| Congenital Heart Disease |
DOI: 10.32604/CHD.2021.014272
ARTICLE
Selexipag as Add-on Therapy for Patients with Pulmonary Arterial Hypertension Associated with Congenital Heart Disease: A Single-Center Retrospective Study
Division of Pediatric Cardiology, Congenital Heart Disease Center, Severance Cardiovascular Hospital, Department of Pediatrics, Yonsei University College of Medicine, Seoul, Korea
*Corresponding Author: Jae Young Choi. Email: cjy0122@yuhs.ac
#These authors contributed equally to this article
Received: 16 September 2020; Accepted: 21 October 2020
Abstract: Purpose: This study examined the efficacy and safety of selexipag in treating pulmonary arterial hypertension (PAH) associated with congenital heart disease (CHD). Materials and Methods: We conducted a retrospective study of patients with CHD-associated PAH, treated with selexipag since December 2017. Thirteen adult patients (mean age, 45.4 years; women, 77%) were treated with selexipag as add-on therapy. Baseline characteristics, World Health Organization functional class, 6-minute walking distance (6MWD) test results, N-terminal pro-B-type natriuretic peptide levels, echocardiographic data, and incidence of side effects were assessed. Results: The majority of patients (12/13, 92.3%) experienced more than one treatment-associated complication; one patient dropped out of the study due to intolerable myalgia. The results of 6MWD test (from 299.2 ± 56.2 m to 363.8 ± 86.5 m, p = 0.039) and tricuspid regurgitation (TR) pressure gradient (from 84.7 ± 20.5 mmHg to 61.6 ± 24.0 mmHg, p = 0.018) improved and remained improved after selexipag treatment in 12 patients. Based on the results of a non-invasive risk assessment, 8 (66.7%) patients showed improvement, 3 (25.0%) showed no interval change, and the status of one patient (8.3%) deteriorated. Moreover, compared to patients treated with a low dosage, patients treated with a medium-to-high dosage showed a greater increase in 6MWD results (88.3 ± 26.4 m vs. 55.3 ± 27.6 m, p = 0.043) and a greater reduction in the TR pressure gradient (−33.7 ± 10.9 mmHg vs. −12.5 ± 12.0 mmHg, p = 0.015). Conclusion: Selexipag is an efficient pulmonary vasodilator as add-on therapy in treating CHD-associated PAH.
Keywords: Selexipag; congenital heart disease; pulmonary arterial hypertension
Pulmonary arterial hypertension (PAH) associated with congenital heart disease (CHD) (PAH-CHD) is one of the most common types of PAH [1–3]. Recent progress in medical and surgical treatment of CHD means that nowadays most affected patients survive into adulthood [4], leading to an increase in the prevalence of PAH-CHD. The prevalence of PAH among adult patients with CHD is estimated to be 5–10%, depending on the underlying shunt size and location and complexity of the CHD [5]. PAH-CHD stratified by shunt lesions consists of heterogeneous subgroups [6,7], as follows: (1) Eisenmenger syndrome, (2) PAH associated with predominant systemic-to-pulmonary shunts (either correctable or not), (3) PAH with small/coincidental defects, and (4) PAH after repair (corrected PAH-CHD). Treatment of PAH-CHD requires a delicate and meticulous approach by PAH-CHD experts, including precise early diagnosis of CHD and confirmation of PAH-CHD, including operability, based on the results of pulmonary vascular resistance and vasoreactive tests. Timely corrective surgery or intervention is imperative in operable PAH-CHD, and staged palliation for further definitive treatment is crucial in the treatment of complex or borderline PAH-CHD. Targeted PAH therapy and suitable supportive care, including diuretics, oxygen supplementation, and the use of appropriate anti-coagulation agents are important at every strategic point in-patient care [7]. Recent guidelines recommend sequential or upfront use of a combination of selective PAH drugs, targeting endothelin, nitric oxide, and prostacyclin pathways [8]. Selexipag (Uptravi, Actelion Pharmaceuticals) is a highly selective, oral, long-acting prostacyclin receptor (IP receptor) agonist [9,10]. It was approved by the Food and Drug Administration in December 2015 and by the European Medicines Agency in May 2016, allowing for marketing of an oral combination therapy, targeting the prostacyclin pathway. A phase III trial (GRIPHON) has shown that, compared to placebo, selexipag reduced the risk of mortality or major complications associated with PAH [11] and improved the results of a 6-minute walking distance test. Thereafter, many studies have demonstrated the clinical efficacy and tolerability in various patient groups, including patients with connective tissue disease [12,13], chronic pulmonary thromboembolism [14], as well as pediatric populations [15]; heterogeneous real-life data also appear to support the clinical trial findings [16]. Regarding treatment of PAH-CHD, post-hoc analysis of the GRIPHON study data has demonstrated that selexipag may delay disease progression and is well-tolerated [17]; however, this trial only included corrected PAH-CHD patients. Thus, the clinical efficacy and safety of selexipag among patients with PAH-CHD requires elucidation. This study examined the efficacy and safety of selexipag in PAH-CHD patients, including patients with Eisenmenger syndrome and partially repaired CHD.
Adult patients (age >18 years), prescribed selexipag, from December 2017 to August 2019 at the Division of Pediatric Cardiology, Congenital Heart Center, Severance Cardiovascular Hospital, a tertiary university hospital, were retrospectively screened and enrolled in this study. Patients with PAH-CHD, previously confirmed by right heart catheterization, were included; patients with idiopathic PAH was excluded. At baseline, all patients were treated with all types of PAH drugs, except prostanoids; selexipag was added to the baseline PAH-targeting drug.
The Institutional Review Board of Severance Hospital (4-2019-1083) approved the study protocol, and this study was performed in accordance with the Declaration of Helsinki; the requirement for acquisition of informed consent was waived owing to the study’s retrospective nature.
Although this study was performed retrospectively, our clinical policy strictly followed the relevant PAH management guidelines [8]. Right heart catheterization was performed using a Berman or Swan-Ganz catheter, and cardiac output was measured by thermodilution. The pulmonary vasoreactivity test was performed using inhaled O2 and iloprost. According to the guidelines on the pulmonary vascular resistance index, the values >8 Wood unit (WU)*m2 were indicative of inoperable cases [8,18]. In patients with borderline PAH (pulmonary vascular resistance index range, 4–8 WU*m2) with an atrial septal defect (ASD), treatment modalities (surgery vs. interventions, and approaches with vs. without fenestration) were discussed and decided on in a multidisciplinary team meeting. When indicated, transcatheter closure of the atrial septal defect was performed, as previously reported. [19,20] Temporary balloon occlusion test was performed for additional information of reversibility of PAH as well as testing for masked left ventricular restriction in patients with ASD.
Except for patients with an atrial septal defect, all patients with CHD had Eisenmenger syndrome. ASD patients with acceptable range of PAH were treated either completely repaired (N = 1) or partially repaired with fenestration (N = 5). However, one patient with uncorrectable PAH was not repaired, and has been treated with PAH drugs and will be analyzed by further catheterization, which is so called “PAH treat and repair strategy” [21]. Selexipag treatment was initiated at a dose of 200 µg twice daily which was increased weekly or biweekly in increments of 200 µg twice daily until unmanageable side effects developed. When unmanageable side effects developed, the dose was decreased by 200 µg twice daily. Up-titration was resumed once side effects had subsided or become tolerable; nevertheless, increases were administered at a slower rate. If intolerable and untreatable side effects recurred, the dose was decreased by 200 µg once or twice daily, and the adjusted dose was regarded as the maintenance dose. We prescribed supportive analgesics (ibuprofen) in all cases as well as antiemetic (metoclopramide) and antidiarrheal drugs (loperamide), as required. All supportive drugs were prescribed at their usual dosage and increased to the maximal dosage, as required and indicated.
Baseline characteristics, hemodynamic and echocardiographic data, World Health Organization functional class, 6-minute walking distance test results, and N-terminal pro-B-type natriuretic peptide (NT-proBNP) levels were assessed. Data from the last available right heart catheterization measurement were adopted as the baseline. Patients were followed up every 1–4 weeks during selexipag dose titration and every 3–6 months thereafter. Side effects of selexipag and World Health Organization functional class were assessed at every clinic visit by the attending physician. The 6-minute walking distance test was performed according to the American Thoracic Society guidelines [22]. Transthoracic echocardiography was performed in supine and left lateral decubitus positions; the basic 2-dimensional and M-mode echocardiography, and continuous wave and tissue Doppler data were obtained. Data on echocardiographic prognostic markers of PAH such as the pressure gradient of tricuspid regurgitation, tricuspid annular plane systolic excursion of the right atrial area, and presence of pericardial effusion were collected. The doppler image of tricuspid regurgitation was obtained well before and after selexipag treatment in all patients, which might be related with patient characteristics who had severe PAH and at least mild tricuspid regurgitation.
Risk assessment was based on non-invasive methods, as defined in the French registry, and involved the following parameters: Low risk was defined as World Health Organization functional class I–II, 6-minute walking distance >440 m, and NT-proBNP <300 ng/L [23,24].
The D’Agostino-Pearson test was used to verify the normality of distribution assumption of continuous variables. Normally distributed continuous variables were reported as means and standard deviations, whereas their non-normally distributed counterparts were expressed as medians with ranges. Categorical variables were expressed as counts (percentages). Changes from baseline to follow-up measurements of variables of interest were assessed using the chi-squared test, paired t-test, or Wilcoxon test, as suitable, and a 2-tailed p-value <0.05 was considered statistically significant. Statistical analysis was performed using SPSS version 25.0 (IBM Corp., Armonk, NY, USA).
A total of 14 patients were treated with selexipag at our department; a single patient with idiopathic PAH was excluded from this study. The baseline characteristics of the enrolled patients are summarized in (Tab. 1). Patients were either World Health Organization functional class II (38.5%) or III (61.5%). All patients were already being treated with PAH drugs; 12 patients were receiving monotherapy, (with an endothelin receptor antagonist and a phosphodiesterase type 5 inhibitor prescribed to 9 and 3 patients, respectively) and 1 was on dual therapy. The median treatment duration was 49 months (range, 3–120 months). There were no differences in sex, age, World Health Organization functional class, or baseline PAH drug use between patients with and without Eisenmenger syndrome (data not shown). No patients died during the study period or were lost to follow-up.
Table 1: Baseline and hemodynamic characteristics of patients (n = 13)
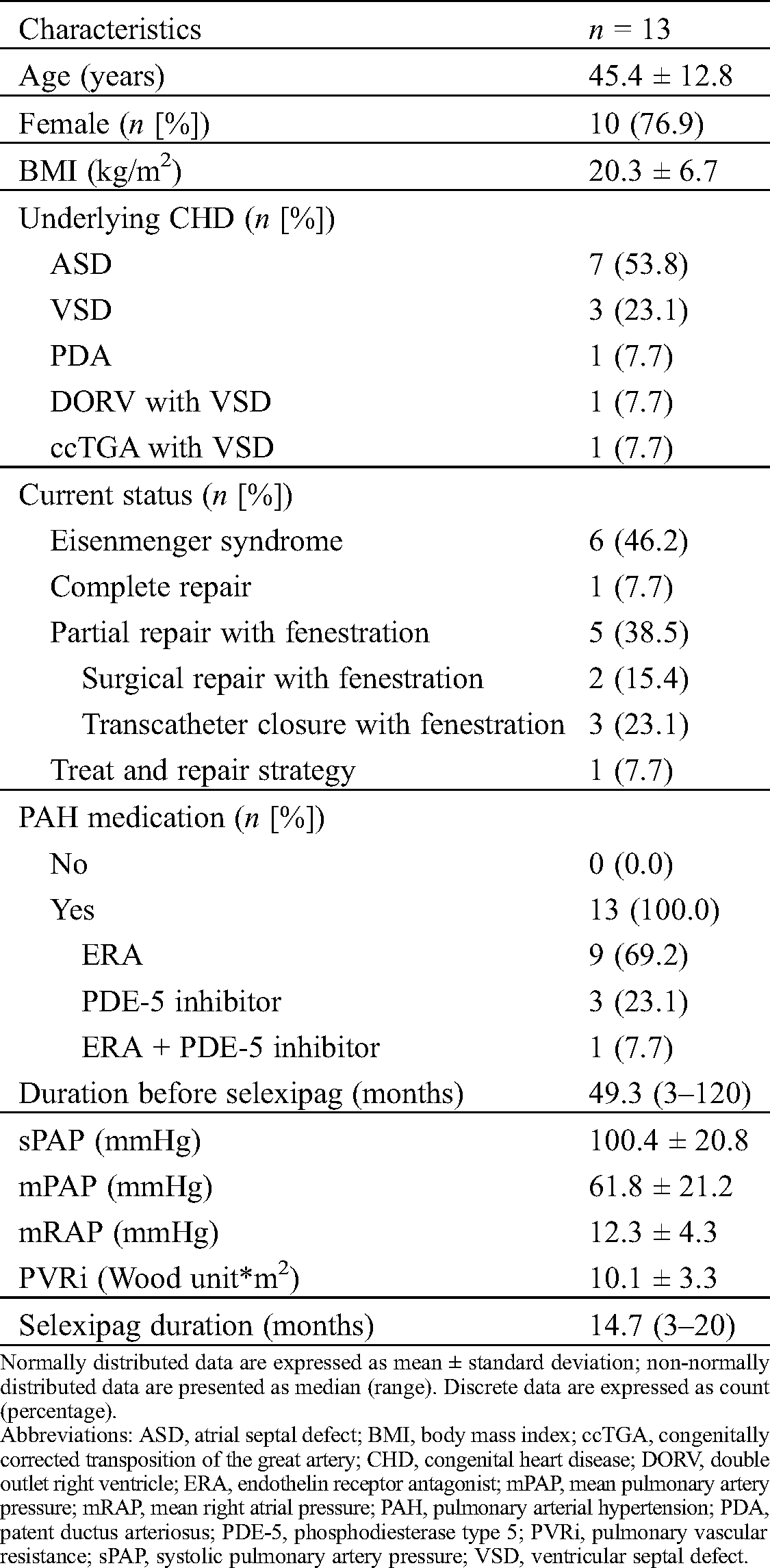
In the present study, no patient reached the maximal maintenance dosage of selexipag (mean, 1166 µg/day), as the majority of patients (92.3%) experienced more than 1 side effect. The maximal maintenance dosage was low (<1000 µg/day) in 41.6% of patients, medium (1000–2000 µg/day) in 50% of patients, and high (>2000 µg/day) in one (8.4%) (Fig. 1a). After adjustment for weight, half of the patients reached a dosage of 20–60 µg/kg/day (Fig. 1b).
In the present study, the most common side effect was musculoskeletal pain, followed by headache, and nausea and diarrhea (Fig. 2). No unexpected side effects were reported. One patient with a ventricular septal defect and Eisenmenger syndrome suffered due to intolerable myalgia, resulting in drug discontinuation and exclusion from the present study’s analysis of efficacy and risk reassessment. One patient, who was previously diagnosed with rheumatic arthritis and had suffered from arthralgia prior to selexipag initiation, could not bear the dosage titration, resulting in a maintenance dosage of 400 µg/day.
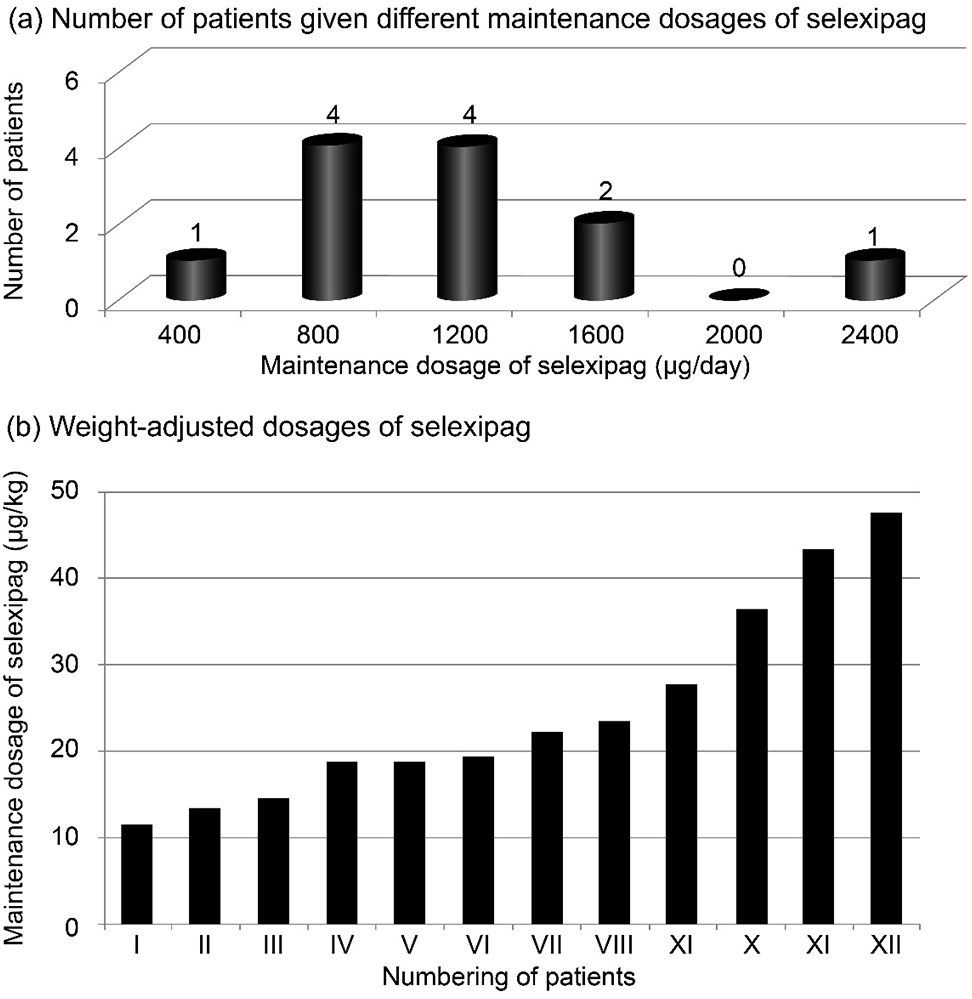
Figure 1: Individual maintenance dosage of selexipag (A) and weight-adjusted dosage (B) (n = 12)
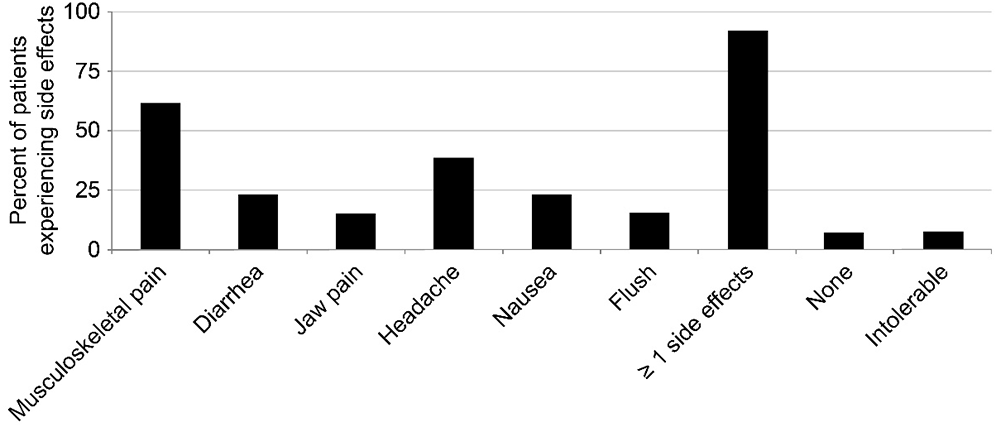
Figure 2: Percentage of patients experiencing different side effects associated with selexipag treatment (n = 13)
Following exclusion of one patient due to selexipag discontinuation, 12 patients in total were included in the efficacy analysis. Follow-up (FU) assessment was performed at 12.3 ± 2.8 months after selexipag initiation. After selexipag treatment, the results of the 6-minute walking distance test increased (from 299.2 ± 56.2 m to 363.8 ± 86.5 m, p = 0.039; Fig. 3a) and the tricuspid regurgitation pressure gradient decreased (from 84.7 ± 20.5 mmHg to 61.6 ± 24.0 mmHg, p = 0.018; Fig. 3b). Concurrently, NT-proBNP levels decreased (from 1580 ± 497 pg/mL to 629 ± 220 pg/mL, p = 0.096; Fig. 3c); however, this change was not statistically significant. Furthermore, World Health Organization functional class improved in 5 patients and remained stable in 7 patients. The tricuspid annular plane systolic excursion and right atrium fractional area change showed no relevant changes.
Figure 3: Changes in clinical parameters after selexipag treatment: (A) 6-minute walking distance (6MWD), (B) tricuspid regurgitation (TR) pressure gradient, and (C) N-terminal pro-B-type natriuretic peptide (NT-proBNP)
Changes in the risk assessment, based on the French registry group [23], between baseline and follow-up are presented in Fig. 4. Among individual patients, 8 (66.7%) showed improvement, 3 (25.0%) showed no change, and 1 (16.7%) showed worsening at the time of risk reassessment. Individual profiles of risk assessment before and after selexipag was described in (Tab. 2). Among 5 patients with Eisenmenger syndrome, 2 showed improvement in 6-minute walking distance scores (n = 1) and NT-proBNP levels (n = 1), whereas the remaining 3 exhibited no change. As for patients without Eisenmenger syndrome, improvement was observed in 6 cases, and a single patient experienced worsening.
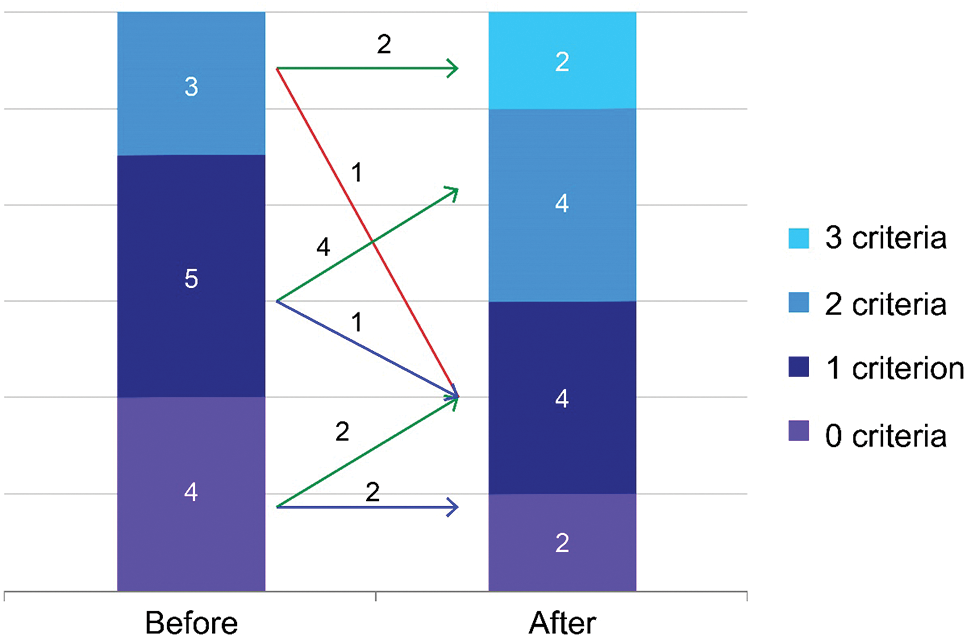
Figure 4: Changes in the assessed risk after selexipag treatment using 3 non-invasive parameters: 6-minute walking distance, World Health Organization functional class, and N-terminal pro-B-type natriuretic peptide. The number of parameters in the low-risk group is counted for each patient. Low risk criteria was defined as World Health Organization functional class I–II, 6-minute walking distance >440 m, and NT-proBNP <300 ng/L, respectively [23]. Green, blue, and red arrows indicate clinical improvement, no interval change, and worsening, respectively
Table 2: Changes of risk assessment in individual patients (N = 12)

Compared to the low dosage group (n = 5), the medium-to-high maintenance dosage group (n = 7) showed a significant change in 6-minute walking distance scores (88.3 ± 26.4 m vs. 55.3 ± 27.6 m, p = 0.043, Fig. 5a) and tricuspid regurgitation pressure gradient (−33.7 ± 10.9 mmHg vs. −12.5 ± 12.0 mmHg, p = 0.015, Fig. 5b). Concurrently, there was no difference in the change in NT-proBNP levels between the groups (Fig. 5c). Nevertheless, the maintenance dosage did not seem to affect the patients’ PAH status, irrespective of the presence of Eisenmenger syndrome (1371 ± 558 in non-Eisenmenger vs. 880 ± 334 in Eisenmenger, p = 0.112).
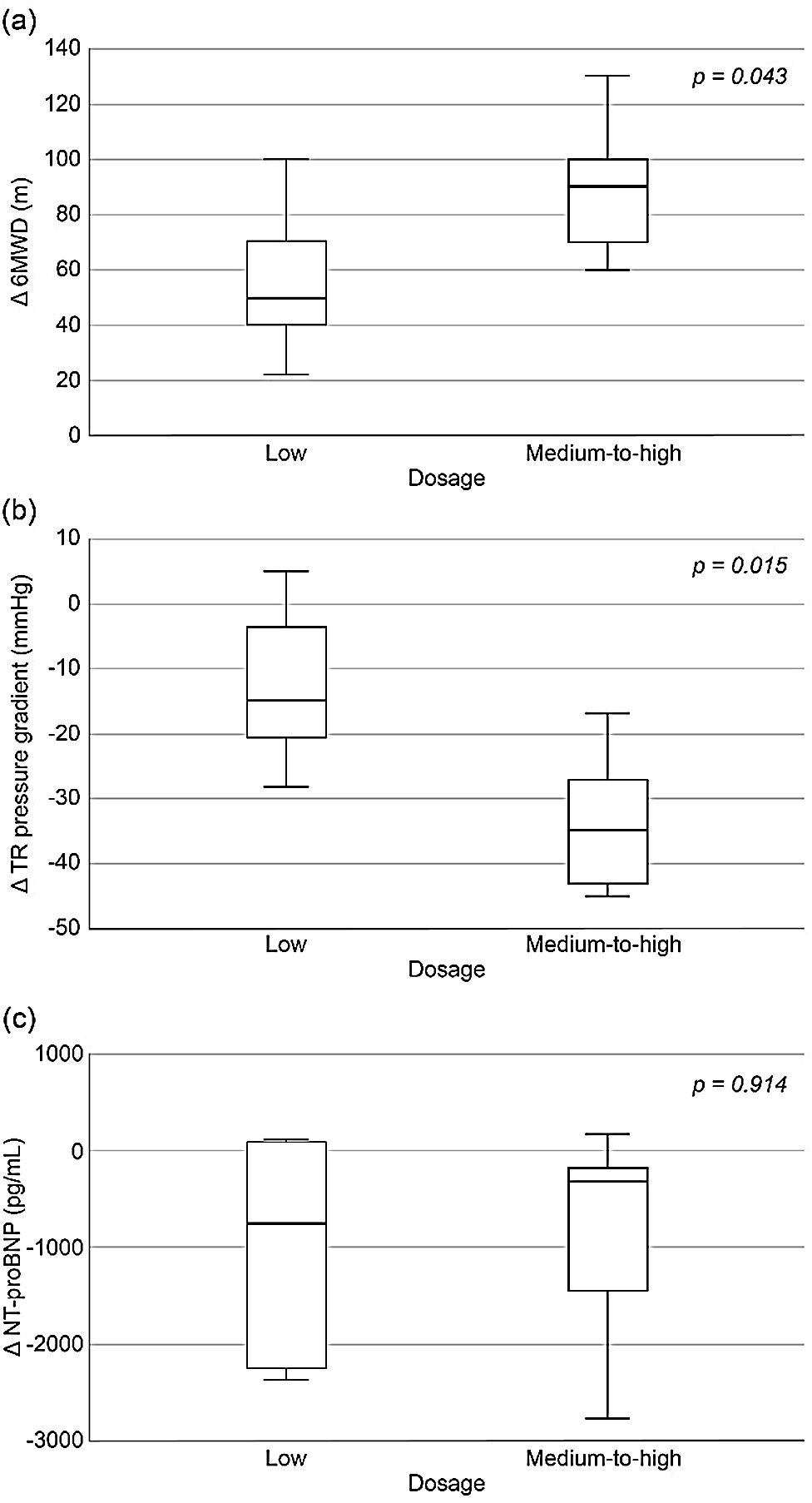
Figure 5: Comparison of changes in parameters before and after selexipag treatment stratified according to maintenance dosage (low vs. medium-to-high): (a) 6MWD, (b) TR pressure gradient, and (c) NT-proBNP
The present study reported real-life outcomes of selexipag treatment among PAH-CHD patients, including patients with Eisenmenger syndrome and partially repaired CHD. Previous studies have reported on the successful initiation of selexipag and functional improvement in 4 patients with Eisenmenger syndrome [25] and complex CHD [15]. In the present study, we have included the assessment of selexipag efficacy at improving or at least stabilizing the status of patients with Eisenmenger syndrome or partially repaired CHD.
In the present study, all patients were alive at the end of the study period, and no life-threatening adverse effects were observed. Side effects observed in the present study were consistent with previous reports [11,16]. As in the previous studies, in the present study, the majority of patients experienced side effects.
The maintenance dose in the present study was lower than that reported in previous studies [11,16]. A separate study involving Japanese patients has reported a relatively lower maintenance dose [26]. These discrepancies in dosage might be related to several factors: (1) There is a lack of specialist nursing care to encourage uptake of this treatment among patients; (2) The cost-benefit ratio, particularly among patients with long-term severe PAH, is inadequate, and the concurrent use of selexipag results in marked relief from PAH symptoms even at a low dose required to control side effects; and (3) The optimal dosage for underweight patients or patients who belong to other ethnic groups requires further research.
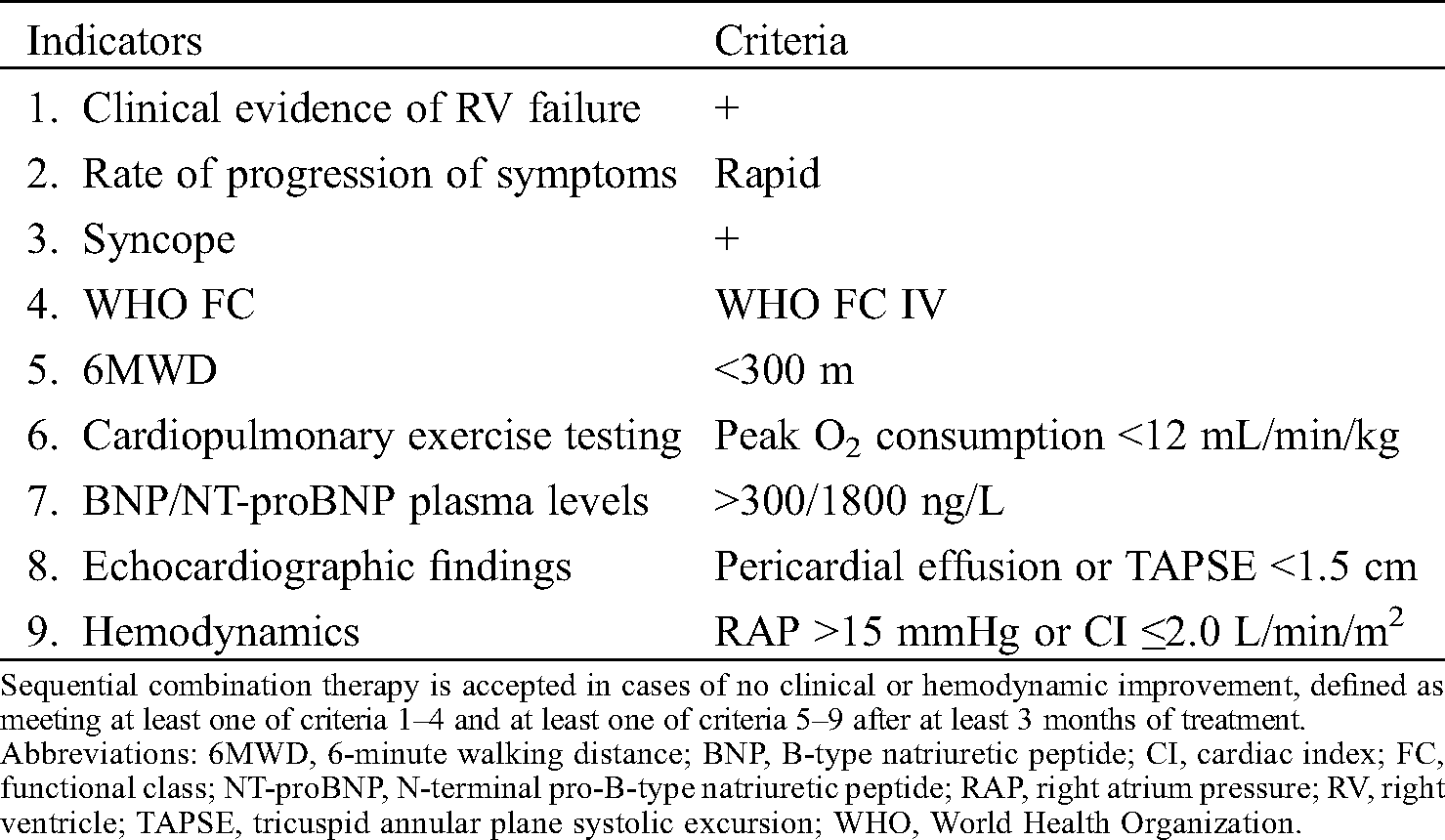
The present study has shown a marked improvement in the 6-minute walking distance test and a reduction in tricuspid regurgitation pressure gradient despite a relatively lower maintenance dosage. This finding might be accounted for by the study sample characteristics. Other studies have performed dose escalation even in low-risk groups, when the patients could not fulfill the low risk criteria [14]. In the present study, we tended to add selexipag to the regimen of patients with extreme PAH, as sequential add-on therapy is approved by the Korean National Health Insurance Review and Assessment Service for cases involving clinical and/or hemodynamic worsening of PAH but not in cases that do not achieve an improvement in clinical or hemodynamic status during risk assessment (Tab. 3). Low baseline 6-minute walking distance scores and high NT-proBNP levels in this study reflect the clinical status of our patients, as a recent study has shown that a high NT-proBNP level is associated with poor clinical outcomes among PAH patients [27].
Table 3: Current reimbursement guideline approved by the Korean National Health Insurance Review and Assessment Service for sequential combination treatment of pulmonary arterial hypertension
A recent post-hoc analysis of the GRIPHON study data has shown the relationship between risk profile and mortality/morbidity among PAH patients [24], demonstrating the benefits of selexipag at improving risk profile even among patients with corrected CHD. The present study adds to the body of clinical evidence, showing the impact of selexipag on improving risk profiles even among patients with Eisenmenger syndrome or partially repaired CHD.
Dose-related efficacy was not evident in the previous real-life study on selexipag; however, it was detected in the present study despite a small number of patients. Nevertheless, this phenomenon should be further investigated in a larger cohort.
This study was a non-randomized retrospective study with some inherent limitations. Moreover, follow-up right heart catheterization data were only available for 3 patients due to the relative shortness of the follow-up period and invasiveness of the test, resulting in right heart catheterization data being excluded from the analysis in the present study. The cardiopulmonary exercise test was performed in selective cases and not included in the present report. Reported clinical improvement might be exaggerated by a combination of a small number of included patients and biases associated with the treatment of high-risk patients. Nevertheless, the clinical efficacy of selexipag as add-on therapy in the present group of high-risk patients is evident as the included high-risk group had severe PAH-CHD despite long-term treatment with PAH drug(s) prior to study enrollment.
The present study contributes to the evidence on the clinical efficacy of selexipag as add-on therapy in the real-life treatment of patients with PAH-CHD, including Eisenmenger syndrome and partially repaired CHD.
Acknowledgement: These authors take responsibility for all aspects of the reliability and freedom from bias of the data presented and their discussed interpretation.
Funding Statement: This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflicts of Interest: The authors have no potential conflicts of interest to disclose.
1. Dimopoulos, K., Wort, S. J., Gatzoulis, M. A., Dimopoulos, K. (2014). Pulmonary hypertension related to congenital heart disease: A call for action. European Heart Journal, 35(11), 691–700. DOI 10.1093/eurheartj/eht437. [Google Scholar] [CrossRef]
2. Jiang, X., Jing, Z. C. (2013). Epidemiology of pulmonary arterial hypertension. Current Hypertension Reports, 15(6), 638–649. DOI 10.1007/s11906-013-0397-5. [Google Scholar] [CrossRef]
3. Ling, Y., Johnson, M. K., Kiely, D. G., Condliffe, R., Elliot, C. A. et al. (2012). Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: Results from the pulmonary hypertension registry of the United Kingdom and Ireland. American Journal of Respiratory and Critical Care Medicine, 186(8), 790–796. DOI 10.1164/rccm.201203-0383OC. [Google Scholar] [CrossRef]
4. Kaemmerer, H., Gorenflo, M., Hoeper, M., Huscher, D., Ewert, P. et al. (2013). Pulmonary arterial hypertension in patients with congenital heart disease: Current issues and health care situation. Deutsche Medizinische Wochenschrift (1946), 138(23), 1247. [Google Scholar]
5. Pascall, E., Tulloh, R. M. (2018). Pulmonary hypertension in congenital heart disease. Future Cardiology, 14(4), 343–353. DOI 10.2217/fca-2017-0065. [Google Scholar] [CrossRef]
6. D'Alto, M., Mahadevan, V. S. (2012). Pulmonary arterial hypertension associated with congenital heart disease. European Respiratory Review, 21(126), 328–337. DOI 10.1183/09059180.00004712. [Google Scholar] [CrossRef]
7. Kaemmerer, H., Apitz, C., Brockmeier, K., Eicken, A., Gorenflo, M. et al. (2018). Pulmonary hypertension in adults with congenital heart disease: Updated recommendations from the Cologne Consensus Conference 2018. International Journal of Cardiology, 272, 79–88. DOI 10.1016/j.ijcard.2018.08.078. [Google Scholar] [CrossRef]
8. Galiè, N., Humbert, M., Vachiery, J. L., Gibbs, S., Lang, I. et al. (2015). ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the european society of cardiology (ESC) and the european respiratory society (ERSEndorsed by: association for european paediatric and congenital cardiology (AEPCinternational society for heart and lung transplantation (ISHLT). European Heart Journal, 37(1), 67–119. [Google Scholar]
9. Kuwano, K., Hashino, A., Asaki, T., Hamamoto, T., Yamada, T. et al. (2007). 2-{4-[(5, 6-Diphenylpyrazin-2-yl)(isopropyl) amino] butoxy}-N-(methylsulfonyl) acetamide (NS-304an orally available and long-acting prostacyclin receptor agonist prodrug. Journal of Pharmacology and Experimental Therapeutics, 322(3), 1181–1188. DOI 10.1124/jpet.107.124248. [Google Scholar] [CrossRef]
10. Kuwano, K., Hashino, A., Noda, K., Kosugi, K., Kuwabara, K. (2008). A long-acting and highly selective prostacyclin receptor agonist prodrug, 2-{4-[(5,6-diphenylpyrazin-2-yl)(isopropyl) amino] butoxy}-N-(methylsulfonyl) acetamide (NS-304ameliorates rat pulmonary hypertension with unique relaxant responses of its active form,{4-[(5, 6-diphenylpyrazin-2-yl)(isopropyl) amino] butoxy} acetic acid (MRE-269on rat pulmonary artery. Journal of Pharmacology and Experimental Therapeutics, 326(3), 691–699. DOI 10.1124/jpet.108.138305. [Google Scholar] [CrossRef]
11. Sitbon, O., Channick, R., Chin, K. M., Frey, A., Gaine, S. et al. (2015). Selexipag for the treatment of pulmonary arterial hypertension. New England Journal of Medicine, 373(26), 2522–2533. DOI 10.1056/NEJMoa1503184. [Google Scholar] [CrossRef]
12. Denton, C. P., Hachulla, É., Riemekasten, G., Schwarting, A., Frenoux, J. M. et al. (2017). Efficacy and safety of selexipag in adults with Raynaud’s phenomenon secondary to systemic sclerosis: A randomized, placebo-controlled, phase II study. Arthritis & Rheumatology, 69(12), 2370–2379. DOI 10.1002/art.40242. [Google Scholar] [CrossRef]
13. Gaine, S., Chin, K., Coghlan, G., Channick, R., Di Scala, L. et al. (2017). Selexipag for the treatment of connective tissue disease-associated pulmonary arterial hypertension. European Respiratory Journal, 50(2), 1602493. DOI 10.1183/13993003.02493-2016. [Google Scholar] [CrossRef]
14. Berlier, C., Schwarz, E. I., Saxer, S., Lichtblau, M., Ulrich, S. (2019). Real-life experience with selexipag as an add-on therapy to oral combination therapy in patients with pulmonary arterial or distal chronic thromboembolic pulmonary hypertension: A retrospective analysis. Lung, 197(3), 353–360. DOI 10.1007/s00408-019-00222-7. [Google Scholar] [CrossRef]
15. Gallotti, R., Drogalis-Kim, D. E., Satou, G., Alejos, J. (2017). Single-center experience using selexipag in a pediatric population. Pediatric Cardiology, 38(7), 1405–1409. DOI 10.1007/s00246-017-1677-7. [Google Scholar] [CrossRef]
16. Barnikel, M., Kneidinger, N., Klenner, F., Waelde, A., Arnold, P. et al. (2019). Real-life data on Selexipag for the treatment of pulmonary hypertension. Pulmonary Circulation, 9(1), 2045894019832199. DOI 10.1177/2045894019832199. [Google Scholar] [CrossRef]
17. Beghetti, M., Channick, R. N., Chin, K. M., Di Scala, L., Gaine, S. et al. (2019). Selexipag treatment for pulmonary arterial hypertension associated with congenital heart disease after defect correction: Insights from the randomised controlled GRIPHON study. European Journal of Heart Failure, 21(3), 352–359. DOI 10.1002/ejhf.1375. [Google Scholar] [CrossRef]
18. Moller, J. H., Patton, C., Varco, R. L., Lillehei, C. W. (1991). Late results (30 to 35 years) after operative closure of isolated ventricular septal defect from 1954 to 1960. American Journal of Cardiology, 68(15), 1491–1497. DOI 10.1016/0002-9149(91)90284-R. [Google Scholar] [CrossRef]
19. Jung, S. Y., Choi, J. Y. (2018). Transcatheter closure of atrial septal defect: principles and available devices. Journal of Thoracic Disease, 10(S24), S2909–S2922. DOI 10.21037/jtd.2018.02.19. [Google Scholar] [CrossRef]
20. Jung, S., Kim, A. Y., Jung, J. W., Choi, J. Y. (2019). Procedural, early and long-term outcomes after percutaneous closure of atrial septal defect: Comparison between large and very large atrial septal defect groups. Korean Circulation Journal, 49(10), 975–986. DOI 10.4070/kcj.2018.0391. [Google Scholar] [CrossRef]
21. Kijima, Y., Akagi, T., Takaya, Y., Akagi, S., Nakagawa, K. et al. (2016). Treat and repair strategy in patients with atrial septal defect and significant pulmonary arterial hypertension. Circulation Journal, 80(1), 227–234. DOI 10.1253/circj.CJ-15-0599. [Google Scholar] [CrossRef]
22. Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories. (2002). ATS statement: Guidelines for the six-minute walk test. American Journal of Respiratory Critical Care Medicine, 166(1), 111–117. DOI 10.1164/ajrccm.166.1.at1102. [Google Scholar] [CrossRef]
23. Boucly, A., Weatherald, J., Savale, L., Jaïs, X., Cottin, V. et al. (2017). Risk assessment, prognosis and guideline implementation in pulmonary arterial hypertension. European Respiratory Journal, 50(2), 1700889. DOI 10.1183/13993003.00889-2017. [Google Scholar] [CrossRef]
24. Sitbon, O., Chin, K. M., Channick, R. N., Benza, R. L., Di Scala, L. et al. (2020). Risk assessment in pulmonary arterial hypertension: Insights from the GRIPHON study. Journal of Heart and Lung Transplantation, 39(4), 300–309. DOI 10.1016/j.healun.2019.12.013. [Google Scholar] [CrossRef]
25. El-Kersh, K., Suliman, S., Smith, J. S. (2018). Selexipag in congenital heart disease-associated pulmonary arterial hypertension and eisenmenger syndrome: First report. American Journal of Therapeutics, 25(6), e714–e715. DOI 10.1097/MJT.0000000000000727. [Google Scholar] [CrossRef]
26. Tanabe, N., Ikeda, S., Tahara, N., Fukuda, K., Hatano, M. et al. (2017). Efficacy and safety of an orally administered selective prostacyclin receptor agonist, selexipag, in Japanese patients with pulmonary arterial hypertension. Circulation Journal, 81(9), 1360–1367. DOI 10.1253/circj.CJ-16-1348. [Google Scholar] [CrossRef]
27. Chin, K. M., Rubin, L. J., Channick, R., Di Scala, L., Gaine, S. et al. (2019). Association of N-terminal pro brain natriuretic peptide and long-term outcome in patients with pulmonary arterial hypertension: Insights from the phase III GRIPHON study. Circulation, 139(21), 2440–2450. DOI 10.1161/CIRCULATIONAHA.118.039360. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |