
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE
NOTCH3 Mutations and CADASIL Phenotype in Pulmonary Arterial Hypertension Associated with Congenital Heart Disease
1
Department of Adult Cardiac Surgery, National Center for Cardiovascular Diseases, Fuwai Hospital, Chinese Academy of Medical
Sciences, Peking Union Medical College, Beijing, China
2
BestNovo (Beijing) Medical Laboratory, Beijing, China
3
Key Laboratory of Pulmonary Vascular Medicine, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy
of Medical Sciences and Peking Union Medical College, Beijing, China
* Corresponding Author: Rui Jiang. Email:
Congenital Heart Disease 2022, 17(6), 675-686. https://doi.org/10.32604/chd.2022.021626
Received 24 February 2022; Accepted 22 June 2022; Issue published 11 October 2022
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: The etiology of pulmonary arterial hypertension associated with congenital heart disease (PAHCHD) is complicated and the phenotype is heterogeneous. Genetic defects of NOTCH3 were associated with cerebral disease and pulmonary hypertension. However, the relationship between NOTCH3 mutations and the clinical phenotype has not been reported in CHD-PAH. Methods: We eventually enrolled 142 PAH-CHD patients from Fuwai Hospital. Whole exome sequencing (WES) was performed to screen the rare deleterious variants of NOTCH3 gene. Results: This PAH-CHD cohort included 43 (30.3%) men and 99 (69.7%) women with the mean age 29.8 ± 10.9 years old. The pathogenic or likely pathogenic mutations of NOTCH3 were identified in five cases. Patients 2, 5, 8 and 11 carried the same NOTCH3 mutation c.1630C > T (pArg544Cys), which is the hot-spot mutation for cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Patient 3 carried the NOTCH3 mutation p.Arg75Gln that has also been reported to be associated with the CADASIL. Patients 2, 5, 8, 11 took the examination of the cerebral magnetic resonance imaging (MRI) and confirmed the phenotype of CADASIL. Conclusions: We first reported the NOTCH3 rare mutations and CADASIL phenotypes in CHD-PAH patients. The NOTCH3 rare variants were with a relatively high positive rate and CADASIL phenotypes were likely enriched in PAH-CHD patients. The preoperative neurological examination might be recommended for PAH-CHD patients to determine the surgical contraindications and reduce intraoperative neurological complications.Keywords
Congenital heart disease (CHD) is the most common birth defect with an incidence of 0.4%–1.5% in the total newborn infants every year. The morbidity and mortality of CHD are the first place of non-infectious diseases of children in the world [1,2]. Approximate 5%–10% of CHD adults develop pulmonary arterial hypertension (PAH) [3]. The etiology of PAH-CHD is complicated, and the phenotype is heterogeneous [4]. PAH stems from an interplay among many detrimental factors, including the sizes of the congenital cardiac defect, the delayed treatment and the defects of genetics or epigenetics factors [5]. The systemic pulmonary shunt leads to the increase of pulmonary blood flow and the blocked decrease of physiological pulmonary vascular resistance. In addition, the increased shear stress of pulmonary vessels leads to the damage of pulmonary vascular endothelium, the proliferation of smooth muscle cells and extracellular matrix and formation of intravascular thrombosis [6].
Genetic variants are closely associated with PAH-CHD [7,8]. Roberts et al. [9] screened the bone morphogenetic protein receptor 2 (BMPR2) gene in 40 adults and 66 children with PAH-CHD and demonstrated that 6% of the subjects have BMPR2 mutations. Liu et al. [10] reported that BMPR2 mutations were significantly more enriched in the CHD patients with PAH than in the CHD patients without PAH. These previous studies demonstrated that the pathogenic variants of BMPR2 might predispose the susceptibility of PAH in CHD patients. However, whether the deleterious variants in other genes influence the development of CHD-PAH remains largely unknown.
NOTCH3 gene is a key member of NOTCH signaling pathway, which is evolutionarily conserved and involved in regulation of cell–cell interactions. NOTCH3 is predominantly expressed in the vascular smooth muscle cells (VSMCs), and is crucial for development of heart, pulmonary and brain vessels [11]. NOTCH3 mutations have been reported previously in PAH patients [12] and were also linked to CADASIL [13]. However, the clinical phenotypes caused by NOTCH3 mutations are diversified, including CADASIL and PAH [11,14]. Up to date, the relationship between NOTCH3 mutations and clinical phenotypes has not been analyzed in CHD-PAH. We used whole exome sequencing to detect the rare variants of NOTCH3 gene in 142 CHD-PAH patients, and we found NOTCH3 mutations associated with the CADASIL phenotype in CHD-PAH patients for the first time.
2.1 Study Population and Clinical Data Collection
Patients who were first diagnosed with PAH-CHD were consecutively enrolled at Fuwai Hospital of Chinese Academy of Medical Sciences in Beijing from October 31, 2018, to October 25, 2019. CHD was diagnosed by echocardiography or cardiac CT scanning and PAH was diagnosed by right heart catheterization (RHC) or echocardiography according to the guideline3. All PAH-CHD patients had a moderately increased pulmonary artery pressure (pulmonary artery systolic pressure greater than 40 mmHg). Seven patients were excluded because they were unwilling to undergo genetic testing (n = 4) or combined left ventricular system lesions with congenital heart disease (n = 2) or acquired heart disease (n = 1). Finally, 142 patients were included in the study. The research proposal was approved by the Ethical Committee of National Center for Cardiovascular Diseases, Fuwai Hospital, CAMS&PUMC (Approval No. 2017-JQ-81425002). All the patients were of Chinese ancestry and provided written informed consent for DNA analysis.
2.2 Whole Exome Sequencing and NOTCH3 Mutations Screening
CHD-PAH patients were screened by whole exome sequencing (WES) to detect the rare variants in the coding regions and the intron/exon boundaries of NOTCH3. The pathogenic genes of pulmonary hypertension and congenital heart disease were also analyzed respectively. We used the QIAamp® DNA Kit (Qiangen, Germany) to extract DNA from whole blood according to standard operating methods. The DNA extracted from all blood samples was tested, and the OD260/280 was between 1.75–1.85, and the concentration was >30 ng/μL. The DNA libraries were prepared based on the protocols of Agilent SureSelect QXT Library Prep Kit (5500-0127). Exome capture was prepared based on the protocols of Agilent SureSelect QXT Target Enrichment for Illumina Multiplexed Sequencing. Then the DNA libraries were mixed with capture probes of targeted regions using the SureSelect Human All Exon V6 kit. Illumina NovaSeq 6000 S2 Reagent Kit (300 cycles) was used for paired-end 2 × 150 bp sequencing on an Illumina NovaSeq 6000 System. A total of thirty-three exons of NOTCH3 gene were detected, which encodes 2321 amino acids. The mutations of NOTCH3 were checked with online database like HGMD (http://www.hgmd.org/) and the 1000 Genome project (https://www.internationalgenome.org/). The variants were confirmed by Sanger sequencing.
The exome sequencing data was processed using the established bioinformatics pipeline [15]. In brief, paired-end reads were mapped and aligned to the human reference genome (version GRCh37/hg19) by BWA-MEM (Burrows-Wheeler Aligner). Picard Mark duplicates was used to identify and flag PCR duplicate reads. Varscan (version 2.4.3) and Samtools (version 1.9) were used to call genetic variants and estimate accuracy of variant calls respectively. To minimize potential technical artifacts, heuristic filters were used and the variants that met any of the following conditions: missingness >10%, minimum read depth ≤15 reads, allele balance ≤20%, genotype quality <30, mappability <1 (based on 150 bp fragments) were excluded. ANNOVAR was used to annotate the variants and aggregate information about allele frequencies (AF) and in silico predictions of deleteriousness. The population AF was obtained from Exome Aggregation Consortium (ExAC), 1000G Genomes Project, NHLBI Exome Sequencing Project (ESP) and gnomAD genome East Asian. Rare variants were defined by AF < 0.5% in four databases. We used REVEL (https://sites.google.com/site/revelgenomics/) as an ensemble method for predicting the pathogenicity of missense variants. The mutation is considered harmful to the protein structure when the REVEL score is greater than 0.5.
Results were presented as percentages, median (interquartile range), or mean (standard error of mean or SD), as indicated. The frequency of rare variants was compared by χ2 test or Fisher exact test. All the statistical testing was 2 sided and considered statistically significant at a level of P < 0.05. All analyses were performed with PASW Statistics, version 18.0 (SPSS Inc., Chicago, IL, USA).
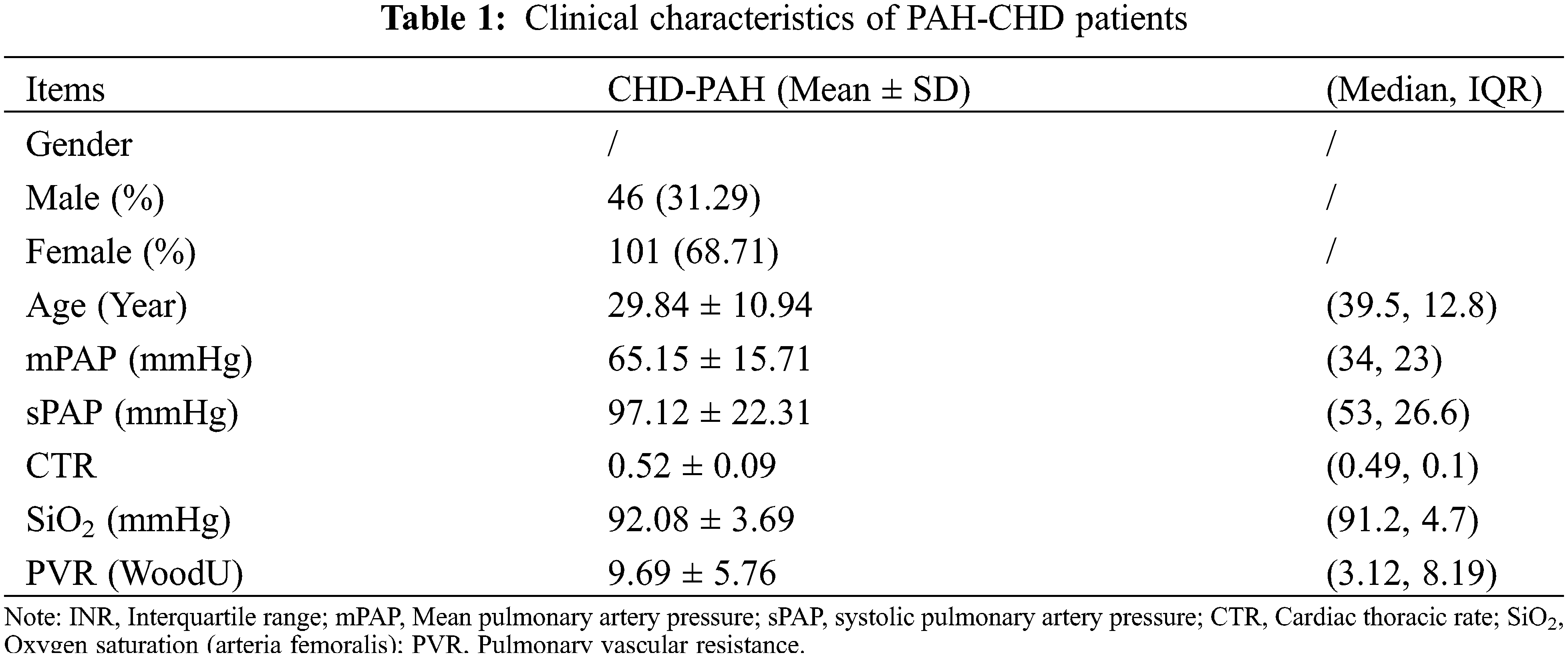
3.1 The Clinical Characteristics of PAH-CHD Patients
Totally 142 PAH-CHD patients were recruited in our study eventually, including 54 atrial septal defects (ASD 38.0%), 66 ventricular septal defects (VSD 46.5%), 7 patent ductus arteriosus (PDA 4.9%), and 15 (10.6%) cases had complex CHD. Forty-three cases (30.3%) were men and 99 cases (69.7%) were women. The mean age was 29.8 ± 10.9 years old. The mean value of mPAP (mean Pulmonary Arterial Pressure) and sPAP (systolic Pulmonary Arterial Pressure) in the cohort were 65.2 ± 15.7mmHg and 97.1 ± 22.3 mmHg, respectively (see Table 1). Furthermore, the patients demonstrated decreased SiO2, which indicated the reduced cardiopulmonary function.
3.2 NOTCH3 Rare Variants of PAH-CHD Patients
Whole exome sequencing was performed in the 142 PAH-CHD patients. Pathogenic or likely pathogenic mutations of PAH and CHD related to pathogenic gene were not found in all patients. We identified 11 (7.7%) patients carrying rare NOTCH3 missense mutants (see Table 2). The full list of PAH and CHD related variants of 11 patients were shown in Table S1. Among these patients, five (3.4%) cases carried the pathogenic or likely pathogenic mutations of NOTCH3. Patients 2, 5, 8 and 11 carried the same mutation NOTCH3 c.1630C>T (pArg544Cys), which has been reported to be a hot-spot mutant in CADASIL [15–17]. In addition, patient 5 also carried c.515G>A, which was with an uncertain significance by ACMG criteria but REVEL indicated harmful to protein structure with a score of 0.893. Patient 3 carried a likely pathogenic mutation c.224G>A (p.Arg75Gln) in NOTCH3, which was also associated with CADASIL. These rare NOTCH3 variants (c.1630C>T, c.224G>A and c.515G>A) were confirmed by Sanger sequencing (Fig. 1). All the variants have an allele frequency of <0.5% in the gnomAD genome East Asian. NOTCH3 rare variants c.224G>A and c.515G>A were found only in the VSD patients. The variant c.1630C>T was not significantly different among the four groups patients (ASD, VSD, PDA and complex CHD) (P = 0.849).
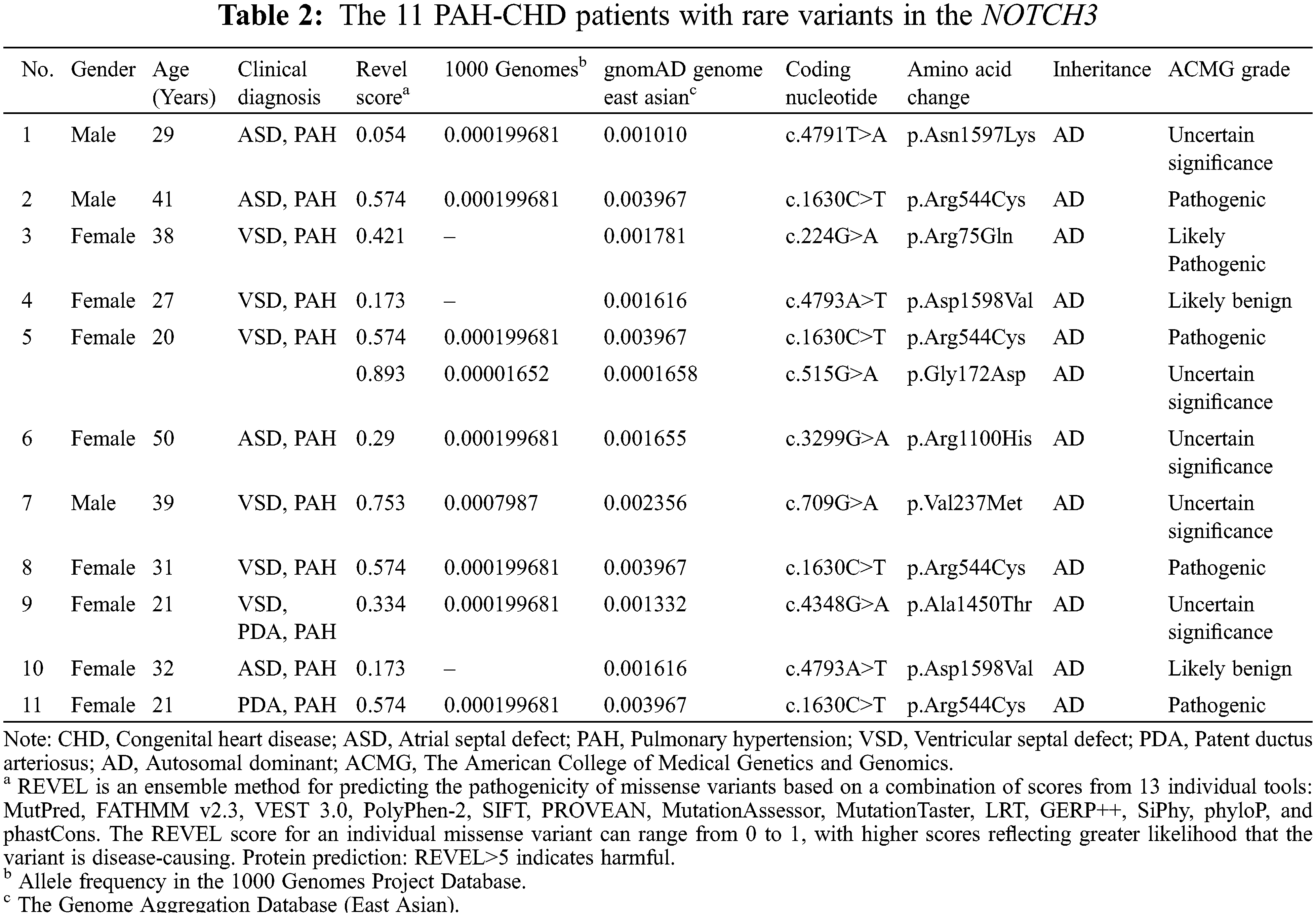
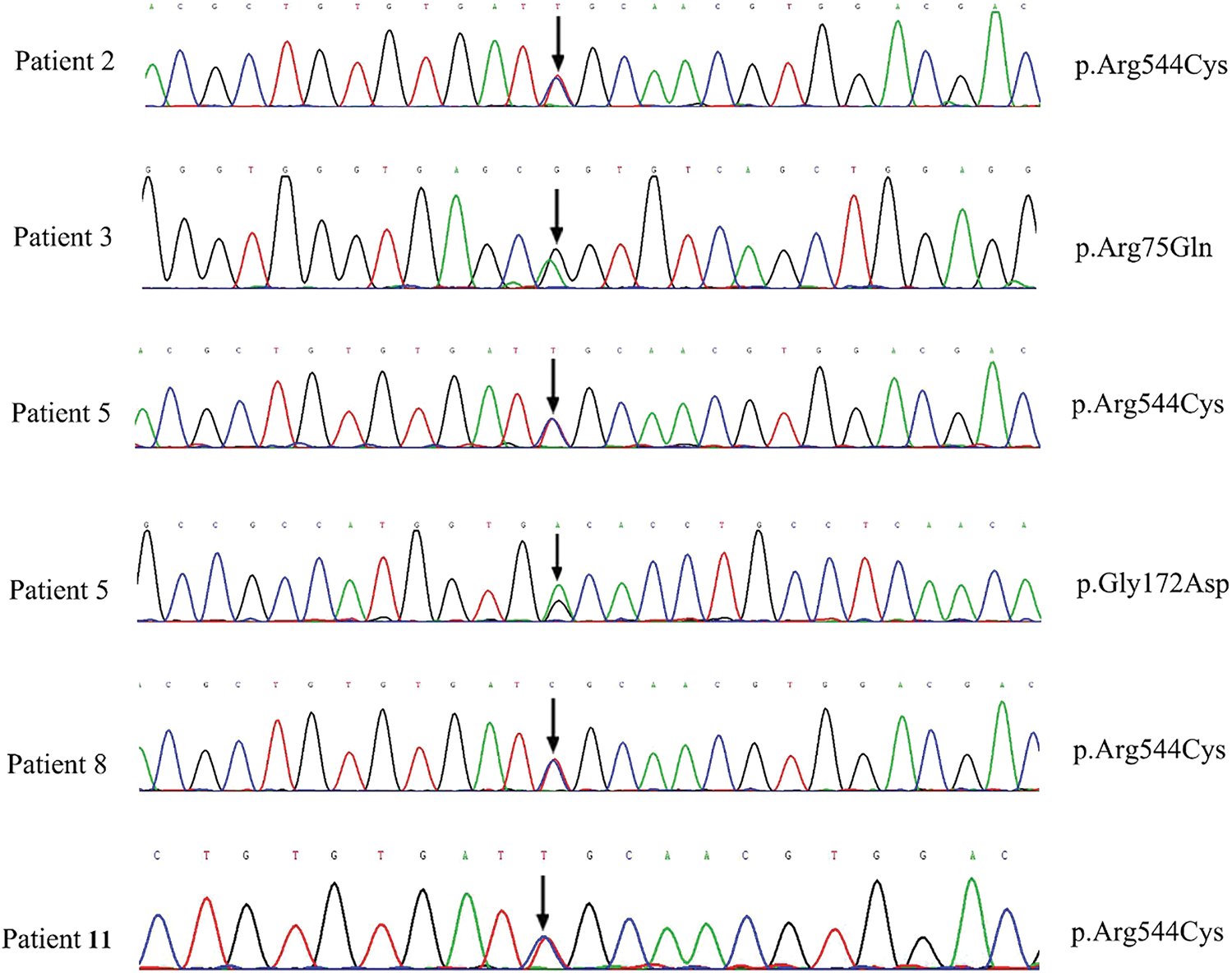
Figure 1: The sanger sequencing results of NOTCH3 c.1630C>T, c.224G>A and c.515G>A carried by five patients
3.3 Clinical Phenotype of Five PAH-CHD Patients
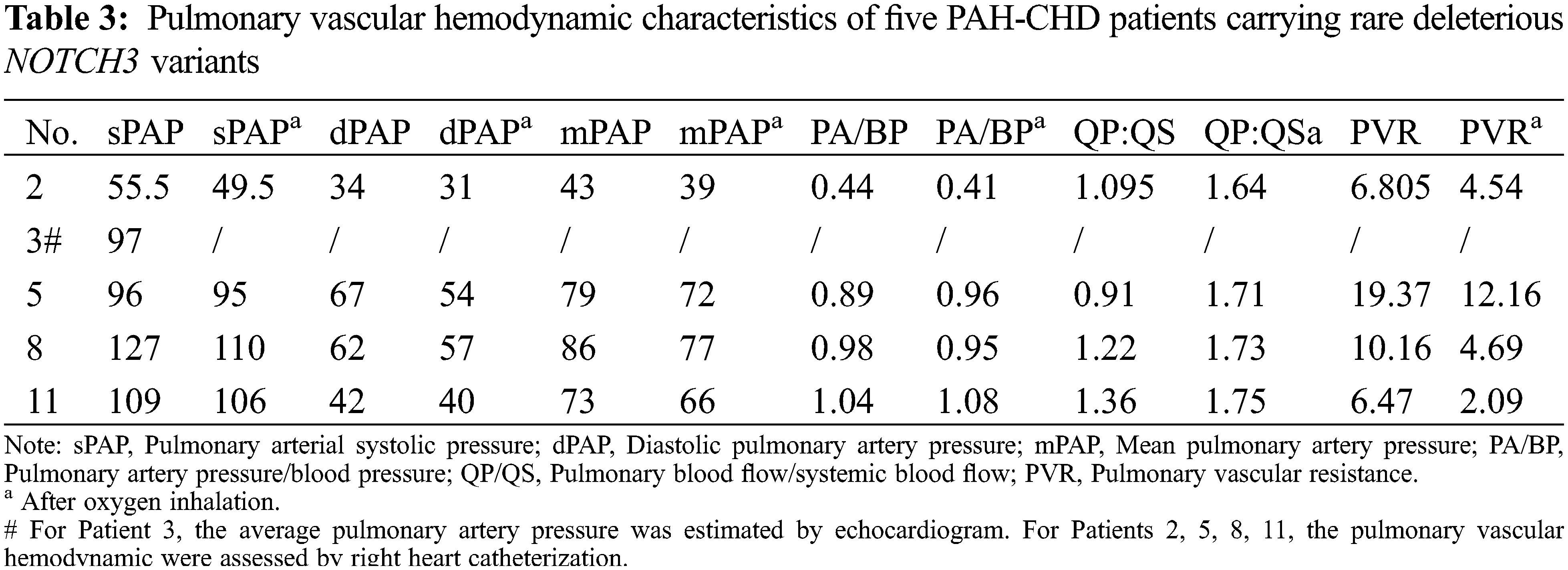
Five patients (ID: 2, 3, 5, 8, 11) carrying the pathogenic variants of NOTCH3 were suspected as CADASIL. To confirm the cerebral pathogenic phenotype, four patients (ID: 2, 5, 8, 11) undertook MRI. Patient 3 who had no neurologic symptoms refused to take the examination because of the short hospitalization time and the lack of MRI equipment in the local community. Although these patients denied suffering from Seizure, migraine and other neurological symptoms, Surprisingly, the cerebral MRI results of four cases showed that the presence of white matter changes, which conformed the CADASIL phenotype (Fig. 2). Patient 2 had a few white matter lesions in bilateral paraventricular and central posterior gyrus (Fig. 2A). Patient 5 had white matter scattered ischemic lesions in the cerebral hemisphere (Fazekas I) (Fig. 2B). Multiple lacunar lesions and mild white matter demyelination changes were presented in the bilateral basal ganglia and semi-oval central of Patient 8 (Fig. 2C). Patient 11 had a few white matter lesions in the left occipital lobe and the right radiating crown (DWML I) (Fig. 2D). The results of pulmonary vascular hemodynamic examination of five patients are shown in Table 3. All of five patients had severe pulmonary hypertension.
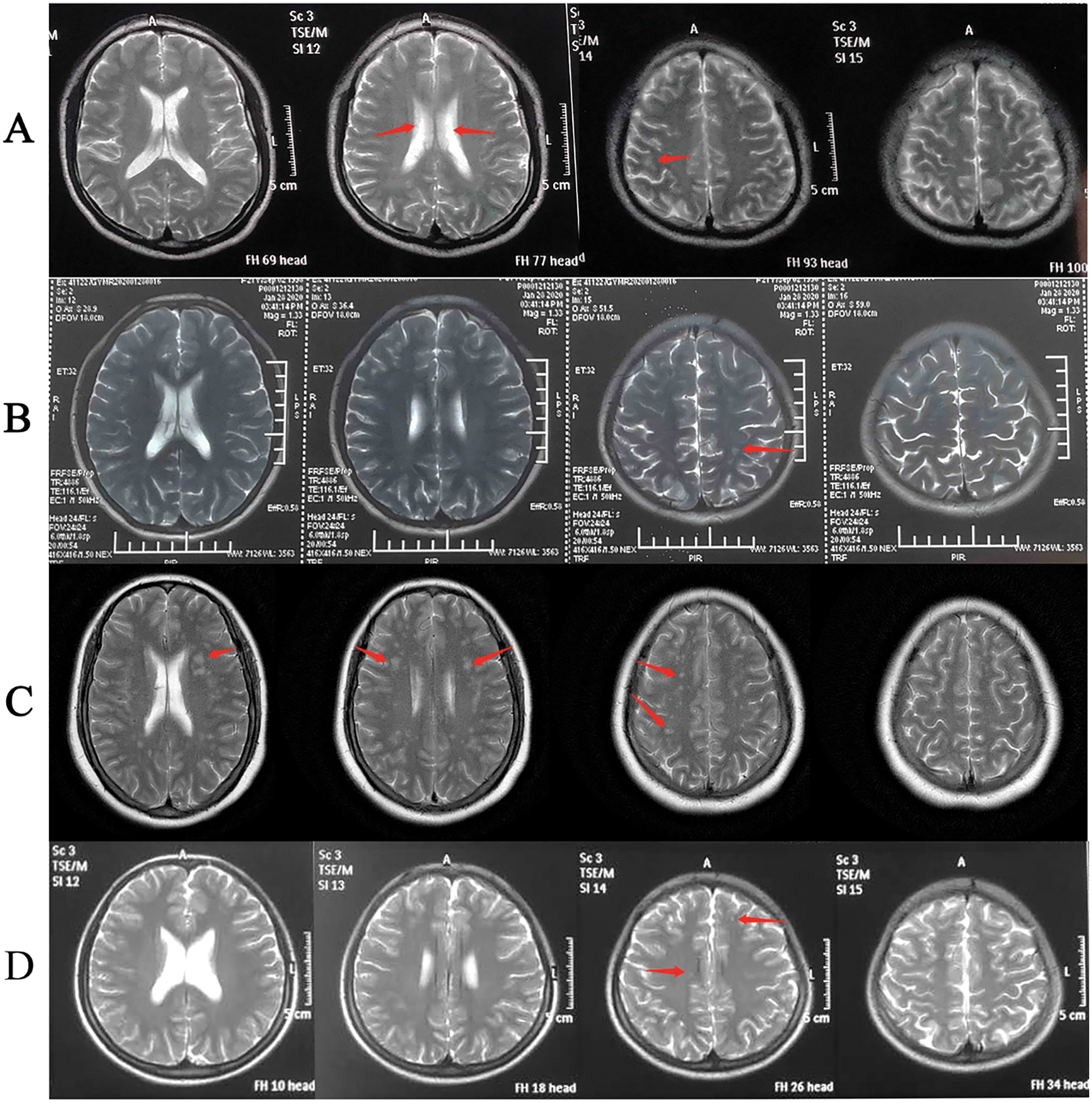
Figure 2: The cerebral MRI of Patients 2,5,8, and 11
To our knowledge, the etiology of PAH-CHD is complicated and affected by many factors, and the phenotype is heterogeneous [4]. Defining the cause is essential for the treatment of PAH-CHD patients [18]. CADASIL is the most prevalent inherited cause of cerebral small-vessel disease, cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy, a disorder linked to mutations in NOTCH3 [19,20]. The pathophysiology of CADASIL mainly includes cerebral infarction, cerebral hemorrhage, and white matte changes, etc. [19,21]. In our study, four out of five patients carried the same NOTCH3 mutation (c.1630C>T, p.Arg544Cys). One patient carried NOTCH3 c.224G>A, which has been reported in patients with a CADASIL phenotype [20]. Moreover, NOTCH3 c.224G>C mutation in the same position has been indicated to be pathogenic in several studies [22–25]. According to American College of Medical Genetics and Genomics (ACMG) guideline [26], NOTCH3 c.224G>A was a likely pathogenic mutation. Patients 2, 5, 8 and 10 displayed CADASIL image characteristics of cerebral MRI (Fig. 2), which conformed to the CADASIL phenotype.
Genetic testing finds that the pathogenic mutation of NOTCH3 can be diagnosed as CADASIL [19]. Except being the well-known pathogenic gene for CADASIL, NOTCH3 was also involved in pulmonary hypertension pathogenesis [27,28]. Although NOTCH3 mutations were found to be pathogenic in CADASIL and PAH diseases, it has been assumed that NOTCH3 has distinct function in these two diseases. However, in our study, there is a possibility that the same NOTCH3 mutation could cause both CADASIL and PAH-CHD pathogenesis or at least predispose to the development of PAH-CHD, which needs to be subsequently confirmed in the large cohort and functional studies. Furthermore, whether PAH-CHD patients need to receive genetic testing before surgery is still elusive. A neurological examination is needed to determine whether CADASIL is present for PAH-CHD patients without genetic testing. The symptoms of CADASIL are affected by age, drug use, and other factors [29]. Therefore, even though PAH-CHD patients have a CADASIL phenotype, they do not necessarily show obvious neurological symptoms clinically.
Three patients with p.Arg544Cys mutation were treated with surgery in this study. No neurological complications and abnormalities were found in the surgical patients, but the MRI results suggested that the CADASIL was indeed present. The reason might be that most patients with PAH-CHD were young, the onset time was not long for cerebrovascular disease and the condition was not serious in our study. Although the number of cases in this study was small, however, 11 (7.7%) PAH-CHD patients had NOTCH3 rare mutations, including 5 (3.5%) patients who were diagnosed with CADASIL which was a relatively high prevalence, indicating that the importance of neurological examination before undergoing cardiac surgery for PAH-CHD patients. In addition, the small number of cases may also be one of the reasons that no obvious neurological complications were observed in PAH-CHD patients.
PAH-CHD patients undergoing the neurological examination before surgery is not a clinical routine requirement [30], unless the patient has a history of neurological symptoms, such as headache, dizziness, or peripheral atherosclerosis that is more serious. In addition, there is a great risk of surgery if the neurological examination is not completed or related interventions are not taken, which has an important impact on the postoperative treatment of PAH-CHD patients with CADASIL. Patients undergoing systemic anticoagulation during cardiac surgery and some patients who require postoperative oral warfarin anticoagulation and blood pressure fluctuations may confer an increased risk of cerebral hemorrhage [31]. Hypoperfusion of cerebral infarction area under cardiopulmonary bypass may also increase the risk of cerebral ischemia and hypoxia [32,33]. What’s more, PAH-CHD patients also have headaches and dizziness due to the presence of hypoxia and the use of targeted drugs to reduce lung pressure [34,35], which will mask the mild symptoms of CADASIL and make it difficult to have obvious clinical prompts.
The results of this study suggest that the CADASIL has a relatively high incidence in CHD-PAH patients. Clinically, attention should be paid to investigate the nervous system problems of CHD-PAH patients, determine whether the patients are contraindicated in surgery, and reduce intraoperative neurological complications, especially for elder patients. The symptoms of headaches and dizziness originally caused by CHD-PAH should also be noted. It is crucial to avoid increasing the risk of surgery due to missed diagnosis of CADASIL in PAH-CHD patients. Nevertheless, the impact of postoperative drug use on CADASIL should also be concerned if surgical treatment is allowed in PAH-CHD patients with some obvious abnormal symptoms of CADASIL.
In this study, we first reported the relationship between NOTCH3 mutations and the CADASIL phenotype in PAH-CHD patients. We found that PAH-CHD patients have CADASIL with a relatively high prevalence. This suggests that PAH-CHD patients should be recommended to undergo a neurological examination before surgery to clarify the surgical contraindications, which is also important for the treatment of patients during and after surgery. However, our research has some limitations. There are no obvious clinical symptoms of CADASIL in the five patients, long-term follow-up observation study is needed to pay attention to the changes in CADASIL symptoms and the effects of cardiac surgical treatment on CADASIL. The sample size of this study is relatively small, and subsequent large sample clinical studies are needed to further confirm the importance of neurological examination before surgery in PAH-CHD patients. In addition, the NOTCH3 gene causing CADASIL is not only related to the pathogenicity of CADASIL and PAH, but also may be associated with the pathogenic of PAH-CHD patients, and the large cohort and functional studies are in progress.
In conclusion, the NOTCH3 rare mutations and CADASIL phenotype in CHD-PAH patients were first reported. The results strongly implicate that the NOTCH3 rare variants and CADASIL phenotype were likely enriched in PAH-CHD patients. Preoperative neurological examination might be recommended for PAH-CHD patients to determine the surgical contraindications and reduce intraoperative neurological complications. However, we have only screened for rare variants of NOTCH3 of small number of patients. Larger cohort studies are needed to confirm the relationship among the NOTCH3, CADASIL phenotype and PAH-CHD.
Acknowledgement: We thank patients for providing clinical examination data. Thanks to BestNovo (Beijing) Medical Laboratory for technical support and bioinformatics analysis of WES data. Professor Rui Jiang is responsible for enrolling patients with clinically characterizes, study design and perform surgery. Xiaojian Wang improved the study design and revised the manuscript. Jiangping Xu, Xiang Feng and Shaoye Wang collected the samples and clinical information. Kaisheng Lai and Zhe Liu analyzed and interpreted the data and wrote the manuscript. All authors contributed to and discussed the results and critically reviewed the manuscript. All authors read and approved the final manuscript.
Authorship: Rui Jiang, Kaisheng Lai and Xiaojian Wang had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the results. Jianping Xu, Xiang Feng and Shaoye Wang collected data and critical revision of the manuscript for important intellectual content. Rui Jiang and Kaisheng Lai drafted of the manuscript. Kaisheng Lai and Zhe Liu detected the samples and conduct data analysis.
Data Availability Statement: Data are available on reasonable request.
Funding Statement: Grant 81425002 from the National Science Fund for Distinguished Young Scholars; Grants 81670052, and 81870050 from the National Natural Science Foundation of China; Grant 2018ZX09711001-003-012 from the Drug Innovation Major Project, CAMS Fund for Key Laboratory of Pulmonary Vascular Medicine (2017PT32016).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Penton, A. L., Leonard, L. D., Spinner, N. B. (2012). Notch signaling in human development and disease. Seminars in Cell & Developmental Biology, 23(4), 450–457. DOI 10.1016/j.semcdb.2012.01.010. [Google Scholar] [CrossRef]
2. de la Pompa, J. L., Epstein, J. A. (2012). Coordinating tissue interactions: Notch signaling in cardiac development and disease. Developmental Cell, 22(2), 244–254. [Google Scholar]
3. Galie, N., Humbert, M., Vachiery, J. L., Gibbs, S., Lang, I. et al. (2016). 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERSEndorsed by: Association for European Paediatric and Congenital Cardiology (AEPCInternational Society for Heart and Lung Transplantation (ISHLT). European Heart Journal, 37(1), 67–119. [Google Scholar]
4. Chen, I. C., Dai, Z. K. (2015). Insight into pulmonary arterial hypertension associated with congenital heart disease (PAH-CHDClassification and pharmacological management from a pediatric cardiological point of view. Acta Cardiologica Sinica, 31(6), 507–515. [Google Scholar]
5. Harries, C., Armstrong, I. (2012). A review of the management of pulmonary arterial hypertension associated with congenital heart disease. European Journal of Cardiovascular Nursing, 11(2), 239–247. DOI 10.1016/j.ejcnurse.2010.10.001. [Google Scholar] [CrossRef]
6. Meng, L., Liu, X., Teng, X., Gu, H., Yuan, W. et al. (2019). Osteopontin plays important roles in pulmonary arterial hypertension induced by systemic-to-pulmonary shunt. FASEB Journal, 33(6), 7236–7251. DOI 10.1096/fj.201802121RR. [Google Scholar] [CrossRef]
7. Brida, M., Gatzoulis, M. A. (2018). Pulmonary arterial hypertension in adult congenital heart disease. Heart, 104(19), 1568–1574. [Google Scholar]
8. Pfarr, N., Fischer, C., Ehlken, N., Becker-Grunig, T., Lopez-Gonzalez, V. et al. (2013). Hemodynamic and genetic analysis in children with idiopathic, heritable, and congenital heart disease associated pulmonary arterial hypertension. Respiratory Research, 14, 3. [Google Scholar]
9. Roberts, K. E., McElroy, J. J., Wong, W. P., Yen, E., Widlitz, A. et al. (2004). BMPR2 mutations in pulmonary arterial hypertension with congenital heart disease. European Respiratory Journal, 24(3), 371–374. DOI 10.1183/09031936.04.00018604. [Google Scholar] [CrossRef]
10. Liu, D., Liu, Q. Q., Guan, L. H., Jiang, X., Zhou, D. X. et al. (2016). BMPR2 mutation is a potential predisposing genetic risk factor for congenital heart disease associated pulmonary vascular disease. International Journal of Cardiology, 211, 132–136. DOI 10.1016/j.ijcard.2016.02.150. [Google Scholar] [CrossRef]
11. Morris, H. E., Neves, K. B., Montezano, A. C., MacLean, M. R., Touyz, R. M. (2019). Notch3 signalling and vascular remodelling in pulmonary arterial hypertension. Clinical Science, 133(24), 2481–2498. DOI 10.1042/CS20190835. [Google Scholar] [CrossRef]
12. Chida, A., Shintani, M., Matsushita, Y., Sato, H., Eitoku, T. et al. (2014). Mutations of NOTCH3 in childhood pulmonary arterial hypertension. Molecular Genetics & Genomic Medicine, 2(3), 229–239. [Google Scholar]
13. Joutel, A., Corpechot, C., Ducros, A., Vahedi, K., Chabriat, H. et al. (1996). Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature, 383(6602), 707–710. [Google Scholar]
14. Hosseini-Alghaderi, S., Baron, M. (2020). Notch3 in development, health and disease. Biomolecules, 10(3485. [Google Scholar]
15. Tang, S. C., Chen, Y. R., Chi, N. F., Chen, C. H., Cheng, Y. W. et al. (2019). Prevalence and clinical characteristics of stroke patients with p.R544C NOTCH3 mutation in Taiwan. Annals of Clinical and Translational Neurology, 6(1), 121–128. [Google Scholar]
16. Lee, J. S., Ko, K., Oh, J. H., Park, J. H., Lee, H. K. (2016). Phenotypic features of cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy subjects with R544C mutation. Dementia and Neurocognitive Disorders, 15(1), 15–19. [Google Scholar]
17. Liu, K., Qin, F., Sun, X., Zhang, Y., Wang, J. et al. (2018). Analysis of the genes involved in mendelian forms of low-renin hypertension in Chinese early-onset hypertensive patients. Journal of Hypertension, 36(3), 502–509. DOI 10.1097/HJH.0000000000001556. [Google Scholar] [CrossRef]
18. Schuuring, M. J., Bouma, B. J., Cordina, R., Gatzoulis, M. A., Budts, W. et al. (2013). Treatment of segmental pulmonary artery hypertension in adults with congenital heart disease. International Journal of Cardiology, 164(1), 106–110. DOI 10.1016/j.ijcard.2011.06.084. [Google Scholar] [CrossRef]
19. Wang, M. M. (2018). CADASIL. Handbook of Clinical Neurology, 148, 733–743. [Google Scholar]
20. Liu, X., Zuo, Y., Sun, W., Zhang, W., Lv, H. et al. (2015). The genetic spectrum and the evaluation of CADASIL screening scale in Chinese patients with NOTCH3 mutations. Journal of the Neurological Sciences, 354(1–2), 63–69. DOI 10.1016/j.jns.2015.04.047. [Google Scholar] [CrossRef]
21. Caplan, L. R. (2015). Lacunar infarction and small vessel disease: Pathology and pathophysiology. Journal of Stroke, 17(1), 2–6. DOI 10.5853/jos.2015.17.1.2. [Google Scholar] [CrossRef]
22. Kim, Y., Choi, E. J., Choi, C. G., Kim, G., Choi, J. H. et al. (2006). Characteristics of CADASIL in Korea: A novel cysteine-sparing Notch3 mutation. Neurology, 66(10), 1511–1516. [Google Scholar]
23. Mizuno, T., Muranishi, M., Torugun, T., Tango, H., Nagakane, Y. et al. (2008). Two Japanese CADASIL families exhibiting Notch3 mutation R75P not involving cysteine residue. Journal of Internal Medicine, 47(23), 2067–2072. DOI 10.2169/internalmedicine.47.1391. [Google Scholar] [CrossRef]
24. Kim, H. J., Kim, H. Y., Paek, W. K., Park, A., Young Park, M. et al. (2012). Amyotrophic lateral sclerosis and frontotemporal lobar degeneration in association with CADASIL. The Neurologist, 18(2), 92–95. DOI 10.1097/NRL.0b013e318247bb2d. [Google Scholar] [CrossRef]
25. Wollenweber, F. A., Hanecker, P., Bayer-Karpinska, A., Malik, R., Bazner, H. et al. (2015). Cysteine-sparing CADASIL mutations in NOTCH3 show proaggregatory properties in vitro. Stroke, 46(3), 786–792. [Google Scholar]
26. Richards, S., Aziz, N., Bale, S., Bick, D., Das, S. et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genetics in Medicine, 17(5), 405–424. [Google Scholar]
27. Gomez, J., Reguero, J. R., Junquera, M. R., Alvarez, C., Moris, C. et al. (2016). Next generation sequencing of the NOTCH3 gene in a cohort of pulmonary hypertension patients. International Journal of Cardiology, 209, 149–150. DOI 10.1016/j.ijcard.2016.02.024. [Google Scholar] [CrossRef]
28. Yang, H., Zeng, Q., Ma, Y., Liu, B., Chen, Q. et al. (2018). Genetic analyses in a cohort of 191 pulmonary arterial hypertension patients. Respiratory Research, 19(1), 87. [Google Scholar]
29. Adib-Samii, P., Brice, G., Martin, R. J., Markus, H. S. (2010). Clinical spectrum of CADASIL and the effect of cardiovascular risk factors on phenotype: Study in 200 consecutively recruited individuals. Stroke, 41(4), 630–634. [Google Scholar]
30. D’Alto, M., Mahadevan, V. S. (2012). Pulmonary arterial hypertension associated with congenital heart disease. European Respiratory Review, 21(126), 328–337. DOI 10.1183/09059180.00004712. [Google Scholar] [CrossRef]
31. Deng, X., Jin, B., Li, S., Li, Y., Zhou, H. et al. (2019). Guideline implementation and early risk assessment in pulmonary arterial hypertension associated with congenital heart disease: A retrospective cohort study. Clinical Respiratory Journal, 13(11), 693–699. DOI 10.1111/crj.13076. [Google Scholar] [CrossRef]
32. Vedel, A. G., Holmgaard, F., Rasmussen, L. S., Langkilde, A., Paulson, O. B. et al. (2018). High-target versus low-target blood pressure management during cardiopulmonary bypass to prevent cerebral injury in cardiac surgery patients: A randomized controlled trial. Circulation, 137(17), 1770–1780. [Google Scholar]
33. Sun, M., Cheng, C., Zhang, R., Xu, X., Wen, L. et al. (2014). Pulmonary arterial hypertension after operation for congenital heart disease: Analysis of baseline clinical characteristics of 122 Chinese patients. Zhonghua Xin Xue Guan Bing Za Zhi, 42(5), 396–399. [Google Scholar]
34. Noel, Z. R., Kido, K., Macaulay, T. E. (2017). Selexipag for the treatment of pulmonary arterial hypertension. American Journal of Health-System Pharmacy, 74(15), 1135–1141. DOI 10.2146/ajhp160798. [Google Scholar] [CrossRef]
35. Beghetti, M., Channick, R. N., Chin, K. M., di Scala, L., Gaine, S. et al. (2019). Selexipag treatment for pulmonary arterial hypertension associated with congenital heart disease after defect correction: Insights from the randomised controlled GRIPHON study. European Journal of Heart Failure, 21(3), 352–359. DOI 10.1002/ejhf.1375. [Google Scholar] [CrossRef]
6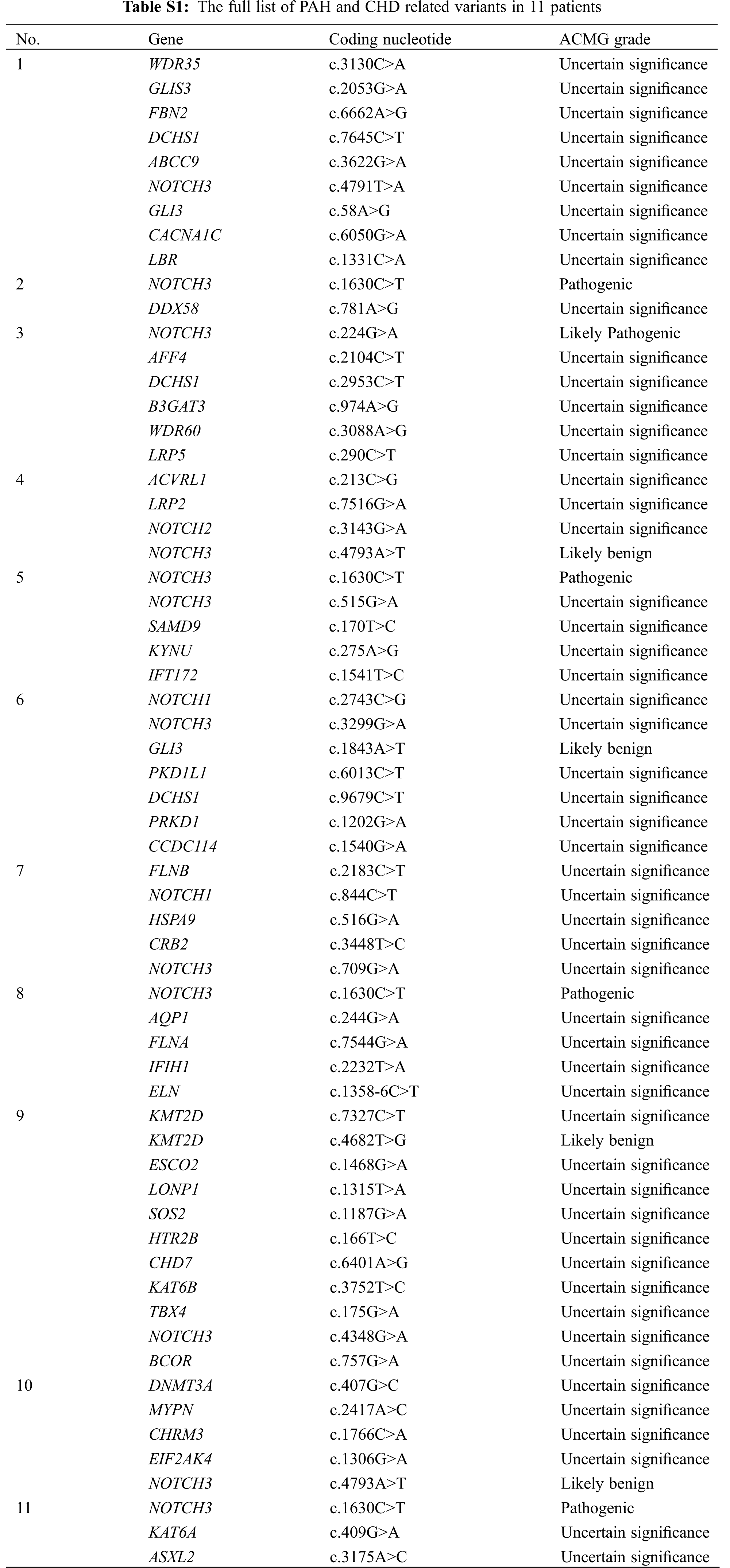
Cite This Article
 Copyright © 2022 The Author(s). Published by Tech Science Press.
Copyright © 2022 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools