| Congenital Heart Disease |
DOI: 10.32604/CHD.2021.015887
ARTICLE
Whole Exome Sequencing Identifies A Novel Pathogenic Bmpr2 Variant in Pulmonary Atresia
1Department of Cardiothoracic Surgery, Shanghai Children’s Hospital, Shanghai Jiaotong University, Shanghai, China
2Molecular Diagnostic Laboratory, Shanghai Children’s Hospital, Shanghai Jiaotong University, Shanghai, China
*Corresponding Authors: Li Shen. Email: shenli@shchildren.com.cn; Rufang Zhang. Email: zhangrf@shchildren.com.cn
Received: 25 January 2021; Accepted: 01 April 2021
#The authors contribute equally
Abstract: Objective: Pulmonary atresia (PA) is a rare type of complex cyanotic congenital heart defect characterized primarily by an undeveloped pulmonary valve or pulmonary artery. Therefore, defining a disease-causing gene mutation in a pulmonary atresia family is a possible method of genetic counseling, future prenatal diagnosis, and therapeutic approaches for pulmonary atresia. Methods: Blood samples were collected from six PA family members, and genomic DNA was extracted using the QIAamp DNA Blood Mini Kit. Gene detection was performed using a second-generation sequencing gene panel. Results: Genetic testing results indicated that a heterozygous mutation originating from maternal inheritance was detected in the BMPR2 gene of the proband’s genomic DNA. The pathogenic gene was c.2804C>T (p. A935V). The mutation was also detected in the genomic DNA of the proband’s elder brother (III-1), but not in other family members. Conclusion: To the best of our knowledge, this is the first study to report the BMPR2 variant responsible for pulmonary atresia. The frequency of the c.2804C>T (p. A935V) mutation detected in this family is extremely low in the normal population (1/246048). The mutation was highly conserved among different species. Sorting intolerant from tolerant (SIFT) predicts it to be a harmful mutation.
Keywords: Pulmonary atresia; gene mutation; BMPR2 gene
Congenital heart disease is the most common form of birth malformation. Pulmonary atresia is a rare type of complex cyanotic congenital heart defect characterized primarily by an undeveloped pulmonary valve or pulmonary artery, thus obstructing blood travel from the heart to pick up oxygen from the lungs. The morbidity of the disease is 1.3%–3.4% of all congenital heart malformations with poor prognosis [1]. Approximately 20% of children with pulmonary atresia with ventricular septal defects die within 2 years of age [2]. During fetal life, the development of prenatal ultrasound technology and a better understanding of the progression of pulmonary atresia have become crucial points for the choice of lifetime medical care, including multiple surgeries or other interventions. However, ultrasound examination could not detect all potentially effected children, especially those with major artery-pulmonary collateral arteries (MAPCAs) [3]. Under normal circumstances, patients do not suffer purely from PA. Usually, PA patients can survive with other heart diseases. These heart diseases create left to right shunts that make it possible for the blood to pick up oxygen from the lungs through natural passages between the heart or the arteries, such as ventricular septum defect, patent ductus arteriosus, and atrial septum defect. Otherwise, patients cannot survive. Accordingly, PA is divided into two categories: pulmonary atresia with ventricular septal defect (PA/VSD) and pulmonary atresia with intact ventricular septum (PA/IVS). The ventricular septal integrity of the PA is also known as right-sided heart dysplasia syndrome. Such patients often lack the right ventricular outflow tract, resulting in poor right ventricular development, which leads to a loss of opportunities for biventricular repair [4]. Therefore, children with PA often develop heart failure and cyanosis. Presently, PA is closely related to the heterozygous deletion of chromosome 22q11.2, and the range of the deletion was from 1437 Mbs to 2706 Mbs [5]. The ratio of deletion mutations among patients is approximately 30% to 40%. In children with PA with tetralogy of Fallot, the incidence of this heterozygous deletion is approximately 13% [6]. If the child has major artery-pulmonary collateral arteries (MAPCAs), the incidence of heterozygous deletion can be as high as 50% [7]. At present, the main treatment for PA is surgery. With the rise of 3D printing technology in recent years, personalized computer modeling of children with 3D printing technology has helped improve the accuracy of the surgical procedure, while significantly reducing intraoperative radiation and contrast load, and partly assists in reducing the complications it causes [8,9]. Although the surgical technique and perioperative management have improved, the prognosis of PA remains poor [10]. Therefore, screening for children with PA in the neonatal or even fetal period through genetic or biomarker methods will greatly increase the detection rate, so children with PA gain early diagnosis and treatment.
At present, several genetic variants have been found to be associated with the pathogenesis of PA, including chromosome copy number variations and single gene mutations. Chromosomal variations mainly include chromosome 22q11.2 heterozygous deletion, chromosome 1q21.1 heterozygous deletion, 16p13.1, 5q14.1, 5q14.1, 10p13 duplication, and 17q13.2 chromosome deletion. The related genes located in these CNVs including MYH11, ABCC6, NDE1, TBX1, DHFR, PDE88, AP3B, ARSB, DMGDH, CUBN, CAMTA2, CHRNE, GP18A, ENO, GJA5, GJA8, and BCL9 [5,11]. These genes were reported to be related to the metabolism of folate and vitamin B12 [12,13]. At the same time, several single gene mutation sites were also found, including GJA5, GJA1, GDF1, and MTHFR. [1]. Bone morphogenetic protein type 2 receptor (BMPR2) is a transforming growth factor-β superfamily receptor that plays diverse roles in various tissues and organs, including the pulmonary vascular endothelium, pulmonary vascular smooth muscle, vasculogenesis, and osteogenesis. So far, BMPR2 and its corresponding type 1 receptors both function in the development or differentiation of embryos, organs, and bones [14]. BMPR2 consists of 1,038 amino acids and functions in various fields such as extracellular, transmembrane, kinase, and C-terminal cytoplasmic domains [15]. Reports have shown that BMPR2 enhances almost every step of vascular development. Variations in the BMPR2 gene lead to various constructions and functions of the gene, and these changes are particularly associated with clinical disorders, including pulmonary arterial hypertension (PAH), cancers, obesity, and metabolic diseases [15–19]. BMPR2 plays important roles in various biological pathways and promotes vascular development; the canonical BMPR2-mediated signaling cascade is reported to be associated with human microvascular endothelial cells (HMVECs), human umbilical vein endothelial cells (HUVECs), and aortic endothelial cells [20].
In the present study, we collected a PA family with two affected and four unaffected members. To uncover novel pathogenic genes or variants, we performed whole exome sequencing of six family members. Using bioinformatics tools, we identified a novel variant at c.2804C>T (p. A935V) in BMPR2 as a disease-causing variant, which may function in heart development. To our knowledge, this is the first study to report BMPR2 as a disease-causing variant of heritable PA.
2.1 Clinical Features of PA Cases
This study collected a PA family from Anhui Province, China, which was admitted to the Department of Cardiothoracic Surgery, Shanghai Children’s Hospital. The symptoms of the PA family are presented in Tab. 1. The family had three generations and two patients. The proband (III-2) was a male infant weighing 3600 g born mature without any specific information about the disease on fetal screening echocardiography. He soon developed symptoms of cyanosis of the face and mouth after birth and was therefore admitted to a local hospital. Since the patient received anti-infective treatment, after which his symptoms were not relieved, he was referred to our hospital. Physical examination revealed cyanosis, wheezing, and shortness of breath, and continuous murmurs could be caught on the precordium. His percutaneous arterial oxygen saturation (SpO2) level in the right arm, measured using pulse oximetry, decreased to < 70% without oxygen uptake. The blood routine suggested a WBC 20.25 × 109/L (normal level: 8.00–12.00 × 109/L). Blood gas analysis indicated CO2 retention and low oxygen saturation. The echocardiogram also showed pulmonary atresia with an intact ventricular septum and patent ductus arteriosus. He was started on prostaglandin immediately after admission for ductal-dependent pulmonary blood flow. According to the medical history, the proband had a history of repeated upper respiratory tract infections and pneumonia. The proband was finally diagnosed with pulmonary atresia with an intact ventricular septum, patent ductus arteriosus, and atrial septal defect, which showed typical symptoms of this disease. He went to the operating room three times, after which he performed well with appropriate oxygen saturation.
Table 1: Symptoms presented in this PA family

Further family history research revealed that the proband’s elder brother (III-1) had similar symptoms (Fig. 1). The proband’s elder brother was diagnosed with pulmonary atresia with a ventricular septum defect based on the present history, relevant examination, and physical examination. The patient was also cured by surgery. Using whole exome sequencing, we found that the proband’s mother carried the mutant gene and presented related symptoms in our hospital. She presented with obesity (BMI = 26.03) and exhaustion after mild exercise or movement. It has been reported that mutations in BMPR2 could be associated with obesity, as well as several cardiovascular-related diseases, all of which mainly present exhaustion after movement.
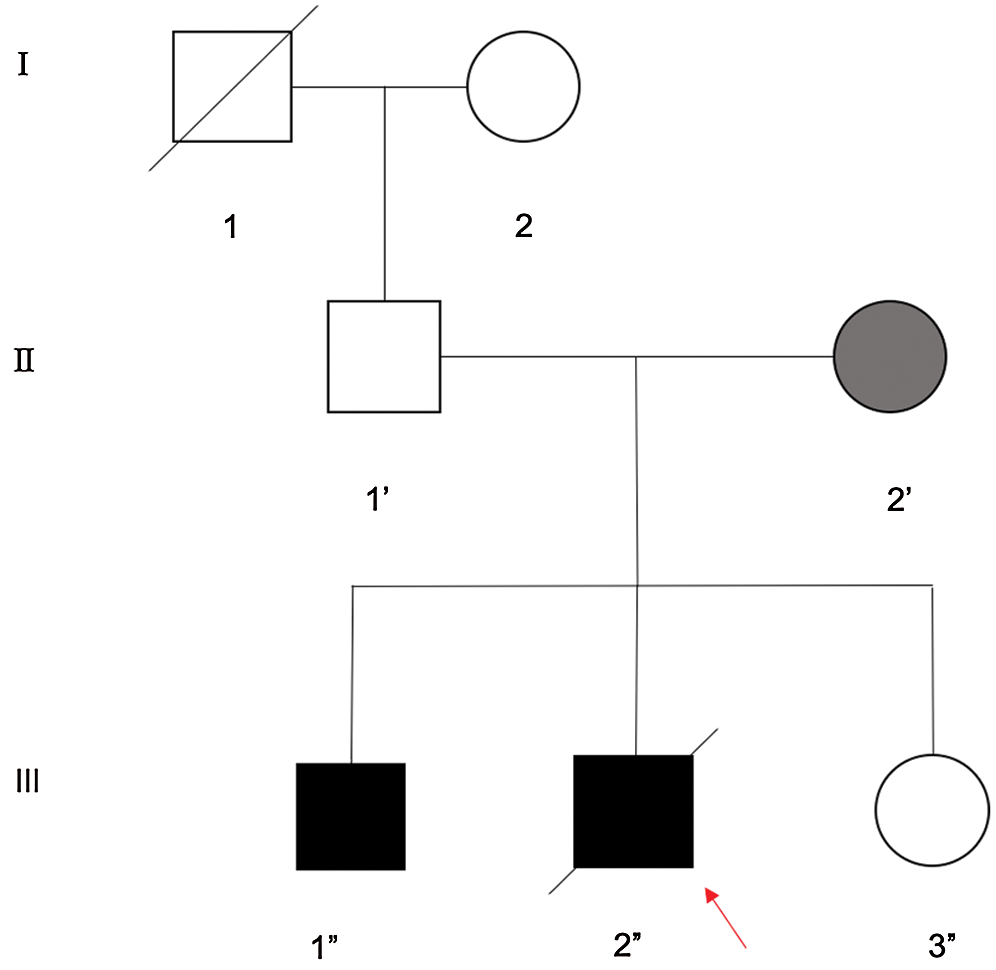
Figure 1: The pedigree of three generations of a PA family. Pedigree of a three-generation, Chinese family with two affected individuals. Squares indicate males and circles represent females. Black and white symbols represent affected and unaffected individuals, respectively. The proband (red arrow) is no longer alive (III-2”)
2.2.1 Diagnostic Criteria of PA Patients with Cardiac Color Doppler
According to the latest clinical guidelines, prenatal PA is mainly diagnosed using four-dimensional color Doppler ultrasound (Philips EPIQ7, Seattle, USA), presenting an abnormal performance. PA can be diagnosed early in the second phase of pregnancy (18–22 weeks) using this method. Such fetal four-dimensional color Doppler ultrasound abnormalities mainly appear in the pulmonary artery, tricuspid valve, and right ventricle. In addition, abnormal reflux between the various arteries of such fetuses can also be detected. Postpartum children are assessed by measuring size and function of the right ventricle, size of the tricuspid valve, and degree of reflux [21,22].
2.2.2 Physical Examination and Imaging Evaluation
Physical examination and imaging assessment were performed by two cardiothoracic surgeons and two imaging specialists in our hospital. Physical examination revealed cyanosis, wheezing, and shortness of breath, and continuous murmurs could be caught on the precordium. The relevant auxiliary examinations before surgery, including CT scan, electrocardiogram, and chest radiography, are presented in Fig. 2. After obtaining written consent and signatures from all participants or their legal guardians, venous blood was collected from six people in the family. The collection and use of patient biological samples was reviewed by the Shanghai Children’s Hospital Ethics Committee (No. 2019R001-E01).
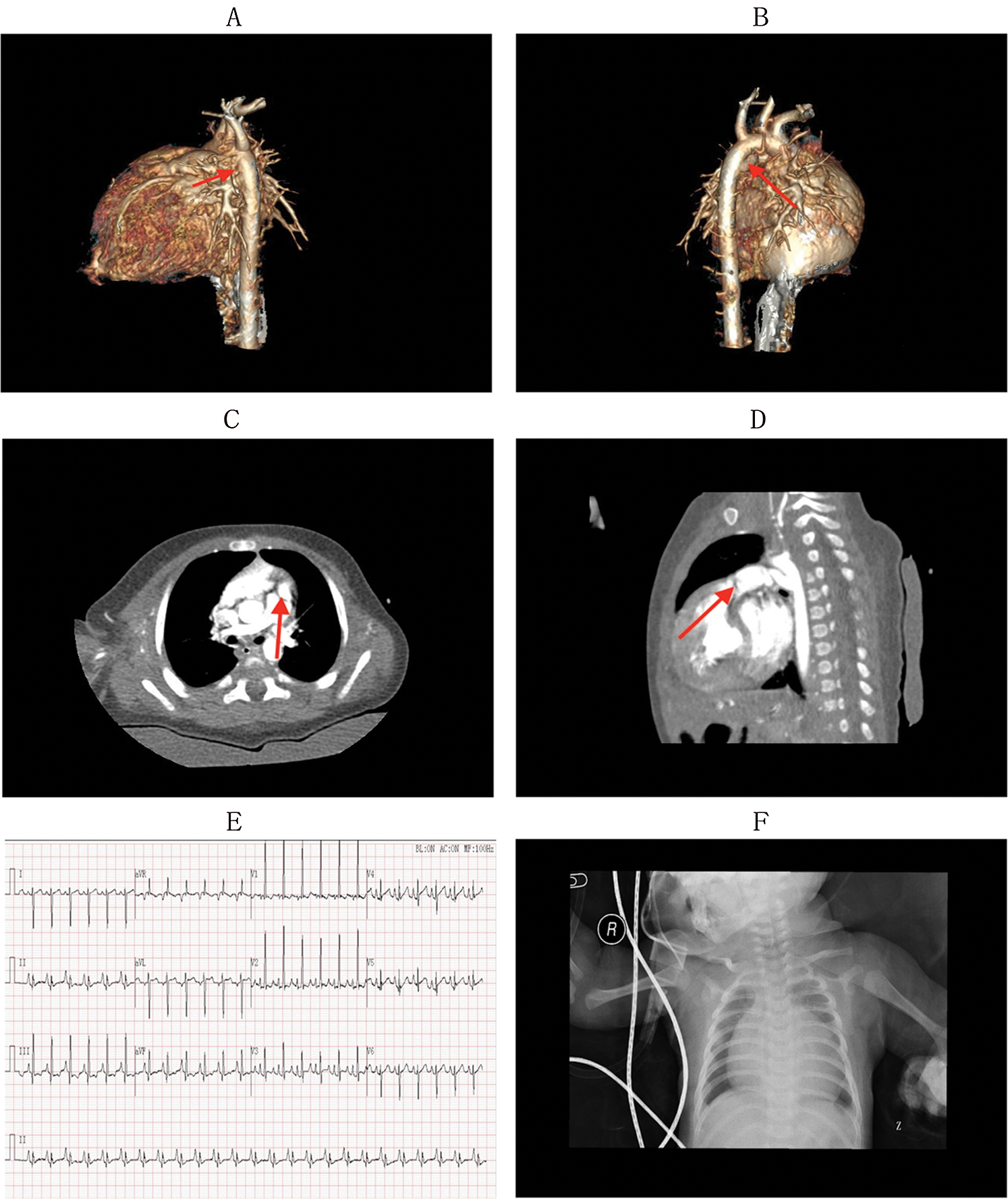
Figure 2: The relevant auxiliary examinations before surgery: CT scan, electrocardiogram, and X-ray of chest
Panels A–D show the CT scan before surgery (A: red arrow shows a narrow pulmonary artery of the proband; B: the red arrow shows the blood supply of the pulmonary artery was the descending aorta; C, D both show pulmonary atresia as well as an undeveloped pulmonary valve). Panel E shows the electrocardiogram of the proband before surgery, which presents an atrial hypertrophy, ventricular hypertrophy, change in ST and T waves. Panel F shows the X-ray of the chest of the proband before surgery, which presents an increased heart shadow. The cardiothoracic ratio of the proband was approximately 75.9%.
2.3 Whole Exome Sequencing and Sanger Sequencing Verification
2.3.1 Extraction of Genomic DNA
Genomic DNA was extracted from 2 mL peripheral blood samples from six family members, according to the manufacturer’s experimental procedure (QIAamp DNA Blood Mini Kit, QIAGEN, Dusseldorf, Germany). DNA degradation and RNA contamination were analyzed using agarose gel electrophoresis DNA purity was detected using a spectrophotometer. DNA concentrations were quantified. Finally, the samples were recorded and stored in a refrigerator at −80°C.
2.3.2 Methods of Whole Exome Sequencing and Identification of Variants
The SeqCap_EZ_ExomeV3 (Nimblegen, Roche, Basel, Switzerland) sequencing capture system was used to capture the entire exome sequencing coding region. Sequencing was performed using a HiSeqTM 2000(Illumina, California, USA), and the results were compared with reference to the human genome GRCh37.p5 (hg19). Ingenuity Variant Analysis (QIAGEN, Dusseldorf, Germany) was used with the variational annotation filtering procedure. Sanger sequencing was performed using a 3500DX Genetic Analyzer (ABI, ThermoFisher, Massachusetts, USA). Comparisons and analyses of the results were performed using Mutation Surveyor 4.0 (SoftGenetics, USA) software.
2.3.3 Sanger Sequencing Verification
To identify pathogenic variants, we evaluated the pathogenicity of the six dominant variants using bioinformatics tools, such as PCR amplification and Sanger sequencing, and mutation detection within the cases, including two patients with PA and five other PA-free relatives. In addition, we verified that this mutation co-segregated with the disease.
After DNA extraction using the method in step 1.3.1, array comparative genomic hybridization (aCGH) experiments were performed using Agilent Human Genome CGH 4 × 180 oligonucleotide arrays (Agilent, Santa Clara, CA, USA). These microarrays contain 180,000 probes with a 13 kb average probe spacing. Arrays were scanned with an Agilent scanner and analyzed with Cytogenomics 3.0.2.11 software (Agilent). The following databases were used for the analysis: DGV (http://dgv.tcag.ca), DECIPHER (https://decipher.sanger.ac.uk/), OMIM (https://www.omim.org), ClinGen (https://www.clinicalgenome.org/), Orphanet (https://www.orpha.net/consor/cgi-bin/index.php), and PubMed (https://www.ncbi.nlm.nih.gov).
In this study, two patients with PA and family members without PA were subjected to whole exome sequencing analysis. First, the standard karyotype of these family members revealed a normal 46, XY karyotype. Array-CGH performed on the proband with PA did not show any chromosomal imbalance. The original sequences obtained by sequencing each sample were finely filtered and subjected to subsequent bioinformatics analysis. The target area coverage was 99.41%, with an average coverage depth of >20×. The whole exome capture was successful, and sequencing quality was reliable. Two patients with PA and the proband’s mother, who was considered to be a carrier of the mutated gene, shared the mutant gene. The proband’s sister, who was considered a normal control, did not have the variation. Therefore, the mutant gene was considered a candidate gene mutation. In order to refine our list of mutations, we screened for mutations that were predicted to be functionally harmful by SIFT and CADD, which were not reported in any database. The CADD score was 22.8. According to the guidelines for genetic variant interpretations (ACMG) by Richards et al. [23], the mutation was variant uncertain significance (VUS). However, in this family, both children gained the disease, and the mother who carried the mutation developed cardiovascular disease symptoms. Furthermore, only one mutation was found in the family that was associated with this disease. Our test identified a novel homozygous missense variant c.2804C>T (p. A935V) in BMPR2. Moreover, the changes in the construction of the gene were proven to be associated with pulmonary diseases such as PAH; therefore, in our opinion, the mutation was associated with the disease.
Whole exome sequencing results demonstrated that the BMPR2 gene mutation may be related to the pathogenesis of PA. A heterozygous mutation in the BMPR2 gene c.2804C>T (p. A935V) was rarely found in the normal gnomAD database (1/246048). Mutation of BMPR2 was found in the blood samples of the two patients with PA and the proband’s mother. In addition, no BMPR2 mutations were found in the blood samples of other family members, who were treated as normal controls. The mutation is shown in Fig. 3.
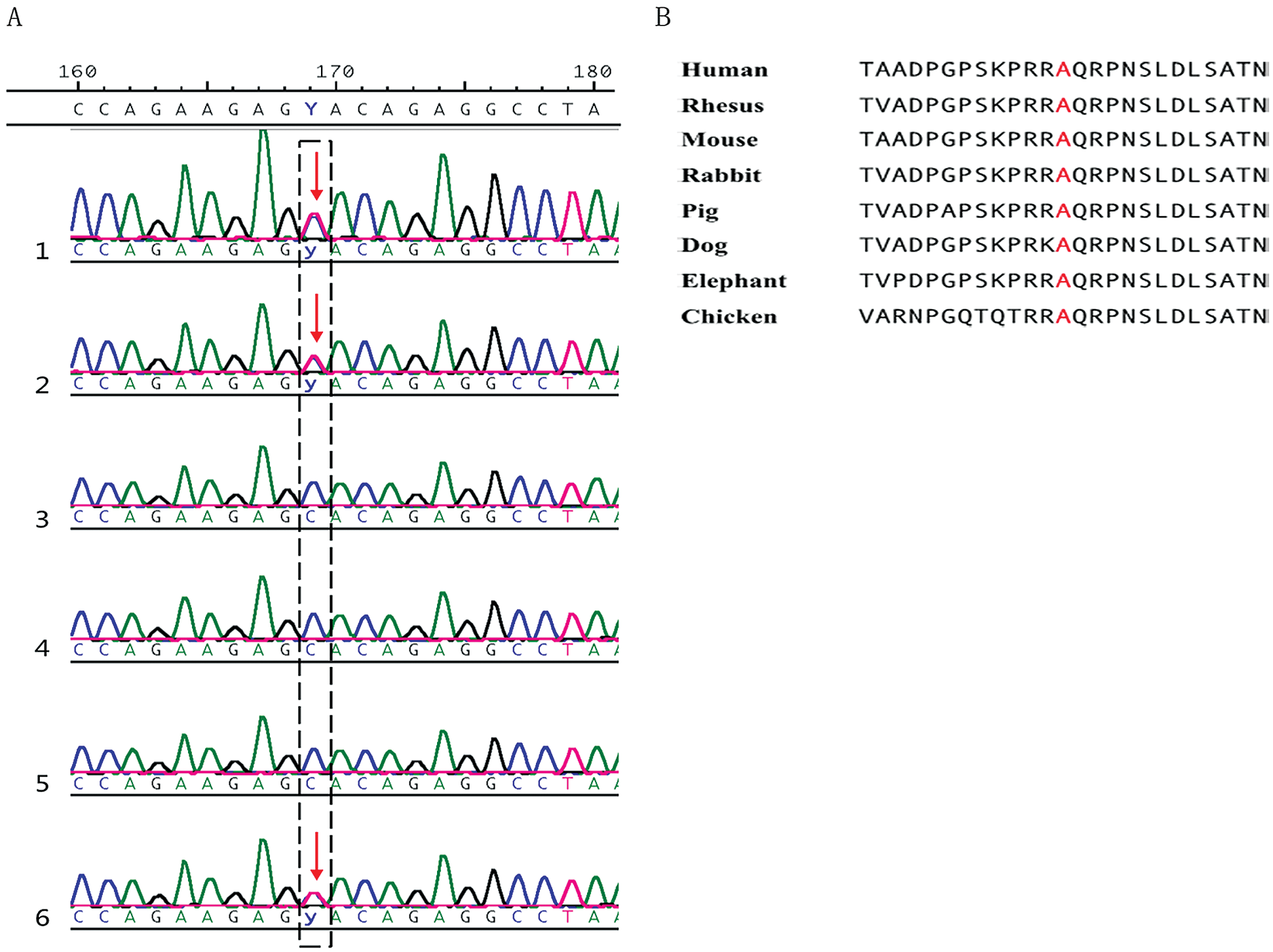
Figure 3: The description of a rare mutation in BMPR2 gene. (A). A mutation was found in the proband, proband’s mother, and proband’s elder brother (Lines 1, 2, and 6). Lines 1 through 6 represent the proband, the proband’s mother, the proband’s father, the proband’s sister, the proband’s grandfather, the proband’s grandmother, and the proband’s elder brother, respectively. (B). The mutation is highly conserved in various species
PA is a common complex cyanotic congenital heart disease. The etiology of PA is complex, and an increasing number of studies have shown that PA is primarily related to genetic factors. In this study, we performed whole exome sequencing of six members of a PA family to identify disease-causing genes. Epidemiological studies have shown that the incidence of congenital heart disease in congenital malformations is 28%, and the incidence of PA is 1.3%–3.4%. In recent years, in addition to the classic MYH6, HAND1, HAND2, and HEY family genes, new genes have been discovered. These genes, including CHD7, COL1A1, COL5A2, FBN2, NOTCH1, NSD1, and TSC2, as well as genes that only remain at the animal level, such as CHD (DGCR2, DAW1, LRP1, and MYH10), control the recessive inheritance of congenital heart diseases [1]. Bone morphogenetic protein (BMP) is a group of proteins that promotes bone formation and acts on cells such as osteoblasts, chondrocytes, and nerve cells that can affect embryonic growth and development [24]. BMP is a multifunctional cytokine belonging to the TGF-β superfamily. When BMP2 binds to the cell surface threonine/serine kinase receptor, it can play a regulatory role. The TGF superfamily and related enzymes transmit signals through two types of heteropolypeptide receptors: BMPR1 (approximately 50–55 kDa) and BMPR2 (approximately 70–80 kDa). BMPR1 can be activated by BMPR2 and stimulates Smads in the cytoplasm. BMPR1 also phosphorylates Smads, such as Smad1, Smad5, and Smad8, which promotes the binding between Smads and Smad4, and, finally, co-Smads participate in the regulation of target genes [25,26].
In recent years, the genetic pathogenesis of PA has been confirmed in several studies. In 2014, Xie et al. first discovered the relationship between copy number variation and PA [5]. These variations include 16p13.1, 22q11.2, 5q14.1, 10p1, and 17p13.2 [5]. There are four rare pathogenic mutations leading to PA, in which repeated mutations of DHFR5q14.1 and CUBN10p13 regulate folate-mediated metabolism, MTHFR regulates the metabolism of folic acid and vitamin B12, and 17q13.2 deletion regulates CAMT2 effects on NKX2.5 [27]. In 2012, Soemedi et al. first proposed that the 1q21.1 mutation is a possible pathogenesis of congenital heart disease [11]. Copy number variation analysis showed that most PA was caused by the GJA5 gene mutation. Similarly, the repetition of chromosomes such as 16p13.1, 5q14.1, 5q14.1, and 10p13 and the deletion of the 17q13.2 chromosome may also result in PA [28]. In recent years, single gene locus research has found that GJA5, GJA1, GDF1, MTHFR, etc. [7,29] may be related to the pathogenesis of PA. In 1995, Rosenzweig et al. first discovered that transfected COS-1 cells, BMP7, and BMP4 (112262) were linked to BMPR2 [30]. The chemical bonds between these connections were only stable in the presence of BMPR1. Most studies have found that mutations in the BMPR2 gene located on chromosome 2q33 are associated with the pathogenesis of most cardiovascular diseases, including familial pulmonary hypertension (PAH) and pulmonary veno-occlusive disease (PVOD) [31]. Recently, Oriaku et al. [32] discovered a novel mutation in BMPR2 (exon 6; c.712C>T; p.Gln238*) in two unrelated patients with different clinical phenotypes. One had pulmonary arterial hypertension and the other pulmonary veno-occlusive disease/pulmonary capillary hemangiomatosis, indicating the variable involvement of pulmonary BMPR2 mutation. Moreover, Lyu et al. [33] compared the features of rare BMPR2 variants in pulmonary arterial hypertension, which showed that the variants in the extracellular ligand-binding domain altered the amino acid electric status and are more pathogenic. Interestingly, it has been reported that the dysfunction of BMPR2 resulting from DNA mutation could alter glucose-supported mitochondrial respiration and impair cellular responses to insulin in cardiomyocytes [34], which is a potential mechanism of BMPR2 mutation in pulmonary disease. Certain deletions of the BMPR2 gene have been shown to trigger atherosclerosis in animal experiments [35]. Some of the diseases caused by genes that interact with BMPR2 include Prader-Willi syndrome caused by the action of NIPA1 [36].
In this study, we performed whole exome sequencing, bioinformatics analysis, and Sanger sequencing for a PA family of three generations. It was found that unreported new mutations in BMPR2 may be related to the pathogenesis of PA in this family. This family demonstrates the genetic basis of PA pathogenesis, which increases the genetic profile of PA. It has been shown that BMPR2 is prominently expressed during vascular development and is associated with the canonical Wnt signaling pathway. In addition to PA, the mutation could also lead to typical cardiovascular diseases, such as PAH and obesity [37]. Both the proband and the proband’s brother developed the disease, and the proband’s mother, who carries the gene mutation, also presents with typical symptoms, including obesity and other cardiovascular disease symptoms. According to these results, the proposed mechanism of the occurrence of PA by BMPR2 mutation is shown in Fig. 4.
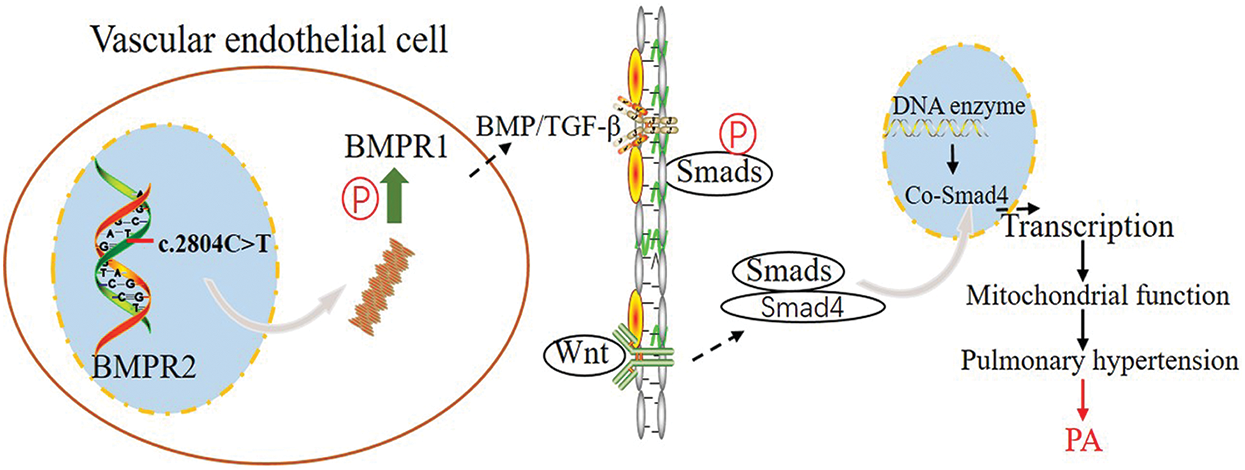
Figure 4: Graphic illustrating the pathways that contribute to PA. Mutated BMPR2 altered BMP signal transduction, further affecting the downstream mechanism of PA. P: Phosphorylation; PA: pulmonary atresia
The study of these genes is either in the screening stage or in the animal experiment stage, but the correlation between them and the pathogenesis of PA is still unclear [35–38]. This study aimed to verify the pathogenesis of PA by performing whole exome sequencing and find candidate mutations in a PA family. In this study, we performed high-throughput second-generation sequencing technology for whole exome sequencing of patient samples. Whole exome sequencing uses target sequence capture technology to capture high-throughput DNA from all exome regions of the genome, which not only can quickly detect the pathogenic genes of rare genetic diseases, but can also be used for common diseases caused by multiple genes. This reveals the genetic pathogenesis of these diseases.
In summary, the clinical manifestations, diagnostic criteria, and treatment options of PA are relatively clear, and the concept that genetic factors play an important role in the pathogenesis of PA is also recognized by most clinicians and researchers. Gene mutations are associated with PA. However, the pathogenesis of PA remains unclear. This study found that in a PA family, the BMP signaling pathway is possibly associated with cardiac development and confirmed that mutations in this gene may be closely related to the pathogenesis of PA in this family. However, it is also necessary to verify the overall prevalence of mutations in the BMPR2 gene in the PA population in a large number of sporadic PA cases and PA families. Further in vivo and in vitro experiments are needed to determine whether the mutation leads to the pathogenesis of PA and its pathogenesis, and to elucidate the association between genotype and phenotype. In addition, this study also suggests that the BMP family may be an important signaling molecule involved in the pathogenesis of PA. The BMP signaling pathway may be a breakthrough in the pathogenesis of PA.
Here, we report a clinical case of rare PA in a Chinese family. This case highlights the importance of clinical diagnosis of PA associated with mutations in BMPR2. Hence, molecular genetic screening of the BMPR2 gene could be used to confirm the clinical diagnosis of PA. Our present study also emphasizes the significance of high-quality genetic analysis by whole exome sequencing in the molecular diagnosis of rare congenital heart diseases with possible phenotypic heterogeneity.
Acknowledgement: We sincerely thank the patient and his family members for their participation. This work was supported by Shanghai Children’s Hospital.
Availability of Data and Materials: Data that support the findings of this study are available from the corresponding author upon reasonable request.
Funding Statement: Chinese National Natural Science Foundation, No. 81371499. The sponsor of this fund is also the corresponding author of this article. The role of each author: MQ: drafting and perfecting the manuscript; XL: genetic analysis; LS, RZ, JL, and JG: patient workup. MQ, XL, LS, RZ, JL, and JG: final approval for the version to be published and agreement to be accountable for all aspects of the work.
Conflicts of Interest: The authors declare that they have no competing interests.
1. Gao, M., He, X., Zheng, J. (2017). Advances in molecular genetics for pulmonary atresia. Cardiology in the Young, 27(2), 207–216. DOI 10.1017/S1047951116001487. [Google Scholar] [CrossRef]
2. Elias, P., Poh, C. L., du Plessis, K., Zannino, D., Rice, K. et al. (2018). Long-term outcomes of single-ventricle palliation for pulmonary atresia with intact ventricular septum: Fontan survivors remain at risk of late myocardial ischaemia and death. European Journal of Cardio-Thoracic Surgery: Official Journal of the European Association for Cardio-thoracic Surgery, 53(6), 1230–1236. DOI 10.1093/ejcts/ezy038. [Google Scholar] [CrossRef]
3. Yang, S. H., Luo, P. H., Tian, X. X., Li, X. Y., Li, X. Q. et al. (2018). Prenatal diagnosis of pulmonary atresia with ventricular septal defect. Journal of Medical Ultrasonics, 45(2), 341–344. DOI 10.1007/s10396-017-0809-2. [Google Scholar] [CrossRef]
4. Gómez, Olga, Martinez, J. M. (2018). Obstetric imaging: fetal diagnosis and care, pp. 373–377.e1. Pulmonary Stenosis and Atresia. [Google Scholar]
5. Xie, L., Chen, J. L., Zhang, W. Z., Wang, S. Z., Zhao, T. L. et al. (2014). Rare de novo copy number variants in patients with congenital pulmonary atresia. PLoS One, 9(5), e96471. DOI 10.1371/journal.pone.0096471. [Google Scholar] [CrossRef]
6. Mercer-Rosa, L., Elci, O. U., Pinto, N. M., Tanel, R. E., Goldmuntz, E. (2018). 22q11.2 deletion status and perioperative outcomes for tetralogy of fallot rare de novo copy number variants in patients with congenital pulmonary atresia with Pulmonary Atresia and Multiple Aortopulmonary Collateral Vessels. Pediatric Cardiology, 39(5), 906–910. DOI 10.1007/s00246-018-1840-9. [Google Scholar] [CrossRef]
7. Guida, V., Ferese, R., Rocchetti, M., Bonetti, M., Sarkozy, A. et al. (2013). A variant in the carboxyl-terminus of connexin 40 alters GAP junctions and increases risk for tetralogy of Fallot. European Journal of Human Genetics: EJHG, 21(1), 69–75. DOI 10.1038/ejhg.2012.109. [Google Scholar] [CrossRef]
8. Sun, X., Meng, Y., You, T., Li, P., Wu, H. et al. (2013). Association of growth/differentiation factor 1 gene polymorphisms with the risk of congenital heart disease in the Chinese Han population. Molecular Biology Reports, 40(2), 1291–1299. DOI 10.1007/s11033-012-2172-0. [Google Scholar] [CrossRef]
9. Capelli, C., Sauvage, E., Giusti, G., Bosi, G. M., Ntsinjana, H. et al. (2018). Patient-specific simulations for planning treatment in congenital heart disease. Interface Focus, 8(1), 20170021. DOI 10.1098/rsfs.2017.0021. [Google Scholar] [CrossRef]
10. He, X., Gao, B., Shi, G., Chen, H., Du, X. et al. (2018). Surgical strategy and outcomes for the delayed diagnosis of pulmonary atresia with intact ventricular septum. Journal of Cardiology, 72(1), 50–55. DOI 10.1016/j.jjcc.2017.12.009. [Google Scholar] [CrossRef]
11. Soemedi, R., Topf, A., Wilson, I. J., Darlay, R., Rahman, T. et al. (2012). Phenotype-specific effect of chromosome 1q21.1rearrangements and GJA5 duplications in 2436 congenital heart disease patients and 6760 controls. Human Molecular Genetics, 21(7), 1513–1520. DOI 10.1093/hmg/ddr589. [Google Scholar] [CrossRef]
12. Kozyraki, R. (2001). Cubilin, a multifunctional epithelial receptor: An overview. Journal of Molecular Medicine-Jmm, 79(4), 161–167. DOI 10.1007/s001090100193. [Google Scholar] [CrossRef]
13. Tsaroucha, A. K., Chatzaki, E., Lambropoulou, M., Despoudi, K., Laftsidis, P. et al. (2008). Megalin and cubilin in the human gallbladder epithelium. Clinical and Experimental Medicine, 8(3), 165–170. DOI 10.1007/s10238-008-0174-y. [Google Scholar] [CrossRef]
14. Zhang, D., Mehler, M. F., Song, Q., Kessler, J. A. (1998). Development of bone morphogenetic protein receptors in the nervous system and possible roles in regulating trkC expression. Journal of Neuroscience, 18(9), 3314–3326. DOI 10.1523/JNEUROSCI.18-09-03314.1998. [Google Scholar] [CrossRef]
15. Kim, M. J., Park, S. Y., Chang, H. R., Jung, E. Y., Munkhjargal, A. et al. (2017). Clinical significance linked to functional defects in bone morphogenetic protein type 2 receptor, BMPR2. BMB Reports, 50(6), 308–317. DOI 10.5483/BMBRep.2017.50.6.059. [Google Scholar] [CrossRef]
16. Orriols, M., Gomez-Puerto, M. C., Ten Dijke, P. (2017). BMP type II receptor as a therapeutic target in pulmonary arterial hypertension. Cellular and Molecular Life Sciences, 74(16), 2979–2995. DOI 10.1007/s00018-017-2510-4. [Google Scholar] [CrossRef]
17. Garcia-Rivas, G., Jerjes-Sanchez, C., Rodriguez, D., Garcia-Pelaez, J., Trevino, V. (2017). A systematic review of genetic mutations in pulmonary arterial hypertension. BMC Medical Genetics, 18(1), 1655. DOI 10.1186/s12881-017-0440-5. [Google Scholar] [CrossRef]
18. Hardwick, J. C., Kodach, L. L., Offerhaus, G. J., van den Brink, G. R. (2008). Bone morphogenetic protein signalling in colorectal cancer. Nature Reviews Cancer, 8(10), 806–812. DOI 10.1038/nrc2467. [Google Scholar] [CrossRef]
19. Kim, H. H., Hyung, W. J., Cho, G. S., Kim, M. C., Han, S. U. et al. (2010). Morbidity and mortality of laparoscopic gastrectomy versus open gastrectomy for gastric cancer: An interim report–A phase III multicenter, prospective, randomized Trial (KLASS Trial). Annals of Surgery, 251(3), 417–420. DOI 10.1097/SLA.0b013e3181cc8f6b. [Google Scholar] [CrossRef]
20. Finkenzeller, G., Hager, S., Stark, G. B. (2012). Effects of bone morphogenetic protein 2 on human umbilical vein endothelial cells. Microvascular Research, 84(1), 81–85. DOI 10.1016/j.mvr.2012.03.010. [Google Scholar] [CrossRef]
21. Todros, T., Paladini, D., Chiappa, E., Russo, M. G., Gaglioti, P. et al. (2003). Pulmonary stenosis and atresia with intact ventricular septum during prenatal life. Ultrasound in Obstetrics and Gynecology, 21(3), 228–233. DOI 10.1002/uog.63. [Google Scholar] [CrossRef]
22. Maeno, Y. V., Boutin, C., Hornberger, L. K., McCrindle, B. W., Cavalle-Garrido, T. et al. (1999). Prenatal diagnosis of right ventricular outflow tract obstruction with intact ventricular septum, and detection of ventriculocoronary connections. Heart, 81(6), 661–668. DOI 10.1136/hrt.81.6.661. [Google Scholar] [CrossRef]
23. Richards, C. S., Bale, S., Bellissimo, D. B., Das, S., Grody, W. W. et al. (2008). ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 10(4), 294–300. DOI 10.1097/GIM.0b013e31816b5cae. [Google Scholar] [CrossRef]
24. Zhao, L., Zhou, Z., Wang, S., Jiao, Q., Wu, J. et al. (2017). A recurrent mutation in bone morphogenetic proteins-2-inducible kinase gene is associated with developmental dysplasia of the hip. Experimental and Therapeutic Medicine, 13(5), 1773–1778. DOI 10.3892/etm.2017.4191. [Google Scholar] [CrossRef]
25. Miyazono, K., Kusanagi, K., Inoue, H. (2001). Divergence and convergence of TGF-beta/BMP signaling. Journal of Cellular Physiology, 187(3), 265–276. DOI 10.1002/jcp.1080. [Google Scholar] [CrossRef]
26. Luo, K. (2017). Signaling cross talk between TGF-β/Smad and other signaling pathways. Cold Spring Harbor Perspectives in Biology, 9(1), a022137. DOI 10.1101/cshperspect.a022137. [Google Scholar] [CrossRef]
27. Song, K., Backs, J., McAnally, J., Qi, X., Gerard, R. D. et al. (2006). The transcriptional coactivator CAMTA2 stimulates cardiac growth by opposing class II histone deacetylases. Cell, 125(3), 453–466. DOI 10.1016/j.cell.2006.02.048. [Google Scholar] [CrossRef]
28. Tomita-Mitchell, A., Mahnke, D. K., Struble, C. A., Tuffnell, M. E., Stamm, K. D. et al. (2012). Human gene copy number spectra analysis in congenital heart malformations. Physiological Genomics, 44(9), 518–541. DOI 10.1152/physiolgenomics.00013.2012. [Google Scholar] [CrossRef]
29. Izumi, K., Lippa, A. M., Wilkens, A., Feret, H. A., McDonald-McGinn, D. M. et al. (2013). Congenital heart defects in oculodentodigital dysplasia: Report of two cases. American Journal of Medical Genetics Part A, 161(12), 3150–3154. DOI 10.1002/ajmg.a.36159. [Google Scholar] [CrossRef]
30. Rosenzweig, B. L., Imamura, T., Okadome, T., Cox, G. N., Yamashita, H. et al. (1995). Cloning and characterization of a human type II receptor for bone morphogenetic proteins. Proceedings of the National Academy of Sciences of the United States of America, 92(17), 7632–7636. DOI 10.1073/pnas.92.17.7632. [Google Scholar] [CrossRef]
31. Girerd, B., Lau, E., Montani, D., Humbert, M. (2017). Genetics of pulmonary hypertension in the clinic. Current Opinion in Pulmonary Medicine, 23(5), 386–391. DOI 10.1097/MCP.0000000000000414. [Google Scholar] [CrossRef]
32. Oriaku, I., LeSieur, M. N., Nichols, W. C., Barrios, R., Elliott, C. G. et al. (2020). A novel BMPR2 mutation with widely disparate heritable pulmonary arterial hypertension clinical phenotype. Pulmonary Circulation, 10(3), 2045894020931315. [Google Scholar]
33. Lyu, Z. C., Wang, L., Lin, J. H., Li, S. Q., Wu, D. C. et al. (2020). The features of rare pathogenic BMPR2 variants in pulmonary arterial hypertension: Comparison between patients and reference population. International Journal of Cardiology, 318, 138–143. DOI 10.1016/j.ijcard.2020.06.068. [Google Scholar] [CrossRef]
34. Hemnes, A. R., Fessel, J. P., Chen, X., Zhu, S., Fortune, N. L. et al. (2020). BMPR2 dysfunction impairs insulin signaling and glucose homeostasis in cardiomyocytes. American Journal of Physiology Lung Cellular and Molecular Physiology, 318(2), L429–L441. DOI 10.1152/ajplung.00555.2018. [Google Scholar] [CrossRef]
35. Lu, H., Daugherty, A. (2015). Atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology, 35(3), 485–491. DOI 10.1161/ATVBAHA.115.305380. [Google Scholar] [CrossRef]
36. Tsang, H. T. H., Edwards, T. L., Wang, X., Connell, J. W., Davies, R. J. et al. (2009). The hereditary spastic paraplegia proteins NIPA1, spastin and spartin are inhibitors of mammalian BMP signalling. Human Molecular Genetics, 18(20), 3805–3821. DOI 10.1093/hmg/ddp324. [Google Scholar] [CrossRef]
37. Schulz, T. J., Tseng, Y. H. (2009). Emerging role of bone morphogenetic proteins in adipogenesis and energy metabolism. Cytokine & Growth Factor Reviews, 20(5–6), 523–531. DOI 10.1016/j.cytogfr.2009.10.019. [Google Scholar] [CrossRef]
38. Jin, S. C., Homsy, J., Zaidi, S., Lu, Q., Morton, S. et al. (2017). Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nature Genetics, 49(11), 1593–1601. DOI 10.1038/ng.3970. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |