
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
CASE REPORT
Life Threatening Broad QRS Tachycardia in an Infant with Conduction Disorder and SCN5A Mutation
1
Department of Pediatric Cardiology and Cardiac Surgery, Bambino Gesù Children’s Hospital Centro Cardiologico Pediatrico del
Mediterraneo, San Vincenzo Hospital, Taormina, Italy
2
Cardiovascular Surgery Department, Rajaie Cardiovascular Medical and Research Center, Iran University of Medical Sciences,
Tehran, Iran
* Corresponding Author: Elio Caruso. Email:
(This article belongs to the Special Issue: Nightmare Case Reports in Congenital Heart Disease)
Congenital Heart Disease 2022, 17(5), 551-556. https://doi.org/10.32604/chd.2022.023711
Received 10 May 2022; Accepted 30 July 2022; Issue published 06 September 2022
 View Full Text
View Full Text Download PDF
Download PDFAbstract
We present the case of an infant admitted to our department for a rapid broad complex tachycardia and cardiovascular collapse. The patient was submitted to genetic testing because of a conduction defect at baseline ECG and family history of gene mutation. A new SCN5A gene mutation variant was found leading to diagnosis of sodium-channel dysfunction arrhythmia.Graphic Abstract

Keywords
We present the case of an infant with a rapid wide complex tachycardia during fever (38.8°C), who presented at baseline ECG sinus bradycardia and conduction disorder related to SCN5A gene mutation inherited from the mother. In recent years some VT episodes in infants or small children have been related to mutations of the SCN5A gene encoding for the cardiac sodium channel. Apart from the children with a baseline or drug-induced typical Brugada ECG pattern, some of these young patients present with an atypical conduction disorder and have been recently labelled as “Brugada-like” children [1].
A previously healthy 8 months old child presented during a febrile illness at the emergency room of our hospital because of pallor, lethargy, hypotension, tachypnea and variation in intensity of the first heart sound. The standard ECG showed a rapid broad complex tachycardia with a heart rate of 300 beats/min (Fig. 1 panel A). A regular sinus rhythm returned during sedation with isoflurano before external defibrillation. The ECG showed sinus bradycardia (72 beats/min), prolonged PR interval (0.20 s), a conduction defect (RBBB + anterior fascicular block-type), wide QRS complex (0.10 s), normal ventricular repolarization, normal QTc interval (Fig. 1 panel B). Immediately his color and peripheral perfusion improved. The echocardiogram showed a structural normal heart with normal coronary arteries. During hospitalization a transesophageal electrophysiological study (EPS) was performed using the method previously defined [2] for evaluation of supraventricular tachycardia (SVT). Written Consent was obtained from patient’s family. Midazolam (0.1 mg/kg) was administered for sedation. 5–45 mA current was transmitted by a FIAB programmable Cardiac Stimulator 8817 for 5–20 ms. Stimulation was started by 10 mA for 10 ms with a heart rate faster than the baseline and was adjusted according to the stimulus threshold. Single and double extrastimuli and rapid atrial pacing in the baseline state and after isoproterenol iv infusion have not been induced SVT. Negative evaluation for metabolic disorders, infectious, and toxicological pathogeneses of VT. Every etiologies of nonspecific QRS duration lengthening, hyperkalemia, drugs, myocardial ischemia, were excluded prior to genetic testing for a channelopathy. Parents refused any type of medications for the child and also invasive EPS. For 7 years follow-up, he remained asymptomatic without any recurrences of tachycardia and an exercise test performed on the treadmill was normal. Since his abnormal basal ECG with conduction defect, normal structural heart, we have been induced to perform genetic testing. The molecular analysis performed at 6 years, showed a missense mutation in the sodium voltage-gated channel α sub-unit 5 (SCN5A) gene (Refseq: NM_198056.2 base substitution: c.5219G > A; Aminoacid Analysis: p.G1740E; heterozygote genotype). At present, this variant is to be considered likely pathogenetic (class 4: ACMG Variant classification), taking into account the consistency with the clinical suspicion, the frequency in the general population and according to the guidelines of the American College of Medical Genetics and Genomics [3].
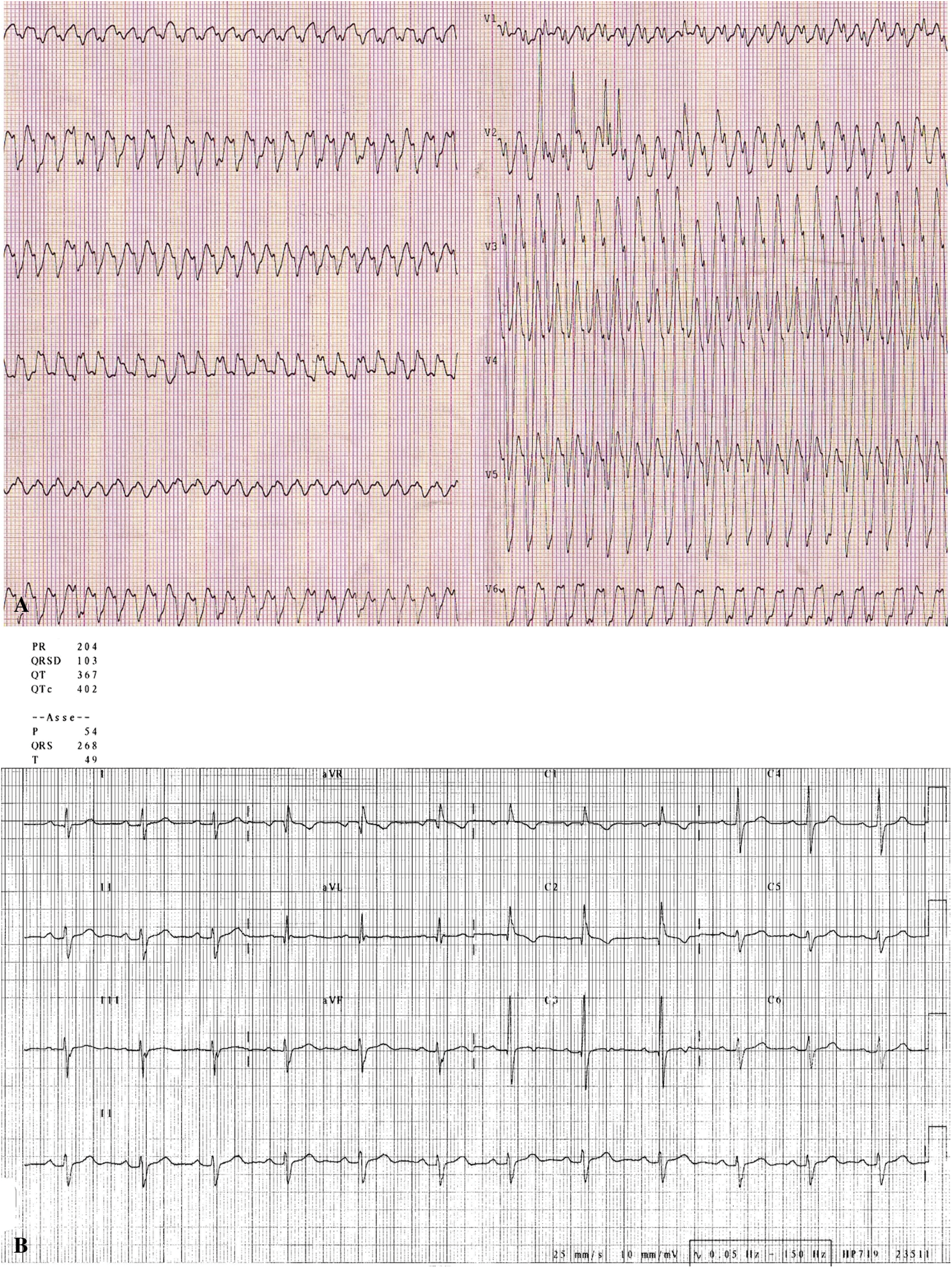
Figure 1: A. ECG shows a rapid broad complex tachycardia with a heart rate of 300 beats/min and AV dissociation. B. ECG shows sinus bradycardia (72 beats/min), prolonged PR interval (0.20 s), a conduction defect (RBBB + anterior fascicular block-type), wide QRS complex (0.10 s), normal ventricular repolarization, normal QTc interval
His mother presented also the same gene mutation but she is asymptomatic until now with a normal baseline ECG (Fig. 2).
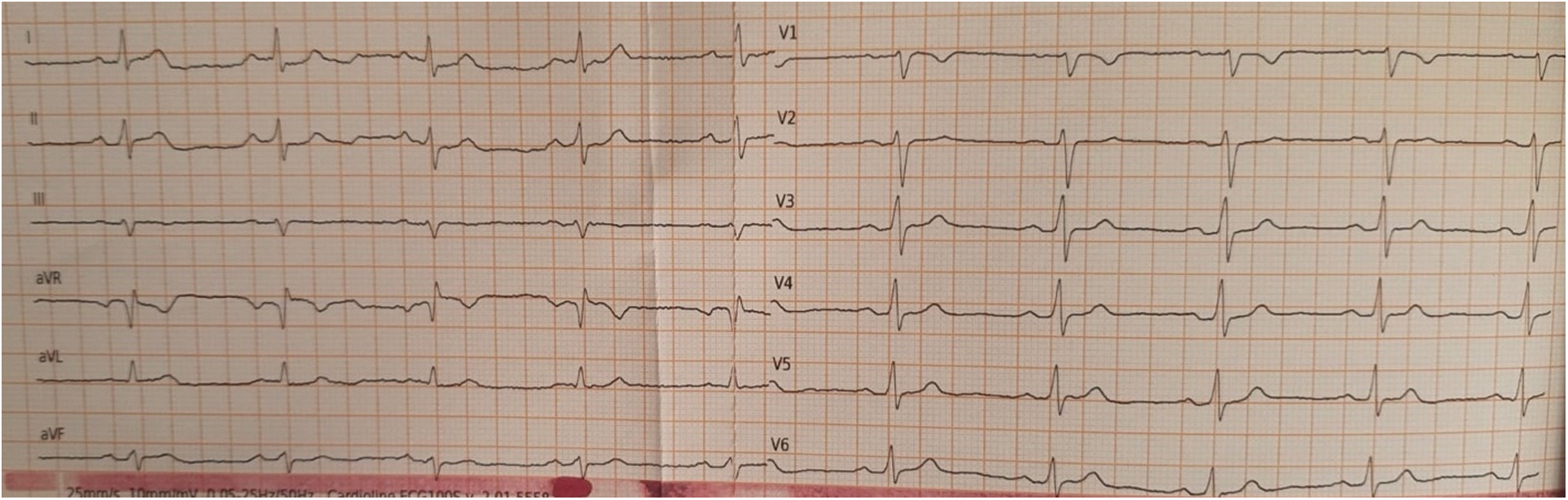
Figure 2: Mother’s ECG shows sinus rhythm at 60 beats/min, normal AV conduction, normal QRS interval and morphology, normal ventricular repolarization, normal QTc interval
Life threatening VT in infants is a rare event. The role of fever as a precipitating factor for ventricular arrhythmias in subjects with sodium channel disorders is well recognized [4–7]. These episodes are often life threatening because of the potential degeneration into ventricular fibrillation. In this scenario a comprehensive clinical evaluation is mandatory for the exclusion of common predisposing conditions such as congenital structural cardiac defects, previous cardiac surgery, cardiomyopathies, metabolic disorder or viral myocarditis. Moreover, it is essential to rule out primary arrhythmic disorders. Sometimes a very fast broad complex tachycardia is difficult to diagnose when not all typical signs of VT are present as “fusion” or “capture” beats or VA dissociation. During VT, dissociation between atrial and ventricular activity during tachycardia is a hallmark of VT but sometimes P waves could be difficult to recognize during a broad QRS tachycardia if heart rate is very fast and have to look other signs or look for not electrocardiographic signs such as variations in jugular pulsations, the loudness of the first heart sound, and changes in systolic blood pressure [8]. Our working diagnosis was viral myocarditis or supraventricular tachycardia. Our case report showed an ECG with heart rate of 300 beats/min with not clear signs of AV dissociation but RS interval > 100 ms in precordial leads [9] and negative concordance was highly suggestive for VT although the basic electrocardiogram showed anterior fascicular block-type. According to the last ESC guideline [10] we checked all signs to do differential diagnosis between VT and atrial flutter: though the AV dissociation was not clear on the ECG (Fig. 1A), QRS wide > 120 ms, regular R-R interval, high ventricular rate, chest lead negative concordance and RS in precordial leads are all suggestive signs of VT. Moreover, in emergency room other colleagues administered adenosine bolus that did not patent atrial flutter and our transesophageal EP study did not induce supraventricular arrhythmia like atrial flutter.
Loss of function mutations in depolarizing channels may present as rapid monomorphic VT, VF, or syncope in infants having an intraventricular conduction delay [1]. Kanter et al. [1] sustained the hypotesys that infants having ventricular arrhythmias and intraventricular conduction delay while in sinus rhythm may have a loss-of-function depolarizing channel mutation, particularly in the normal structural and functional heart. They consider these clinical phenotype to be Brugada-like, because there are differences from classical adult Brugada syndrome (BrS) and from many reports of childhood BrS.
The first gene to be linked to the Brugada syndrome is SCN5A, the gene encoding for the α-subunit of the voltage-gated cardiac sodium channel (Nav1.5) [11]. To date, nearly a quarter of BrS patients were found to be SCN5A variants carriers and more than 300 SCN5A variants were BrS-related mutations including missense variants, non-sense variants, nucleotide insertion/deletions, and splice site variants. BrS-related SCN5A variants are usually loss-of-function variants and mainly located in the region between DI and DII, intracellular connection between DIII and DIV region, P ring and D-terminal of DIII region including SCN5A polymorphism [11–13].
The p.G1740E variant is to be considered likely pathogenetic according to ACMG Variant classification (class 4). No functional in vitro studies are reported, and the variant is not described in the scientific literature as a mutation. Several in silico computational algorithms (SIFT, PolyPhen-2, Align-GVGD) support a deleterious effect of the variant on the gene or on the gene product (conserved regions, evolutionary line, impact on splicing). The p.G1740E variant is located in the DIV-S5/S6 domain believed to be critical for the function of the SCN5A protein and where other pathogenic or possibly pathogenic variants have been reported. In particular, a variant is known (p.G1740R) which affects the same amino acid residue reported in association with Brugada Syndrome [13–15].
Above ECG signs during tachycardia, not inducibility of SVT at EPS and its onset during fever, genetic results about SCN5A mutation led us to hypothesize a diagnosis of VT.
In recent years some authors have identified a subgroup of children presenting with rapid VT also showing intraventricular conduction delay at the baseline ECG [16]. In such a group of patient genetic testing commonly identify mutations of the SCN5A or CACNB2 gene encoding for the cardiac sodium and calcium channel, respectively. We did a “Brugada like” diagnosis.
In conclusion we add one more case of very rare syndrome in children called phenotype “Brugada like”.
As SCN5A is the main gene related to Brugada Syndrome, we support the hypotesis that the above clinical scenario of fever, ventricular tachycardia and conduction disorder at baseline ECG, could be related to cardiac sodium-channel dysfunction due to a “Brugada like” phenotype associated to SCN5A G1740E variant.
This case report confirm that genetic testing is mandatory in infants with baseline conduction disorder and associated wide QRS tachycardia. Although their intermediate-term follow-up is mostly favourable, additional life-threatening arrhythmias may occur. Follow-up outpatient appointments include clinical evaluation 24 h ECG monitoring once a month. Moreover, we suggest aggressively treat fevers with ibuprofen and acetaminophen. We describe a first case of a broad complex tachycardia diagnosed as a monomorphic VT during fever in a child with SCN5A mutation with a long term follow up without medical therapy.
Authorship: The authors confirm contribution to the paper as follows: study conception and design: E. Caruso, A. Di Pino; data collection: S. Farruggio; analysis and interpretation of results: Paolo Guccione, Mohammadrafie Khorgami; draft manuscript preparation: E. Caruso. All authors reviewed the results and approved the final version of the manuscript.
Funding Statement: The authors received no specific funding for this study.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Kanter, R. J., Pfeiffer, R., Hu, D., Barajas-Martinez, H., Carboni, M. P. et al. (2012). Brugada-like syndrome in infancy presenting with rapid ventricular tachycardia and intraventricular conduction delay. Circulation, 125(1), 14–22. DOI 10.1161/CIRCULATIONAHA.111.054007. [Google Scholar] [CrossRef]
2. Benson jr, D. W., Dunnigan, A., Benditt, D. G., Pritzker, M. R., Thompson, T. R. (1983). Transesophageal study of infant supraventricular tachycardia: Electrophysiologic characteristics. The American Journal of Cardiology, 52(8), 1002–1006. DOI 10.1016/0002-9149(83)90520-9. [Google Scholar] [CrossRef]
3. Richards, S., Aziz, N., Bale, S., Bick, D., Das, S. et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genetics in Medicine, 17(5), 405–424. DOI 10.1038/gim.2015.30. [Google Scholar] [CrossRef]
4. Mok, N. S., Priori, S. G., Napolitano, C., Chan, N. Y., Chahine, M. et al. (2003). A newly characterized SCN5A mutation underlying Brugada syndrome unmasked by hyperthermia. Journal of Cardiovascular Electrophysiology, 14(4), 407–411. DOI 10.1046/j.1540-8167.2003.02379.x. [Google Scholar] [CrossRef]
5. Porres, J. M., Brugada, J., Urbistondo, V., García, F., Reviejo, K. et al. (2002). Fever unmasking the Brugada syndrome. Pacing and Clinical Electrophysiology, 25(11), 1646–1648. DOI 10.1046/j.1460-9592.2002.01646.x. [Google Scholar] [CrossRef]
6. Amin, A. S., Meregalli, P. G., Bardai, A., Wilde, A. A., Tan, H. L. (2008). Fever increases the risk for cardiac arrest in the Brugada syndrome. Annals of Internal Medicine, 149(3), 216–218. DOI 10.7326/0003-4819-149-3-200808050-00020. [Google Scholar] [CrossRef]
7. Silva, M. A., Elias Neto, J., Futuro, G., Merçon, E. S., Vasconcelos, D. et al. (2021). Life-threatening ventricular arrhythmia induced by atrial tachycardia in a child with an SCN5A mutation. Arritmia ventricular potencialmente fatal induzida por taquicardia atrial em uma criança com mutação de SCN5A. Arquivos Brasileiros de Cardiologia, 117(suppl 1), 19–22. [Google Scholar]
8. Wellens, H. J. (2001). Electrophysiology: Ventricular tachycardia: Diagnosis of broad QRS complex tachycardia. Heart, 86(5), 579–585. DOI 10.1136/heart.86.5.579. [Google Scholar] [CrossRef]
9. Brugada, P., Brugada, J., Mont, L., Smeets, J., Andries, E. W. (1991). A new approach to the differential diagnosis of a regular tachycardia with a wide QRS complex. Circulation, 83(5), 1649–1659. DOI 10.1161/01.CIR.83.5.1649. [Google Scholar] [CrossRef]
10. Brugada, J., Katritsis, D. G., Arbelo, E., Arribas, F., Bax, J. J. et al. (2019). ESC guidelines for the management of patients with supraventricular tachycardia the task force for the management of patients with supraventricular tachycardia of the european society of cardiology (ESC). European Heart Journal, 41(5), 655–720. DOI 10.1093/eurheartj/ehz467. [Google Scholar] [CrossRef]
11. Antzelevitch, C., Patocskai, B. (2016). Brugada syndrome: Clinical, genetic, molecular, cellular, and ionic aspects. Current Problems in Cardiology, 41(1), 7–57. DOI 10.1016/j.cpcardiol.2015.06.002. [Google Scholar] [CrossRef]
12. Li, W., Yin, L., Shen, C., Hu, K., Ge, J. et al. (2018). SCN5A variants: Association with cardiac disorders. Frontiers in Physiology, 9, 1372. DOI 10.3389/fphys.2018.01372. [Google Scholar] [CrossRef]
13. Kapplinger, J. D., Tester, D. J., Alders, M., Benito, B., Berthet, M. et al. (2010). An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm, 7(1), 33–46. DOI 10.1016/j.hrthm.2009.09.069. [Google Scholar] [CrossRef]
14. Priori, S. G., Napolitano, C., Gasparini, M., Pappone, C., Della Bella, P. et al. (2002). Natural history of Brugada syndrome: Insights for risk stratification and management. Circulation, 105(11), 1342–1347. DOI 10.1161/hc1102.105288. [Google Scholar] [CrossRef]
15. Meregalli, P. G., Tan, H. L., Probst, V., Koopmann, T. T., Tanck, M. W. et al. (2009). Type of SCN5A mutation determines clinical severity and degree of conduction slowing in loss-of-function sodium channelopathies. Heart Rhythm, 6(3), 341–348. DOI 10.1016/j.hrthm.2008.11.009. [Google Scholar] [CrossRef]
16. Chockalingam, P., Rammeloo, L. A., Postema, P. G., Hruda, J., Clur, S. A. et al. (2011). Fever-induced life-threatening arrhythmias in children harboring an SCN5A mutation. Pediatrics, 127(1), e239–e244. DOI 10.1542/peds.2010-1688. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2022 The Author(s). Published by Tech Science Press.
Copyright © 2022 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools