| Congenital Heart Disease |
DOI: 10.32604/CHD.2021.016548
ARTICLE
When the Blood Pressure Misleads You: A Diagnostic Conundrum in an Unusual Case of Coarctation
1Department of Pediatrics, Division of Cardiology, University of British Columbia, Vancouver, Canada
2Division of Pediatric Cardiothoracic Surgery, University of British Columbia, Vancouver, Canada
3Department of Pediatric Radiology, British Columbia Children’s Hospital, Vancouver, Canada
*Corresponding Author: Shubhayan Sanatani. Email: ssanatani@cw.bc.ca
Received: 24 March 2021; Accepted: 25 May 2021
Abstract: A 4-month-old previously healthy baby was found to be in congestive heart failure with LV dysfunction and a right aortic arch with severe coarctation, undetectable by blood pressure measurements. A cardiac CT and central blood pressure led to the diagnosis of a unique anatomic variant of aortic coarctation. Once diagnosed the patient underwent surgery with an uncomplicated recovery.
Keywords: Aorta; coarctation; right arch; congenital heart disease
Abbreviations
| CoA: | Coarctation |
| LV: | Left ventricle |
| CT: | Computed tomography |
| ER: | Emergency room |
| OR: | Operative room |
A previously healthy 4-month-old girl presented to the emergency department with a recent history of difficulty breathing and reduced oral intake. On examination, the child was tachycardic with a gallop rhythm, tachypneic with reduced peripheral perfusion, and had weak peripheral pulses.
Born at term after an uneventful pregnancy with normal antenatal scans. Her perinatal and postnatal course were unremarkable. Parents reported no concerns with feeding or breathing prior to the ER visit.
The differential diagnosis included structural heart disease and myocarditis/cardiomyopathy. Tachycardia-induced cardiomyopathy was unlikely, given presenting sinus rhythm. Structural heart disease considerations included valvular aortic stenosis, coarctation, and anomalous left coronary artery from the pulmonary artery. There was no evidence of a valvular lesion and the coronaries were normal.
Chest X-ray (Fig. 1) demonstrated cardiomegaly with pulmonary edema. The electrocardiogram demonstrated T wave inversion in the left lateral leads. Troponin I was 0.48 ug/L (normal high <0.05) and BNP was >5000 ng/L (normal high <37). Echocardiography revealed severe left ventricular dysfunction from qualitative and quantitative assessment with an ejection fraction of 12%, with a dilated left ventricle (LVd z-score +5) and a right aortic arch. Echo imaging of the arch was challenging, however, there was evidence of right arch coarctation on the two-dimensional images and a mild diastolic tail on Doppler interrogation. There was a left sided vessel supplying the descending aorta with laminar flow. Despite these findings, there was no upper/lower limb blood pressure gradient and there was no brachiofemoral difference in palpation of the peripheral pulses. An estimate of the LV systolic pressure, obtained from the mitral regurgitation Doppler jet, when compared to a non-invasive peripheral systolic blood pressure, demonstrated a pressure difference of 50 mmHg between the LV and the peripheral blood pressure measured in the extremities.
Cardiac CT (Figs. 2–4) identified an ascending aorta that gave rise to dilated common carotid arteries and an extremely tortuous and stenotic right aortic arch. The right subclavian artery originated beyond the area of severe arch stenosis with an additional area of narrowing at its origin. Collateral vessels were seen in the right side of the neck. A long and relatively narrow right-sided dorsal aortic root passed in a caudal direction where it was joined by an equally long but larger left-sided dorsal aortic root reflecting ductal flow in utero. The dorsal roots fused below the level of the carina. The descending aorta was also right-sided but crossed to the left at the level of the diaphragm. The left-sided dorsal root remnant supplied an aberrant left subclavian artery, a dilated left vertebral artery, and collateral vessels in the left side of the neck. There was no evidence of a double aortic arch.
Both the right subclavian artery and aberrant left subclavian artery were distal to the coarctation, making the measurement of extremity blood pressures unhelpful and falsely reassuring in determination of an arch gradient. Only the carotid arteries were directly supplied from the ascending aorta. Simultaneous invasive measurements of pressures in the carotid and femoral arteries in the OR revealed a 60 mmHg arch gradient.
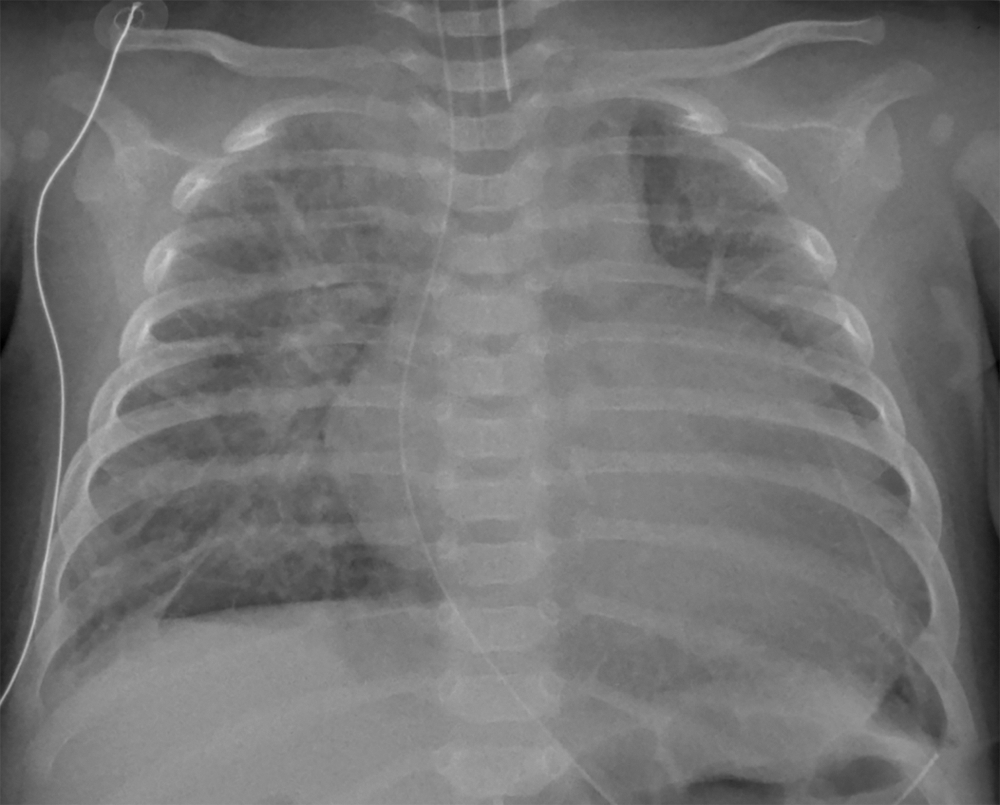
Figure 1: Chest X-ray on admission. Cardiomegaly with marked pulmonary interstitial edema and right basal linear atelectasis
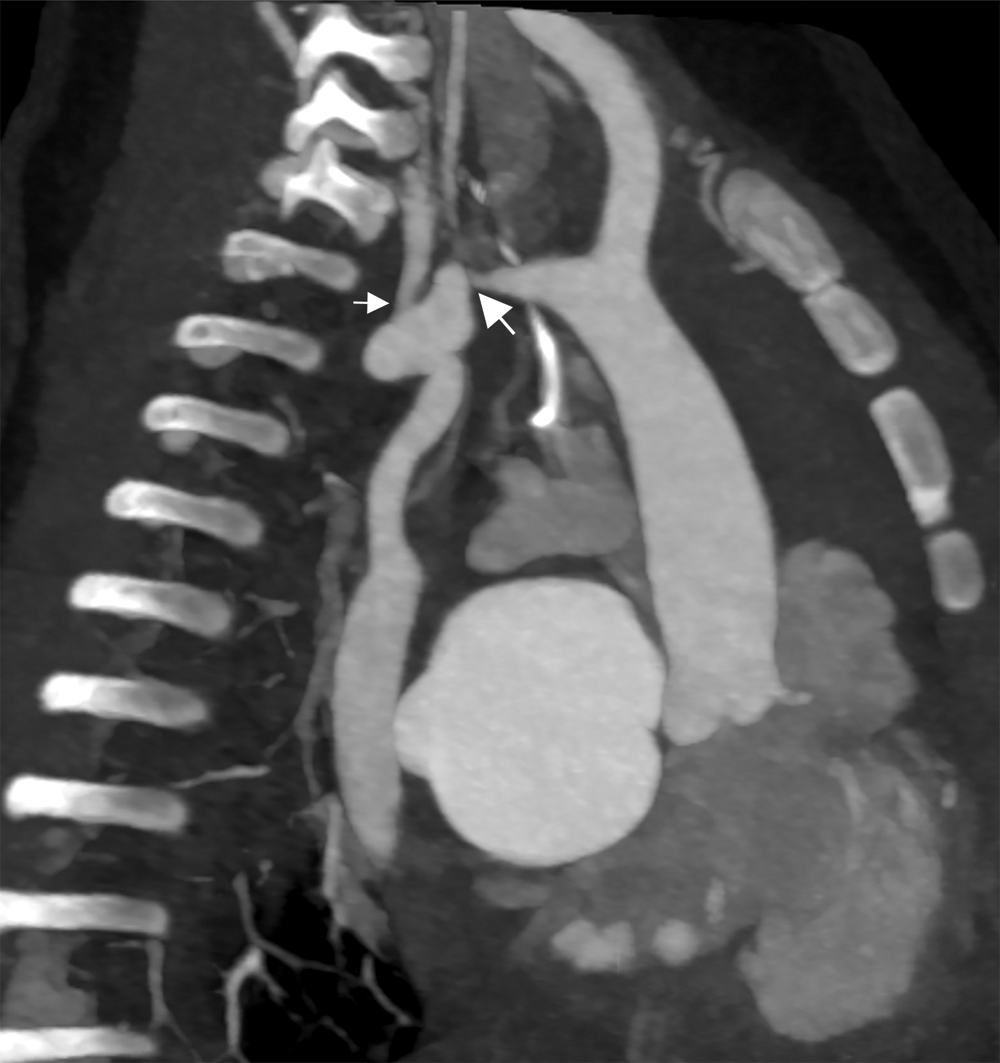
Figure 2: Cardiac CT—Sagittal oblique maximum intensity projection multiplanar reconstruction (MIP/MPR) on admission. Extremely tortuous and stenotic right aortic arch. Severe discrete coarctation (large arrow). The right subclavian artery originates beyond the area of severe arch stenosis with an additional area of narrowing at its origin (small arrow)
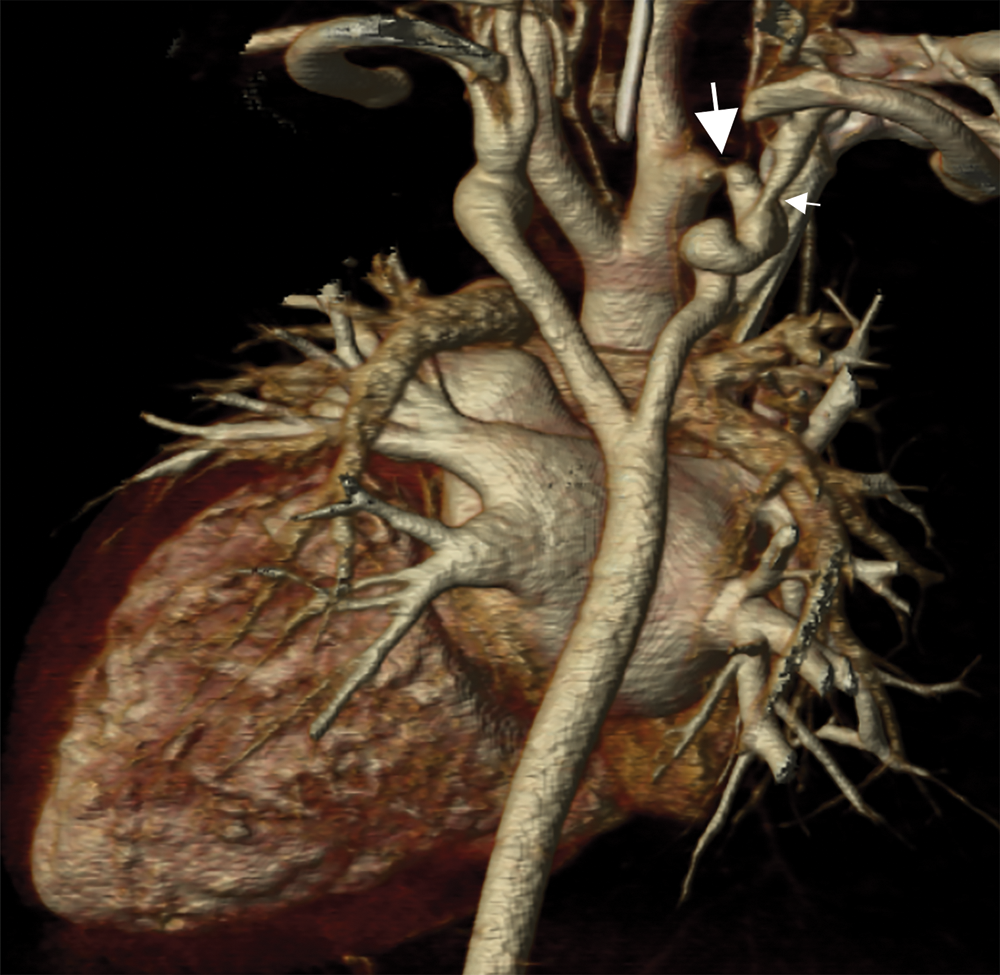
Figure 3: A coronal 3D reconstruction viewed from posterior, shows stenosis of the right sided arch (large arrow), and stenosis of the proximal right subclavian artery (small arrow). The dorsal aortic roots are long and the left sided dorsal root is larger than the right indicating ductal flow in utero
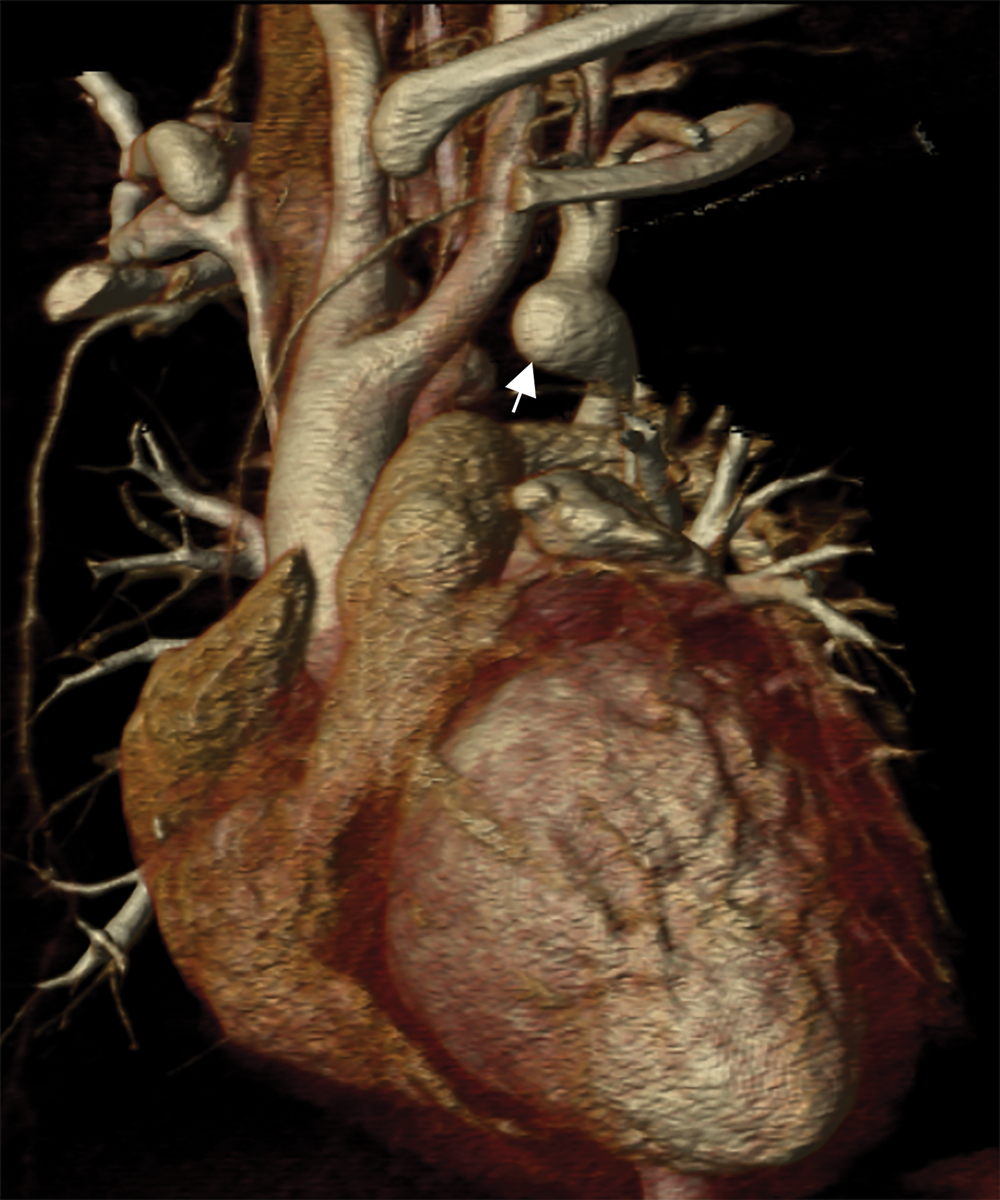
Figure 4: An oblique coronal 3D reconstruction viewed from anterior. The ascending aorta gives rise to the two dilated common carotid arteries. A large ductal remnant is present (arrow). The left subclavian is being supplied by the left-sided dorsal root remnant
To avoid the use of cardiopulmonary bypass in a baby with severe LV dysfunction, surgical management consisted of anastomosis of the left-sided dorsal aorta to the left common carotid artery to re-establish continuity between the ascending and descending aorta (Figs. 5 and 6 from the follow up CT). After completion of this anastomosis, there was a persistent 25 mmHg gradient present between the right common carotid artery and the femoral artery, which was attributed to the small size of the dorsal aorta. Therefore, the entire left-sided dorsal aorta was incised from its origin to its point of confluence with the right-sided dorsal aortic root and patch enlargement was performed. Direct inspection of the dorsal aorta revealed a macroscopically abnormal vessel. Postoperative transesophageal echocardiography showed improved left ventricular function.
The clinical suspicion and diagnosis of coarctation of the aorta depends primarily on the palpation of peripheral pulses, with differential blood pressures lending further support to the diagnosis. In rare cases, unusual arch anatomy can negate the standard diagnostic modalities. For example, when both subclavian arteries arise distal to the coarctation, pulses and blood pressure will be reduced but symmetric in both arms and legs. Another example than the one we describe is when the usual left sided juxtaductal coarctation involves the origin of the left subclavian artery and the right subclavian artery is aberrant [1,2]. A pulse discrepancy will only be apparent on comparison of the carotid pulses with the brachial or femoral pulses [3]. In an infant, qualitative assessment of the carotids is not routinely done.
To our knowledge, this type of complexity in the aortic arch malformation has only been described in patients with PHACE syndrome [4,5] (Posterior fossa abnormalities, Hemangiomas, Arterial abnormalities, Cardiac abnormalities, Eye abnormalities, Sternal cleft) and arterial tortuosity syndrome. Our case did not fulfil the criteria for PHACE syndrome [4]. Other vessels were not abnormal in morphology as expected in the arterial tortuosity syndrome [6]. This case is consistent with an isolated aortic arch abnormality, albeit a very complex one.
This case presented a diagnostic challenge on several levels. Firstly, the initial suspected diagnosis of typical coarctation did not fit the clinical scenario as the child’s pulses were weak but symmetrically weak in all the extremities. Although there were two dimensional and Doppler echocardiographic evidence of a coarctation, we could not identify the origin of the subclavian arteries adequately, impeding full comprehension of the lesion. Additionally, our initial invasive measurement of arm and leg blood pressures were falsely reassuring against coarctation. Ultimately, the combination of cross-sectional imaging and direct pressure measurement in the carotid artery provided us with definitive answers.
The patient’s postoperative course was uneventful. She was extubated on post-operative day 1 and transferred to the cardiac ward on post-operative day 2. She was discharged from the hospital on post-operative day 8 on anti-failure medication that was gradually weaned. At 3-month follow-up, the extended reconstruction is irregular and mildly narrowed with no evidence of recurrent obstruction and the right-sided dorsal root remnant has decreased in size following the repair (Figs. 5 and 6).
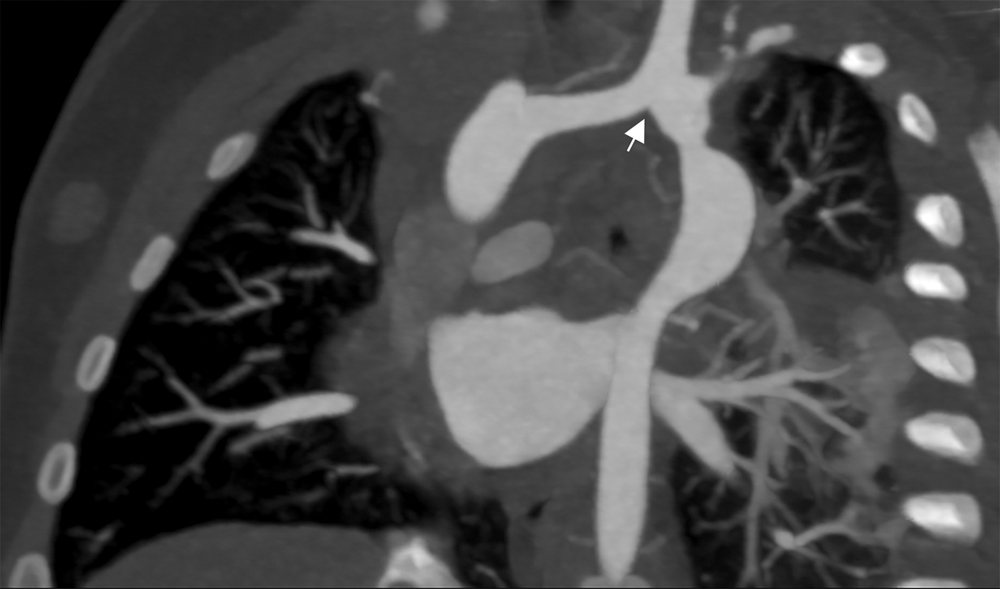
Figure 5: Cardiac CT coronal oblique view 3 months post-operatively. The reconstructed left-sided aortic arch consists of an anastomosis of the left-sided dorsal remnant to the left carotid artery after excision of the ductal remnant (arrow). Irregular left-sided dorsal root course with mild narrowing
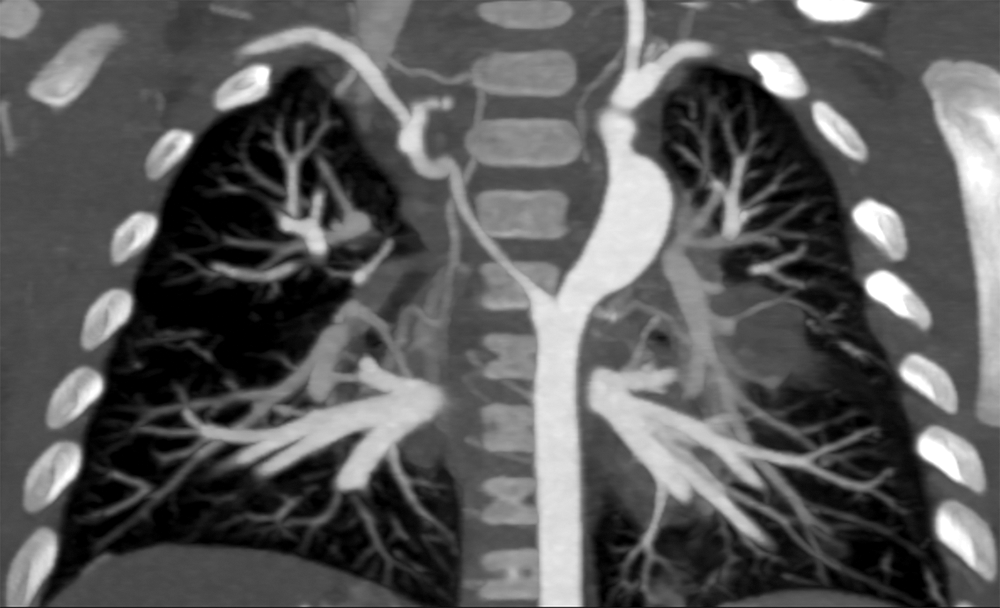
Figure 6: Cardiac CT coronal view 3 months post-operatively. Regression in size of the right-sided dorsal root. Reconstructed left-sided dorsal root is somewhat irregular with a point of mild narrowing
We report a case of unusual arch anatomy with coarctation of the aorta and severe LV dysfunction with both subclavian arteries distal to the coarctation and equal blood pressure in the extremities.
Acknowledgement: We would like to thank Dr. Jim Potts for his contribution in editing the images.
Ethics Statement: Permission for publication was granted from the patient’s guardian over the phone.
Funding Statement: The authors received no specific funding for this study.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Le Rous, B. T., Williams, M. A. (1968). An unusual aortic coarctation. Thorax, 23(6), 640–644. [Google Scholar]
2. Clagett, O. T., Kirklin, J. W., Edwards, J. E. (1954). Anatomic variations and pathologic changes in coarctation of the aorta: A study of 124 cases. Surgery, Gynecology & Obstetrics, 98(1), 103–114. [Google Scholar]
3. Phillips, R., Culham, G. (1997). Coarctation of the aorta. Stanley Baum’s, Abrams’ angiography, pp. 434–440. Philadelphia, USA: Little Brown & Co. [Google Scholar]
4. Garzon, M. C., Epstein, L. G., Heyer, G. L., Frommelt, P. C., Orbach, D. B. et al. (2016). PHACE syndrome: Consensus-derived diagnosis and care recommendations. Journal of Pediatrics, 178, 24–33. [Google Scholar]
5. Caragher, S. P., Scott, J. P., Siegel, D. H., Mitchell, M. E., Frommelt, P. C. et al. (2016). Aortic arch repair in children with PHACE syndrome. Journal of Thoracic and Cardiovascular Surgery, 152(3), 709–717. [Google Scholar]
6. Beyens, A., Albuisson, J., Boel, A., Al-Essa, M., Al-Manea, W. et al. (2018). Arterial tortuosity syndrome: 40 new families and literature review. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 20(10), 1236–1245. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |