| Congenital Heart Disease |
DOI: 10.32604/CHD.2021.015167
ARTICLE
High Prevalence of Genetic Alterations in Infantile-Onset Cardiomyopathy
1Department of Pediatrics, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
23billion, Inc., Seoul, Korea
3Genome Research Center for Birth Defects and Genetic Diseases, Asan Institute for Life Sciences, Asan Medical Center, Seoul, Korea
4Division of Cardiology, Department of Pediatrics, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
5Division of Neonatology, Department of Pediatrics, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
6Medical Genetics Center, Asan Medical Center Children’s Hospital, University of Ulsan College of Medicine, Seoul, Korea
*Corresponding Authors: Jae Suk Baek. Email: lipton79@hanmail.net; Beom Hee Lee. Email: bhlee@amc.seoul.kr
Received: 27 November 2020; Accepted: 23 December 2020
#These authors contributed equally to this work
Abstract: Background and Method: The genetic cause of infantile-onset cardiomyopathy is rarely investigated. Here, we conducted whole exome sequencing (WES) and mitochondrial DNA (mtDNA) sequencing in eight patients with infantile-onset cardiomyopathy to identify genetic variations. Result: Among these patients, two (25%) had dilated cardiomyopathy (DCMP), two (25%) had left ventricular non-compaction (LVNC), and four (50%) had hypertrophic cardiomyopathy (HCMP). Except four patients identified prenatally, the remaining patients presented at a median age of 85.5 days. WES identified genetic variants in a total of seven (87.5%) patients and mtDNA sequencing in the other case. TPM1 and MYH7 variants were identified in the two patients with DCMP; MYH11 and MYLK2 variants in the two patients with LVNC; HRAS, BRAF, and MYH7 variants in three patients with HCMP; and MT-ND1 variant in one patient with HCMP having high blood lactic acid levels. Among the eight variants, four were classified as pathogenic or likely-pathogenic according to the American College of Medical Genetics (ACMG) guidelines, and the remaining were identified as variants of unknown significance (VUSs). Three pathogenic mutations were de novo, whereas four (likely-pathogenic or VUSs) were inherited from a respective parent, excluding one variant where parental testing was unavailable, questioning whether these inherited variants are disease-causing. Three patients died before 3 months of age. Conclusion: Genomic studies, such as WES with additional mtDNA sequencing, can identify a genetic variant in high proportions of patients with infantile-onset cardiomyopathy. The clinical implication of the parentally inherited variant needs to be assessed in a larger patient and family cohort with a longitudinal follow-up.
Keywords: Cardiomyopathy; whole exome sequencing; infantile-onset
Cardiomyopathies are structural and functional heart muscle disorders that affect ventricular systolic function, diastolic function, or both. The incidence of cardiomyopathy is approximately one per 100,000 in children less than 18 years old [1,2]. Some studies have shown that cardiomyopathy below the age of one was four-to-eight times higher in incidence than that in the population over one year of age [1,3]. Cardiomyopathy in children has serious complications and high mortality rates despite the recent development of mechanical cardiopulmonary support and is the major causative disease of pediatric heart transplantation [4]. In particular, infantile cardiomyopathy that develops before 1 year of age, including congenital cardiomyopathy, has a worse prognosis than late-onset cardiomyopathy [5,6].
Generally, the most common cause of childhood cardiomyopathy is idiopathic [7]. However, recent developments in genetic testing have identified genetic alterations in a significant substantial number of previously classified idiopathic cardiomyopathies [8]. Compared to adults, pediatric patients have fewer comorbidities, such as arteriosclerosis and diabetes, and have relatively stronger associations with genetic alterations. Indeed, several studies have been conducted to identify genetic causes in pediatric patients with cardiomyopathy [9,10].
As more than 60 genes are associated with cardiomyopathy, multi-gene testing using next-generation sequencing techniques, such as panel-gene testing, is an appropriate diagnostic method. However, panel-gene testing cannot identify genetic causes outside the list of genes in the designed panel. Moreover, the time, cost, and effectiveness of a genomic study must be considered; whole exome sequencing (WES) is considered one of the fastest diagnostic tools in cardiomyopathy [4,11]. However, to our knowledge, no studies have been conducted to characterize infantile-onset cardiomyopathy with high mortality rates and genetic background.
Therefore, the aim of the present study was to identify variants by performing WES and additional mitochondrial DNA (mtDNA) sequencing in patients with infantile-onset cardiomyopathy. By assessing the pathogenicity of the variant and its existence in the parents, we investigated the association of the identified variant with the disease in detail.
2.1 Subjects and Clinical Evaluation
WES was conducted in patients with cardiomyopathy diagnosed at Asan Medical Center Children's Hospital, Seoul, Korea between August 2018 and June 2020. Among them, eight patients, including four with congenital cardiomyopathy suspected at prenatal examination, were infantile-onset and included in this study. Detailed demographics and clinical characteristics of the patients were reviewed, including age, initial presentations, gender, family history, laboratory results, radiological results, and genetic test results.
Informed consent was obtained from the parents of the patients for the genetic test. This study was approved by the Institutional Review Board for Human Research of Asan Medical Center (IRB numbers 2018-0574 and 2018-0180).
2.2 Definitions of Cardiomyopathies
• Dilated cardiomyopathy (DCMP): Left ventricular fractional shortening (FS) or ejection fraction (EF) > 2 SD below the normal mean for age and Left ventricular end-diastolic dimension (LVEDD) or volume > 2 SD above the normal mean for body surface area [4,12].
• Left ventricular non-compaction (LVNC): Prominent LV trabeculations with deep recesses communicating with the LV cavity and a thin, compacted epicardial layer [13].
• Hypertrophic cardiomyopathy (HCMP): Left ventricular posterior wall thickness at end diastole >2 SD above the normal mean for body surface area [14].
2.3 Variant Annotation and Interpretation by WES Analysis
WES was performed using genomic DNA isolated from patient’s whole blood or a buccal swab sample. All exons of all human genes (approximately 22,000) were captured using a Twist Human Core Exome Kit (Twist Bioscience, San Francisco, CA, USA). The captured genomic regions were sequenced using a NovaSeq 6000 platform (Illumina, San Diego, CA, USA). Raw genome sequencing data analysis included alignment to the reference sequence (NCBI genome assembly GRCh37; accessed February 2009). The mean depth of coverage was 100-fold with 99.2% higher coverage than 10-fold. The pathogenicity of each variant according to the American College of Medical Genetics (ACMG) guidelines [15] and relevant patient phenotypes were assessed using the automated variant interpretation system EVIDENCE [16]. Candidate variants based on EVIDENCE were reviewed and selected by medical geneticists. Sanger sequencing of the variant identified via exome sequencing was performed for patients and their parents.
2.4 Whole Mitochondrial DNA Sequencing
Genomic DNA was isolated from the peripheral blood using a PUREGENE DNA isolation kit (Gentra, Minneapolis, MN, USA). The amplifications by PCR were performed in 30 cycles. After verifying that the single-specific PCR product was amplified, DNA sequencing was performed using the same primers used in PCR and BigDye Terminatore V3.1 Cycle Sequencing Ready reaction kit (Applied Biosystems, Foster city, CA, USA) according to the manufacturer’s instructions. Electrophoresis and analysis of the reaction mixtures were conducted with an ABI 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
The patient’s clinical data are summarized in Tab. 1. There were three male and five female patients. Two of eight patients (25%) had DCMP (patient Nos. 1, 2), two (25%) had LVNC (patient Nos. 3, 4), four (50%) had HCMP (patient Nos. 5–8). Four patients were suspected as cardiomyopathy prenatally, and the other four were identified at 85.5 [interquartile range (IQR) 73.5–138.3] days of life.
Table 1: Clinical data of patients with infantile-onset cardiomyopathy
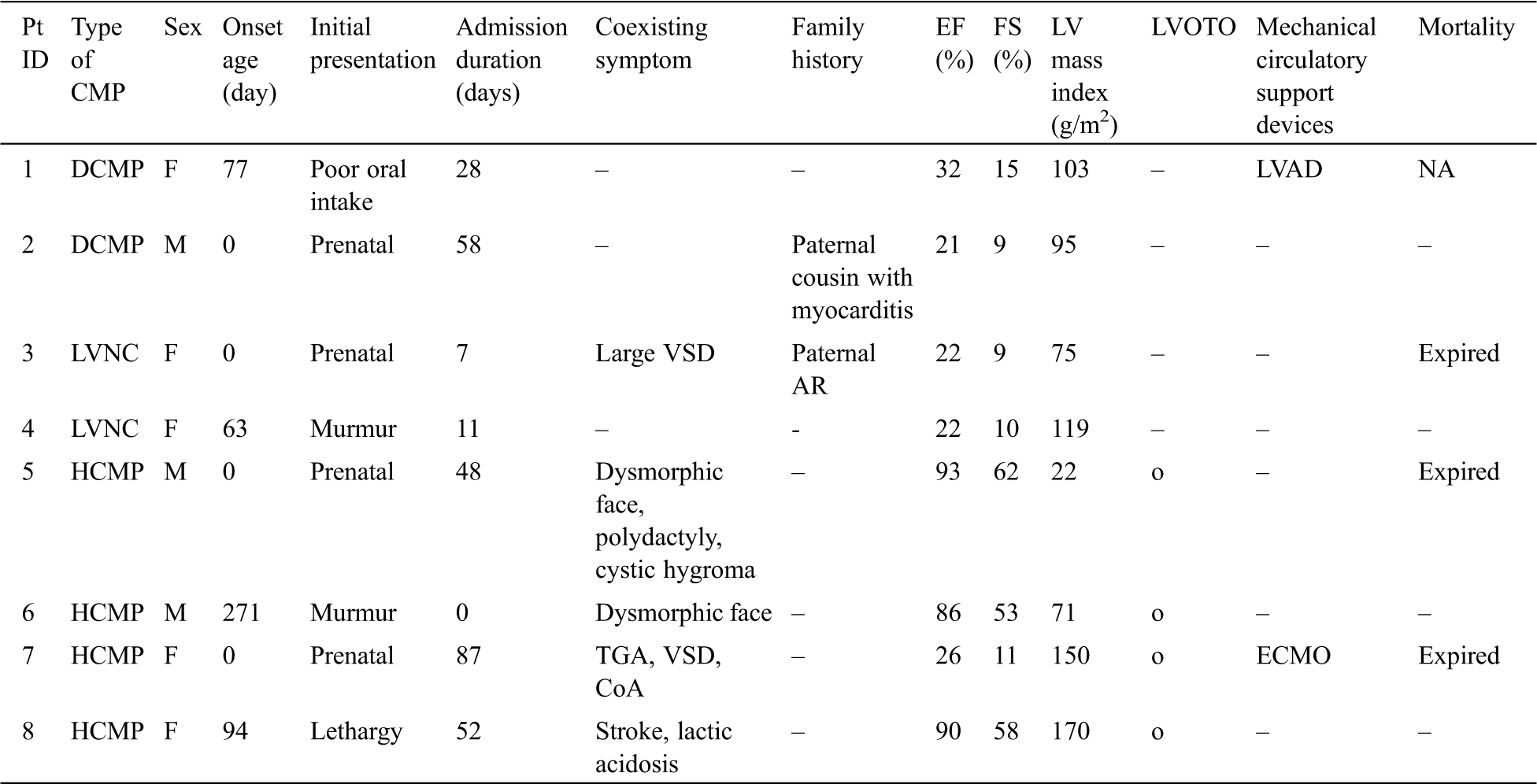
The presenting signs in those diagnosed later were heart murmur (two patients), lethargy (one patient), and poor oral intake (one patient). All patients were hospitalized in the intensive care unit, and their median duration of admission was 48 (IQR 19.5–55) days. Two patients (25%) were provided mechanical circulatory support; three (37.5%) died at 48 [27.5–67.5] days of life.
Five patients had coexisting symptoms. Structural abnormalities in the heart were found in two patients—patient No. 3 had a large ventricular septal defect (VSD) with a diameter of 12 mm and patient No. 7 had transposition of great arteries, VSD, and coarctation of aorta. Multiple extracardiac anomalies with dysmorphic face were found in two patients—patient No. 5 had macrocephaly, cystic hygroma, epicanthal fold, proptosis, low set ears, micrognathia, widely spaced nipples, and undescended left testis and patient No. 6 had fragile hair, low set ears, and a webbed neck. Patient No. 8 showed symptoms of metabolic diseases, such as hemorrhagic stroke and severe lactic acidosis [>15 mmol/L (ref. 0.56–1.390)]. The median (IQR, reference range) of EF and FS for the two patients with DCMP was 27% (24–29%, ref. > 50) and 12.5% (11–14%, ref. > 28), respectively; the median (IQR, reference range) LV mass index for four patients with HCMP was 110 g/m2 (59–155 g/m2, ref. >95), and all four had a degree of left ventricular outflow tract obstruction (LVOTO).
The echocardiography images representing each phenotype are summarized in Fig. 1. Patient No. 1 with DCMP shows dilated left ventricle (Figs. 1A, 1B), Patient No. 4 with LVNC shows non-compacted excessive trabeculation (Figs. 1C, 1D), Patient No. 7 with HCMP shows severe thickening of the ventricular wall (Figs. 1E, 1F). The electrocardiogram representing each phenotype is shown in Fig. 2. Left ventricular hypertrophy was observed in patient No. 1 with DCMP and patient No. 4 with LVNC (Figs. 2A, 2B), and biventricular hypertrophy and wall strain pattern were observed in patient No. 7 with HCMP (Fig. 2C).
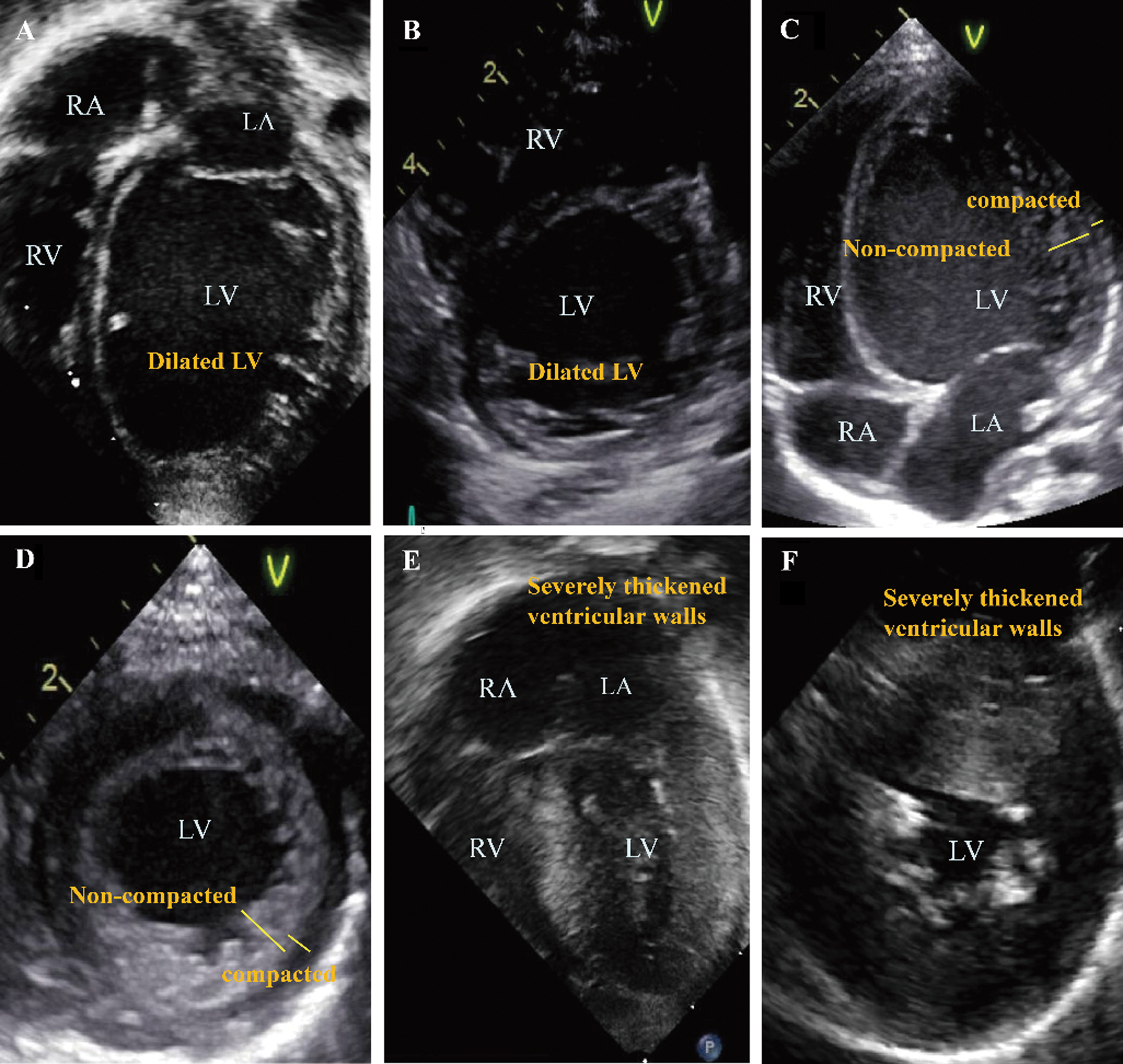
Figure 1: Echocardiographic 4-chamber and parasternal short axis images for different phenotypes. (A, B) An infant with a dilated cardiomyopathy (patient No. 1), (C, D) A neonate with a left ventricular non-compaction cardiomyopathy (patient No. 4), (E, F) A neonate with a hypertrophic cardiomyopathy (patient No. 7). Abbreviations: RA, right atrium; LA, left atrium; RV, right ventricle; LV, left ventricle
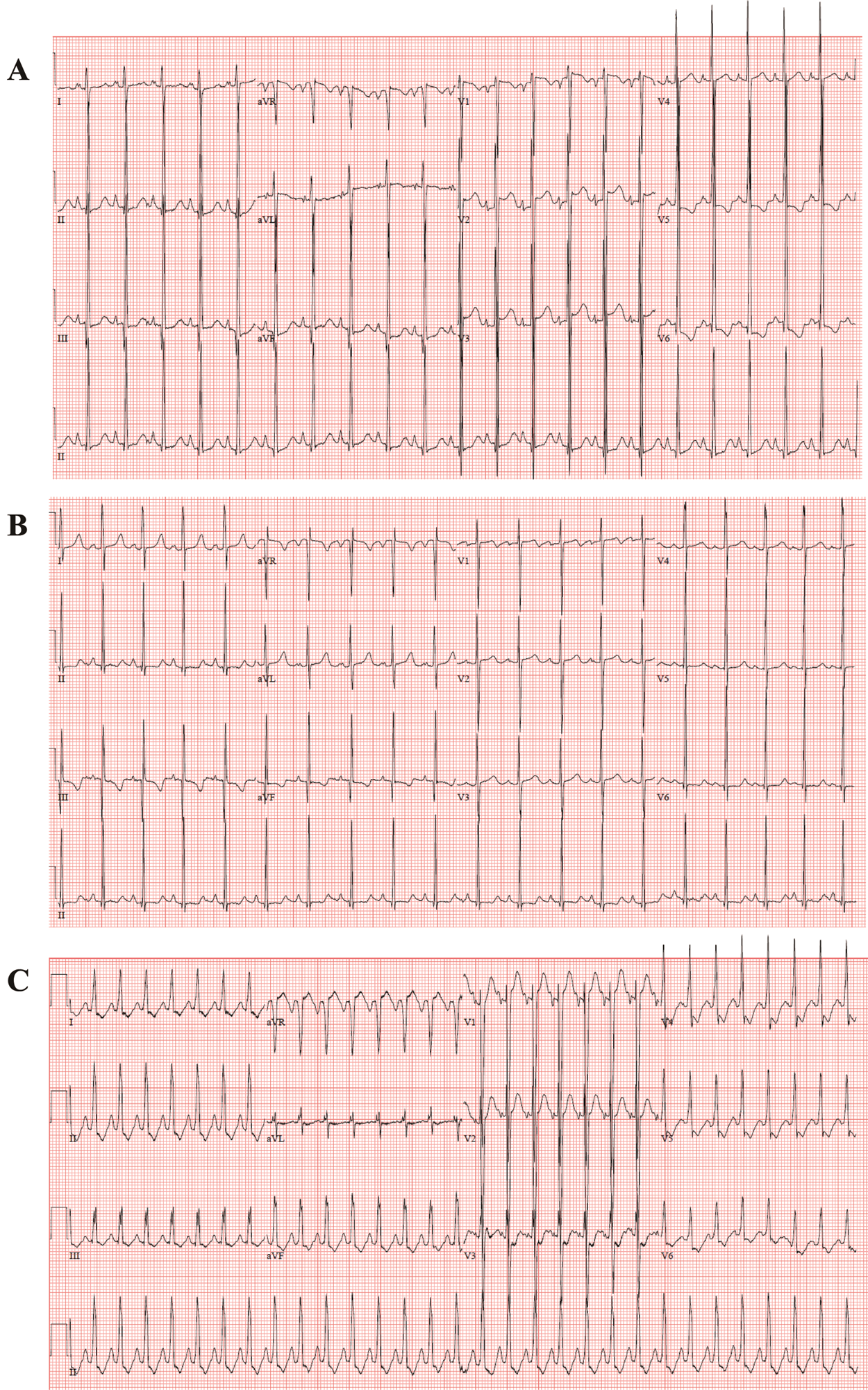
Figure 2: Electrocardiogram for different phenotypes. (A, B) An infant with a dilated cardiomyopathy (patient No. 1), (C, D) A neonate with a left ventricular non-compaction cardiomyopathy (patient No. 4), (E, F) A neonate with a hypertrophic cardiomyopathy (patient No. 7)
WES identified a genetic variation in seven out of eight patients. A detailed analysis of the genetic variation is summarized in Fig. 3 and Tab. 2. WES was carried out, and common variants corresponding to 98% with a minor allele frequency >5% were filtered out. Only variants matching the disease reported to date were screened and, among them, likely-benign, benign, and non-coding variants with low evidence according to the ACMG guidelines [15] were excluded. Lastly, the final candidate genetic variant was selected by a medical geneticist considering the relationship between gene and patient phenotypes.
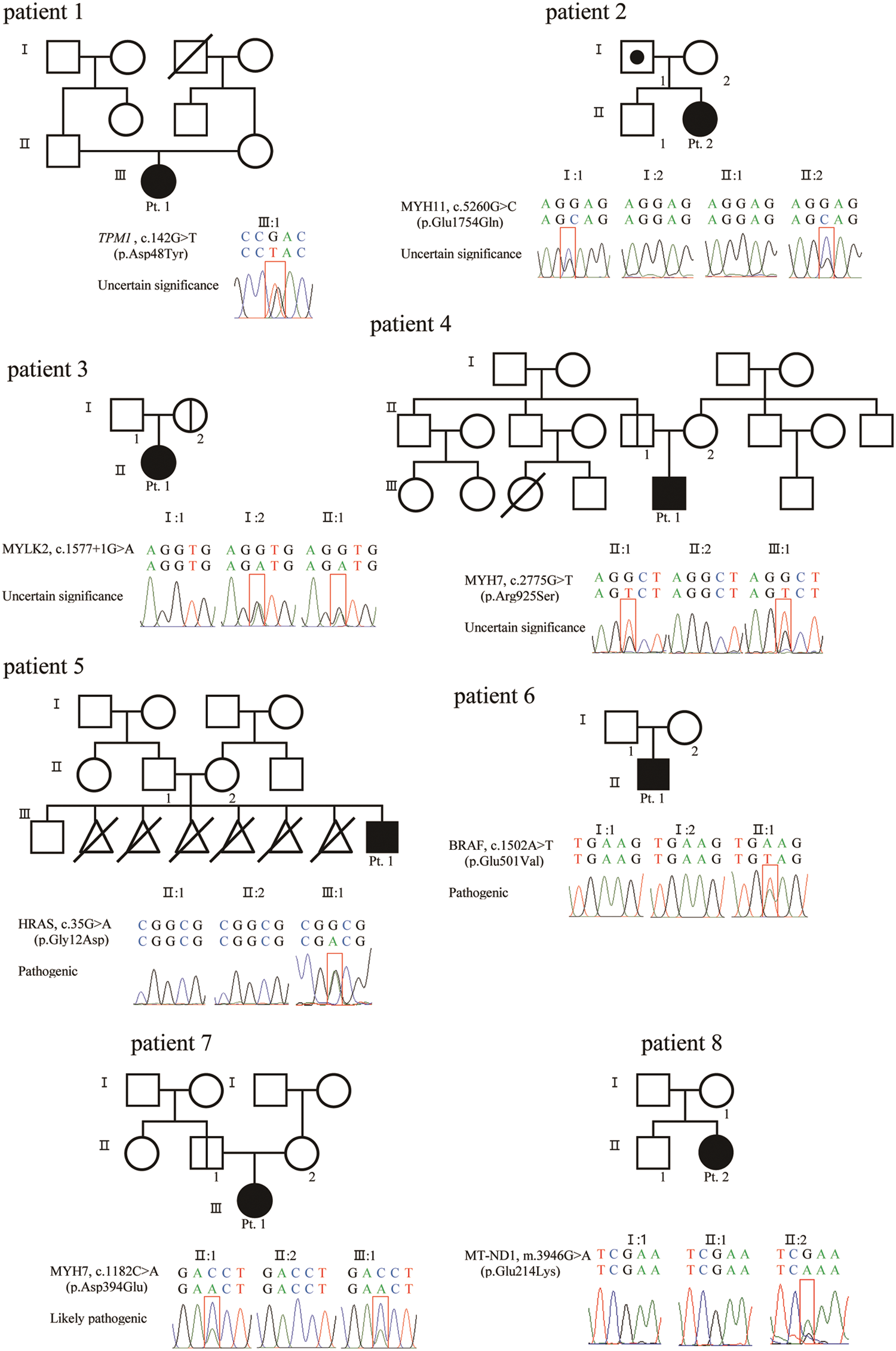
Figure 3: Sanger sequence of variant and pedigree for patients and parents
Table 2: Genetic variations found in patients with infantile-onset cardiomyopathy
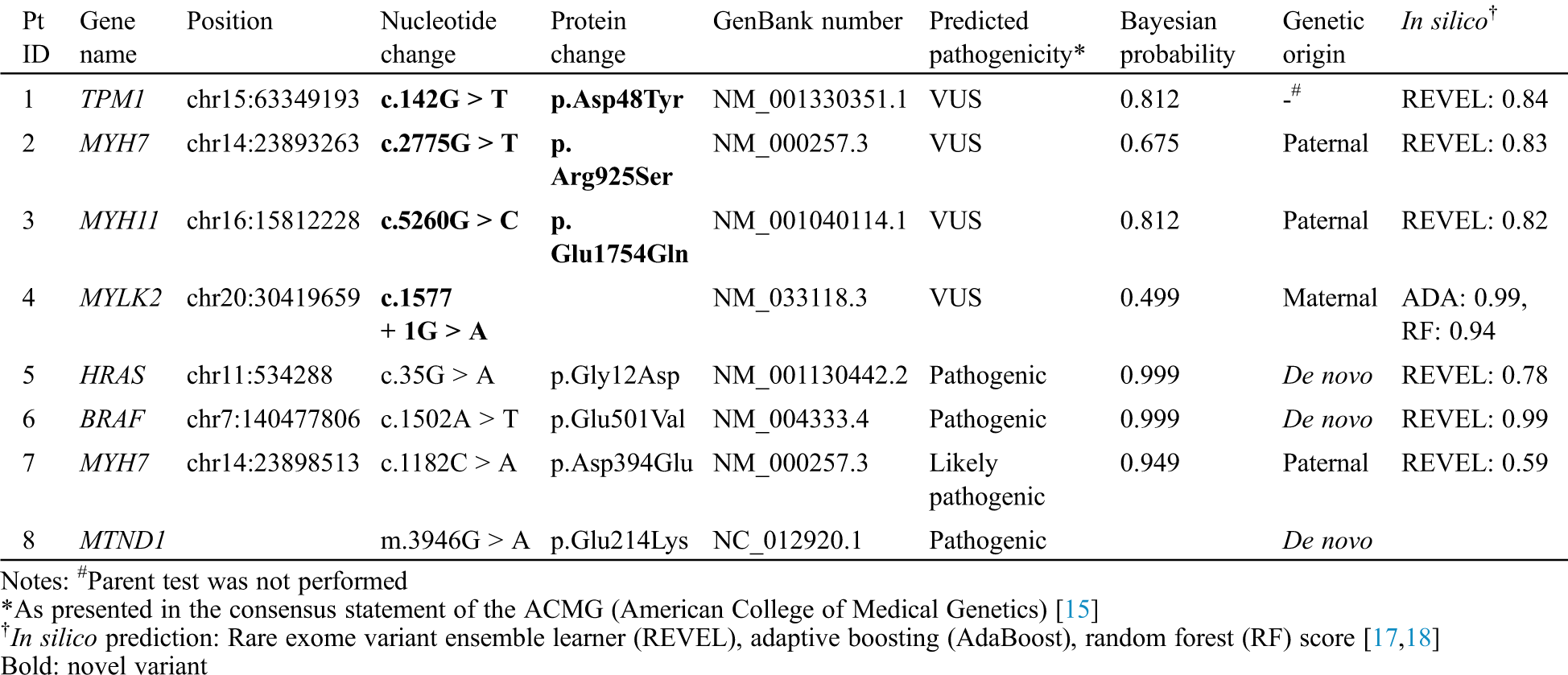
In the two patients with DCMP, genetic variations were identified in the genes TPM1, which is responsible for DCMP1Y (OMIM 611878, www.omim.org), and MYH7, which is responsible for DCMP1S (OMIM 160760). According to the ACMG guidelines [15], two variants, c.142G > T (p. Asp48Tyr) in TPM1 and c.2775G > T (p. Arg925Ser) in MYH7, were classified as variants of unknown significance (VUSs). Parental tests were available in patient 2, which was inherited from an asymptomatic parent.
In the two patients with LVNC, the MYH11 variant for aortic aneurysm, familial thoracic 4 (AAT4) (OMIM 160745) and the MYLK2 variant for cardiomyopathy, hypertrophic 1 (CMH1) (OMIM 606566) were identified. According to the ACMG guidelines [15], two variants, c.5260G > C (p. Glu1754Gln) in MYH11 and c.1577 + 1G > A in MYLK2, were classified as VUSs. All mutations were inherited from one parent; the father of patient No. 3 had aortic regurgitation, and the parent of patient No. 4 was asymptomatic.
In the four patients with HCMP, the genetic variations were identified in the genes HRAS, which is responsible for Costello syndrome (OMIM 190020), BRAF, responsible for Cardiofaciocutaneous syndrome (CFC, OMIM 164757), and MYH7, responsible for cardiomyopathy, hypertrophic 1 CMH1 (OMIM 160760). According to the ACMG guidelines [15], one variant, c.1182C > A (p. Asp394Glu) in MYH7 was classified as likely-pathogenic and the other two variants, c.35G > A (p. Gly12Asp) in HRAS and c.1502A > T (p. Glu501Val) in BRAF, were classified as pathogenic. Parental tests were available for the three patients; the HRAS and BRAF variants were de novo, and the MYH7 variant was inherited from an asymptotic parent. However, WES did not identify a mutation in patient No. 8, but mtDNA sequencing found a pathogenic mutation, m.3946G > A (p. Glu214Lys), in MT-ND1, which has been reported in patients with mitochondrial myopathy, encephalopathy, lactic acidosis, stroke-like episodes (MELAS; OMIM 540000). No relevant mutation was found in the mother’s mtDNA sequencing.
The clinical courses of the subjects are summarized in Tab. 3. Patient No. 1 was lost to follow-up after 1 month of life because of distance contraint from home. Among the seven patients with a follow-up period [median (IQR), 6 (1–10) months], one (12.5%, patient No. 4) is recovering heart function, three (37.5%, patient Nos. 2, 6, and 8) have stationary heart function with medications, and three (37.5%, patient Nos. 3, 5, and 7) had died; these were all identified prenatally. Median (IQR) survival duration was 48 (27.5–67.5) days of life (Fig. 4).
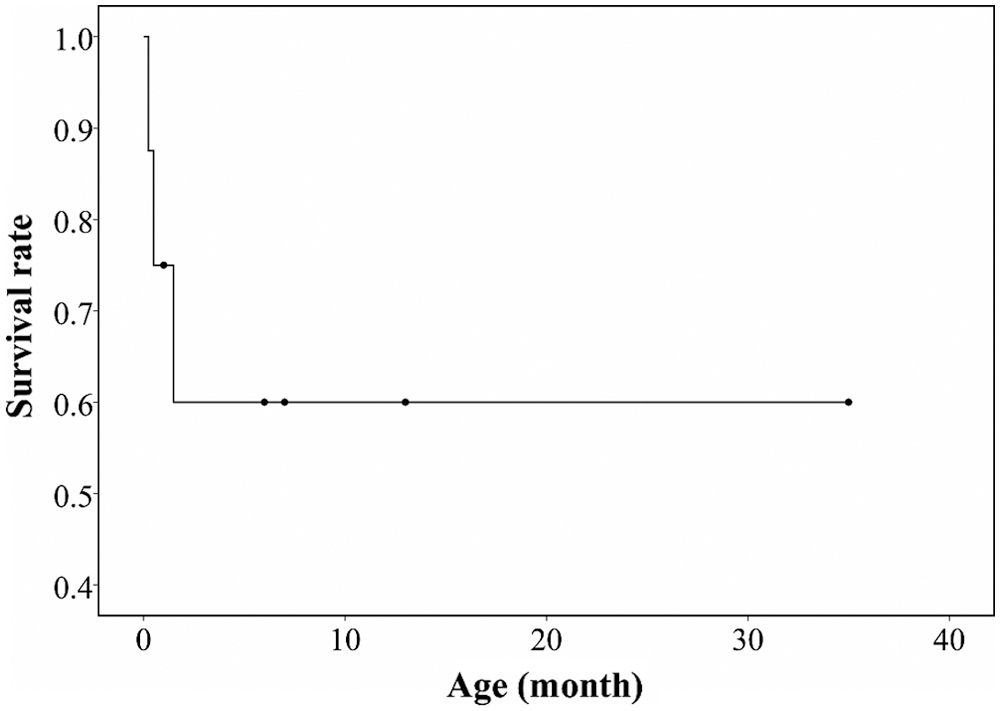
Figure 4: Kaplan-Meier survival graph of the patients with infantile-onset cardiomyopathy
Table 3: Clinical courses of patients with infantile-onset cardiomyopathy
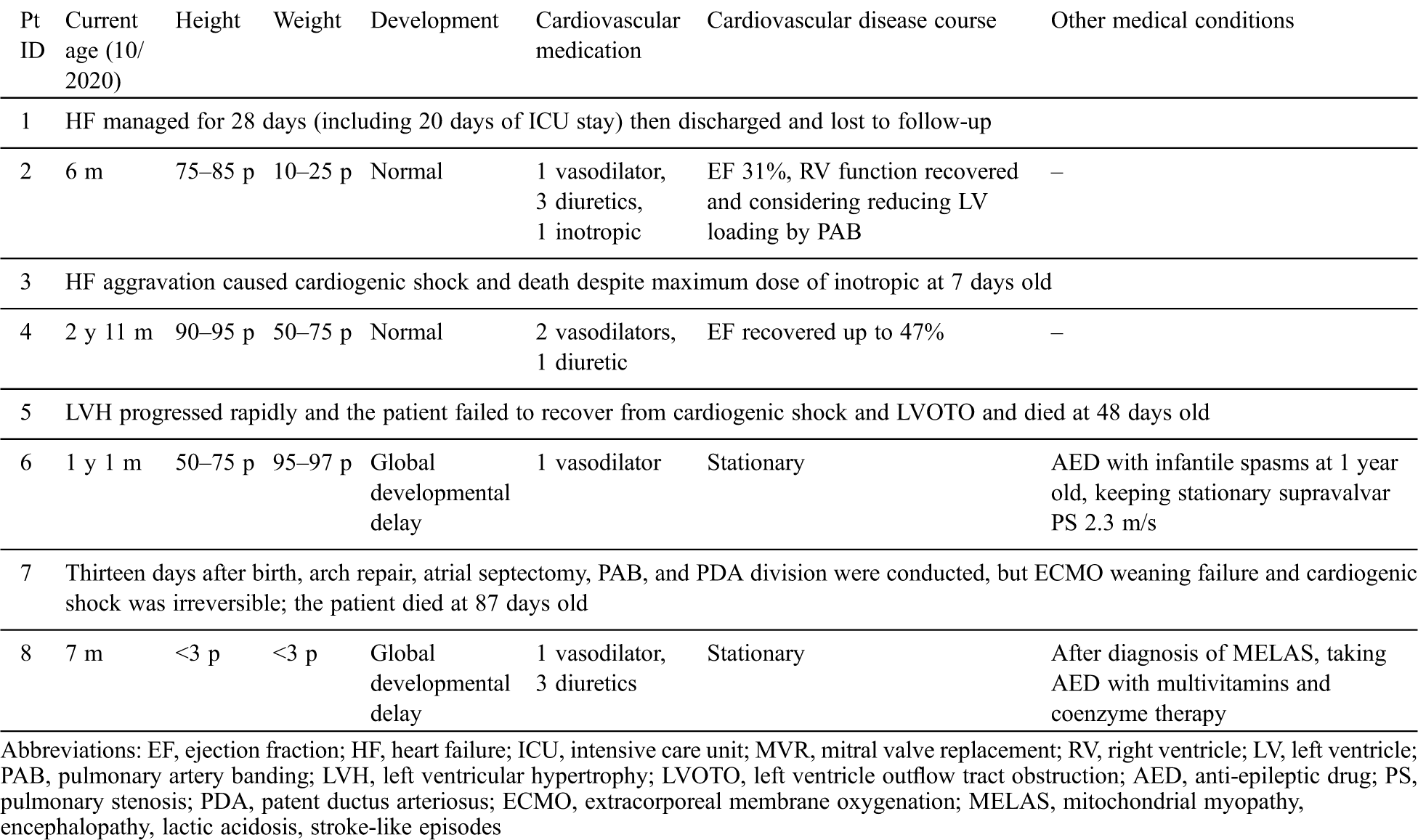
Among the two patients with DCMP, patient No. 1 was lost from follow-up and patient No. 2 had a persistently low cardiac output of 31% in EF but had normal growth and development until 6 months of age.
Among the two patients with LVNC, patient No. 3 died 1 week after birth due to progressive heart failure, and patient No. 4 is now recovering heart function to EF 47%, showing normal development and growth until 35 months of age.
Among the four patients with HCMP, patient No. 5 with Costello syndrome had progressive LVOTO and died due to cardiogenic shock at 48 days of life and patient No. 7 had an open-heart operation and died from failure of extracorporeal membrane oxygenation weaning at 86 days of life. Patient No. 6 with CFC has a similar level of hypertrophy and LVOTO until 13 months of age and is currently taking an anti-epileptic drug and undergoing rehabilitation for epilepsy and global development delay (case 17 in a previous study by Lee et al. [19]). Patient No. 8 with MELAS is being treated with vasodilator, diuretics, and anti-epileptic drug for seizure. At 7 months of age, she was below the third percentile in height and weight. She could make eye contact but could not control her head. Cardiac hypertrophy and LVOTO were stationary, but the blood lactic acid levels could be decreased to 3.7 mmol/L upon treatment with multivitamins, coenzymes, and arginine.
In this study, all patients with infantile-onset cardiomyopathy were identified based on variants associated with cardiomyopathy via WES and additional mtDNA sequencing. Kindel et al. [8] identified genetic etiology in 76% of pediatric patients with cardiomyopathy, and a comparable or somewhat higher frequency was found in our study. Moreover, in our study, all the genetic variants found in HCMP were pathogenic or likely-pathogenic, whereas the variants in DCMP or LVNC were VUSs. It has been the general consensus that genetic alteration is a major contributing factor to the development of infantile-onset cardiomyopathy. Pediatric patients with cardiomyopathy generally survive up to a median age of three, and the 1year rate of death is 31% [3]. The higher mortality and earlier death age observed in the present study [3/8 (37.5%), 48 (27.5–67.5) days] indicate that infantile-onset cardiomyopathy may hold a severe clinical course, which is attributed to the genetic factors.
In general, the phenotype suggested by a genetic defect identified in each of the patients was consistent with that of the affected patient. Genetic variants in patients with DCMP and HCMP in this study have been found in similar phenotypes in previous studies [9,20]. However, genetic variants found in patients with LVNC (MYH11 and MYLK2) have not been reported as being directly related to the disease phenotype. The MYLK2 variants have been previously described as being associated with HCMP [21]. Meanwhile, in a previous study, the MYLK2 variant was associated with LVNC, as with our patient No. 4 [22]. The MYH11 variant in patient No. 3 was associated with the development of aortic aneurysm, patent ductus arteriosus, or HCMP [23]. During the human embryonic stage, the development of the coronary artery is important for the disappearance of the sinusoids and transformation of the spongy myocardium into compact musculature. Therefore, it is possible that the MYH11 variant disturbs this transformation of fetal cardiac musculature by altering the myosin heavy-chain formation in the fetal blood vessels [24,25]. LVNCs might be considered a distinct subtype in the spectrum of cardiomyopathy but is still classified as a sub-trait or unclassified cardiomyopathy, given that LVNCs are found in other types of cardiomyopathy (DCMP or HCMP) [26,27]. As most patients have LVNC with other cardiomyopathies rather than as an isolated phenotype, the causative association of a gene to the development of LVNC is not yet clear [22,28]. Therefore, further experience with more cases of isolated LVNC or LVNC without DCMP or HCMP is required to assess this causative association.
In this study, one patient (patient No. 8) was diagnosed with MELAS through whole mtDNA sequencing, although WES did not find pathogenic variants in her nuclear DNA. Cardiomyopathy is one of the most characteristic features of mitochondrial disease, and her high blood lactic acid level was an additional pathognomonic finding to suspect a mitochondrial disease [20,29]. Indeed, infantile cardiomyopathy can be the first manifestation in some patients with mitochondrial disease [30]. Therefore, mtDNA sequencing should be considered in addition to or even before WES in patients with infantile-onset cardiomyopathy and high blood lactic acid level.
Importantly, one of the greatest challenges in the interpretation of genetic testing results in cardiomyopathy is that many parents share the variant found in their affected child but they do not have any subjective, relevant symptoms [31,32]. In this study, the parents carrying the same variant of their respective child were mostly asymptomatic (3/4, 75%). As most of the hereditary cardiomyopathies are inherited in an autosomal-dominant manner, a varying degree of penetrance and expressivity is expected, which can be affected by genomic, epigenetic, or environmental factors [33]. Not only RNA content and DNA sequence but also methylation status, chromatin structure and accessibility, protein composition, cell history, microenvironment, and cellular states in situ can affect the phenotype of subcellular, cellular, and tissue scales and finally that of the system scales [34,35]. In particular, TPM1, MYLK2, and MYH7 variants, found in the asymptomatic parents, were reported to confer a variable degree of penetrance and expressivity [36–38].
As subclinical cardiomyopathy is likely to progress gradually, a single evaluation is not sufficient to determine whether the asymptomatic carrier is not affected by cardiomyopathy; thus, the identified variant is not the causative variant. Rather, assessing their cardiac function on a regular basis for the long-term should be considered. There are no defined guidelines for these genotype-positive/phenotype-negative patients. However, those parameters would be helpful to predict the cardiomyopathy in the early stage, such as left ventricular global longitudinal strain, peak left atrial longitudinal strain that can be measured by cardiac magnetic resonance imaging, or three-dimensional echocardiography [39–43].
As an increasing number of subjects become involved in genomic studies such as WES, a major concern is the interpretation of variants identified in each subject. Importantly, according to the ACMG guidelines, the variants in the genes associated with cardiomyopathy should be considered to inform the subject regardless of the primary medical condition for which the genomic study was required [44]. In this respect, prediction for the pathogenicity of each identified variant is very important to interpret the causality of each variant. The population frequency, location of a variant, characteristics of a variant (frameshift, splicing, nonsense, and missense) and in silico analysis tools, such as REVEL, ada, and rf score, have been used to determine pathogenicity in accordance with ACMG guidelines (pathogenic, likely-pathogenic, VUS, likely-benign, and benign) [15–18,45,46]. In this study, only two of seven nuclear DNA variants, classified as pathogenic according to the ACMG guidelines, were de novo, whereas all the likely-pathogenic variants or VUSs were found in either parent. Among them, only two families with VUSs have relevant clinical symptoms or a family history. Therefore, the question remains that these VUSs might not be responsible for the development of CMP, and how much importance a physician should place on the variant, especially VUS, during genetic counseling for the family with the affected patient. In particular, as most parents are of reproductive age, whether to perform prenatal genetic testing for the next pregnancy based on the variant identified in the affected patient must be cautiously approached. To obtain a more relevant solution, larger scale genomic data with longitudinal clinical data of the asymptomatic but variant-carrying family members are required.
There are several limitations to this study. Due to the small number of patients, it was impossible to perform subgroup analysis among the patients with infantile cardiomyopathy. In addition, as mentioned earlier, the interpretation of the pathogenicity of each variant was limited by the observation of the same variant in asymptomatic family members. Due to the variable and mostly short-term follow-up periods, it was difficult to describe the long-term outcome of infantile cardiomyopathy in association with the genetic findings, which must be re-evaluated in a larger cohort with longer-term evaluation. Lastly, multiple variants in different genes can be found in a single patient [47], but in our study, only a single genetic variant was filtered as responsible for the patient phenotype. As we applied the artificial intelligence-based pipeline for the evaluation of genome data [16], the criteria for the filtration might have missed some low impact variants, which would have contributed to the development of cardiomyopathy together with the variant identified in each patient.
Genomic studies, such as WES, can identify a genetic variant in high proportions of patients with infantile-onset cardiomyopathy. However, due to a wide range of penetrance and expressivity, the interpretation of each variant’s pathogenicity is limited in most cases.
Data Sharing: Not applicable.
Author Contribution: JP: Drafted and revised the manuscript critically for important intellectual content and substantially contributed to the interpretation of data. GHS, MK: Substantially contributed to the analysis and interpretation of data. YL, YC, J-KK, Y-HK, JJY, EA-RK, EJ, BSL: Substantially contributed to the acquisition of data. JSB, BHL: Designed the study, revised the manuscript critically for important intellectual content, and approved the final version to be published.
Funding Statement: This work was supported by an Institute for Information and Communications Technology Promotion (IITP) grant funded by the Korean government (MSIT) (2018-0-00861, Intelligent SW Technology Development for Medical Data Analysis).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Lipshultz, S. E., Sleeper, L. A., Towbin, J. A., Lowe, A. M., Orav, E. J. et al. (2003). The incidence of pediatric cardiomyopathy in two regions of the United States. New England Journal of Medicine, 348(17), 1647–1655. DOI 10.1056/NEJMoa021715. [Google Scholar] [CrossRef]
2. Nugent, A. W., Daubeney, P. E., Chondros, P., Carlin, J. B., Cheung, M. et al. (2003). The epidemiology of childhood cardiomyopathy in Australia. New England Journal of Medicine, 348(17), 1639–1646. DOI 10.1056/NEJMoa021737. [Google Scholar] [CrossRef]
3. Towbin, J. A., Lowe, A. M., Colan, S. D., Sleeper, L. A., Orav, E. J. et al. (2006). Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA, 296(15), 1867–1876. DOI 10.1001/jama.296.15.1867. [Google Scholar] [CrossRef]
4. Lipshultz, S. E., Law, Y. M., Asante-Korang, A., Austin, E. D., Dipchand, A. I. et al. (2019). Cardiomyopathy in children. Classification and diagnosis. A scientific statement from the American heart association. Circulation, 140(1), 1090. [Google Scholar]
5. Fesslova, V., Mongiovì, M., Pipitone, S., Brankovic, J., Villa, L. (2010). Features and outcomes in utero and after birth of fetuses with myocardial disease. International Journal of Pediatrics, 2010(5), 1–9. DOI 10.1155/2010/628451. [Google Scholar] [CrossRef]
6. Colan, S. D., Lipshultz, S. E., Lowe, A. M., Sleeper, L. A., Messere, J. et al. (2007). Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children. Findings from the Pediatric Cardiomyopathy Registry. Circulation, 115(6), 773–781. [Google Scholar]
7. Wilkinson, J. D., Landy, D. C., Colan, S. D., Towbin, J. A., Sleeper, L. A. et al. (2010). The pediatric cardiomyopathy registry and heart failure. Key results from the first 15 years. Heart Failure Clinics, 6(4), 401–413. DOI 10.1016/j.hfc.2010.05.002. [Google Scholar] [CrossRef]
8. Kindel, S. J., Miller, E. M., Gupta, R., Cripe, L. H., Hinton, R. B. et al. (2012). Pediatric cardiomyopathy. Importance of genetic and metabolic evaluation. Journal of Cardiac Failure, 18(5), 396–403. DOI 10.1016/j.cardfail.2012.01.017. [Google Scholar] [CrossRef]
9. Lee, T. M., Hsu, D. T., Kantor, P., Towbin, J. A., Ware, S. M. et al. (2017). Pediatric Cardiomyopathies. Circulation Research, 121(7), 855–873. DOI 10.1161/CIRCRESAHA.116.309386. [Google Scholar] [CrossRef]
10. Towbin, J. A. (2014). Inherited cardiomyopathies. Pediatric Cardiology, 78(10), 2347–2356. [Google Scholar]
11. Dai, G., Pu, Z., Cheng, X., Yin, J., Chen, J. et al. (2019). Whole-exome sequencing reveals novel genetic variation for dilated cardiomyopathy in pediatric Chinese patients. Pediatric Cardiology, 40(5), 950–957. DOI 10.1007/s00246-019-02096-1. [Google Scholar] [CrossRef]
12. Lang, R. M., Bierig, M., Devereux, R. B., Flachskampf, F. A., Foster, E. et al. (2005). Recommendations for chamber quantification. A report from the American society of echocardiography’s guidelines and standards committee and the chamber quantification writing group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. Journal of the American Society of Echocardiography, 18(12), 1440–1463. DOI 10.1016/j.echo.2005.10.005. [Google Scholar] [CrossRef]
13. Arbustini, E., Weidemann, F., Hall, J. L. (2014). Left ventricular noncompaction. Journal of the American College of Cardiology, 64(17), 1840–1850. DOI 10.1016/j.jacc.2014.08.030. [Google Scholar] [CrossRef]
14. Grenier, M. A., Osganian, S. K., Cox, G. F., Towbin, J. A., Colan, S. D. et al. (2000). Design and implementation of the North American Pediatric Cardiomyopathy Registry. American Heart Journal, 139(2), s86–s95. DOI 10.1067/mhj.2000.103933. [Google Scholar] [CrossRef]
15. Richards, S., Aziz, N., Bale, S., Bick, D., Das, S. et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–423. DOI 10.1038/gim.2015.30. [Google Scholar] [CrossRef]
16. Seo, G. H., Kim, T., Choi, I. H., Park, J. Y., Lee, J. et al. (2020). Diagnostic yield and clinical utility of whole exome sequencing using an automated variant prioritization system, EVIDENCE. Clinical Genetics, 98(6), 562–570. DOI 10.1111/cge.13848. [Google Scholar] [CrossRef]
17. Jian, X., Boerwinkle, E., Liu, X. (2014). In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Research, 42(22), 13534–13544. DOI 10.1093/nar/gku1206. [Google Scholar] [CrossRef]
18. Ioannidis, N. M., Rothstein, J. H., Pejaver, V., Middha, S., McDonnell, S. K. et al. (2016). REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. American Journal of Human Genetics, 99(4), 877–885. DOI 10.1016/j.ajhg.2016.08.016. [Google Scholar] [CrossRef]
19. Lee, Y., Choi, Y., Seo, G. H., Kim, G. H., Choi, I. H. et al. (2020). Clinical and molecular spectra of BRAF-associated RASopathy. Journal of Human Genetics, 2020. DOI 10.1038/s10038-020-00852-3. [Google Scholar] [CrossRef]
20. Hsu, Y. R., Yogasundaram, H., Parajuli, N., Valtuille, L., Sergi, C. et al. (2016). MELAS syndrome and cardiomyopathy: linking mitochondrial function to heart failure pathogenesis. Heart Failure Reviews, 21(1), 103–116. DOI 10.1007/s10741-015-9524-5. [Google Scholar] [CrossRef]
21. Davis, J. S., Hassanzadeh, S., Winitsky, S., Lin, H., Satorius, C. et al. (2001). The overall pattern of cardiac contraction depends on a spatial gradient of myosin regulatory light chain phosphorylation. Cell, 107(5), 631–641. DOI 10.1016/S0092-8674(01)00586-4. [Google Scholar] [CrossRef]
22. Vershinina, T., Fomicheva, Y., Muravyev, A., Jorholt, J., Kozyreva, A. et al. (2020). Genetic spectrum of left ventricular non-compaction in paediatric patients. Cardiology, 145(11), 746–756. DOI 10.1159/000510439. [Google Scholar] [CrossRef]
23. Hajj, H., Dagle, J. M. (2012). Genetics of patent ductus arteriosus susceptibility and treatment. Seminars in Perinatology, 36(2), 98–104. DOI 10.1053/j.semperi.2011.09.019. [Google Scholar] [CrossRef]
24. Swärd, K., Krawczyk, K. K., Morén, B., Zhu, B., Matic, L. et al. (2019). Identification of the intermediate filament protein synemin/SYNM as a target of myocardin family coactivators. American Journal of Physiology–Cell Physiology, 317(6), C1128–C1142. DOI 10.1152/ajpcell.00047.2019. [Google Scholar] [CrossRef]
25. Freedom, R. M., Yoo, S. J., Perrin, D., Taylor, G., Petersen, S. et al. (2005). The morphological spectrum of ventricular noncompaction. Cardiology in the Young, 15(4), 345–364. DOI 10.1017/S1047951105000752. [Google Scholar] [CrossRef]
26. Sedaghat-Hamedani, F., Haas, J., Zhu, F., Geier, C., Kayvanpour, E. et al. (2017). Clinical genetics and outcome of left ventricular non-compaction cardiomyopathy. European Heart Journal, 38(46), 3449–3460. DOI 10.1093/eurheartj/ehx545. [Google Scholar] [CrossRef]
27. Elliott, P., Andersson, B., Arbustini, E., Bilinska, Z., Cecchi, F. et al. (2007). Classification of the cardiomyopathies: A position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. European Heart Journal, 29(2), 270–276. DOI 10.1093/eurheartj/ehm342. [Google Scholar] [CrossRef]
28. Ichida, F. (2020). Left ventricular noncompaction—risk stratification and genetic consideration. Journal of Cardiology, 75(1), 1–9. DOI 10.1016/j.jjcc.2019.09.011. [Google Scholar] [CrossRef]
29. Zhang, D., Li, Y., Heims-Waldron, D., Bezzerides, V., Guatimosim, S. et al. (2018). Mitochondrial cardiomyopathy caused by elevated reactive oxygen species and impaired cardiomyocyte proliferation. Circulation Research, 122(1), 74–87. DOI 10.1161/CIRCRESAHA.117.311349. [Google Scholar] [CrossRef]
30. Repp, B. M., Mastantuono, E., Alston, C. L., Schiff, M., Haack, T. B. et al. (2018). Clinical, biochemical and genetic spectrum of 70 patients with ACAD9 deficiency: is riboflavin supplementation effective? Orphanet Journal of Rare Diseases, 13(1), 980. DOI 10.1186/s13023-018-0784-8. [Google Scholar] [CrossRef]
31. Ho, C. Y. (2010). Genetics and clinical destiny: improving care in hypertrophic cardiomyopathy. Circulation, 122(23), 2430–2440. DOI 10.1161/CIRCULATIONAHA.110.978924. [Google Scholar] [CrossRef]
32. Paldino, A., De Angelis, G., Merlo, M., Gigli, M., Dal Ferro, M. et al. (2018). Genetics of dilated cardiomyopathy: clinical implications. Current Cardiology Reports, 20(10), 13. DOI 10.1007/s11886-018-1030-7. [Google Scholar] [CrossRef]
33. Lehner, B. (2013). Genotype to phenotype: lessons from model organisms for human genetics. Nature Reviews Genetics, 14(3), 168–178. DOI 10.1038/nrg3404. [Google Scholar] [CrossRef]
34. Camp, J. G., Platt, R., Treutlein, B. (2019). Mapping human cell phenotypes to genotypes with single-cell genomics. Science, 365(6460), 1401–1405. DOI 10.1126/science.aax6648. [Google Scholar] [CrossRef]
35. Cooper, D. N., Krawczak, M., Polychronakos, C., Tyler-Smith, C., Kehrer-Sawatzki, H. (2013). Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Human Genetics, 132(10), 1077–1130. DOI 10.1007/s00439-013-1331-2. [Google Scholar] [CrossRef]
36. Wang, J., Guo, R. Q., Guo, J. Y., Zuo, L., Lei, C. H. et al. (2018). Investigation of myocardial dysfunction using three-dimensional speckle tracking echocardiography in a genetic positive hypertrophic cardiomyopathy Chinese family. Cardiology in the Young, 28(9), 1106–1114. DOI 10.1017/S1047951118000860. [Google Scholar] [CrossRef]
37. Alpert, N. R., Mohiddin, S. A., Tripodi, D., Jacobson-Hatzell, J., Vaughn-Whitley, K. et al. (2005). Molecular and phenotypic effects of heterozygous, homozygous, and compound heterozygote myosin heavy-chain mutations. American Journal of Physiology-Heart and Circulatory Physiology, 288(3), H1097–H1102. DOI 10.1152/ajpheart.00650.2004. [Google Scholar] [CrossRef]
38. Deacon, D. C., Happe, C. L., Chen, C., Tedeschi, N., Manso, A. M. et al. (2019). Combinatorial interactions of genetic variants in human cardiomyopathy. Nature Biomedical Engineering, 3(2), 147–157. DOI 10.1038/s41551-019-0348-9. [Google Scholar] [CrossRef]
39. Paldino, A., De Angelis, G., Ferro, M. D., Faganello, G., Porcari, A. et al. (2020). High prevalence of subtle systolic and diastolic dysfunction in genotype-positive phenotype-negative relatives of dilated cardiomyopathy patients. International Journal of Cardiology, 324, 108–114. DOI 10.1016/j.ijcard.2020.09.036. [Google Scholar] [CrossRef]
40. Piras, P., Torromeo, C., Evangelista, A., Esposito, G., Nardinocchi, P. et al. (2019). Non-invasive prediction of genotype positive-phenotype negative in hypertrophic cardiomyopathy by 3D modern shape analysis. Experimental Physiology, 104(11), 1688–1700. DOI 10.1113/EP087551. [Google Scholar] [CrossRef]
41. Brouwer, W. P., van Dijk, S. J., Stienen, G. J., van Rossum, A. C., van der Velden, J. et al. (2011). The development of familial hypertrophic cardiomyopathy: from mutation to bedside. European Journal of Clinical Investigation, 41(5), 568–578. DOI 10.1111/j.1365-2362.2010.02439.x. [Google Scholar] [CrossRef]
42. Germans, T., Rüssel, I. K., Götte, M. J., Spreeuwenberg, M. D., Doevendans, P. A. et al. (2010). How do hypertrophic cardiomyopathy mutations affect myocardial function in carriers with normal wall thickness? Assessment with cardiovascular magnetic resonance. Journal of Cardiovascular Magnetic Resonance, 12(1), 1308. DOI 10.1186/1532-429X-12-13. [Google Scholar] [CrossRef]
43. Fatkin, D., Yeoh, T., Hayward, C. S., Benson, V., Sheu, A. et al. (2011). Evaluation of left ventricular enlargement as a marker of early disease in familial dilated cardiomyopathy. Circulation: Cardiovascular Genetics, 4(4), 342–348. DOI 10.1161/CIRCGENETICS.110.958918. [Google Scholar] [CrossRef]
44. Kalia, S. S., Adelman, K., Bale, S. J., Chung, W. K., Eng, C. et al. (2017). Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0A policy statement of the American College of Medical Genetics and Genomics. Genetics in Medicine, 19(2), 249–255. DOI 10.1038/gim.2016.190. [Google Scholar] [CrossRef]
45. Matthijs, G., Souche, E., Alders, M., Corveleyn, A., Eck, S. et al. (2016). Guidelines for diagnostic next-generation sequencing. European Journal of Human Genetics, 24(1), 2–5. DOI 10.1038/ejhg.2015.226. [Google Scholar] [CrossRef]
46. Jennings, L. J., Arcila, M. E., Corless, C., Kamel-Reid, S., Lubin, I. M. et al. (2017). Guidelines for validation of next-generation sequencing–based oncology panels. Journal of Molecular Diagnostics, 19(3), 341–365. DOI 10.1016/j.jmoldx.2017.01.011. [Google Scholar] [CrossRef]
47. Bales, N. D., Johnson, N. M., Judge, D. P., Murphy, A. M. (2016). Comprehensive versus targeted genetic testing in children with hypertrophic cardiomyopathy. Pediatric Cardiology, 37(5), 845–851. DOI 10.1007/s00246-016-1358-y. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |