Acknowledgement: The authors would like to thank the Research Management Office (RMC), Universiti Tun Hussein Onn Malaysia and Ministry of Higher Education, Malaysia for supporting this research under the Tier-1 Research Grant, vote no. H938.
Funding Statement: This research is supported by Tier-1 Research Grant, vote no. H938 by Research Management Office (RMC), Universiti Tun Hussein Onn Malaysia and Ministry of Higher Education, Malaysia.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. N. G. Nguyen, “DNA sequence classification by convolutional neural network,” Journal of Biomedical Science & Engineering, vol. 9, no. 5, pp. 280–286, 2016. [Google Scholar]
2. K. Chao, Basic concepts of DNA, proteins, genes and genomes, in Graduate Institute of Biomedical Electronics & Bioinformatics, National Taiwan University, Taipei, Taiwan, pp. 1–5, 2006. [Google Scholar]
3. M. Hapudeniya, “Artificial neural networks in bioinformatics,” Sri Lanka Journal of Bio-Medical Informatics, vol. 1, no. 2, pp. 104, 2010. [Google Scholar]
4. H. Hasic, E. Buza and A. Akagic, “A hybrid method for prediction of protein secondary structure based on multiple artificial neural networks,” in 40th Int. Convention on Information and Communication Technology, Electronics and Microelectronics (MIPRO), IEEE, pp. 1195–1200, 2017. [Google Scholar]
5. Z. Xing, J. Pei and E. Keogh, “A brief survey on sequence classification,” ACM SIGKDD Explorations Newsletter, vol. 12, no. 1, pp. 40–48, 2010. [Google Scholar]
6. N. A. Kassim and A. Abdullah, “Classification of DNA sequences using convolutional neural network approach,” Faculty of Computing, Universiti Teknologi Malaysia (UTMMalaysia, vol. 2, pp. 1–6, 2017. [Google Scholar]
7. M. A. Siddiquee and H. Tasnim, “A comprehensive study of decision trees to classify DNA sequences,” University of New Mexico, vol. 1, no. 1, pp. 1–4, 2018. [Google Scholar]
8. C. H. Wu, J. W. Mclarty and E. Science, “Book review neural networks and genome informatics,” Computational Chemistry, vol. 25, no. 4, pp. 427–428, 2001. [Google Scholar]
9. N. M. Nawi, M. Z. Rehman and A. Khan, “Countering the problem of oscillations in bat-BP gradient trajectory by using momentum,” Lecture Notes in Electrical Engineering, vol. 285, pp. 103–110, 2014. [Google Scholar]
10. N. M. Nawi, F. Hamzah, N. A. Hamid, M. Z. Rehman, M. Aamir et al., “An optimized back propagation learning algorithm with adaptive learning rate,” International Journal of Advance Science and Engineering Information Technology, vol. 7, no. 5, pp. 1693, 2017. [Google Scholar]
11. J. Eickholt and J. Cheng, “DNdisorder: Predicting protein disorder using boosting and deep networks,” BMC Bioinformatics, vol. 14, no. 1, pp. 107, 2013. [Google Scholar]
12. W. Kartous, A. Layeb and S. Chikhi, “A new quantum cuckoo search algorithm for multiple sequence alignment,” Journal of Intelligent Systems, vol. 23, no. 3, pp. 261–275, 2014. [Google Scholar]
13. A. Dakhli, W. Bellil and C. Ben Amar, “Wavelet neural networks for DNA sequence classification using the genetic algorithms and the least trimmed square,” Procedia Computer Science, vol. 96, pp. 418–427, 2016. [Google Scholar]
14. S. Shadab, Md T. A. Khan, N. Neezi, S. Adilina and S. Shatabda, “DeepDBP: Deep neural networks for identification of DNA-binding proteins,” Informatics in Medicine Unlocked, vol. 19, pp. 100318, 2020. [Google Scholar]
15. H. Gunasekaran, K. Ramalakshmi, A. Rex Macedo Arokiaraj, S. Deepa Kanmani, C. Venkatesan et al., “Analysis of DNA sequence classification using CNN and hybrid models,” Computational and Mathematical Methods in Medicine, vol. 2021, 2021. [Google Scholar]
16. X. S. Yang, “A new metaheuristic Bat-inspired algorithm,” in Nature Inspired Cooperative Strategies for Optimization (NICSO 2010Studies in Computational Intelligence, Springer, Berlin, Heidelberg, vol. 284, pp. 65–74, 2010. [Google Scholar]
17. I. Fister, X. S. Yang, S. Fong and Y. Zhuang, “Bat algorithm: Recent advances,” in 15th IEEE Int. Sym. on Computational Intelligence and Informatics, Budapest, Hungary, pp. 163–167, 2014. [Google Scholar]
18. J. H. Lin, C. Chao-Wei, Y. Chorng-Horng and T. Hsien-Leing, “A chaotic Levy flight bat algorithm for parameter estimation in nonlinear dynamic biological systems,” Journal of Computer Information Technology, vol. 2, no. 2, pp. 56–63, 2012. [Google Scholar]
19. N. M. Nawi, M. Z. Rehman and A. Khan, “The effect of bat population in Bat-Bp algorithm,” Lecture Notes in Electrical Engineering, vol. 291, pp. 295–302, 2014. [Google Scholar]
20. X. Shan, K. Liu and P. L. Sun, “Modified bat algorithm based on lévy flight and opposition based learning,” Scientific Programming, vol. 2016, no. 2, pp. 1–13, 2016. [Google Scholar]
21. N. M. Nawi, M. Z. Rehman, A. Khan, H. Chiroma and T. Herawan, “A modified bat algorithm based on gaussian distribution for solving optimization problem,” Journal of Computational and Theoretical Nanoscience, vol. 13, no. 1, pp. 706–714, 2016. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |