
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

DNA Methylation Variation Is Identified in Monozygotic Twins Discordant for Congenital Heart Diseases
1 Guangdong Provincial People’s Hospital, Southern Medical University, Guangzhou, 510100, China
2 Department of Cardiovascular Surgery, Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangdong Provincial Clinical Research Center for Child Health, Guangzhou, 510623, China
3 Department of Pediatric Cardiology, Guangdong Cardiovascular Institute, Guangdong Provincial Key Laboratory of South China Structural Heart Disease, Guangzhou, 510100, China
4 Clinical Physiology Laboratory, Institute of Pediatrics, Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangdong Provincial Key Laboratory of Research in Structural Birth Defect Disease, Guangzhou, 510623, China
5 Department of Echocardiogram, Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangdong Provincial Clinical Research Center for Child Health, Guangzhou, 510623, China
6 Department of Pediatrics, Washington University School of Medicine, St. Louis, MO, 63130, USA
7 Clinical Biological Resource Bank of Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangzhou, 510623, China
* Corresponding Authors: Jia Li. Email: ; Zhiwei Zhang. Email:
Congenital Heart Disease 2024, 19(2), 247-256. https://doi.org/10.32604/chd.2024.052583
Received 07 April 2024; Accepted 28 April 2024; Issue published 16 May 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Aims: Multiple genes and environmental factors are known to be involved in congenital heart disease (CHD), but epigenetic variation has received little attention. Monozygotic (MZ) twins with CHD provide a unique model for exploring this phenomenon. In order to investigate the potential role of Deoxyribonucleic Acid (DNA) methylation in CHD pathogenesis, the present study examined DNA methylation variation in MZ twins discordant for CHD, especially ventricular septal defect (VSD). Methods and Results: Using genome-wide DNA methylation profiles, we identified 4004 differentially methylated regions (DMRs) in 18 MZ twin pairs discordant for CHD, and 2826 genes were identified. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis revealed a list of CHD-associated pathways. To further investigate the role of DNA methylation in VSD, data from 7 pairs of MZ twins with VSD were analyzed. We identified 1614 DMRs corresponding to 1443 genes associated with arrhythmogenic right ventricular cardiomyopathy, cyclic guanosine monophosphate-protein kinase G (cGMP-PKG) signaling pathway by KEGG analysis, and cell-cell adhesion, calcium ion transmembrane transport by GO analysis. A proportion of DMR-associated genes were involved in calcium signaling pathways. The methylation changes of calcium signaling genes might be related to VSD pathogenesis. Conclusion: CHD is associated with differential DNA methylation in MZ twins. CHD may be etiologically linked to DNA methylation, and methylation of calcium signaling genes may be involved in the development of VSD.Graphic Abstract
Keywords
Congenital heart diseases (CHD) affecting approximately 0.8% of live infants are among the most common birth defects [1]. Despite significant advances in treatment, CHD remains a leading cause of morbidity and mortality in infants and children. There are numerous genes known to be related to cardiac development, and the most common form of CHD is ventricular septal defect (VSD) [2,3] the etiology and pathological mechanisms remain mostly unknown.
DNA methylation is a common epigenetic modification, largely occurring at the carbon-5 position of CpG dinucleotides (5 mC) [4]. Through the activity of DNA methyltransferases (DNMTs), DNA methylation is mitotically heritable and important for gene expression control. Differential methylation was localized to promoter-associated GpG islands, which are established regulatory regions of downstream genes [5]. Abnormal patterns of DNA methylation are linked to various human diseases [6–9], including CHD [10,11]. Previous studies have shown that NOX5, PRDM16, and NDRG2 are regulated by DNA hypermethylation or hypomethylation in VSD [12–14]. Maternal dietary intake of folic acid also affects the methylation status of VSD-associated genes [15], which demonstrates that VSD pathogenesis may be influenced by DNA methylation.
Monozygotic (MZ) twins share nearly identical genetic material but can differ phenotypically. In recent years, epigenetic studies have become more common, epigenetic modification has been known as the main cause of individual differences in MZ twins [16–18]. A variety of congenital defects have been detected in MZ twin pairs with discordant DNA methylation profiles, including congenital cataracts, congenital renal agenesis, and non-syndromic cleft lip [19–21]. In previous studies, MZ twins have been found to have different DNA methylation patterns, and these differences may be associated with phenotypic discordance [22–24].
The role of DNA methylation in MZ discordant pairs for CHD remains less well understood. In the present study, we investigated DNA methylation variations in MZ twins who were discordant for CHD, particularly for VSD. Our study identified differentially methylated regions (DMRs) associated with CHD status using genome-wide methylation profiling. We further investigated the functional significance of these DMRs and their potential role in CHD pathogenesis.
2.1 Ethics Statement and Samples
The Ethics Committee of the Guangzhou Women and Children Medical Center approved this study (Ethical Review Number [2021] No. 260B00). The samples were taken from monozygotic twin children at the Guangzhou Women and Children’s Medical Center between 2020 and 2023, with informed consent obtained from their parents. One of the twins was diagnosed with CHD by echocardiography, and the other as control with no heart malformations. Peripheral blood samples were collected from each twin using standard venipuncture techniques. The collected whole blood was centrifuged to obtain a cellular precipitate and the samples were stored at −80°C until DNA extraction.
2.2 DNA Extraction and Bisulfite Treatment
Blood and Tissue DNA Kit (Tiangen Biochemical, DP705) was used to extract genomic DNA from cellular precipitates. Nanodrop and agarose gel electrophoresis were used to determine DNA concentration and purity 1.5 μg of genomic DNA was bisulfite treated in accordance with the manufacturer’s instructions using Zymo Research’s EZ DNA methylation kit (Irvine, USA). Polymerase Chain Reaction (PCR) products without methyl groups were used to control the quality of the bisulfite conversion. PCR amplification was carried out to obtain the final DNA library. After the library construction is completed, use Qubit for preliminary quantification and dilute the concentration of library construction to 1 ng/μl. Then, detect the insertion fragment length of the library by Agilent 5400 (Agilent Technologies Inc., California, USA), and after meeting expectations, and library is subsequently sequenced in the Illumina Novaseq platform instrument (Illumina Corporation, San Diego, USA).
2.3 Reduced Representation Bisulfite Sequencing (RRBS) Data Analysis
We aligned RRBS sequencing reads against the UCSC Genome Browser (University of California Santa Cruz, Santa Cruz, CA, USA) mm10 reference genome, classified methylation status using Bismark (Babraham Institute, Cambridge CB22 3AT, UK), and identified differently methylated regions (DMRs) using Fisher’s exact test implemented in the DSS r package. The criteria for selecting DMRs are as follows: The methylation level difference between patients and controls should be at least 0.2. The q-value (adjusted p-value) should be ≤0.05. On the DAVID bioinformatics server (https://david.ncifcrf.gov), DMRs were mapped to genes based on their proximity to promoter regions. GO and KEGG pathway analyses were conducted.
GraphPad Prism 5.0 (GraphPad Software Inc., San Diego, USA) was used for all non-bioinformatic statistical analyses. If the samples followed a normal distribution, either the t-test or one-way ANOVA (Analysis of Variance) was used. However, if the variances were not equal, or if a certain sample did not follow a normal distribution, Statistical analyses were performed using the Kruskal-Wallis rank sum test. p < 0.05 and p < 0.01, respectively, was taken as significance. For bioinformatics analysis, Benjamin’s correction procedure used FDR (False Discovery Rate) p < 0.05. Only statistically significant gene sets were used for GO analysis and KEGG analysis.
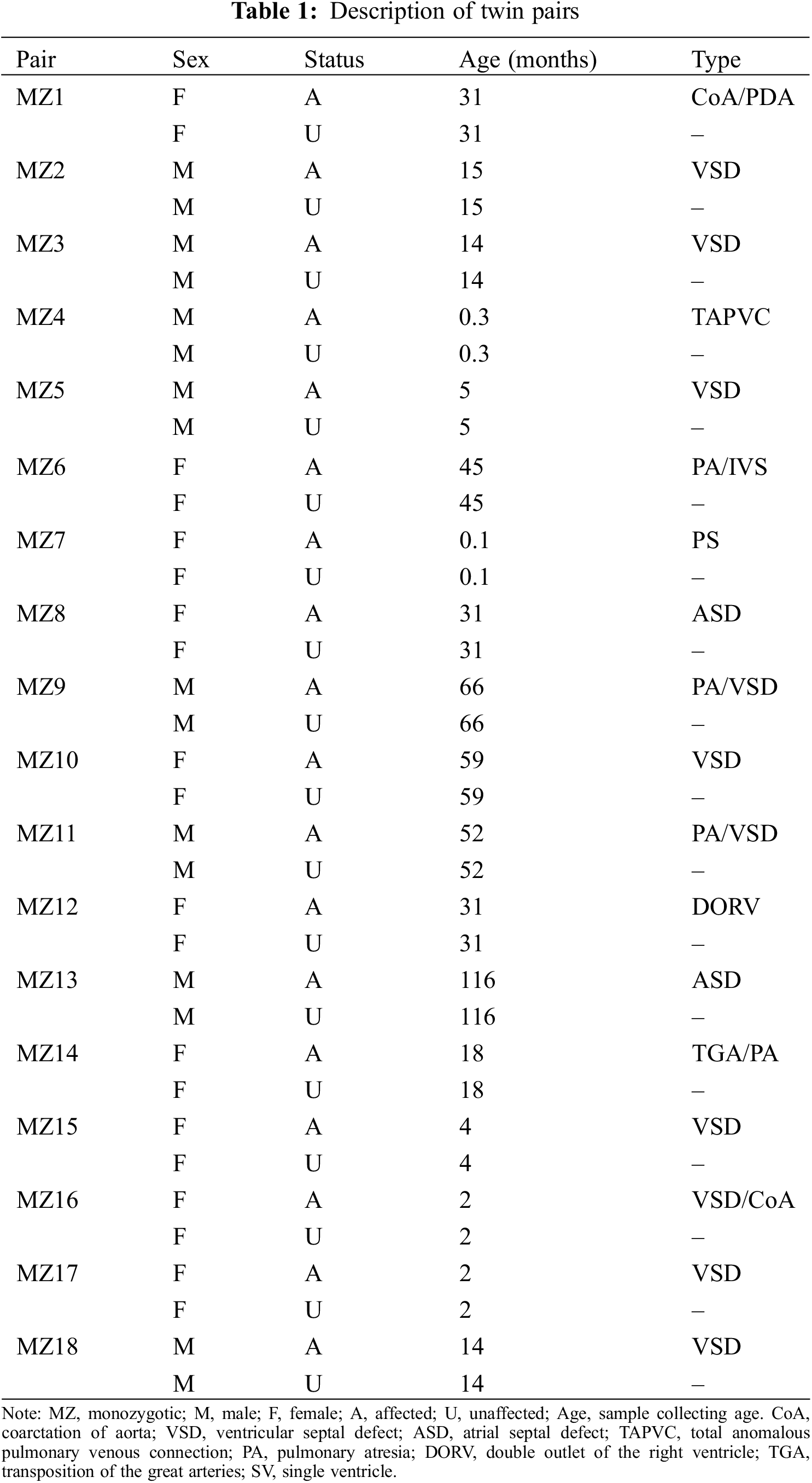
4.1 Characteristic of Monozygotic Twin Pairs
Table 1 shows the characteristics of 18 CHD-discordant monozygotic twin pairs (18 CHD cases and 18 unaffected siblings) that were DNA methylated. The types of CHD include coarctation of aorta (CoA), ventricular septal defect (VSD), atrial septal defect (ASD), total anomalous pulmonary venous connection (TAPVC), pulmonary atresia (PA), double outlet of the right ventricle (DORV), transposition of the great arteries (TGA) and single ventricle (SV).
4.2 DNA Methylation Profiles of CHD
Based on RRBS, DNA methylation profiles were highly correlated among individuals, indicating that the DNA methylation levels of individuals were conserved. Co-twins showed a higher correlation between their methylation patterns than unrelated individuals, indicating low within-pair variation in methylation (Fig. 1A, r for twins = 0.999, and r for unrelated individuals = 0.97). We analyzed the methylation levels of gene regions across various samples by heat-map and Methylation levels of gene regions were not significantly different across individuals (Fig. 1B).
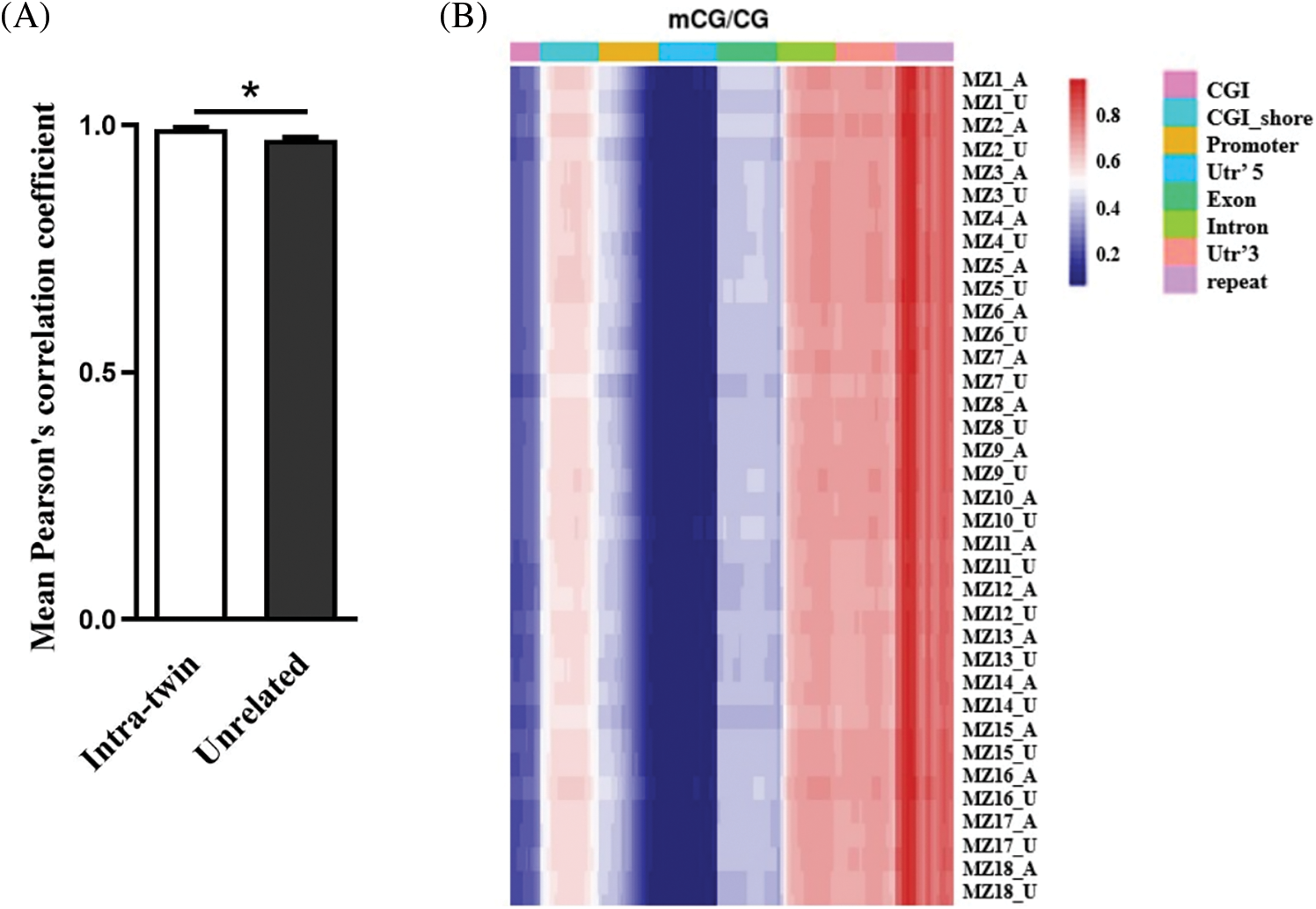
Figure 1: MZ twins for CHD are highly correlated with DNA methylation. (A) Pearson’s correlation coefficients comparing intra-twin and unrelated methylation levels, *p < 0.05. (B) Heat-map of methylation levels of gene region
4.3 Differentially Methylated Regions (DMRs) of CHD
Based on systematic regional analyses, we identified DMRs in 18 MZ CHD discordant twin pairs ranging in number from 138401 in MZ13 to 188746 in MZ4 (Fig. 2A). Introns and intergenic regions of gene bodies were highly enriched with DMRs, but the overall distribution pattern between groups did not differ significantly. To further investigate the relationship between DNA methylation and CHD, we identified a total of 4004 DMRs for all groups, the DMRs were annotated to a total of 2826 genes. GO analysis of DMR-associated genes identified terms such as actin cytoskeleton organization, regulation of small GTPase-mediated signal transduction, and modulation of synaptic transmission (Fig. 2B). KEGG analysis showed that pathways including axon guidance, efferocytosis, and adherents junction were related to DMRs-associated genes (Fig. 2C).
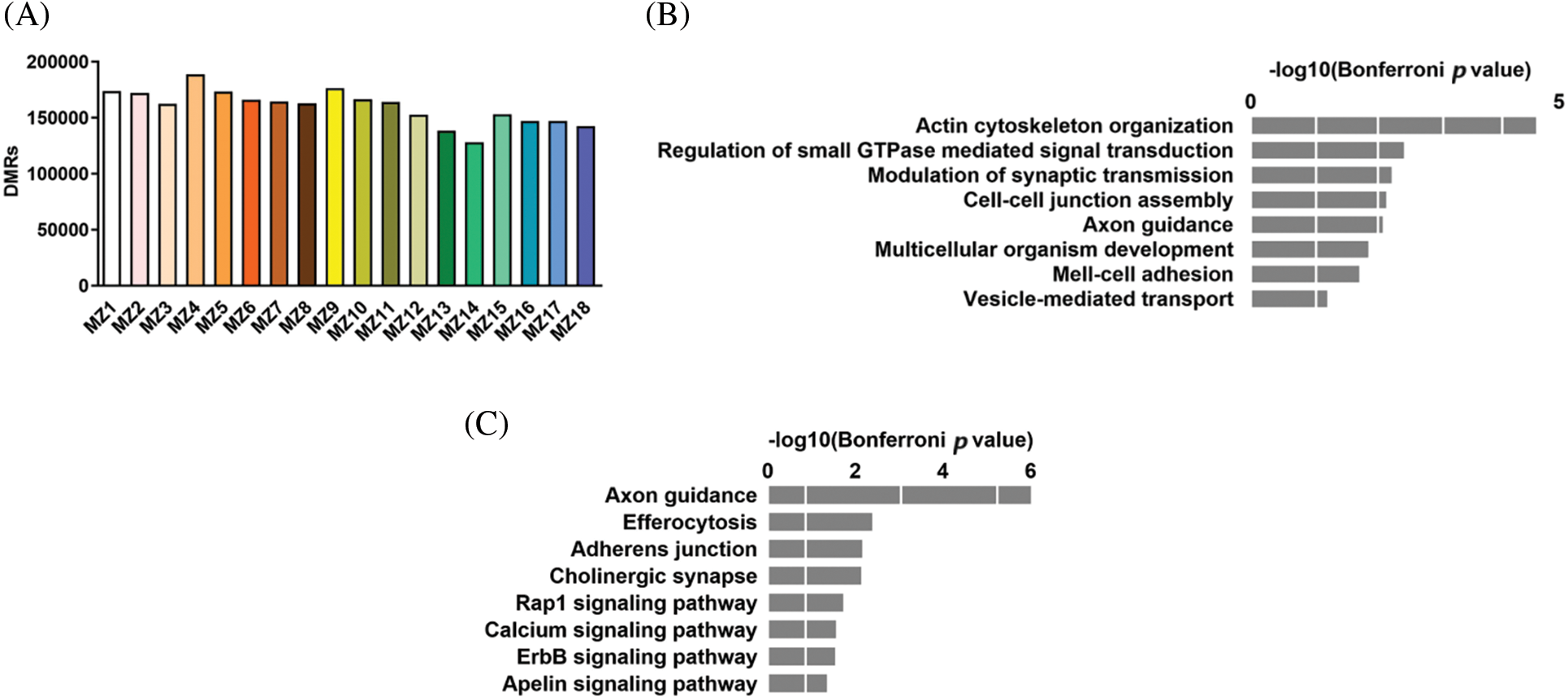
Figure 2: Differentially methylated regions (DMRs) in the MZ CHD discordant twins. (A) Number of DMRs in MZ discordant twins. (B) GO analysis of DMRs associated genes. (C) KEGG analysis of DMRs associated genes
4.4 Differentially Methylated Regions (DMRs) of VSD
To understand the role of DNA methylation in different types of CHD, we first analyzed DMRs in 7 pairs of MZ twins with VSD. VSD was diagnosed by echocardiography without other complex cardiac malformations. We identified a total of 1614 DMRs for all groups, the DMRs were annotated to a total of 1443 genes. The identified DMRs were highly enriched in introns and intergenic regions. Fig. 3A shows that DMR-associated genes were highly related to the binding of calcium ions to membranes, cell-cell adhesion, and establishment of cell polarity. KEGG analysis showed that pathways including arrhythmogenic right ventricular cardiomyopathy, cGMP-PKG signaling pathway, cellular senescence, and Rap1 signaling pathway were related to DMRs-associated genes (Fig. 3B).
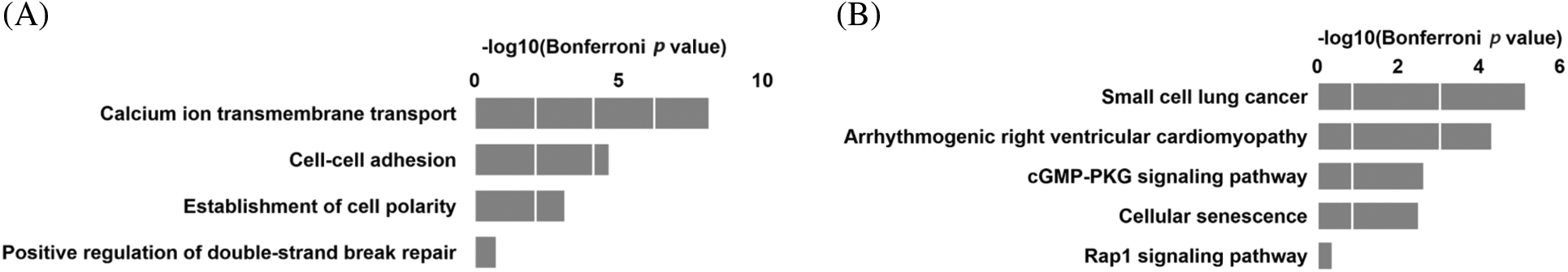
Figure 3: Differentially methylated regions (DMRs) in the VSD discordant twins. (A) GO analysis of DMRs associated genes in the VSD discordant twins. (B) KEGG analysis of DMRs associated genes in the VSD discordant twins
The MZ twin study provides a unique opportunity for studying epigenetic variation. The advantage of using MZ twin samples in epigenetic studies was that MZ twins have an almost identical genetic makeup, sharing 100% of their DNA. This genetic similarity allows researchers to study the effects of environmental factors on gene expression and epigenetic modifications without confounding genetic differences. According to the MZ twin discordance, environmental and epigenetic factors play an important role in the etiology of the disorder. Epigenetic modifications contribute to various biological processes and diseases [25,26]. Cardiac development and function are under the regulation of complicated mechanisms including epigenetic modifications [27–29], and aberrant alteration of epigenetics can induce heart diseases such as CHD [30]. DNA methylation of cardiomyocytes is dynamic during development and disease, which plays a role in cardiomyocyte maturation and adaptation [31]. Our study demonstrates that phenotypic discordance may be related to differences in DNA methylation in MZ twins that were discordant for CHD.
Here, CHD is not associated with global changes in DNA methylation status, as shown by the similar average genomic methylation levels between CHD and non-CHD individuals. The dynamic changes in DNA methylation profiles during individual development remain relatively stable [32]. However, DNA methylation changes at individual gene loci can indeed occur, and these changes result in altered expression of genes with important biological functions and disease [33,34]. Different types of CHD have different levels of methylation of imprinted genes. The methylation of imprinted genes is associated with CHD [35].
We found that DMRs associated genes were significantly enriched for AMPK signaling pathway, efferocytosis, and axon guidance. And GO terms directly related to actin cytoskeleton organization, regulation of small GTPase mediated signal transduction, and modulation of synaptic transduction. Previous studies have shown that axon guidance and adherents junction are involved in the development of cardiovascular disease [36,37]. However, further experiments are needed to verify how methylation changes in DMRs related genes lead to congenital occurrence.
The most common form of CHD is VSD [2,3], and epigenetic factors play an important role in this condition. To further investigate the role of DNA methylation in VSD, data from MZ twin pairs with VSD were analyzed. DMRs associated genes were highly related to arrhythmogenic right ventricular cardiomyopathy and cGMP-PKG signaling pathway. Calcium signaling pathways were involved in a majority of DMR-associated genes, such as ANXA2, ATP2A3, and CACNA2D3. Calcium signaling pathways play a key physiological and pathological role in heart function [38]. Calcium-handling genes are important for cardiomyocyte contraction, and mutations in these genes can lead to arrhythmias [39]. In cardiomyocytes, calcium also modulates transcription through a complex signaling network [40]. In animal studies, calcium signaling was vital for heart development. Disruption of NFATC1 caused embryonic lethality and ventricular septal malformation in mice [41]. Calreticulin-deficient mice were rescued from developmental defects by overexpressing activated calcineurin [42]. In humans, mutations in calcium-signaling genes were linked to CHD patients [43]. We speculate methylation changes of calcium signaling genes may related to VSD occurrence, but further experiments are needed to verify.
As a study, we included 18 pairs of twins with different affection statuses and phenotypes, but given the complexity of CHDs, this is not a significant limitation. We did not further analyze the DNA methylation profiles in other CHD except VSD. In future experiments, we will continue to increase the number of cases and analyze the DNA methylation profiles of different CHD. Furthermore, blood DNA methylation profiles were obtained due to the difficulty in obtaining heart samples, and epigenetic markers can vary according to cell type. Blood DNA has significantly enriched pathways associated with the immune response, whereas heart tissue DNA relates to muscle contractions and cardiomyopathies. It demonstrates that tissues differ in their methylation levels. The findings provided evidence that DNA methylation, at a global and very distinct level, plays a pivotal role in CHD, despite its limitations.
A difference in DNA methylation has been found among MZ twins discordant with CHD. DNA methylation may play an important role in the etiology of CHD, and methylation of calcium signaling genes may contribute to VSD development. Our findings suggest that environmental and stochastic factors may play a role in the etiology of epigenetic differences and the subsequent discordant CHD phenotypes. It is therefore imperative that further research be undertaken into the possible variations occurring in twin pregnancies that might impart these epigenetic changes between otherwise genetically identical twins.
Acknowledgement: We thank all the researchers in the group for their participation in the study, as well as Ms. Liu Xiaoyun from Sun Yat-sen University Cancer Hospital and Dr. Wang Fei from Guangzhou Women and Children’s Medical Centre for their contribution in the bioinformatic analysis.
Funding Statement: China’s National Natural Science Foundation provided funding for this study (81900222), Guangzhou Science and Technology Program (SL2022A04J01269, 202201020646), and Guangzhou Health Science and Technology Program (20211A010026).
Author Contributions: The authors confirm contribution to the paper as follows: conceptualization, data curation, investigation, and writing—review and editing: Shuliang Xia; conceptualization, data curation, and writing—review and editing: Huikang Tao; data curation and investigation: Shixin Su, Jinqing Feng; conceptualization, data curation, investigation: Xinxin Chen; formal analysis and investigation: Li Ma, Fengxiang Li; data curation and formal analysis: Bei Gao; ultrasound diagnosis and evaluation: Jianru Li, Xumei Liu; data curation: Lei Pi. conceptualization, data curation, investigation, supervision: Jia Li, Zhiwei Zhang. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding authors. Because of the difficulty in collecting cases of phenotypically discordant identical twins, this study will continue and the raw data cannot be made public at this time.
Ethics Approval: A review and approval of the research involving human subjects was obtained from Guangzhou Women and Children’s Medical Center (Ethical Review Number: [2021] No. 260B00). The participant’s legal guardian or next of kin provided written informed consent to the publication of any potentially identifiable images or data.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Stallings EB, Isenburg JL, Aggarwal D, Lupo PJ, Oster ME, Shephard H, et al. Prevalence of critical congenital heart defects and selected co-occurring congenital anomalies, 2014–2018: a U.S. population-based study. Birth Defects Res. 2022;114(2):45–56. doi:10.1002/bdr2.v114.2. [Google Scholar] [CrossRef]
2. Nayak S, Patel A, Haddad L, Kanakriyeh M, Varadarajan P. Echocardiographic evaluation of ventricular septal defects. Echocardiogr. 2020;37(12):2185–93. doi:10.1111/echo.v37.12. [Google Scholar] [CrossRef]
3. Zhao L, Chen L, Yang T, Wang T, Zhang S, Chen L, et al. Birth prevalence of congenital heart disease in China, 1980–2019: a systematic review and meta-analysis of 617 studies. Eur J Epidemiol. 2020;35(7):631–42. doi:10.1007/s10654-020-00653-0. [Google Scholar] [PubMed] [CrossRef]
4. Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16(1):6–21. doi:10.1101/gad.947102. [Google Scholar] [PubMed] [CrossRef]
5. Pepin ME, Drakos S, Ha CM, Tristani-Firouzi M, Selzman CH, Fang JC, et al. DNA methylation reprograms cardiac metabolic gene expression in end-stage human heart failure. Am J Physiol Heart Circ Physiol. 2019;317(4):H674–84. doi:10.1152/ajpheart.00016.2019. [Google Scholar] [PubMed] [CrossRef]
6. Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43(8):768–75. doi:10.1038/ng.865. [Google Scholar] [PubMed] [CrossRef]
7. Esteller M, Fraga MF, Guo M, Garcia-Foncillas J, Hedenfalk I, Godwin AK, et al. DNA methylation patterns in hereditary human cancers mimic sporadic tumorigenesis. Hum Mol Genet. 2001;10(26):3001–7. doi:10.1093/hmg/10.26.3001. [Google Scholar] [PubMed] [CrossRef]
8. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3(6):415–28. doi:10.1038/nrg816. [Google Scholar] [PubMed] [CrossRef]
9. Younesian S, Yousefi AM, Momeny M, Ghaffari SH, Bashash D. The DNA methylation in neurological diseases. Cells. 2022;11(21):3439. doi:10.3390/cells11213439. [Google Scholar] [PubMed] [CrossRef]
10. Joshi RO, Kukshal P, Chellappan S, Guhathakurta S. The study of expression levels of DNA methylation regulators in patients affected with congenital heart defects (CHDs). Birth Defects Res. 2022;114(7):228–37. doi:10.1002/bdr2.v114.7. [Google Scholar] [CrossRef]
11. Serra-Juhé C, Cuscó I, Homs A, Flores R, Torán N, Pérez-Jurado LA. DNA methylation abnormalities in congenital heart disease. Epigenetics. 2015;10(2):167–77. doi:10.1080/15592294.2014.998536. [Google Scholar] [PubMed] [CrossRef]
12. Wijnands KP, Chen J, Liang L, Verbiest MM, Lin X, Helbing WA, et al. Genome-wide methylation analysis identifies novel CpG loci for perimembranous ventricular septal defects in human. Epigenomics. 2017;9(3):241–51. doi:10.2217/epi-2016-0093. [Google Scholar] [PubMed] [CrossRef]
13. Zhu C, Yu ZB, Chen XH, Ji CB, Qian LM, Han SP. DNA hypermethylation of the NOX5 gene in fetal ventricular septal defect. Exp Ther Med. 2011;2(5):1011–5. doi:10.3892/etm.2011.294. [Google Scholar] [PubMed] [CrossRef]
14. Zhu C, Yu ZB, Chen XH, Pan Y, Dong XY, Qian LM, et al. Screening for differential methylation status in fetal myocardial tissue samples with ventricular septal defects by promoter methylation microarrays. Mol Med Rep. 2011;4(1):137–43. [Google Scholar] [PubMed]
15. González-Peña SM, Calvo-Anguiano G, Martínez-de-Villarreal LE, Ancer-Rodríguez PR, Lugo-Trampe JJ, Saldivar-Rodríguez D, et al. Maternal folic acid intake and methylation status of genes associated with ventricular septal defects in children: case-control study. Nutrients. 2021;13(6):2071. doi:10.3390/nu13062071. [Google Scholar] [PubMed] [CrossRef]
16. Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102(30):10604–9. doi:10.1073/pnas.0500398102. [Google Scholar] [PubMed] [CrossRef]
17. Kaminsky ZA, Tang T, Wang SC, Ptak C, Oh GH, Wong AH, et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet. 2009;41(2):240–5. doi:10.1038/ng.286. [Google Scholar] [PubMed] [CrossRef]
18. Tan Q, Christiansen L, Von Bornemann Hjelmborg J, Christensen K. Twin methodology in epigenetic studies. J Exp Biol. 2015;218(1):134–9. doi:10.1242/jeb.107151. [Google Scholar] [PubMed] [CrossRef]
19. Jin M, Zhu S, Hu P, Liu D, Li Q, Li Z, et al. Genomic and epigenomic analyses of monozygotic twins discordant for congenital renal agenesis. Am J Kidney Dis. 2014;64(1):119–22. doi:10.1053/j.ajkd.2014.01.423. [Google Scholar] [PubMed] [CrossRef]
20. Wei T, Sun H, Hu B, Yang J, Qiao C, Yan M. Exome sequencing and epigenetic analysis of twins who are discordant for congenital cataract. Twin Res Hum Genet. 2015;18(4):393–8. doi:10.1017/thg.2015.34. [Google Scholar] [PubMed] [CrossRef]
21. Young JI, Slifer S, Hecht JT, Blanton SH. DNA methylation variation is identified in monozygotic twins discordant for non-syndromic cleft lip and palate. Front Cell Dev Biol. 2021;9:656865. doi:10.3389/fcell.2021.656865. [Google Scholar] [PubMed] [CrossRef]
22. Mohandas N, Bass-Stringer S, Maksimovic J, Crompton K, Loke Y, Walstab J, et al. Epigenome-wide analysis in newborn blood spots from monozygotic twins discordant for cerebral palsy reveals consistent regional differences in DNA methylation. Clin Epigenetics. 2018;10:25. doi:10.1186/s13148-018-0457-4. [Google Scholar] [PubMed] [CrossRef]
23. Ribel-Madsen R, Fraga MF, Jacobsen S, Bork-Jensen J, Lara E, Calvanese V, et al. Genome-wide analysis of DNA methylation differences in muscle and fat from monozygotic twins discordant for type 2 diabetes. PLoS One. 2012;7(12):e51302. doi:10.1371/journal.pone.0051302. [Google Scholar] [PubMed] [CrossRef]
24. Webster AP, Plant D, Ecker S, Zufferey F, Bell JT, Feber A, et al. Increased DNA methylation variability in rheumatoid arthritis-discordant monozygotic twins. Genome Med. 2018;10(1):64. doi:10.1186/s13073-018-0575-9. [Google Scholar] [PubMed] [CrossRef]
25. Wu H, Eckhardt CM, Baccarelli AA. Molecular mechanisms of environmental exposures and human disease. Nat Rev Genet. 2023;24(5):332–44. doi:10.1038/s41576-022-00569-3. [Google Scholar] [PubMed] [CrossRef]
26. Yang M, Luo H, Yi X, Wei X, Jiang D. The epigenetic regulatory mechanisms of ferroptosis and its implications for biological processes and diseases. Med Comm. 2023;4(3):e267. [Google Scholar]
27. Burridge PW, Sharma A, Wu JC. Genetic and epigenetic regulation of human cardiac reprogramming and differentiation in regenerative medicine. Annu Rev Genet. 2015;49:461–84. doi:10.1146/genet.2015.49.issue-1. [Google Scholar] [CrossRef]
28. Illi B, Ciarapica R, Capogrossi MC. Chromatin methylation and cardiovascular aging. J Mol Cell Cardiol. 2015;83:21–31. doi:10.1016/j.yjmcc.2015.02.011. [Google Scholar] [PubMed] [CrossRef]
29. Greco CM, Condorelli G. Epigenetic modifications and noncoding RNAs in cardiac hypertrophy and failure. Nat Rev Cardiol. 2015;12(8):488–97. doi:10.1038/nrcardio.2015.71. [Google Scholar] [PubMed] [CrossRef]
30. Balasubramanian R, Vuppalapati S, Avanthika C, Jhaveri S, Peddi NC, Ahmed S, et al. Epidemiology, genetics and epigenetics of congenital heart diseases in twins. Cureus. 2021;13(8):e17253. [Google Scholar] [PubMed]
31. Shi H, Chen S, Meng FW, Ossip DJ, Yan C, Li D, et al. Epigenome-wide DNA methylation profiling in comparison between pathological and physiological hypertrophy of human cardiomyocytes. Front Genet. 2023;14:1264382. doi:10.3389/fgene.2023.1264382. [Google Scholar] [PubMed] [CrossRef]
32. Shi H, Chen S, Meng FW, Ossip DJ, Yan C, Li D. Dynamics of DNA methylation and DNMT expression during gametogenesis and early development of scallop patinopecten yessoensis. Mar Biotechnol. 2019;21(2):196–205. doi:10.1007/s10126-018-09871-w. [Google Scholar] [PubMed] [CrossRef]
33. Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447(7143):425–32. doi:10.1038/nature05918. [Google Scholar] [PubMed] [CrossRef]
34. Shahrzad S, Bertrand K, Minhas K, Coomber B. Induction of DNA hypomethylation by tumor hypoxia. Epigenetics. 2007;2(2):119–25. doi:10.4161/epi.2.2.4613. [Google Scholar] [PubMed] [CrossRef]
35. Chang S, Wang Y, Xin Y, Wang S, Luo Y, Wang L, et al. DNA methylation abnormalities of imprinted genes in congenital heart disease: a pilot study. BMC Med Genomics. 2021;14(1):4. doi:10.1186/s12920-020-00848-0. [Google Scholar] [PubMed] [CrossRef]
36. Jenkins EL, Caputo M, Angelini GD, Ghorbel MT. Chronic hypoxia down-regulates tight junction protein ZO-2 expression in children with cyanotic congenital heart defect. ESC Heart Fail. 2016;3(2):131–7. doi:10.1002/ehf2.12081. [Google Scholar] [PubMed] [CrossRef]
37. Sanchez-Castro M, Pichon O, Briand A, Poulain D, Gournay V, David A, et al. Disruption of the SEMA3D gene in a patient with congenital heart defects. Hum Mutat. 2015;36(1):30–3. doi:10.1002/humu.2015.36.issue-1. [Google Scholar] [CrossRef]
38. Bare DJ, Yue L, Ai X. A special issue on calcium dynamics of the heart: remodeling of ion channels and regulatory pathways. Pflugers Arch. 2021;473(3):313–6. doi:10.1007/s00424-021-02532-3. [Google Scholar] [PubMed] [CrossRef]
39. Landstrom AP, Dobrev D, Wehrens XHT. Calcium signaling and cardiac arrhythmias. Circ Res. 2017;120(12):1969–93. doi:10.1161/CIRCRESAHA.117.310083. [Google Scholar] [PubMed] [CrossRef]
40. Pereira L, Ruiz-Hurtado G, Rueda A, Mercadier JJ, Benitah JP, Gómez AM. Calcium signaling in diabetic cardiomyocytes. Cell Calcium. 2014;56(5):372–80. doi:10.1016/j.ceca.2014.08.004. [Google Scholar] [PubMed] [CrossRef]
41. de la Pompa JL, Timmerman L, Takimoto H, Yoshida H, Elia A, Samper E, et al. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature. 1998;392(6672):182–6. doi:10.1038/32419. [Google Scholar] [PubMed] [CrossRef]
42. Terrar DA. Calcium signaling in the heart. Adv Exp Med Biol. 2020;1131:395–443. doi:10.1007/978-3-030-12457-1. [Google Scholar] [CrossRef]
43. Izarzugaza JMG, Ellesøe SG, Doganli C, Ehlers NS, Dalgaard MD, Audain E, et al. Systems genetics analysis identifies calcium-signaling defects as novel cause of congenital heart disease. Genome Med. 2020;12(1):76. doi:10.1186/s13073-020-00772-z. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools