DOI:10.32604/cmc.2021.017769
| Computers, Materials & Continua DOI:10.32604/cmc.2021.017769 | |
| Article |
DTLM-DBP: Deep Transfer Learning Models for DNA Binding Proteins Identification
1Department of Computer Engineering, Faculty of Engineering, Arab Academy of Science and Technology, Egypt
2Centre for Artificial Intelligence & Robotics, Malaysia-Japan International Institute of Technology, Universiti Teknologi Malaysia
*Corresponding Author: Uswah Khairuddin. Email: uswah.kl@utm.my
Received: 10 January 2021; Accepted: 16 March 2021
Abstract: The identification of DNA binding proteins (DNABPs) is considered a major challenge in genome annotation because they are linked to several important applied and research applications of cellular functions e.g., in the study of the biological, biophysical, and biochemical effects of antibiotics, drugs, and steroids on DNA. This paper presents an efficient approach for DNABPs identification based on deep transfer learning, named “DTLM-DBP.” Two transfer learning methods are used in the identification process. The first is based on the pre-trained deep learning model as a feature’s extractor and classifier. Two different pre-trained Convolutional Neural Networks (CNN), AlexNet 8 and VGG 16, are tested and compared. The second method uses the deep learning model as a feature’s extractor only and two different classifiers for the identification process. Two classifiers, Support Vector Machine (SVM) and Random Forest (RF), are tested and compared. The proposed approach is tested using different DNA proteins datasets. The performance of the identification process is evaluated in terms of identification accuracy, sensitivity, specificity and MCC, with four available DNA proteins datasets: PDB1075, PDB186, PDNA-543, and PDNA-316. The results show that the RF classifier, with VGG-Net pre-trained deep transfer learning features, gives the highest performance. DTLM-DBP was compared with other published methods and it provides a considerable improvement in the performance of DNABPs identification.
Keywords: DNABPs; deep transfer learning; AlexNet 8; VGG 16; SVM; RF
Deoxyribonucleic Acid (DNA) represents the cell blueprint that contains the main information that codes all organisms. DNA can perform its functions with the help of thousands of proteins, which are called DNA binding proteins (DNABPs). DNABPs have several jobs, such controlling protein production, regulating cell growth and storing DNA in the nucleus. DNABPs play an important role in the structural composition of DNA. In addition, they regulate and control different cellular processes such as the transcription, replication, recombination, repair and modification of DNA.
DNABPs identification is considered a major challenge of genome annotation because they have several linked cellular functions. The identification process may include: identifying the DNABPs (positive sample) from the non-DNABPs (negative sample) [1], identifying the single-stranded DNABPs from the double-stranded DNABPs [2], or identifying the DNABPs from the Ribonucleic acid-binding proteins (RNABPs) [3–5]. In this paper, the identification process is formulated as a binary classification problem to identify DNABPs and non-DNABPs. DNABPs are the proteins that have DNA binding domains and they generally interact with the major groove of B-DNA. Non-DNABPs, on the other hand, are the structural proteins within the chromosomes.
Several experimental technical methods can be used for identifying DNABPs, but they are time-consuming and expensive [6]. Therefore, there is a significant need to find a suitable and efficient computational method for replacing these experimental methods. Recently, several computational and statistical methods have been proposed for DNABPs identification, but most of these methods cannot provide the invaluable knowledge base for DNABPs identification. With the advancements in machine and deep learning techniques over recent years, several methods based on machine and deep learning have been presented.
Zhu et al., proposed a method for DNABPs identification based on the position-specific scoring matrices (PSSM) and co-occurrence matrix. The results achieved an accuracy of 97.06% for Yeast dataset, 98.95% for Human dataset, and 89.69% for H.Pylori dataset [7]. The PSSM with SVM (PSFM-DT) tested by Xu et al. [8] achieved an accuracy of 79.96% for PDB1075 dataset, and 79.96% for PDB186 dataset. In addition, the PSSM with RF tested by Waris et al. [9] achieved an accuracy of 92.3% for their tested dataset. Chowdhury et al., proposed a method (iDNAProt-ES) for DNABPs identification by extracting the structural and evolutionary features that feed the SVM predictor. The results achieved an accuracy of 90.18% for the jack-knife dataset [10]. Xu used the random forest for DNABPs identification. The results achieved an accuracy of 85.57 for the jack-knife dataset [11].
Zhang et al., proposed a method for DNABPs identification by combining the position-specific frequency matrix and the distance-bigram transformation (PSFM-DBT). The results achieved an accuracy of 81.02% for PDB1075 dataset, and 80.65% for PDB186 dataset [12]. Zhang et al., made features with a fusion of evolutionary, structural, and physicochemical features for DNABPs identification, and used the binary firefly optimization for removing the redundant features. The results achieved an accuracy of 91% for the DNA dataset, and 0.80.9% for PDB186 dataset [13]. Ma et al., proposed a method for DNABPs identification based on selecting the hybrid features using the random forest. The results achieved an accuracy of 89.56% for Mainsett dataset [14].
Moreover, Shen et al., used the multi-scale local average blocks approach for DNABPs identification. The results achieved an accuracy of 91.80% for PDNA-543 dataset, 92.06% for PDNA-41 dataset, 90.23% for PDNA-316 dataset, and 77.6% for PDNA-52 dataset [15]. Krishna et al., proposed a DNABPs identification (DNA-Prot) method by incorporating the evolutionary features into the pseudo-amino acid composition. The results achieved an accuracy of 81.83% for DNA-Prot dataset, and 61.42% for DNA binder dataset [16]. This method was modified by adding the grey model and named iDNA-Prot [17]. Fu et al., applied the same method on the jack-knife test and independent test, which achieved an accuracy of 89.77% and 88.71%, respectively [18]. Wei et al. [19] used RF in the training, called the Method by Local-DPP model. Moreover, Liu et al. [20] used SVM and called it iDNAPro-PseAAC. This method was improved through dimension reduction by Liu et al. [21] and renamed iDNA-Pro-dis. The concept of Pse-AAC was applied in other models called DNABinder [22], PseDNA-Pro [23], and DPP-PseAAC [24]. Biological information was added by Zaman et al. [25] and named (HMMBinder).
Szilagyi et al. presented a method for DNABPs identification (DNABIND) based on the amino acid proportions in the sequence of the protein. The results achieved an accuracy of 67.70% for PDB186 dataset [26]. Gao et al. presented a threading-based method for DNABPs identification (DNA-Threader). The results achieved an accuracy of 59.7% for PDB186 dataset [27]. Szilagyi et al. presented a DNABPs identification method (DNABIND) based on hybrid feature selection using RF and Gaussian naive Bayes (DBPPred). The results achieved an accuracy of 76.90% for PDB186 dataset [28].
Zhang et al., proposed a DNABPs identification method using bootstrap multiple CNN. The results achieved an accuracy of 90.77% for PDNA-543 dataset and 91.04% for PDNA-316 dataset [29]. They used the long short-term memory and CNN. The results achieved an accuracy of 81.83% for DNA-Prot dataset, and 89.19% for Chip-seq dataset [30]. Liu et al., proposed a method for DNABPs identification by combining the auto-cross covariance with ensemble learning (iDNA-KACC). The results achieved an accuracy of 75.16% for the tested dataset [31]. Qu et al., proposed a method for DNABPs identification using mixed feature representation methods. The results achieved an accuracy of 77.43% for PDB1075 dataset, and 81.58% for PDB186 dataset [32]. Hu et al. [33] combined the sequence features with multiple SVMs and named the method TargetDNA. Si et al. [34] presented a meta-based DNABPs identification and named it MetaDBSite.
The main contribution of this paper is the testing and adaptation of pre-trained deep transfer learning models for DNABPs sequence identification. The paper presents a novel approach for DNABPs identification using deep transfer learning. In this approach, two transfer learning methods were tested and compared; in the first method, the pre-trained deep CNN (AlexNet 8 or VGG 16) learning model was used as a feature’s extractor and classifier. In the second method, the deep learning model was used as a feature’s extractor only, while the classifier was either the SVM or RF. The proposed approach was tested using different DNA proteins datasets. The performance of the identification process was evaluated in terms of identification accuracy, sensitivity, specificity, and MCC with four available DNA proteins datasets: PDB1075, PDB186, PDNA-543, and PDNA-316 datasets. The results show that the RF classifier with VGG-Net pre-trained deep transfer learning features produced the highest performance. DTLM-DBP was compared with the other published methods and found to represent a considerable improvement in the performance of DNABPs identification. The remainder of the paper is organized as follows: the second section will present the proposed methodology, the third section gives the results, and the conclusion will be given in the last section.
The general block diagram of the DNABPs identification process in this paper is shown in Fig. 1. Two transfer learning methods were carried out. In the first method, the protein sequences were adapted to CNN models using 1D convolutions layers, then one of the pre-trained deep CNN learning models was used as a feature’s extractor and classifier. In the second method, the deep learning model was used as a feature’s extractor only, while the classifier was either the SVM or RF. More details about each block will be presented in the following subsections.
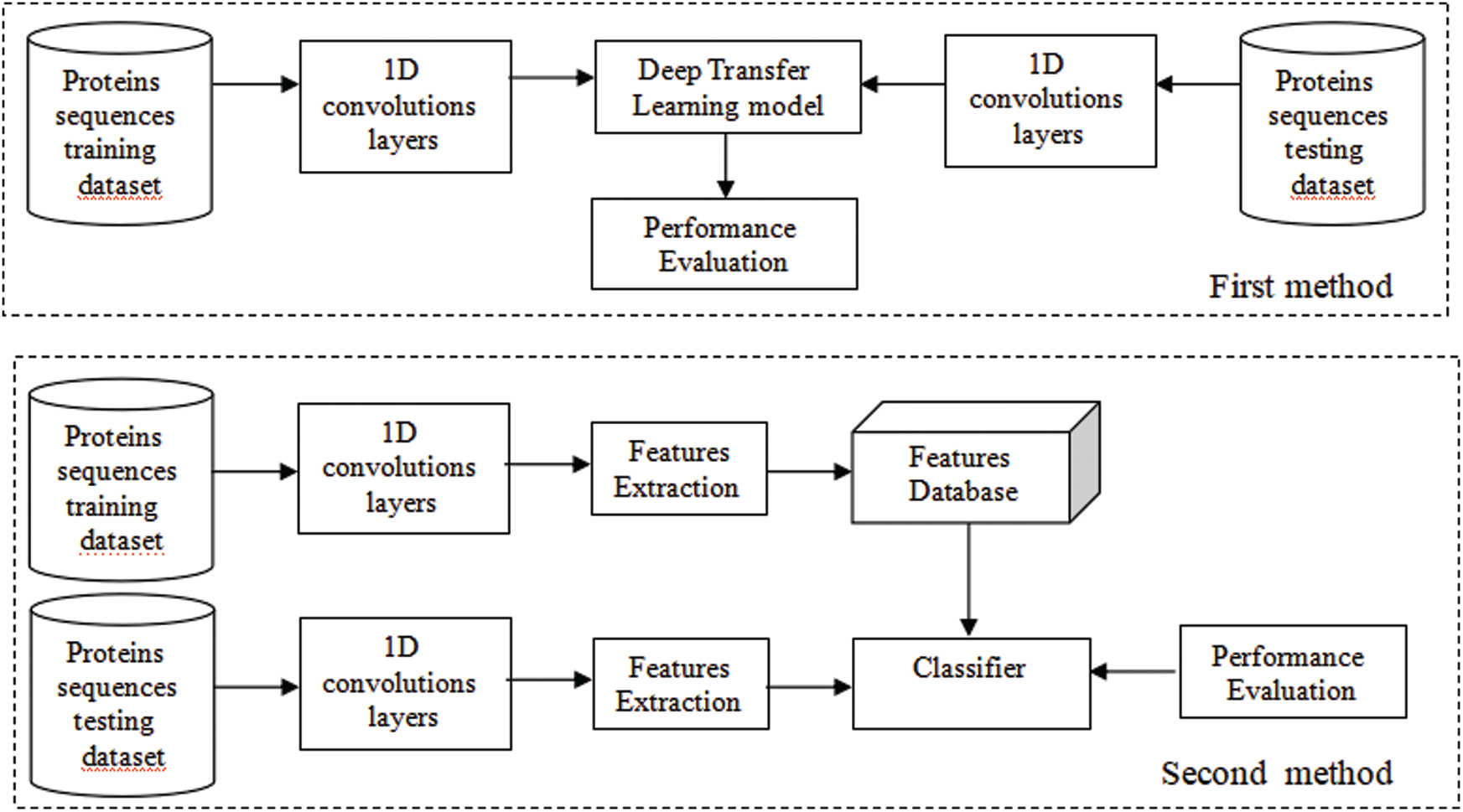
Figure 1: DNABPs identification process
There are several publicly available protein sequences datasets, most of which were collected from the protein data bank (PDB). The researchers collected the sequences data from the PDB website by searching for words such as ‘DNA binding,’ ‘DNA protein’ and other related terms, Then, certain processing procedures were undertaken to avoid the inclusion of redundant data, and finally, the obtained datasets were used in the research. To guarantee the reliability of the proposed approach and for performance evaluation comparison purposes, pre-collected publicly available datasets were used that had been used by several researchers in the literature.
The experimental work was implemented on four different DNABPs datasets: PDB1075, PDB186, PDNA-543, and PDNA-316 datasets. PDB1075 dataset was collected by Liu et al. [21], and included 1,075 protein samples; 525 samples were positive DNABPs and 550 samples were negative non-DNABPs. PDB186 dataset was collected by Lou et al. [28], and included 186 protein samples; 93 samples were positive DNABPs and 93 samples were negative non-DNABPs. PDNA-543 dataset was collected by Hu et al. [33], and included 144,544 protein samples; 9,549 samples were positive DNABPs and 134,995 samples were negative non-DNABPs. PDNA-316 dataset was collected by Si et al. [34], and included 72,718 protein samples; 5,609 samples were positive DNABPs and 67,109 samples were negative non-DNABPs.
2.2 Deep Transfer Learning Models
In this paper, two pre-trained deep transfer learning models, AlexNet and VGG-Net, were adapted for the identification of DNABPs sequences. The model architecture of each training model will be presented. These two models had been selected from the large number of pre-trained deep learning transfer models because, according to the literature, they are the most successful models in terms of the identification process, while their architectures are simple and contain different numbers of convolution layers.
2.2.1 AlexNet-8 Pre-Trained Deep Transfer Learning Model
AlexNet-8 is a CNN that is 8 layers deep, and was introduced by Krizhevsky et al. [35]. The number of parameters in AlexNet-8 is 60 million and the number of neurons is 650,000. It consists of 8 layers (5 convolutional and 3 fully connected), as shown in the model architecture in Fig. 2 [36]. The first and second convolutional layers are followed by normalization and a max-pooling layer, the third and fourth convolutional layers are connected directly, and the last convolutional layer is followed by a max-pooling layer. The output of the convolutional layer passes through a series of two fully connected layers, in which the second fully connected layer is fed into the SoftMax classifier.
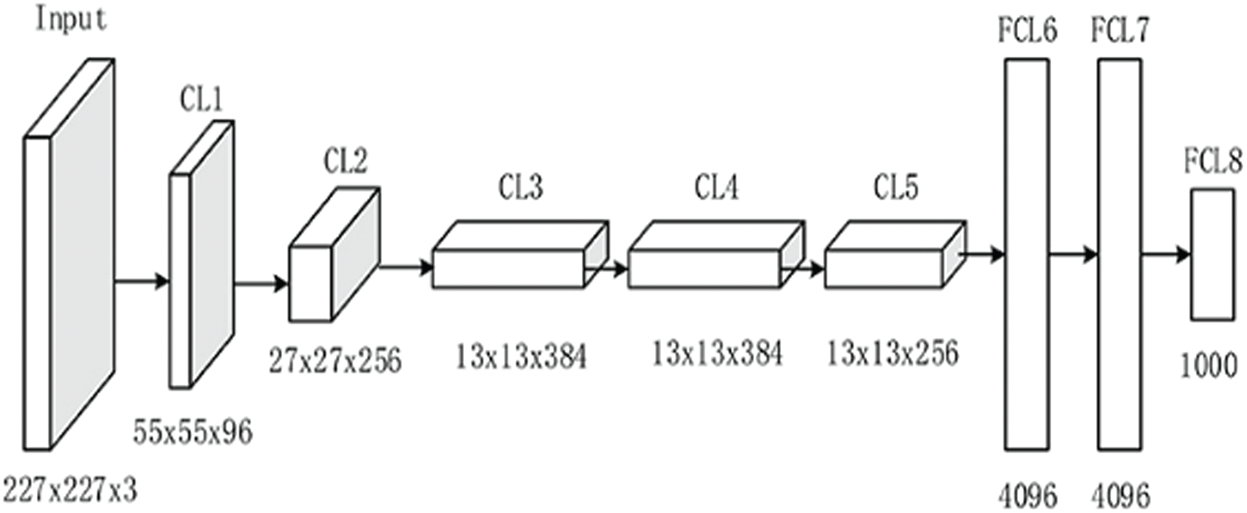
Figure 2: AlexNet-8 model architecture
2.2.2 VGG-16 Pre-Trained Deep Transfer Learning Model
VGG-16 is a CNN model which is 16 layers deep, and was introduced by Simonyan and Zisserman in 2014 [37,38]. According to the literature, VGG-16 offers a considerable improvement over AlexNet in several applications because it is rich with several feature representations that can be used for a wide range of applications. The VGG-16 model architecture is shown in Fig. 3 [39]. It consists of a 16-layer network comprised of convolutional layers.
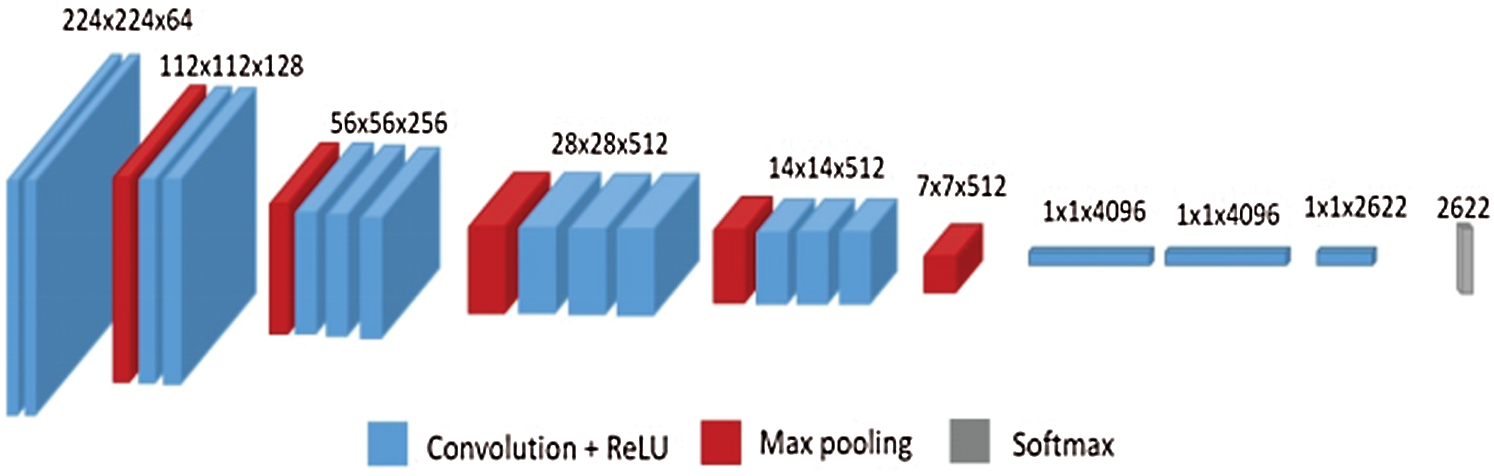
Figure 3: VGG-16 model architecture
The DNA proteins identification process is mainly a binary classification problem between two classes. The first class is the DNABPs that have DNA binding domains and interact with the DNA. The second class is the non-DNABPs, such as the structural proteins within the chromosomes. Several classifiers are suitable for binary classification; the most commonly used classifiers for DNA proteins identification are SVM [8,22,33] and RF [14,16,17,28]. In this paper, the two classifiers were used and compared.
SVM is a set of related supervised-learning models introduced by Cortes et al. [40]. It minimizes the identification error and maximizes the geometric margin. SVMs are the most suitable binary linear identification methods [40–43]. SVM works for two-class problems by separating the data by a separating hyperplane, as shown in Fig. 4.
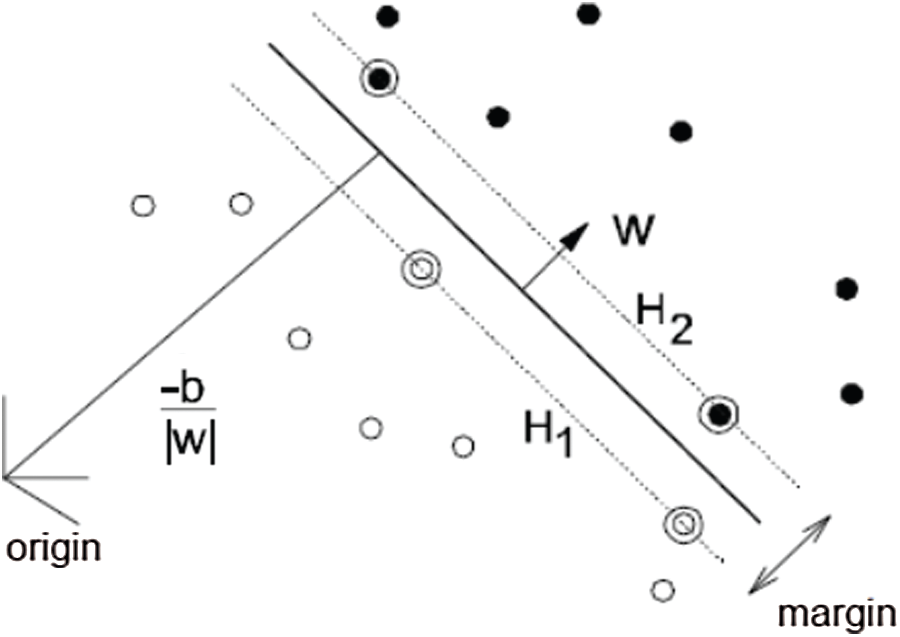
Figure 4: SVM separating hyperplanes
In Fig. 4, consider that the training sequences are represented by {
The primal Lagrange is given as [44]:
where
For minimizing LP with respect to w, b, using the conditions:
Using Eqs. (3)–(5), the dual Lagrangian will be:
The mapping of training vectors xi into the higher dimensional space uses a function called kernel function
Linear kernel:
Polynomial kernel:
RBF kernel:
Sigmoid kernel:
where
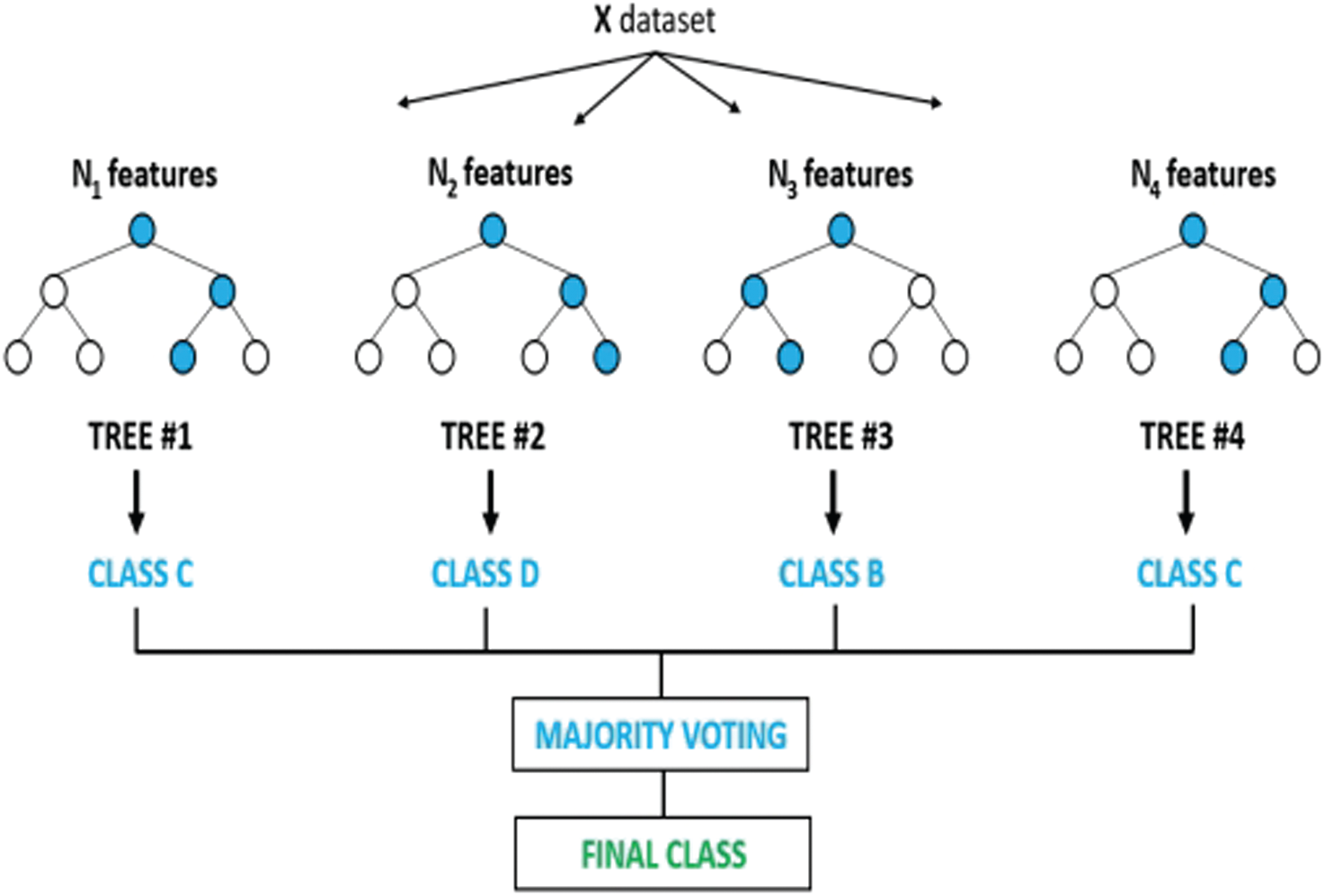
Figure 5: RF algorithm
The DNA protein sequences identification was carried out using the SVM Matlab Toolbox with different kernel functions: linear, polynomial, RBF, and sigmoid kernel. DNABPs identification using SVM can be carried out in two steps. The first step is building the identification simulation model, while the second step is the feature matching for the model performance evaluation. In the modelling step, the features related to the DNA protein sequences are stored. When a tested sequence arrives, its features are matched with the stored features in the model and the identification decision is taken based on the matching process.
RF is a tree collection introduced by Ho [45]; each tree is grown through a subset of all the possible attributes of the input features vectors [46]. It constructs the decision ensemble in random trees based on the input features, and the final identification decision is obtained by combining the results from the trees via voting, as shown in Fig. 5.
3.1 Performance Evaluation Metrics
The performance of the DNA protein sequences identification system is normally evaluated using wide performance metrics, such as identification accuracy, sensitivity, specificity, and Matthew’s correlation coefficient. These metrics can be calculated using four parameters obtained from the testing of the identification system with a certain dataset. The system tests the DNA protein sequences if it is a DNABP (positive sample) or non-DNABP (negative sample). For each DNABP testing, if the test result is positive, this means that the system identifies it as correct (True), and accumulating the positive true results for all the tested protein sequences in the dataset gives the Tp number. If the test result is negative, this means that the system identifies it as incorrect (False) and the accumulation gives the Fn number. For each non-DNABP testing, if the test result is positive, this means that the system identifies it as incorrect (False), and the accumulation gives the Fp number. If the test result is negative, this means that the system identifies it as correct (True) and the accumulation gives the Tn number. Using these four numbers, it is possible to calculate:
1. Accuracy
2. Sensitivity
3. Specificity
4. Matthew’s correlation coefficient
The accuracy, the sensitivity and the specificity are percentages, while the MCC ranges from −1 to +1; the perfect classifier should give 100% for the three first parameters and +1 MCC.
3.2 Deep Transfer Learning Models
This section presents the results of the first DNABPs identification method, which is based on the pre-trained deep transfer learning models as the features extractor and classifier. Two pre-trained deep transfer learning models, AlexNet and VGG-Net, were tested and compared in terms of identification accuracy, sensitivity, specificity, and MCC for the four examined DNA proteins datasets, as shown in Tab. 1.
Table 1: Performance comparison between deep transfer learning models

The results in Tab. 1 show that the VGG-Net 16 pre-trained deep transfer learning model gives higher performance than AlexNet. This may be because the 16-layer VGGnet is deeper than the 8-layer AlexNet, and the VGGnet is rich with several feature representations.
This section presents the results of the second DNABPs identification method, which is based on the pre-trained deep transfer learning models as the features extractor. The classifier is one of the two different classifiers (SVM or RF) used for the identification process. The identification accuracy, sensitivity, specificity, and MCC for the four examined DNA proteins datasets are as shown in Tab. 2.
The results in Tab. 2 show that the RF classifier with VGG-Net pre-trained deep transfer learning features gives the highest performance compared to the other approaches.
3.4 Performance Comparison with Existing Methods
The performance of the proposed DNABPs identification method (DTLM-DBP) was compared with the other published methods for the four available DNA proteins datasets: PDB1075, PDB186, PDNA-543, and PDNA-316 datasets. For PDB1075 dataset, DTLM-DBP was compared with DNAbinder [22], DNA-Prot [16], iDNA-Prot [17], iDNA-Prot-dis [21], PSSM-DT [8], PseDNA-Pro [23], iDNAPro-PseAAC [20], PSFM-DBT [12], Mixed Feature [32], Local-DPP [19], iDNAProt-ES [10], HMMBinder [25], iDNA-KACC [31], and DPP-PseAAC [24], as shown in Tab. 3.
The results in Tab. 3. show that the proposed method gives a better performance than the other published methods.
For PDB186 dataset, DTLM-DBP was compared with DNABIND [26], DNAbinder [22], DNA-Threader [27], DNA-Prot [16], DBPPred [28], iDNA-Prot [17], iDNA-Prot-dis [21], PSSM-DT [8], iDNAPro-PseAAC [20], Mixed Feature [32], PseDNA-Pro [23], iDNAProt-ES [10], PSFM-DBT [12], Local-DPP [19], HMMBinder [25], DPP-PseAAC [24], and iDNA-KACC-EL [31], as shown in Tab. 4. The results show the superiority of the proposed method over the other published methods.
Table 2: Performance comparison between SVM and RF classifiers

Table 3: Comparison of DTLM-DBP with previous methods for PDB 1075 dataset

Table 4: Comparison of DTLM-DBP with previous methods for PDB 186 dataset

For PDNA-543 dataset, DTLM-DBP was compared with TargetDNA [33], EC-RUS [15], and Bootstrap [30], as shown in Tab. 5.
Table 5: Comparison of DTLM-DBP with previous methods for PDNA-543 dataset

For PDNA-316 dataset, DTLM-DBP was compared with MetaDBSite [34], TargetDNA [33], EC-RUS [15], and Bootstrap [30], as shown in Tab. 6.
The results confirmed the efficacy and viability of the proposed method for different datasets.
Table 6: Comparison of DTLM-DBP with previous methods for PDNA-316 dataset

The paper presented an efficient new approach for DNABPs identification based on deep transfer learning “DTLM-DBP.” The protein sequences were adapted to CNN models using 1D convolutions layers, then the VGG-NET 16 pre-trained deep transfer learning models were used as a feature’s extractor. Finally, the RF classifier was used for sequence features matching. DTLM-DBP was tested using different DNA proteins datasets and compared with the other published DNABPs identification methods, and it has provided a considerable improvement in the performance of DNABPs identification.
Acknowledgement: We would like to acknowledge Universiti Teknologi Malaysia for their support via the 2020-2021 Industry-International Incentive Grant which funded this publication.
Funding Statement: This paper was funded under the 2020–2021 Industry-International Incentive Grant by Universiti Teknologi Malaysia (Grant Number: Q.K130000.3043.02M12) which was granted to U. Khairuddin, F. Behrooz and R. Yusof.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. S. Shadab, M. T. A. Khan, N. A. Neezi, S. Adilina and S. Shatabda, “DeepDBP: Deep neural networks for identification of DNA-binding proteins,” Informatics in Medicine Unlocked, vol. 19, no. 2, pp. 100318, 2020. [Google Scholar]
2. C. Tan, T. Wang, W. Yang and L. Deng, “PredPSD: A gradient tree boosting approach for single-stranded and double-stranded DNA binding protein prediction,” Molecules, vol. 25, no. 1, pp. 1–16, 2020. [Google Scholar]
3. J. Qiu, M. Bernhofer, M. Heinzinger, S. Kemper, T. Norambuena et al., “ProNA2020 predicts protein-DNA, protein-RNA, and protein–protein binding proteins and residues from sequence,” Journal of Molecular Biology, vol. 432, no. 7, pp. 2428–2443, 2020. [Google Scholar]
4. J. Zhang, Q. Chen and B. Liu, “DeepDRBP-2L: A new genome annotation predictor for identifying DNA binding proteins and RNA binding proteins using convolutional neural network and long short-term memory,” IEEE/ACM Transactions on Computational Biology and Bioinformatics, 2019. [Google Scholar]
5. A. Trabelsi, M. Chaabane and A. Ben-Hur, “Comprehensive evaluation of deep learning architectures for prediction of DNA/RNA sequence binding specificities,” Bioinformatics, vol. 35, no. 14, pp. 269–277, 2019. [Google Scholar]
6. Z. Zhao, Y. Xu and Y. Zhao, “SXGBsite: Prediction of protein-ligand binding sites using sequence information and extreme gradient boosting,” Genes, vol. 10, pp. 9651–96519, 2019. [Google Scholar]
7. H. J. Zhu, Z. H. You, W. L. Shi, S. K. Xu, T. H. Jiang et al., “Improved prediction of protein–protein interactions using descriptors derived from PSSM via gray level co-occurrence matrix,” IEEE Access, vol. 7, pp. 49456–49465, 2019. [Google Scholar]
8. R. Xu, J. Zhou, H. Wang, Y. He, X. Wang et al., “Identifying DNA-binding proteins by combining support vector machine and PSSM distance transformation,” BMC Systems Biology, vol. 9, no. Suppl. 1, pp. S10, 2015. [Google Scholar]
9. M. Waris, K. Ahmad, M. Kabirand and M. Hayat, “Identification of DNA binding proteins using evolutionary profiles position specific scoring matrix,” Neurocomputing, vol. 199, no. 1, pp. 154–162, 2016. [Google Scholar]
10. S. Y. Chowdhury, S. Shatabda and A. Dehzangi, “iDNAProt-ES: Identification of DNA-binding proteins using evolutionary and structural features,” Scientific Reports, vol. 7, pp. 1–14, 2017. [Google Scholar]
11. R. Xuab, J. Zhoua, B. Liu, Y. Hee, Q. Zouf et al., “Identification of DNA-binding proteins by incorporating evolutionary information into pseudo amino acid composition via the top-n-gram approach,” Journal of Biomolecular Structure and Dynamics, vol. 33, no. 8, pp. 1720–1730, 2015. [Google Scholar]
12. J. Zhang and B. Liu, “PSFM-DBT: Identifying DNA-binding proteins by combing position specific frequency matrix and distance-bigram transformation,” International Journal of Molecular Sciences, vol. 18, pp. 1–16, 2017. [Google Scholar]
13. J. Zhang, B. Gao, H. Chai, Z. Ma and G. Yang, “Identification of DNA-binding proteins using multi-features fusion and binary firefly optimization algorithm,” BMC Bioinformatics, vol. 17, no. 1, pp. 323, 2016. [Google Scholar]
14. X. Ma, J. Guo and X. Sun, “DNABP: Identification of DNA-binding proteins based on feature selection using a random forest and predicting binding residues,” PLOS ONE, vol. 11, no. 12, pp. e0167345, 2016. [Google Scholar]
15. C. Shen, Y. Ding, J. Tang, J. Song and F. Guo, “Identification of DNA-protein binding sites through multi-scale local average blocks on sequence information,” Molecules, vol. 22, pp. 1–20, 2017. [Google Scholar]
16. K. Kumar, G. Pugalenthi and P. N. Suganthan, “DNA-Prot: Identification of DNA binding proteins from protein sequence information using random forest,” Journal of Biomolecular Structure & Dynamics, vol. 26, no. 6, pp. 679–686, 2009. [Google Scholar]
17. W. Z. Lin, J. A. Fang, X. Xiao and K. C. Chou, “iDNA-Prot: Identification of DNA binding proteins using random forest with grey model,” PloS One, vol. 6, no. 9, pp. e24756, 2011. [Google Scholar]
18. X. Z. Fu, W. Zhu, B. Liao, L. Cai, L. Peng et al., “Improved DNA-binding protein identification by incorporating evolutionary information into the Chou’s PseAAC,” IEEE Access, vol. 6, pp. 66545–66556, 2018. [Google Scholar]
19. L. Wei, J. Tang and Q. Zou, “Local-DPP: An improved DNA-binding protein prediction method by exploring local evolutionary information,” Information Sciences, vol. 384, pp. 135–144, 2017. [Google Scholar]
20. B. Liu, S. Wang and X. Wang, “DNA binding protein identification by combining pseudo amino acid composition and profile-based protein representation,” Scientific Reports, vol. 5, no. 1, pp. 15479, 2015. [Google Scholar]
21. B. Liu, J. Xu, X. Lan, R. Xu, J. Zhou et al., “iDNA-Prot—dis: Identifying dna-binding proteins by incorporating amino acid distance-pairs and reduced alphabet profile into the general pseudo amino acid composition,” PloS One, vol. 9, no. 9, pp. e106691, 2014. [Google Scholar]
22. M. Kumar, M. M. Gromiha and G. P. Raghava, “Identification of DNA-binding proteins using support vector machines and evolutionary profiles,” BMC Bioinformatics, vol. 8, no. 1, pp. 463, 2007. [Google Scholar]
23. B. Liu, J. Xu, S. Fan, R. Xu, J. Zhou et al., “PSEDNA-pro: DNA-binding protein identification by combining chou’s pseaac and physicochemical distance transformation,” Molecular Informatics, vol. 34, no. 1, pp. 8–17, 2015. [Google Scholar]
24. M. S. Rahman, S. Shatabda, S. Saha, M. Kaykobad and M. S. Rahman, “DPP-PSEAAC: A DNA binding protein prediction model using chou’s general pseaac,” Journal of Theoretical Biology, vol. 452, no. 1, pp. 22–34, 2018. [Google Scholar]
25. R. Zaman, S. Y. Chowdhury, M. A. Rashid, A. Sharma, A. Dehzangi et al., “DNA-binding protein prediction using hmm profile-based features,” BioMed Research International, vol. 2017, pp. 1–10, 2017. [Google Scholar]
26. A. Szilágyi and J. Skolnick, “Efficient prediction of nucleic acid binding function from low-resolution protein structures,” Journal of Molecular Biology, vol. 358, no. 3, pp. 922–933, 2006. [Google Scholar]
27. M. Gao and J. Skolnick, “A threading-based method for the prediction of DNA binding proteins with application to the human genome,” PLOS Computational Biology, vol. 5, no. 1, pp. e1000567, 2009. [Google Scholar]
28. W. C. Lou, X. Q. Wang, F. Chen, Y. X. Chen, J. Bo et al., “Sequence based prediction of DNA-binding proteins based on hybrid feature selection using random forest and gaussian naïve bayes,” PLos One, vol. 9, no. 1, pp. e86703, 2014. [Google Scholar]
29. Y. Zhang, S. Qiaoc, S. Jia, N. Hand, D. Liuc et al., “Identification of DNA-protein binding sites by bootstrap multiple convolutional neural networks on sequence information,” Engineering Applications of Artificial Intelligence, vol. 79, no. 4, pp. 58–66, 2019. [Google Scholar]
30. Y. Zhang, S. Qiao, S. Ji and Y. Li, “DeepSite: Bidirectional LSTM and CNN models for predicting DNA-protein binding,” International Journal of Machine Learning and Cybernetics, vol. 11, no. 4, pp. 841–851, 2020. [Google Scholar]
31. B. Liu, S. Wang, Q. Dong, S. Li and X. Liu, “Identification of DNA-binding proteins by combining auto-cross covariance transformation and ensemble learning,” IEEE Transactions on NanoBioscience, vol. 15, no. 4, pp. 328–334, 2016. [Google Scholar]
32. K. Qu, K. Han, S. Wu, G. Wang and L. Wei, “Identification of DNA-binding proteins using mixed feature representation methods,” Molecules, vol. 22, no. 10, pp. 1602, 2017. [Google Scholar]
33. J. Hu, Y. Li, M. Zhang, X. Yang, H. B. Shen et al., “Predicting protein-DNA binding residues by weightedly combining sequence-based features and boosting multiple SVMs,” IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 14, no. 6, pp. 1389–1398, 2017. [Google Scholar]
34. J. Si, Z. Zhang, B. Lin and B. Huang, “MetaDBSite: A meta approach to improve protein DNA-binding sites prediction,” BMC Systems Biology, vol. 5, no. Suppl 1, pp. S7, 2011. [Google Scholar]
35. A. Krizhevsky, I. Sutskever and G. E. Hinton, “Magenet classification with deep convolutional neural networks,” in Advances in Neural Information Processing Systems. New York, NY, USA: Curran Associates, Inc., pp. 1097–1105, 2012. [Google Scholar]
36. S. H. Wang, S. Xie, X. Chen, D. S. Guttery, C. Tang et al., “Alcoholism identification based on an AlexNet transfer learning model,” Front Psychiatry, vol. 10, pp. 205, 2019. [Google Scholar]
37. K. Simonyan and A. Zisserman, “Very deep convolutional networks for large-scale image recognition,” in Proc. ICLR, San Diego, CA, USA, pp. 1–14, 2015. [Google Scholar]
38. S. Khatoon, M. M. Hasan, A. Asif, M. Alshmari and Y. K. Yap, “Image-based automatic diagnostic system for tomato plants using deep learning,” Computers, Materials & Continua, vol. 67, no. 1, pp. 595–612, 2021. [Google Scholar]
39. T. Y. Heo, K. M. Kim, H. K. Min, S. M. Gu, J. H. Kim et al., “Development of a deep-learning-based artificial intelligence tool for differential diagnosis between dry and neovascular age-related macular degeneration,” Diagnostics, vol. 10, no. 5, pp. 1–10, 2020. [Google Scholar]
40. C. Cortes and V. Vapnik, “Support-vector networks,” Machine Leaming, vol. 20, pp. 273–297, 1995. [Google Scholar]
41. H. Kasban, “Fingerprint’s verification based on their spectrum,” Neurocomputing, vol. 171, no. 1, pp. 910–920, 2016. [Google Scholar]
42. A. Abozaid, A. Haggag, H. Kasban and M. Eltokhy, “Multimodal biometric scheme for human authentication technique based on voice and face recognition fusion,” Multimedia Tools and Applications, vol. 78, no. 12, pp. 16345–16361, 2019. [Google Scholar]
43. J. S. H. Al-bayatiand and B. B. Üstündağ, “Fused and modified evolutionary optimization of multiple intelligent systems using ANN, SVM approaches,” Computers, Materials & Continua, vol. 66, no. 2, pp. 1479–1496, 2021. [Google Scholar]
44. V. N. Vapnik, Statistical Learning Theory. New York, USA: Wiley, 1998. [Google Scholar]
45. T. K. Ho, “Random decision forests,” in Proc. ICDAR, NW Washington, DC, USA, pp. 278–282, 1995. [Google Scholar]
46. P. Calhoun, M. J. Hallett, X. Su, G. Cafri, R. A. Levine et al., “Random forest with acceptance-rejection trees,” Computational Statistics, vol. 35, no. 3, pp. 983–999, 2020. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |