| Congenital Heart Disease |
DOI: 10.32604/CHD.2021.015831
ARTICLE
miR-208a Promotes Apoptosis in H9c2 Cardiomyocytes by Targeting GATA4
1Department of Medical Genetics, School of Life Sciences, China Medical University, Shenyang, China
2Department of Laboratory Medicine, The People’s Hospital of Liaoning Province, Shenyang, China
3Department of Laboratory Medicine, The People’s Hospital of China Medical University, Shenyang, China
4Department of Pediatrics, The First Affiliated Hospital of China Medical University, Shenyang, China
*Corresponding Author: Kailai Sun. Email: sunkailai@163.com
Received: 19 January 2021; Accepted: 26 February 2021
Abstract: Background: microRNAs are crucial for cardiovascular development and are associated with congenital heart disease (CHD). Recent studies have shown that microRNAs play a role in heart development and is closely related to CHD. The present study investigated the underlying mechanism of microRNA-208a (miR-208a) in “simple” CHD. Material and Methods: Reverse transcription-quantitative PCR (RT-qPCR) demonstrated miR-208a expression levels in children with CHD (n = 27) compared with normal controls (n = 29), in cardiomyocytes from embryo 10 (E10) to post-birth (P7) and organs in adult rats in healthy rats. Apoptosis of H9c2 cells after transfection with miR-208a detected by TUNEL assay. B-cell lymphoma (Bcl)-2, an anti-apoptotic gene, was detected by RT-qPCR, as well as Gata4. After 48h overexpression of miR-208a, GATA4 was detected via western blotting. Dual luciferase reporting system was used to identify the binding sites of miR-208a to Gata4. Results: Expression of miR-208a was upregulated in the CHD group via the control group (p < 0.01). At P7, miR-208a had the highest expression (p < 0.01), and which was highest in myocardiocytes via other organs or tissues (p < 0.01) in adult rats. The number of apoptotic cells increased significantly post-transfection with miR-208a (p < 0.01), while decreased with the miR-208a inhibitor via the control group (p < 0.01). Compared with the control group, there was no significant difference in the expression level of Bcl-2 after miR-208a overexpression (p > 0.05). The present study proved that miR-208a binds directly to the 3´-UTR of Gata4 at site 1,363-1,369 bp. Expression of GATA4 decreased after miR-208a overexpression (p < 0.01), but increased following transfection with a miR-208a inhibitor via the control group (p < 0.05). Conclusions: Our study demonstrated that miR-208a downregulates Bcl-2 by directly targeting GATA4, which may cause CHD. miR-208a may become a new biomarker or therapeutic target for CHD in the future.
Keywords: Simple CHD; miR-208a; GATA4; apoptosis; Bcl-2
“Simple” CHD involves only cardiovascular malformations and no congenital anomalies in other systems. It accounts for 70%–80% of the total number of CHD cases [1]. Genetic factors play important roles in CHD pathogenesis, accounting for 55%–65% of cases in 2008 [2–5]. Deletion of Dicer in cardiomyocytes [6] and in vascular smooth muscle [7], as well as deletion of DiGeorge syndrome critical region gene 8 (Dgcr 8) [8] leads to death during pregnancy and post-natal events, which suggests that microRNA (miRNA or miR) is essential for cardiomyocyte development. Dicer [6,9] can specifically cleave and phosphorylate long non-coding RNA and act on miRNA. DGCR 8 is an important protein involved in the synthesis and maturation of miRNA, and regulates the generation of miRNA, which in turn affects the regulatory effect of miRNA on genes [8].
miRNAs are a class of small endogenous non-coding RNAs. They negatively regulate expression of target genes at the post-transcriptional level to achieve gene silencing by binding to the 3'-UTR [10]. It has been suggested that miRNA is closely associated with atrioventricular septal defects (ASD), ventricle formation, arrhythmia, hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM) and remodeling after myocardial damage [4,9,11–13]. miR-208a encoded by intron 29 of myosin heavy chain 6 (Myh 6) and co-transcribed with its host gene. Upregulation of miR-208a in myocardial cells leads to cardiac hypertrophy [14]. However, whether it plays a role in CHD and what its target gene is remains to be elucidated. According to the database TargetScan GATA4 may be a target gene of miR-208a. GATA4 is involved in embryogenesis and in myocardial differentiation and function. Numerous studies have demonstrated that mutations in this gene have been associated with CHD [15,16]. Low GATA4 expression in embryonic cardiomyocytes can cause myocardial hypoplasia, and even abortion [17]. Considering the model of miRNAs regulating target genes, miRNA(s) may participate in CHD [18] pathogenesis by regulating the expression of GATA4, which indicates that mir-208a plays a role in CHD development through regulation of GATA4.
In the present study, overexpression of miR-208a promoted H9c2 cardiomyocyte apoptosis by targeting GATA4. Above all, miR-208a may play a critical role in CHD pathogenesis.
2.1 Ethical Approval of the Study Protocol
The study protocol was approved by the Ethics Committee of the General Hospital of Shenyang Military Region Shenyang, China.
Plasma from 27 children with CHD (12 males and 15 females) and 29 normal controls (13 males and 16 females) without CHD were provided by the General Hospital of Shenyang Military Region. The characteristics of these two groups are presented in Tab. 1. The diagnosis was confirmed by color Doppler ultrasound and/or surgery. All subjects were aged 1–18 years. All human samples were anti-coagulated with ethylenediamine tetra-acetic acid and centrifuged immediately at 1,600 × g for 15 min to separate the plasma.
Table 1: Clinical findings of patients with CHD and controls

2.2.2 Organ and Tissue Acquisition from Rats
Adult Wistar rats were purchased from the Experimental Animal Center of China Medical University, and the experimental process complied with animal protection regulations. 90 to 110-day-old male rats and 80 to 100-day-old female rats were caged together overnight. The next morning, the vaginal secretions of female rats were observed using a light microscope. If sperm were observed, it was considered to be E0.5 pregnant. Pregnant at E10 and E14 rats were sacrificed and the embryos were removed, at last the heart tissue of fetal rats were removed. Hearts of newborn and P7 rats were removed also. The heart, lung, liver, skeletal muscle, small intestine and stomach of adult rats were harvested too after rats were euthanized.
2.3.1 miRNA Extraction, Reverse Transcription (RT) and RT-qPCR
miRNAs in plasma were extracted using an miRNA extraction kit (Tiangen, Beijing, China). Stem-loop RT was performed with a reverse transcription kit (TaKaRa, Dalian, China) using the RT primer 5'-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGACAAGCTT-3' in a 20 μl reaction system following the program: 16°C for 30 min, 42°C for 30 min and 85°C for 5 min. Expression of miR-208a was analyzed using the ABI Prism 7500 system following amplification with Power SYBR® Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) in a 20 μl reaction system following the program: 95°C for 30 sec, 40 cycles of 95°C for 5 sec and 60°C for 34 sec, 9°C for 15 sec, 60°C for 60 sec, and 95°C for 5 sec. The internal reference was U6. PCR products were confirmed by 1.5% agarose gel electrophoresis. Each sample was repeated three times and an average value was taken. The primers sequences are listed in Tab. 2.
Table 2: PCR primers for the genes

2.3.2 Organ and Tissue Acquisition from Rats, miRNA Extraction, Reverse Transcription and RT-qPCR
The miRNA of tissues and organs extracted using TRIzol® reagent (Invitrogen, Carlsbad, CA, USA). After homogenization, 0.2 ml chloroform was added and vortexed for 15 s. Centrifugation was performed at 9,000 × g for 15 min at 4°C, then 0.5 ml of isopropanol was added to the supernatant and mixed well. Centrifugation was performed at 6,500 × g at 4°C for 12 min and 1% of 75% DEPC-treated ethanol was added to rinse. Further centrifugation was performed for 4 min at 2,000 × g for 5 min and dissolved in 0.01% DEPC-treated water. Total RNA was stored at –70°C. The processes of miRNA reverse transcription and miR-208a in real-time PCR protocol were the same as those aforementioned, but the reverse transcription primer was 5'-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGGCTTTTTG -3' instead. Other primers are listed in Tab. 2.
2.3.3 Construction of miR-208a Eukaryotic Expression Vector
Rat cDNA was obtained using TRIzol®. Pre-miR-208a was amplified by PCR (upstream 191 bp; downstream 101 bp; full-length 378 bp). The primers were 5'-AGGGATCCACCATGGGCTGCT-3'(BamHI) and 5'-CTGAATTCCCAACACCCCCTGC-3'(EcoRI), underline indicating the restriction sites. A 25 μl reaction system was treated using the following program: 94°C for 60 sec, 35 cycles of 98°C for 5 sec, 72°C for 5 sec and 58°C for 5 sec; 72°C for 10 min, and 4°C for use. Then, 2.0 μl of the DNA fragment was ligated with 2.0 μl of pMD18T SIMPLE vector and subsequently transformed, enriched, sequenced and extracted. After double-digestion of the plasmid with BamHI and EcoRI, the DNA fragment was ligated with eukaryotic expression vector pcDNA 3.1(+) (Promega, Fitchburg, WI, USA) to construct the miR-208a expression vector.
2.3.4 Cell Culture and Gene Transfection
H9c2 cells (Biosciences Cell Resource Center, Shanghai, China) were cultured in high-glucose Dulbecco’s modified Eagle’s medium with 5% CO2 and saturated humidity at 37°C. Cells were transfected as three groups: i) miR-208a group, cells were transfected with miR-208a expression vector. 10 μl Lipofectamine® 2000 (Invitrogen, Carlsbad, CA, USA) and 6 μg miR-208a vector were transfected together; ii) Inhibitor group, 10 μl Lipofectamine® 2000 and transfected with 80 nmol miR-208a inhibitor (GenePharma, Shanghai, China); iii) Control group, treated with Lipofectamine® 2000 only. The procedure according to the manufacturer’s instructions of Lipofectamine® 2000.
2.3.5 Construction of GATA4 Wide-3′-UTR-pGL3 and GATA4 del-3′-UTR-pGL3 Vector
The Gata4 wide-3'-UTR was amplified by PCR. The primers were 5'-TAGCTAGCACCCCCTTCCCTCTTC-3'(Nhe I) and 5'-CAGCTAGCACCATTTGATCCAC-3'(Nhe I). A 25 μl reaction system was treated the following program: 94°C for 30 sec, 35 cycles of 94°C for 30 sec, 55°C for 30 sec, and 72°C for 90 sec; 72°C for 10 min, and 4°C for ready. DNA fragment was ligated with pGL3-control (Promega, Fitchburg, WI, USA), double-digested of the plasmid with Xba I to construct GATA4 wide-3′-UTR-pGL3 vector. Using a Quick-Change kit (Stratagene, LJ, CA, USA), the Gata4 del-3’-UTR-pGL3 vector was constructed as follows. The primers were 5'-CCTCCACAACCCGTTAACATTAGGTGAAATGGCT-3' and 5'-AGCCATTTCACCTAATGTTAACGGGTTGTGGAGG-3'. A 25 μl reaction system was treated using the following program: 95°C for 120 sec, 35 cycles of 95°C for 20 sec, 60°C for 10 sec, and 6°C for 150 sec; 72°C for 10 min, and 4°C for use. In del-3′-UTR-pGL3 vector, the site at 1,363-1,369 bp of GATA4 3′-UTR was deleted.
2.3.6 Dual Luciferase Reporter System
When cells reached 50%–60% conluency, 2 μg reporter plasmid pGL3-control and 0.1 μg control plasmid pRL-TK (Promega, Fitchburg, WI, USA) were transfected using Lipofectamine® 2000. A total of 100 μl LAR II was added to the fluorimeter tube together with 20 μl cell lysate, then the luminescence apparatus was put in, luciferase activity was measured in the pGL4, which was read as M1. Next, 100 μl Stop&Glo reagent to stop the reaction. Determination of Renilla fluorescence activity in the internal reference plasmid pRL-TK, reading as M2. M1/M2 was the relative activity of luciferase.
The parameters of late apoptosis were measured using a TUNEL assay (Biyuntian, Shanghai, China). At 72 h post-transfection, H9c2 cells were fixed with 4% formaldehyde solution for 25 min at 4°C, rinsed three times with PBS at room temperature, and treated with 0.2% Triton X-100. After incubation buffer was added cells were incubated for 60 min at 37°C. Then, cells were immersed with 3 ml of 2 × sodium chloride-sodium citrate buffer. The reaction was terminated 15 min later. Unbound fluorescein-12-dUTP was washed away with PBS. After addition of 3 ml of propidium iodide (1 μg/ml) was added for 5 min later, and cells were counterstained with hematoxylin and eosin. Finally, TUNEL-positive cells were counted using a microscope.
2.3.8 Expression of Gata4, Bcl-2 and miR-208a from H9c2 Cells Detected RT-qPCR
After transfection for 48h, H9c2 cells were harvested. Total RNA was extracted from transfected cells using TRIzol 48h post-transfection and then reverse-transcribed using a RT-qPCR kit (TaKaRa, Dalian, China). Gata4 and Bcl-2 were detected using qPCR, Gadph was used as an internal reference. A 20-μl reaction system was treated using the following program: 95°C for 30 s, 40 cycles of 95°C for 5 s and 60°C for 34 s, 95°C for 15 s, 60°C for 1 min, 95°C for 15 s, and 60°C for 15 s. The protocol of miR-208a detection was the same as that used for the rat cells. Primers are listed in Tab. 2.
2.3.9 GATA4 Detected via Western Blotting
At 48 h post-transfection, proteins were extracted with Whole Protein Extraction Kit (KeyGen Biotech, Nanjing, Jiangsu, China). Proteins were separated via 10% SDS-PAGE and blotted onto PVDF membranes. The membranes were soaked in Tris-buffered saline. Following the addition of GATA4 antibody (1:500; sc-9053, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and alkaline phosphatase-labeled goat anti-rabbit secondary antibody (1:4,000; ZN1981-EUL; BalbBiomart, Bejing, China), PVDF membranes were developed and photographed. GAPDH (1:500; AB-P-R001, GoodHere, Hangzhou, Zhejiang, China) was used as the internal reference.
Data are presented as the mean ± standard deviation. The clinical data of the two groups were compared by the rank sum test. Data were analyzed via Kruskal-Wallis and Student’s t-test. SPSS v19.0 (SPSS, Inc., Chicago, USA.) and Graphpad Prism 7 were used for analyses. p < 0.05 was considered to indicate a statistically significant result.
Expression of miR-208a in cardiomyocytes in the CHD group is elevated compared with that in the control group. The present study examined the expression levels of miR-208a in plasma samples from 27 patients with CHD and 29 normal individuals in order to validate the miR-208a participate in CHD. As demonstrated by real-time PCR, miR-208a was upregulated in the CHD group when compared with the control group, suggesting that miR-208a plays a role in CHD (p < 0.01; Fig. 1).
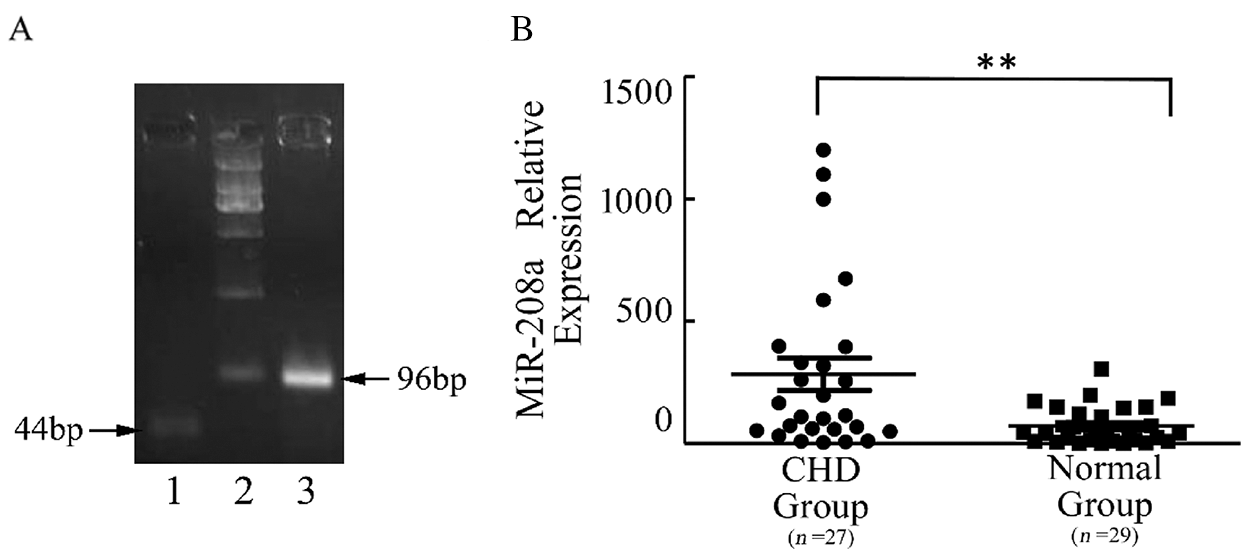
Figure 1: miR-208a expression in plasma of children with CHD and normal group. (A): miRNAs were extracted from plasma, and the reverse-transcribed miR-208a was confirmed by gel electrophoresis after real-time PCR. miR-208a was 44 bp in length. U6 was the internal reference and 96 bp in length. (B): As revealed by real-time PCR, there was a significant difference in miR-208a expression in plasma between CHD Group and Normal Group. Normal Group: control. Lane 1: miR208a; Lane 2: represents DL2000; Lane 3: U6. The dots represent the expression level of miR-208a in CHD Group and the squares represent the expression level in the control. ** p < 0.01 vs. control
Expression of miR-208a shows a temporal dependence in myocardiocytes development and specificity in cardiomyocytes. Expression of miR-208a increased from E10 to P7 and decreased after adulthood (Fig. 2). At P7, miR-208a had the highest expression (p < 0.01; Fig. 2B). In addition, the expression level of miR-208a was highest in myocardiocytes when compared with other organs or tissues (p < 0.01; Fig. 2C). This evidence indicated that miR-208a is essential for the development and function of myocardiocytes.
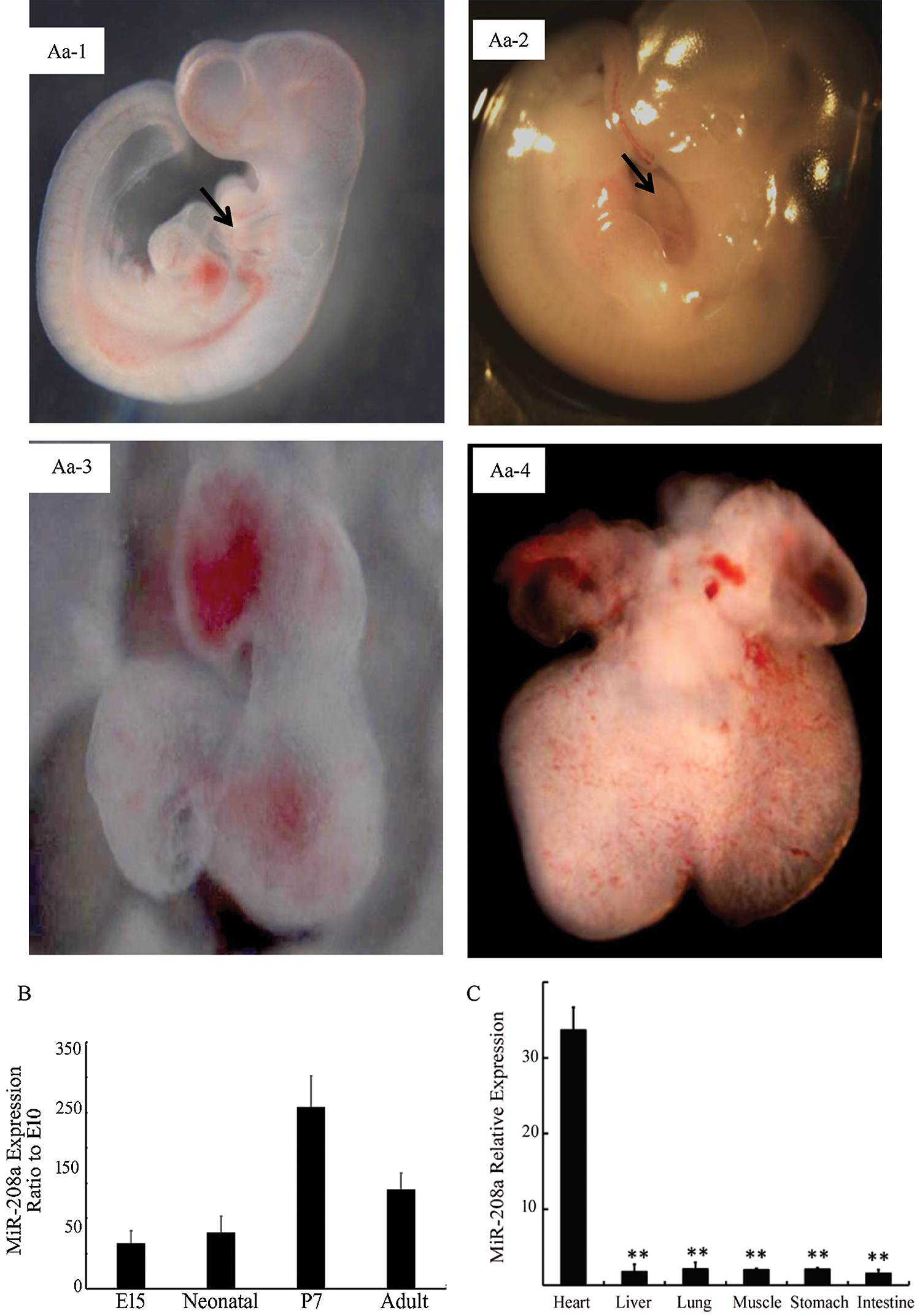
Figure 2: Expression of miR-208a in the heart of rat at different life stages and in various organs or tissues after adulthood. (Aa-1): E10 rat embryo, 32×, arrow for the cardiomyocytes. (Aa-2): E14 rat embryo, 25×, arrow for the cardiomyocytes. (Aa-3): E10 rat cardiomyocytes, 90×. (Aa-4): E14 rat cardiomyocytes, 50×. (B): Relative miR-208a expression ratio to E10 in cardiomyocytes of different embryo stage detected by real-time PCR. miR-208a expression increased gradually in the embryo period and to a peak at P7, but decreased after born. (C): Expression of miR-208a was at the highest in heart, though hardly expressed in other organs or tissues. ** p < 0.01 vs. Heart
miR-208a overexpression promotes apoptosis and expression of bcl-2 decreased. H9c2 cells underwent transfection in three transfection groups, miR-208a, miR-208a inhibitor and control in order to investigate the biological function of miR-208a. Cell apoptosis was promoted in cells with miR-208a compared with the control group, as measured via TUNEL assay. miR-208a inhibitor significantly inhibited cells apoptosis. The number of apoptotic cells increased significantly at 72 h post-transfection in the miR-208a group compared with the control group (p < 0.01). The number of apoptotic cells decreased 72 h after transfection with the miR-208a inhibitor when compared with the control group (p < 0.01). These results suggested that miR-208a promoted apoptosis (Fig. 3A).
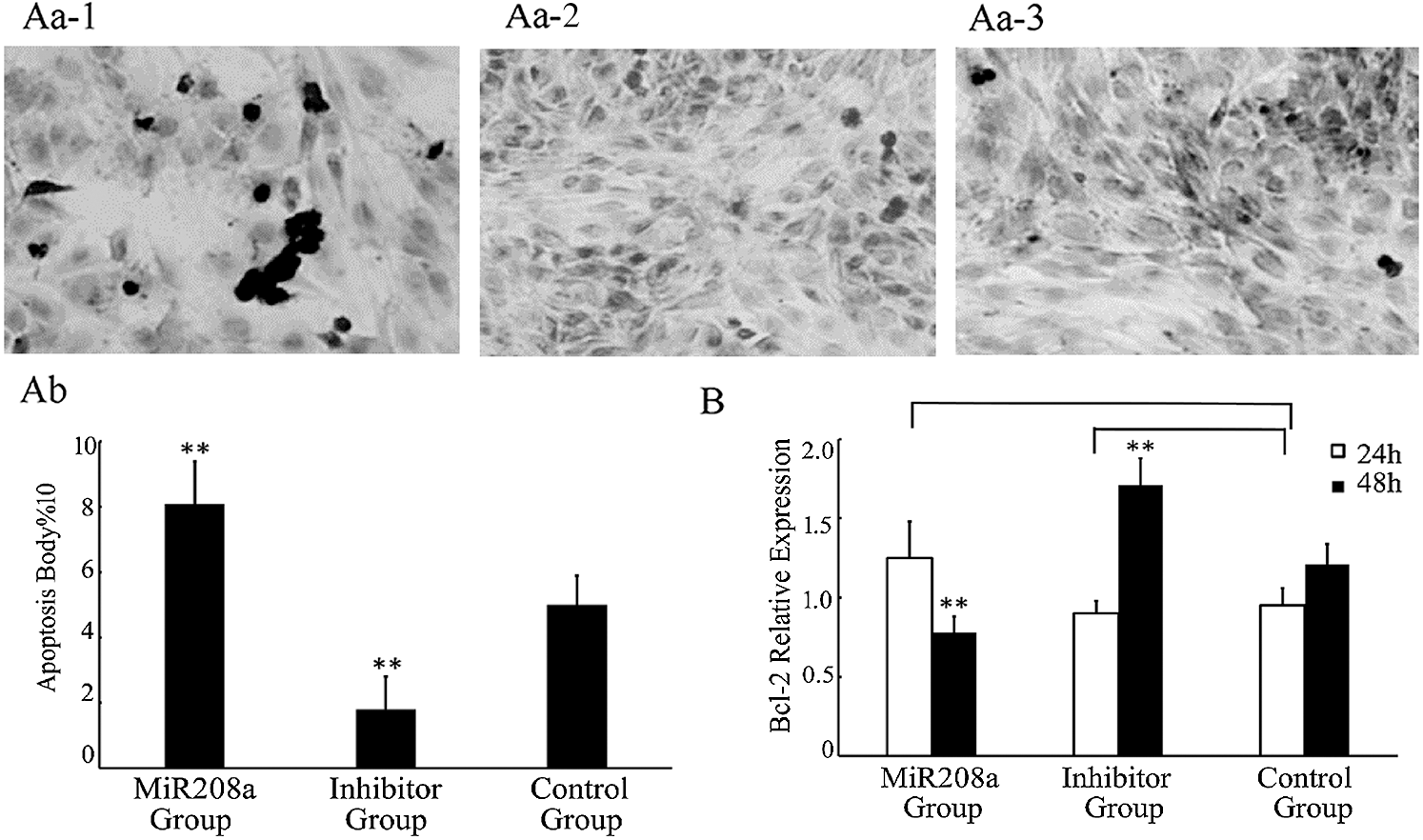
Figure 3: miR-208a promotes apoptosis by target bcl-2. (A): Apoptosis of H9c2 cells after transfection with miR-208a detected by TUNEL assay. Cells stained after transfection (400×). (Aa-1): MiR208a Group: Transfected with miR-208a; arrows indicate apoptotic cells. (Aa-2): Inhibitor Group: Transfected with miR-208a inhibitor. (Aa-3): Control Group: No transfection. (Ab): Statistical analysis of apoptotic cells. The number of apoptotic cells was increased significantly in MiR208a Group as compared with the Control Group; and the number of apoptotic cells was decreased after transfection with miR-208a inhibitor as compared with the Control Group. (B): The expression of bcl-2 determined by real-time PCR after transfection. Compared with Control Group, there was a significant decrease detected in MiR208a Group (p < 0.01), while a significant decrease detected in Inhibitor Group after 48 h (p < 0.01). After 24 h, either in MiR208a Group or Inhibitor Group, the expression of bcl-2 was no significant compared with the Control Group (p > 0.05). “MiR208a Group”: Transfected with miR-208a vector; “Inhibitor Group”: Transfected with miR-208a inhibitor; “Control Group”: Without transfection. ** p < 0.01, ns: p > 0.05 vs. Control Group
Bcl-2, an anti-apoptosis gene, was analyzed after the gene was transfected at 24 h and 48 h by real-time PCR. Compared with the control group, there was no significant difference in the expression level of Bcl-2 24 h after miR-208a overexpression (p > 0.05), but a significant decrease of expression at 48 h was noted (p < 0.01). There was no significant difference in the expression of Bcl-2 24 h after transfection with miR-208a inhibitor (p > 0.05), but a significant increase at 48 h was documented (p < 0.01; Fig. 3B). These results indicate that miR-208a may promote cell apoptosis by regulating Bcl-2, but there was no evidence to prove that miR-208a regulates Bcl-2 directly.
GATA4 is a target gene of miR-208a. From the bioinformatics analysis with Targetscan (http://www.targetscan.org), miR-208a binds directly to the 3´-UTR of GATA4. The present study deleted the predicted binding site (1,363-1,369 bp) as del-3´-UTR (Fig. 4A). In the Dual Luciferase reporter system, miR-208a decreased the luciferase activity together with wide-Gata4-3´-UTR, but had no effect on the luciferase activity of del- Gata4-3´-UTR (p < 0.01; Fig. 4B). RT-qPCR and western blotting analysis were performed in order to further measure the effect of miR-208a on GATA4 expression (Figs. 4C and 4D). Expression of GATA4 decreased after miR-208a overexpression (p < 0.01), but increased following transfection with a miR-208a inhibitor when compared with the control group (p < 0.05) (Fig. 4D). These findings suggested that miR-208a regulates GATA4 expression by directly targeting the GATA4 3´-UTR region negatively.

Figure 4: Gata4 is a direct target of miR-208a. (Aa-1): Predicted binding site miR-208a to Gata 4 3´-UTR. (Aa-2): Sequence fragment of Gata 4 3´-UTR. The box showed the target predicted biding site of miR-208a. (Aa-3): The arrow showed that the predicted biding site was deleted. (B): Pre-miR-208a expression vector transfected H9c2 cells with Gata4 wide-3´-UTR and del-3´-UTR respectively. The luciference of MiR208a Group was significantly lower than Control Group. (C): Gata4 expression decreased in MiR208a Group, but increased in Inhibitor Group after 208a transfected 48 h vs. Control Group. (D): Western blotting analysis of GATA4 expression levels in H9c2 cells. A significant decrease was detected at 48 h by transfected with miR-208a (p < 0.05). There was a significant increase 48 h after transfection with miR-208a inhibitor. “MiR208a Group”: Transfected with miR-208a vector; “Inhibitor Group”: Transfected with miR-208a inhibitor. “Control Group”: Without transfection. ** p < 0.01, *: p < 0.05, ns: p > 0.05
Studies have shown that miRNA play an important role in cardiomyocyte fate by suppressing their target genes [11,13,19–26]; particularly in cell apoptosis, resulting in the development of diseases [27–31]. miR-208a promotes apoptosis in ischemic cardiomyocytes to alter cardiac function [32]. miR-208a, encoded by intron 29 of Myh6 [14,33], is a highly conserved sequence in precursor and mature forms [34] that encodes α-cardiac muscle myosin heavy chain (α-MHC) [14,23,33]. These findings demonstrate that miRNA is critical for cardiovascular development and cardiovascular diseases. However, little is currently known about the function of miRNAs, or miR-208a in CHD.
Therefore, the present study analyzed whether miR-208a plays a role in CHD pathophysiology. Circulating miRNAs have been proven to be associated with cardiovascular diseases, particularly in CHD [18], which may become new biomarkers and therapeutic targets of the diseases [35–39]. Thus, the expression of circulating miR-208a in pediatric patients with CHD was detected first in the present study. The CHD group included patients in ASD, ventricular septal defect (VSD), F4 (F4Tetralogy of Fallot), double-outlet right ventricle, atrioventricular septal defect (AVSD), patent ductus arterious (PDA), aortic stenosis and patent foramen ovale (PFO). The expression of circulating miR-208a was much higher in the CHD group than in the control group, which suggested that miR-208a may play in atrioventricular development of the heart, even lead to CHD. Li et al. demonstrated that 8 circulating miRNAs with VSD were down- or upregulated [40], but miR-208a was not mentioned. The reason may be that the expression of the miR-208 family showed prominent chamber specificity and was expressed abundantly in the left atria [38]. While the present study did not group the expression of miR-208a in different types of CHD, the impact of miR-208a on different types of CHD will be the focus on our future research.
To further clarify the possible role of miR-208a in heart development, we conducted animal experiments in rats. The present study selected two key time points of the embryonic period: E10 (cardiac tube looped to right and early chamber formatted) and E14 (separation of atrium and outflow tract formed) [25] in rat. The cardiomyocytes were obtained from the two time points, as well as newborn, P7, and adult rats in order to analyze the expression of miR-208a. The expression of miR-208a significantly increased following age, and peaked to P7, which also showed myocardial expression specificity in adult rats, and has been shown to be positively correlated in previous studies [14,33]. The present study implied that miR-208a may be associated with heart cardiomyocyte pathophysiology and even play a key role in heart development.
Then, it was revealed that apoptotic bodies were increased in H9c2 cells when miR-208a was overexpressed, and Bcl-2, an anti-apoptosis gene, was downregulated. From predicted databases (www.targetscan.com), miR-208a would not target Bcl-2 but GATA4 directly. Studies showed that GATA4 decreased apoptosis [41] by upregulating Bcl-2 [42]. To the best of our knowledge, it is not known whether miR-208a regulates Gata4 directly and decreases Bcl-2. GATA4, as an important transcription factor, plays a role in cardiovascular development. Mutation in the gene, mutation in its 3´UTR and SNPs variants, and abnormal expression of GATA4 can all lead to CHD [17,43,44]. Wang et al. (34) demonstrated that miR-208a negatively regulated GATA4, although the binding site was not noticed. GATA4 level was elevated while the transcript level was unchanged in a miR-208a -/- mouse heart [33]. In the present study, dual fluorescence reporter experiments suggested that miR-208a negatively regulated Gata4 by binding the site of 3´UTR at 1363-1369 bp, which is predicted as a “seed region”. Overexpression of miR-208a inhibited the expression of GATA4 tested by western blotting. Therefore, the present study speculated that miR-208a plays a role in apoptosis by negatively regulating expression of GATA4, then downregulated bcl-2. Dysregulated expression of miR-208a during embryonic cardiovascular development may lead to uncontrolled expression of one or more target genes, then cause consequent malformation and dysfunction of the heart, thereby resulting in CHD.
In conclusion, the present study implied that: i) Circulating miR-208a expression is upregulated in patients with CHD, ii) miR-208a directly regulates GATA4 expression in cardiomyocytes at the site of Gata4 3´UTR 1363-1369 bp, and iii) miR-208a promotes H9c2 cell apoptosis by downregulating GATA4, while the latter is a positive factor of Bcl-2 expression. These findings suggest that miR-208a may be a new biomarker or therapeutic target for CHD.
We propose that miR-208a, as a potential therapeutic target and mechanistic biomarker for CHD, may be one of non-invasive treatment for CHD.
Acknowledgement: The author would like to thank Prof. Kailai Sun, Prof. Gungrong Qiu and Dr. Hongkun Jiang very much for their construction comments.
Funding Statement: The work was supported by the National Science Foundation of China (No. 8170131).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Lozano, R., Naghavi, M., Foreman, K., Lim, S., Shibuya, K. et al. (2012). Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study. Lancet, 380(9859), 2095–2128. [Google Scholar]
2. Olson, E. N. (2002). A genetic blueprint for growth and development of the heart. Harvey Lectures, 98, 41–64. [Google Scholar]
3. Bruneau, B. G. (2002). Transcriptional regulation of vertebrate cardiac morphogenesis. Circulation, 90(5), 509–519. [Google Scholar]
4. Zhao, Y., Samal, E., Srivastava, D. (2005). Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature, 436(7048), 214–220. DOI 10.1038/nature03817. [Google Scholar] [CrossRef]
5. Gunning, P., O’Neill, G., Hardeman, E. (2008). Tropomyosin-based regulation of the actin cytoskeleton in time and space. Physiological Reviews, 88(1), 1–35. [Google Scholar]
6. Chen, J. F., Murchison, E. P., Tang, R., Callis, T. E., Tatsuguchi, M. et al. (2008). Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proceedings of the National Academy of Sciences of the United States of America, 105(6), 2111–2116. DOI 10.1073/pnas.0710228105. [Google Scholar] [CrossRef]
7. Albinsson, S., Suarez, Y., Skoura, A., Offermanns, S., Miano, J. M. et al. (2010). MicroRNAs are necessary for vascular smooth muscle growth, differentiation, and function. Arteriosclerosis, Thrombosis, and Vascular Biology, 30(6), 1118–1126. DOI 10.1161/ATVBAHA.109.200873. [Google Scholar] [CrossRef]
8. Rao, P. K., Toyama, Y., Chiang, H. R., Gupta, S., Bauer, M. et al. (2009). Loss of cardiac microRNA-mediated regulation leads to dilated cardiomyopathy and heart failure. Circulation Research, 105(6), 585–594. DOI 10.1161/CIRCRESAHA.109.200451. [Google Scholar] [CrossRef]
9. da Costa Martins, P. A., Bourajjaj, M., Gladka, M., Kortland, M., van Oort, R. J. et al. (2008). Conditional dicer gene deletion in the postnatal myocardium provokes spontaneous cardiac remodeling. Circulation, 118(15), 1567–1576. DOI 10.1161/CIRCULATIONAHA.108.769984. [Google Scholar] [CrossRef]
10. Brennecke, J., Stark, A., Russell, R. B., Cohen, S. M. (2005). Principles of microRNA-target recognition. PLoS Biology, 3(3), e85. DOI 10.1371/journal.pbio.0030085. [Google Scholar] [CrossRef]
11. Deacon, D. C., Nevis, K. R., Cashman, T. J., Zhou, Y., Zhao, L. et al. (2010). The miR-143-adducin3 pathway is essential for cardiac chamber morphogenesis. Development, 137(11), 1887–1896. DOI 10.1242/dev.050526. [Google Scholar] [CrossRef]
12. Satoh, M., Minami, Y., Takahashi, Y., Tabuchi, T., Nakamura, M. (2010). Expression of microRNA-208 is associated with adverse clinical outcomes in human dilated cardiomyopathy. Journal of Cardiac Failure, 16(5), 404–410. DOI 10.1016/j.cardfail.2010.01.002. [Google Scholar] [CrossRef]
13. Small, E. M., Olson, E. N. (2011). Pervasive roles of microRNAs in cardiovascular biology. Nature, 469(7330), 336–342. DOI 10.1038/nature09783. [Google Scholar] [CrossRef]
14. van Rooij, E., Quiat, D., Johnson, B. A., Sutherland, L. B., Qi, X. et al. (2009). A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Developmental Cell, 17(5), 662–673. DOI 10.1016/j.devcel.2009.10.013. [Google Scholar] [CrossRef]
15. Zhang, W., Li, X., Shen, A., Jiao, W., Guan, X. et al. (2008). GATA4 mutations in 486 Chinese patients with congenital heart disease. European Journal of Medical Genetics, 51(6), 527–535. DOI 10.1016/j.ejmg.2008.06.005. [Google Scholar] [CrossRef]
16. Shaker, O., Omran, S., Sharaf, E., Hegazy, G. A., Mashaly, M. et al. (2017). A novel mutation in exon 1 of GATA4 in Egyptian patients with congenital heart disease. Turkish Journal of Medical Sciences, 47(1), 217–221. DOI 10.3906/sag-1605-166. [Google Scholar] [CrossRef]
17. Lavallee, G., Andelfinger, G., Nadeau, M., Lefebvre, C., Nemer, G. et al. (2006). The Kruppel-like transcription factor KLF13 is a novel regulator of heart development. EMBO Journal, 25(21), 5201–5213. DOI 10.1038/sj.emboj.7601379. [Google Scholar] [CrossRef]
18. Smith, T. R. C., Caputo, M., Emanueli, C. (2015). MicroRNAs in congenital heart disease. Annals of Translational medicine, 3(21), 333. [Google Scholar]
19. Alvarez-Garcia, I., Miska, E. A. (2005). MicroRNA functions in animal development and human disease. Development, 132(21), 4653–4662. DOI 10.1242/dev.02073. [Google Scholar] [CrossRef]
20. Topkara, V. K., Mann, D. L. (2011). Role of microRNAs in cardiac remodeling and heart failure. Cardiovascular Drugs and Therapy, 25(2), 171–182. DOI 10.1007/s10557-011-6289-5. [Google Scholar] [CrossRef]
21. Chen, J. F., Mandel, E. M., Thomson, J. M., Wu, Q., Callis, T. E. et al. (2006). The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nature Genetics, 38(2), 228–233. DOI 10.1038/ng1725. [Google Scholar] [CrossRef]
22. Zhang, J. S., Zhao, Y., Lv, Y., Liu, P. Y., Ruan, J. X. et al. (2017). miR-873 suppresses H9C2 cardiomyocyte proliferation by targeting GLI1. Gene, 626, 426–432. [Google Scholar]
23. Gupta, M. K., Rao, T. N. (2014). Hearty miR-363 controls HAND1 in cardiac cell specification. Stem Cell Research & Therapy, 5(4), 89. DOI 10.1186/scrt478. [Google Scholar] [CrossRef]
24. Chiavacci, E., Dolfi, L., Verduci, L., Meghini, F., Gestri, G. et al. (2012). MicroRNA 218 mediates the effects of Tbx5a over-expression on zebrafish heart development. PLoS One, 7(11), e50536. DOI 10.1371/journal.pone.0050536. [Google Scholar] [CrossRef]
25. Bruneau, B. G. (2008). The developmental genetics of congenital heart disease. Nature, 451(7181), 943–948. DOI 10.1038/nature06801. [Google Scholar] [CrossRef]
26. Gomase, V. S., Parundekar, A. N. (2009). microRNA: Human disease and development. International Journal of Bioinformatics Research and Applications, 5(5), 479–500. [Google Scholar]
27. Bostjancic, E., Jerse, M., Glavac, D., Zidar, N. (2015). miR-1, miR-133a/b, and miR-208a in human fetal hearts correlate to the apoptotic and proliferation markers. Experimental Biology and Medicine, 240(2), 211–219. DOI 10.1177/1535370214546268. [Google Scholar] [CrossRef]
28. Chen, Z., Zhang, S., Guo, C., Li, J., Sang, W. (2017). Downregulation of miR-200c protects cardiomyocytes from hypoxia-induced apoptosis by targeting GATA-4. International Journal of Molecular Medicine, 39(6), 1589–1596. DOI 10.3892/ijmm.2017.2959. [Google Scholar] [CrossRef]
29. Tu, J. J., Yang, Y. Z., Albert Cheung, H. H., Chen, Z. J., Chan, W. Y. (2017). Conserved miR-10 family represses proliferation and induces apoptosis in ovarian granulosa cells. Scientific Reports, 7(1), 73. DOI 10.1038/srep41304. [Google Scholar] [CrossRef]
30. Wang, X., Yang, C., Liu, X., Yang, P. (2018). Ghrelin alleviates angiotensin II-Induced H9c2 apoptosis: Impact of the miR-208 family. Medical Science Monitor, 24, 6707–6716. DOI 10.12659/MSM.908096. [Google Scholar] [CrossRef]
31. Zhou, Y. L., Sun, Q., Zhang, L., Li, R. (2018). miR-208b targets Bax to protect H9c2 cells against hypoxia-induced apoptosis. Biomedicine & Pharmacotherapy, 106(16), 1751–1759. DOI 10.1016/j.biopha.2018.07.141. [Google Scholar] [CrossRef]
32. Tony, H., Meng, K., Wu, B., Yu, A., Zeng, Q. et al. (2015). MicroRNA-208a dysregulates apoptosis genes expression and promotes cardiomyocyte apoptosis during ischemia and its silencing improves cardiac function after myocardial infarction. Mediators of Inflammation, 479123. [Google Scholar]
33. Callis, T. E., Pandya, K., Seok, H. Y., Tang, R. H., Tatsuguchi, M. et al. (2009). MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. Journal of Clinical Investigation, 119(9), 2772–2786. DOI 10.1172/JCI36154. [Google Scholar] [CrossRef]
34. Xu, C. C., Han, W. Q., Xiao, B., Li, N. N., Zhu, D. L. et al. (2008). Differential expression of microRNAs in the aorta of spontaneously hypertensive rats. Sheng Li Xue Bao, 60(4), 553–560. [Google Scholar]
35. Corsten, M. F., Dennert, R., Jochems, S., Kuznetsova, T., Devaux, Y. et al. (2010). Circulating MicroRNA-208b and MicroRNA-499 reflect myocardial damage in cardiovascular disease. Circulation: Cardiovascular Genetics, 3(6), 499–506. DOI 10.1161/CIRCGENETICS.110.957415. [Google Scholar] [CrossRef]
36. Fichtlscherer, S., Zeiher, A. M., Dimmeler, S. (2011). Circulating microRNAs: Biomarkers or mediators of cardiovascular diseases? Arteriosclerosis, Thrombosis, and Vascular Biology, 31(11), 2383–2390. DOI 10.1161/ATVBAHA.111.226696. [Google Scholar] [CrossRef]
37. Fung, E. C., Butt, A. N., Eastwood, J., Swaminathan, R., Sodi, R. (2019). Circulating microRNA in cardiovascular disease. Advances in Clinical Chemistry, 91, 99–122. [Google Scholar]
38. Kakimoto, Y., Tanaka, M., Kamiguchi, H., Hayashi, H., Ochiai, E. et al. (2016). MicroRNA deep sequencing reveals chamber-specific miR-208 family expression patterns in the human heart. International Journal of Cardiology, 211, 43–48. DOI 10.1016/j.ijcard.2016.02.145. [Google Scholar] [CrossRef]
39. Pirola, C. J., Gianotti, T. F., Castano, G. O., Sookoian, S. (2013). Circulating MicroRNA-122 signature in nonalcoholic fatty liver disease and cardiovascular disease: A new endocrine system in metabolic syndrome. Hepatology, 57(6), 2545–2547. [Google Scholar]
40. Li, D., Ji, L., Liu, L., Liu, Y., Hou, H. et al. (2014). Characterization of circulating microRNA expression in patients with a ventricular septal defect. PLoS One, 9(8), e106318. DOI 10.1371/journal.pone.0106318. [Google Scholar] [CrossRef]
41. Kim, Y., Ma, A. G., Kitta, K., Fitch, S. N., Ikeda, T. et al. (2003). Anthracycline-induced suppression of GATA-4 transcription factor: Implication in the regulation of cardiac myocyte apoptosis. Molecular Pharmacology, 63(2), 368–377. DOI 10.1124/mol.63.2.368. [Google Scholar] [CrossRef]
42. Kobayashi, S., Lackey, T., Huang, Y., Bisping, E., Pu, W. T. et al. (2006). Transcription factor gata4 regulates cardiac BCL2 gene expression in vitro and in vivo. FASEB Journal, 20(6), 800–802. DOI 10.1096/fj.05-5426fje. [Google Scholar] [CrossRef]
43. Garg, V., Kathiriya, I. S., Barnes, R., Schluterman, M. K., King, I. N. et al. (2003). GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature, 424(6947), 443–447. [Google Scholar]
44. Pulignani, S., Vecoli, C., Sabina, S., Foffa, I., Ait-Ali, L. et al. (2016). 3'UTR SNPs and haplotypes in the GATA4 gene contribute to the genetic risk of congenital heart disease. Revista espanola de cardiologia (English Editions), 69(8), 760–765. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |